

Abstract
The highest global prevalence rates for human immunodeficiency virus and acquired immune deficiency syndrome have been recorded in southern Africa; in the United States, individuals of African descent are disproportionately affected by human immunodeficiency virus infection. Human immunodeficiency virus–infected individuals with African ancestry are also estimated to have a 17-fold or greater risk for developing human immunodeficiency virus–associated nephropathy in comparison with their counterparts of non-African descent. Several recent studies have implicated genetic alleles that are more frequent in populations of African descent and increase the risk of human immunodeficiency virus infection and the risk of human immunodeficiency virus–associated neuropathy (HIVAN). The supposition that persons of African descent are more susceptible to human immunodeficiency virus infection because of an underlying genetic predisposition is not supported by available evidence. However, strong, replicated data show that the increased risk for human immunodeficiency virus–associated nephropathy, as well as other major forms of kidney disease in individuals of African descent, is due in part to MYH9 (myosin, heavy chain 9, non-muscle) renal disease susceptibility alleles that are very frequent throughout sub-Saharan Africa but are infrequent or absent in non-Africans. Selection, drift, and demographic events shape the allelic architecture of the human genome: it is expected that these events will be reflected in geographic-specific differentiation in allele frequencies for a small subset of alleles that may be associated with either increased or reduced risk for complex and infectious diseases.
Keywords: acquired immune deficiency syndrome (AIDS), health disparities, human immunodeficiency virus (HIV), human immunodeficiency virus–associated nephropathy (HIVAN), genetic alleles, genetic predisposition, myosin, heavy chain 9, non-muscle (MYH9), nephropathy, chromosome 22 renal susceptibility alleles
Health disparities in the United States and worldwide are well documented for human immunodeficiency virus (HIV) infection, the development of acquired immune deficiency syndrome (AIDS), and AIDS-related deaths.1–3 Although many of the inequities in HIV disease burden are due to nongenetic factors that include risk behaviors, access to health care, and socioeconomic status, host genetics may also play a role in geographic-specific or racial differences in disease susceptibility. Over the last decade, there has been an accumulation of data indicating that 12% to 15% of the variation in HIV progression and viral load is due to host genetic factors.4,5 However, our knowledge of host genetic factors affecting AIDS is biased toward factors prevalent in whites. Because AIDS and HIV were first identified in North America and Europe and because of the early demographics of HIV infection, with the very first infections in these regions occurring in men who have sex with men and a short time later in men with hemophilia, most genetic association studies, including the more recent genome-wide association studies, have been focused on European or European American men, who are not at all representative of the current HIV global epidemic: more than half of HIV-infected persons live in sub-Saharan Africa, and of these, more than half are women.1
The Joint United Nations Program on HIV/AIDS reported that in 2007, of the 33 million people living with HIV infection, more than half (68%) were in sub-Saharan Africa, and 61% were women.1 Eight countries in southern African account for 35% of HIV infections and 38% of AIDS-related deaths.1 In the United States, HIV infection disproportionately affects blacks. Although blacks represent only 12% to 13% of the US population, nearly half (48%) of the 1.1 million persons with HIV infection share African ancestry. Blacks in the United States also account for a higher proportion of cases at all stages from infection to death with AIDS.2 The small number of natural history and treatment cohort studies enrolling black or Hispanic subjects for genetic studies has limited the investigation of genetic predictors for HIV-1 infection and progression in these populations. Indeed, the major genome-wide association studies to date have enrolled European or European American participants.5–9
Associations between HIV infection and a spectrum of renal glomerular diseases were first recognized in the mid 1980s. In 1984, Rao et al.10 reported that HIV-infected patients were presenting with proteinuria in the nephrotic range followed by rapid progression to end-stage renal disease (ESRD), which was mainly attributable to focal and segmental glomerulosclerosis (FSGS). In European, Hispanic, and Asian patients, the most common form of HIV-associated glomerular disease is immune complex gomerulopnephritis,11,12 whereas patients of African descent most frequently develop HIV-associated collapsing glomerulopathy, which is also known as human immunodeficiency virus–associated nephropathy (HIVAN). In 1995, HIVAN was the leading cause of ESRD in men of African descent between 20 and 64 years of age.13 HIVAN is estimated to account for up to 30% of patients in the United States entering dialysis because of end-stage kidney disease as a result of FSGS.14
Notably, HIVAN is rarely diagnosed in people of European descent. The risk of developing HIVAN for individuals of African descent is at least 18-fold higher in comparison with the risk for their white counterparts; over 90% of persons with HIVAN are of African descent.14 This predilection for FSGS has also been observed in HIV-infected people of African descent in Africa, Brazil, and France.15–17 A recent review of the spectrum of kidney diseases in regions of Africa with high HIV prevalence rates also reports an unusually high prevalence of kidney disease.18 Although no systematic study has been performed with standardized criteria for HIV-associated kidney disease, kidney disease among HIV-1–infected persons has been reported to range from 6% in South Africa to 38% in Nigeria.18,19 Like HIVAN in developed nations, African ancestry is a risk predictor of HIVAN in sub-Saharan Africa.18,19 Because of the clustering of kidney diseases with different etiologies (eg, HIVAN, hypertension-associated ESRD, and diabetes-associated ESRD) in African American families and the overall increased rates of many kidney diseases in African Americans, it has been hypothesized that renal disease susceptibility alleles may be more frequent in persons of African origin or descent.20
The dispersal of humans out of Africa 100,000 years ago was followed by a rapid expansion of human populations from a relatively small effective population size. This had the effect of limiting allele and haplotype diversity in non-African populations with respect to African populations: the extent of human genetic variation outside Africa is a subset of the genetic variation observed within Africa. There are very few fixed differences among the world’s subpopulations: genetic diversity in the world’s population is largely attributable to geographic differentials in allele frequencies.21 The extent of genetic variation and diversity among continental, ethnic, and tribal groups reflects ancient, ongoing, and recent selective pressures from pathogens and environmental factors as well as historical and demographic events, such as migration and founder effects, on a background of random genetic drift.22–24 The events shaping our human genome are complex, but they leave their footprints in the allelic and haplotype structure of our genomes: it should not be surprising that human continental populations, isolated subpopulations, and racial/ethnic groups may reflect this variation in differential genetic susceptibility to a spectrum of human pathogens and diseases.25
Here we consider the role of host genetic factors in population-specific differences in susceptibility to HIV infection and progression and in the development of HIVAN in African and European Americans. Because most AIDS-restricted genes have been extensively reviewed elsewhere, the focus of this commentary is a small subset of genes that are highly stratified between populations of European and African descent, that is, those with moderate to high allele frequencies (>10%) in one population but very low allele frequencies in the other; these may be expected to have large population-attributable risks that may provide a genetic basis for disparate risk between individuals of African descent and individuals of European descent. Rare polymorphisms with large effect sizes or common alleles with small effect sizes would not be expected to contribute substantially to HIV-related health disparities.
CHEMOKINE (C-C MOTIF) RECEPTOR 5 Δ32, HUMAN IMMUNODEFICIENCY VIRUS ACQUISITION, AND PROGRESSION TO ACQUIRED IMMUNE DEFICIENCY SYNDROME
The primary coreceptor used by transmissible strains of HIV to enter cells and establish infections is chemokine (C-C motif) receptor 5 (CCR5). In 1996, a series of articles reported that a 32–base pair deletion in the CCR5 gene (CCR5 Δ32) prevented HIV infection in cells from individuals homozygous for the mutated gene.26–29 The mutation introduced a premature stop codon that truncated the CCR5 protein and effectively prevented its cell surface expression. The absence of CCR5 due to this deletion provided nearly complete resistance to HIV infection in those carrying 2 copies of the mutated gene. CCR5 was the first HIV restriction gene discovered, and it remains to date the only mutation that protects against HIV acquisition. Although subsequent studies have identified a handful of individuals homozygous for CCR5 Δ32 who have become HIV-infected, these infection events are extremely rare. Most HIV isolates from CCR5 [delta]32-homozygous infected individuals have been found to use an alternative coreceptor, CXCR4, to establish infection.30 These studies have established the importance of CCR5 as the primary coreceptor for transmissible strains of HIV.
Surveys of the population distribution of the CCR5 Δ32 mutation have revealed that the deletion mutation is restricted to populations of northern European descent or European admixture.31 The frequencies of CCR5 Δ32 are distributed across Europe in a north-to-south cline, with the highest frequencies in northern Europe (15% in Scandinavia, 10% in France, England, and Germany, and 5% in Italy, Greece, and Turkey), and it is rare to absent elsewhere in the world.31,32 Estimates of the age of CCR5 Δ32 suggest a relatively recent origin.31 It has been speculated that the CCR5 Δ32 mutation rose to its current frequency of approximately 10% in the European population by recent selection, possibly by a pathogen such as Variola vaccinia (smallpox) or Yersina pestis (black plague).33
Although homozygosity for CCR5 Δ32 provides nearly absolute protection against HIV acquisition, this protection is afforded to only about 1% of northern Europeans homozygous for CCR5 Δ32, and this is too small a fraction to account for differences in infection rates among African and European individuals. Heterozygosity for CCR5 Δ32 has been shown in many studies to delay the onset of AIDS by 2 or 3 years in untreated persons, and this protective effect continues after the initiation of antiretroviral therapy.29,34–36 Carriers of CCR5 Δ32 tend to have a lower HIV viral copy number; the HIV copy number of the transmitting partner or mother is positively correlated with HIV transmission.36 It is conceivable that lower levels of HIV due to CCR5 Δ32 or other polymorphisms in the CCR5 gene that affect CCR5 expression and protein levels may decrease HIV transmission efficiency and thereby affect the dynamics of HIV transmission in a population-specific manner.37 Approximately 20% of Europeans are predicted to carry at least 1 CCR5 Δ32 allele.
DUFFY ANTIGEN CHEMOKINES AND HIV ACQUISITION
The Duffy antigen receptor for chemokines (DARC) is a chemokine receptor primarily expressed on the surface of red blood cells. DARC has been extensively investigated because it is the cell surface receptor used by the malarial pathogen Plasmodium vivax to enter and infect red blood cells.38 DARC provides classic evidence for the influence of human pathogens on natural selection.39,40 Outside Africa, P. vivax is the most frequent cause of malaria in humans, but within Africa, malarial infections from P. vivax are extremely rare. A single base pair mutation in the DARC gene promoter (−46T→C) disrupts an erythrocyte transcription factor binding site required for DARC gene expression in erthrocytes.41 As a result, individuals homozygous for the −46 C allele express no DARC receptor on their red blood cells and are resistant to P. vivax malaria. The DARC −46 C mutation is one of only a few mutations discovered that reaches near fixation (a >90% allele frequency in sub-Saharan Africa) in 1 continental population and is virtually absent in all other human populations. With an allele frequency of >90% in much of the malarial belt, most sub-Saharan Africans (≈81%) do not express DARC on their cell surfaces and are therefore resistant to P. vivax malaria.
DARC promiscuously absorbs a number of pro-inflammatory chemokines to the surfaces of red blood cells, including those that bind to the CCR5 coreceptor and inhibit HIV binding to CCR5 surface receptors.42,43 The DARC cell surface receptor has also been reported to bind HIV to the surfaces of erythrocytes that may act to facilitate the transfer of HIV to susceptible cells.44,45 Given the critical role of the chemokine/chemokine receptor nexus in HIV infection and the complete absence of the DARC receptor on red blood cells of most Africans, DARC is a highly plausible candidate gene for a role in modifying HIV acquisition and pathogenesis. He et al.44 recently reported that individuals who do not express DARC on their cell surfaces are more likely to acquire HIV infection but survive longer after HIV infection. These observations were based on a genetic association study enrolling African American men serving in the US military. The study was notable because the authors reported that the DARC null phenotype was associated with a 40% increase in the odds of acquiring HIV; this was based on the observation that the rate of the −46C/C genotype (abrogating DARC gene expression) was 70% in the HIV-positive group versus 60% in normal controls without HIV infection. The authors concluded that the DARC null genotype might account for 11% of the HIV-1 burden in Africa and increase the risk of HIV infection by 40%.44 If correct, these results would suggest that individuals of African descent have a genetic vulnerability to HIV-1 acquisition; such a finding could have tremendous implications for public health policies aimed at stemming the tide of new HIV infections.
Such a finding warrants careful consideration and validation. African Americans are an admixed group with various degrees of African and European ancestry. It is estimated that 15% to 20% of the genes in African Americans in the United States derive from the European population.46 Stratification between HIV-infected cases and HIV-uninfected controls must be carefully controlled to avoid a population substructure that may create spurious or inflated genetic associations between alleles and an outcome.47,48 This is critical when a gene that is highly frequent in one population is rare or absent in a second population, such as the case with the DARC −46C null mutation. Four independent groups were unable to replicate the findings of He et al.44 in individuals of African descent enrolled in United States–based natural history cohorts or in black Africans living in Durban, South Africa.49–52 These results are summarized in Figure 1. These investigations varied somewhat in design and methods, but none of the studies provided support for He et al.’s finding that DARC was associated with either susceptibility to HIV infection or with the rate of progression to AIDS. For example, in Winkler et al.’s study of African American injecting drug users enrolled in a natural history cohort,50 the frequencies of the −46 C/C genotype for the null phenotype were almost identical (63.4% versus 63.5%) in the cases and controls.
Fig 1.
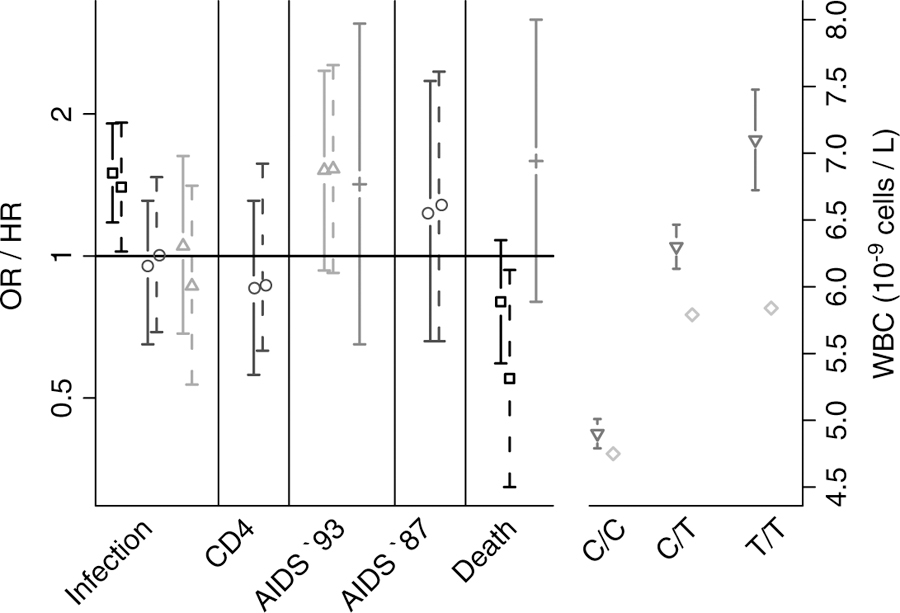
OR and HR values with 95% confidence intervals for associations of DARC with human immunodeficiency virus infection: time to CD4 <200; time to AIDS-defining condition (AIDS ‘93) or CD4 <200; and death from an AIDS-related illness. Crude and adjusted OR/HR values are represented by solid and dashed bars, respectively (left panel). Mean WBCs with 95% confidence intervals, where available, are also shown for each DARC genotype (right panel). The following studies are cited: (□) He et al.,44 (○) Winkler et al.,50 (∆) Walley et al.,49 (+) Horne et al.,52 (◊) Julg et al.,51 and () Nalls.53 Abbreviations: AIDS, acquired immune deficiency syndrome; DARC, Duffy antigen receptor for chemokines; HR, hazard ratio; OR, odds ratio; WBC, white blood cell count.
The DARC null phenotype is, however, associated with neutrophil levels.51,53,54 Approximately 25% to 50% of persons of African descent have persistent benign ethnic neutropenia, with low leukocyte and neutrophil counts below the normalized range for populations of European descent. Leukopenia with neutropenia was first described in 1941 when a physician found that black sharecroppers had lower mean white blood cell counts than a comparable group of white labors.55 This common condition is labeled benign ethnic neutropenia and is not known to be associated with an increased risk for repeated or severe bacterial infections. Recently, it was discovered that white blood counts attributable to neutropenia are associated with DARC expression. Interestingly, African American individuals carrying either 1 or 2 copies of the European-type −46T allele have a normal white blood cell count range, and this indicates that the −46T allele is dominant over the African-type −46C null allele. On the other hand, individuals who inherit 2 DARC null alleles (−46C/C) have lower neutrophil counts. The alternative European-type (−46T) and African-type (−46C) alleles are highly predictive of the neutrophil count (Figure 1).54
In summary, these studies show that the DARC null genotype is a predictor of the benign ethnic neutropenia frequently observed in persons of African descent. On the other hand, there is no convincing evidence that the DARC null genotype is associated with an increased risk of HIV acquisition in people of African descent or origin.
HIV-ASSCOAITED NEPHROPATHY AND MYH9
Kidney disease represents a major health disparity for people of African origin or ancestry; the lifetime risk of ESRD is 2.5% for European Americans and 7.5% for African Americans.56 In 1994, the National Institutes of Health FSGS study began enrolling African and European Americans with biopsy-proven FSGS and HIVAN with collapsing glomerulosclerosis to investigate the genetic correlates to these syndromes; both syndromes are more frequent in people of African ancestry.14 Because African Americans are admixed and on average have 15% to 20% European ancestry, their chromosomes are a mosaic of European and African segments. Through the use of a panel of ancestry informative markers that are evenly distributed across the genome and have large allele frequency differences (30% on average) between Europeans and African Yoruba, the ancestral origin of chromosomal segments can be determined with statistical methods.57,58 Admixture mapping provides a powerful method for mapping susceptibility genes for diseases/traits (eg, kidney disease) that show differential risks (odds ratio ≥2) between 2 discrete, historically isolated populations that have undergone recent admixture.
In this case, because persons of African descent are more likely to develop HIVAN and FSGS, we hypothesized that the risk variation associated with these syndromes would be located on an African-derived chromosomal segment. With a genome-wide admixture scan, MYH9, which encodes the molecular motor non-muscle myosin heavy chain IIA, was identified as a susceptibility gene for HIVAN as well as FSGS and nondiabetic ESRD.59,60 Although many MYH9 single-nucleotide polymorphisms (SNPs) were strongly associated with HIVAN and FSGS, alleles for 3 highly correlated SNPs in intron 23 (rs4821480, rs2032487, and rs4821481) and 1 in intron 14 (rs3752462) composed an extended (E) haplotype block that was more informative than any single SNP for predicting HIVAN or FSGS. Both the risk alleles and the risk E-1 haplotype that they define are extremely common in African Americans (>60%) and rare in European Americans (<4%). In our study of 58 individuals with HIVAN, 76% carried 2 copies of the risk E-1 haplotype, and 24% carried 1 copy of the risk E-1 haplotype (Figure 2). In contrast, in the hypernormal control group comprising African Americans with HIV-1 infection for 8 or more years with normal kidney function, only 35% of the participants carried 2 copies of the risk haplotype. Recent studies indicate that MYH9 risk alleles contribute to major forms of kidney disease, including diabetic ESRD and ESRD attributed to hypertension.61,62 However, we do not yet understand the mechanism leading to MYH9 renal disease. To date, no coding region mutations have been found that account for the associations. We speculate that 1 or more noncoding SNPs alter expression or slice patterns.
Fig 2.
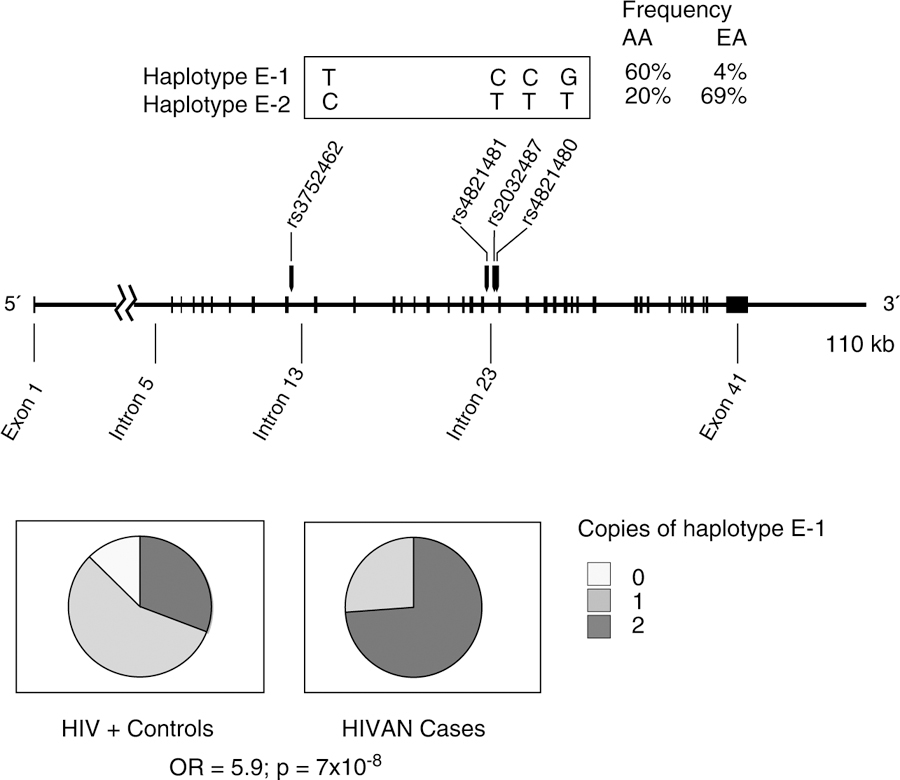
The E haplotype block with the alleles indicated for the most common E haplotypes in Africans (E-1) and Europeans (E-2). SNPs tagging the haplotype block are indicated; however, using only SNPs rs4821481 and rs3732462 is sufficient for inferring the E-1 haplotype. The frequencies of the 3 genotypes for the MYH9 E-1 haplotype (rs4821480, rs2032487, rs4821481, and rs3752462), the most frequent haplotype containing the 4 SNPs, are shown for AAs with biopsy-proven HIVAN cases and hypernormal controls infected with HIV for 8 or more years with normal kidney function. Odds ratios and P values for the recessive model comparing focal and segmental glomerulosclerosis cases and controls are shown. Abbreviations: AA, African American; E, extended; EA, European American; HIV, human immunodeficiency virus; HIVAN, human immunodeficiency virus–associated nephropathy; OR, odds ratio; SNP, single-nucleotide polymorphism.
The MYH9 risk alleles and E-1 haplotype are remarkable in being very frequent and having very large effect sizes. The attributable risk of HIVAN for MYH9 E-1 haplotype carriers is 100%; attributable risk is a measure of how much of the disease would be eliminated if the exposure (eg, the MYH9 risk factor) were not present. These findings have been replicated and provide strong evidence that MYH9 genetic variation contributes to the excess burden of not only HIVAN but also other major forms of kidney disease in individuals of African descent; they also provide a genetic basis for a major US and global health disparity.58,59,60,61
GENES THAT MODIFY HUMAN IMMUNODEFICIENCY VIRUS
Since the discovery of CCR5 Δ32 over a decade ago, a growing number of genes have been identified that modify HIV/AIDS. These genes and their effects have been reviewed extensively elsewhere.4,6 Many of the genes that are predicted to influence HIV acquisition are immune response genes [e.g., human leukocyte antigen (HLA)], intrinsic or innate antiretroviral genes [e.g., killer immunoglobin-like receptors (KIR)], or members of the chemokine receptor nexus. Figure 3 provides the population frequencies for the minor alleles for many of the SNPs or genetic variants that are reported to affect HIV progression, HIV levels, or HIV acquisition; data from the International HapMap Project and the Single Nucleotide Polymorphism Database of the National Center for Biotechnology Information have been used to provide allele frequencies for West Africans (Yoruba from Ibadan, Nigeria) and northern Europeans from Utah. The allele frequencies for these 2 groups are representative of allele frequencies observed in African Americans and European Americans. It is immediately apparent that the majority of these polymorphisms that have been implicated in HIV pathogenesis show quite different allele frequencies between the 2 continental groups. Despite these differences in allele frequencies between African and European groups, there appears to be no major genetic predisposition to faster progression to AIDS between individuals of European descent and individuals of African descent. A comparison of self-reporting African American injecting drug users from Baltimore and European American men who have sex with men from 4 US cities indicate no significance differences in AIDS-free survival (Winkler, unpublished data) between these disparate groups with vastly different access to health care and lifestyles. Similarly, mother-to-infant transmission rates are similar among ethnic/racial groups, and this suggests that there is no major population-specific genetic predictor of HIV transmission and acquisition.63
Fig 3.
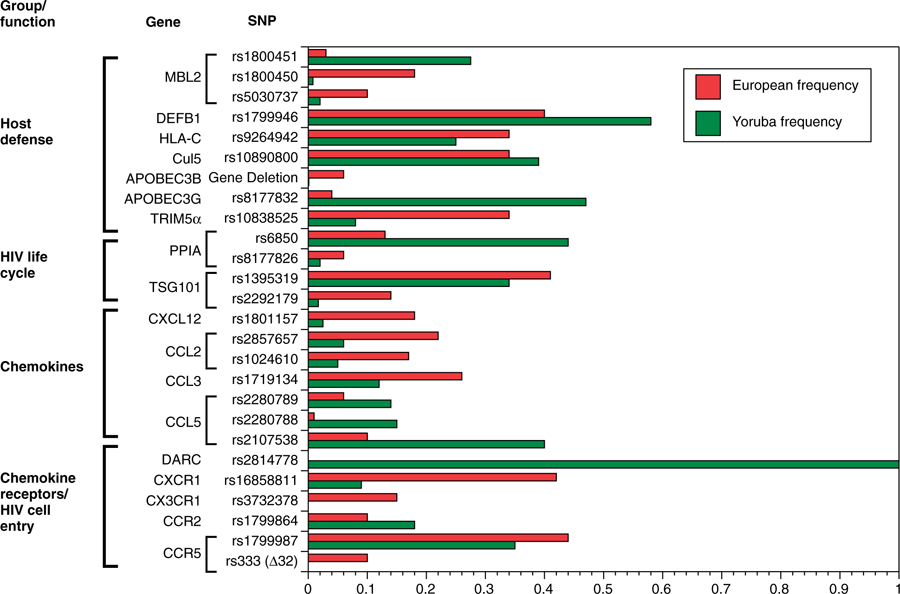
Allele frequencies for gene polymorphisms associated with HIV-1 disease6 in Yoruba from Ibadan, Nigeria and in northern Europeans from Utah. HapMap frequencies were available from the Single Nucleotide Polymorphism Database of the National Center for Biotechnology Information. Abbreviations: APOBEC3, apolipoprotein B messenger RNA–editing enzyme catalytic polypeptide 3; CCL, chemokine (C-C motif) ligand; CCR, chemokine (C-C motif) receptor; Cul5, cullin 5; CXCL12, chemokine (C-X-C motif) ligand 12; CX3CR1, chemokine (C-X3-C motif) receptor 1; CXCR1, cysteine-X-cysteine receptor 1; DARC, Duffy antigen receptor for chemokines; DEFB1, defensin beta 1; HIV, human immunodeficiency virus; HLA-C, human leukocyte antigen C; MBL2, mannose-binding lectin (protein C) 2; PPIA, peptidylprolyl isomerase A (cyclophilin A); TRIM5α, tripartite motif-containing 5 alpha; TSG101, tumor susceptibility gene 101.
CONCLUSION
Since the discovery of CCR5 Δ32 as a major determinant of resistance to HIV infection, considerable effort has been expended to identify genetic variation that predicts infection, pathogenesis, and disease outcomes. The discovery of such genes may provide needed insights into pathways used by the virus for completion of its lifecycle and provide targets for therapeutic interventions. Genetic dissection of host defense genes may also point the way to more effective vaccines or biological modifiers to augment natural immune surveillance. However, it is essential that well-designed studies be carried out in diverse populations because people of color carry much of the burden of HIV/AIDS throughout the world. An understanding of the genetic correlates of HIV disease in all ethnic/racial groups is crucial if the goals of personalized medicine, improved therapeutic interventions, and informed public health policies are to be realized.
ACKNOWLEDGMENT
The author thanks Dr. George Nelson, Dr. Bert Gold, Dr. Ping An, and Dr. Jeffrey Kopp for insightful discussions and the participants who made these studies possible. This project has been funded in whole or in part with federal funds from the National Cancer Institute (National Institutes of Health) under contract HHSN26120080001E. This research was supported by the Intramural Research Program of the Center for Cancer Research (National Cancer Institute, National Institutes of Health).
The contents of this publication do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
DISCLOSURES
Potential conflict of interest: Nothing to report.
REFERENCES
- 1.2008 Report on the Global AIDS Epidemic. Geneva, Switzerland: Joint United Nations Programme on HIV/AIDS; 2008. [Google Scholar]
- 2.Centers for Disease Control and Prevention. Cases of HIV infection and AIDS in the United States and dependent areas, 2007. http://www.cdc.gov/hiv/topics/surveillance/resources/reports/2007report/default.htm. Accessed January 2010.
- 3.Sutton MY, Jones RL, Wolitski RJ, et al. A review of the Centers for Disease Control and Prevention’s response to the HIV/AIDS crisis among blacks in the United States, 1981–2009. Am J Public Health 2009; 99(suppl 2): S351–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet 2004; 36: 565–574. [DOI] [PubMed] [Google Scholar]
- 5.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science 2007; 317: 944–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fellay J Host genetics influences on HIV type-1 disease. Antivir Ther 2009; 14: 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Clerc S, Limou S, Coulonges C, et al. Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03). J Infect Dis 2009; 200: 1194–1201. [DOI] [PubMed] [Google Scholar]
- 8.Limou S, Le Clerc S, Coulonges C, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02). J Infect Dis 2009; 199: 419–426. [DOI] [PubMed] [Google Scholar]
- 9.Dalmasso C, Carpentier W, Meyer L, et al. Distinct genetic loci control plasma HIV-RNA and cellular HIV-DNA levels in HIV-1 infection: the ANRS Genome Wide Association 01 study. PLoS One 2008; 3: e3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao TK, Filippone EJ, Nicastri AD, et al. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med 1984; 310: 669–673. [DOI] [PubMed] [Google Scholar]
- 11.Cantor ES, Kimmel PL, Bosch JP. Effect of race on expression of acquired immunodeficiency syndrome-associated nephropathy. Arch Intern Med 1991; 151: 125–128. [PubMed] [Google Scholar]
- 12.Praditpornsilpa K, Napathorn S, Yenrudi S, et al. Renal pathology and HIV infection in Thailand. Am J Kidney Dis 1999; 33: 282–286. [DOI] [PubMed] [Google Scholar]
- 13.Winston JA, Burns GC, Klotman PE. The human immunodeficiency virus (HIV) epidemic and HIV-associated nephropathy. Semin Nephrol 1998; 18: 373–377. [PubMed] [Google Scholar]
- 14.Kopp JB, Winkler C. HIV-associated nephropathy in African Americans. Kidney Int Suppl 2003; 83: S43–S49. [DOI] [PubMed] [Google Scholar]
- 15.Lopes GS, Marques LP, Rioja LS, et al. Glomerular disease and human immunodeficiency virus infection in Brazil. Am J Nephrol 1992; 12: 281–287. [DOI] [PubMed] [Google Scholar]
- 16.Nochy D, Glotz D, Dosquet P, et al. Renal disease associated with HIV infection: a multicentric study of 60 patients from Paris hospitals. Nephrol Dial Transplant 1993; 8: 11–19. [DOI] [PubMed] [Google Scholar]
- 17.Pakasa M, Mangani N, Dikassa L. Focal and segmental glomerulosclerosis in nephrotic syndrome: a new profile of adult nephrotic syndrome in Zaire. Mod Pathol 1993; 6: 125–128. [PubMed] [Google Scholar]
- 18.Fabian J, Naicker S. HIV and kidney disease in sub-Saharan Africa. Nat Rev Nephrol 2009; 5: 591–598. [DOI] [PubMed] [Google Scholar]
- 19.Naicker S, Han TM, Fabian J. HIV/AIDS–dominant player in chronic kidney disease. Ethn Dis 2006; 16: S2–56–60. [PubMed] [Google Scholar]
- 20.Satko SG, Sedor JR, Iyengar SK, Freedman BI. Familial clustering of chronic kidney disease. Semin Dial 2007; 20: 229–236. [DOI] [PubMed] [Google Scholar]
- 21.Coop G, Pickrell JK, Novembre J, et al. The role of geography in human adaptation. PLoS Genet 2009; 5: e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hancock AM, Witonsky DB, Gordon AS, et al. Adaptations to climate in candidate genes for common metabolic disorders. PLoS Genet 2008; 4: e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 2008; 9: 403–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reed FA, Tishkoff SA. African human diversity, origins and migrations. Curr Opin Genet Dev 2006; 16: 597–605. [DOI] [PubMed] [Google Scholar]
- 25.Tishkoff SA, Kidd KK. Implications of biogeography of human populations for ‘race’ and medicine. Nat Genet 2004; 36: S21–S7. [DOI] [PubMed] [Google Scholar]
- 26.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996; 273: 1856–1862. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Paxton WA, Wolinsky SM, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med 1996; 2: 1240–1243. [DOI] [PubMed] [Google Scholar]
- 28.Liu R, Paxton WA, Choe S, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996; 86: 367–377. [DOI] [PubMed] [Google Scholar]
- 29.McDermott DH, Beecroft MJ, Kleeberger CA, et al. Chemokine RANTES promoter polymorphism affects risk of both HIV infection and disease progression in the Multicenter AIDS Cohort Study. AIDS 2000; 14: 2671–2678. [DOI] [PubMed] [Google Scholar]
- 30.O’Brien TR, Winkler C, Dean M, et al. HIV-1 infection in a man homozygous for CCR5 delta 32. Lancet 1997; 349: 1219. [DOI] [PubMed] [Google Scholar]
- 31.Stephens JC, Reich DE, Goldstein DB, et al. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet 1998; 62: 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinson JJ, Chapman NH, Rees DC, et al. Global distribution of the CCR5 gene 32-basepair deletion. Nat Genet 1997; 16: 100–103. [DOI] [PubMed] [Google Scholar]
- 33.Galvani AP, Slatkin M. Evaluating plague and smallpox as historical selective pressures for the CCR5-Δ32 HIV-resistance allele. Proc Natl Acad Sci U S A 2003; 100: 15276–15279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996; 273: 1856–1862. [DOI] [PubMed] [Google Scholar]
- 35.Hendrickson SL, Jacobson LP, Nelson GW, et al. Host genetic influences on highly active antiretroviral therapy efficacy and AIDS-free survival. J Acquir Immune Defic Syndr 2008; 48: 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ometto L, Zanchetta M, Mainardi M, et al. Co-receptor usage of HIV-1 primary isolates, viral burden, and CCR5 genotype in mother-to-child HIV-1 transmission. AIDS 2000; 14: 1721–1729. [DOI] [PubMed] [Google Scholar]
- 37.Schliekelman P, Garner C, Slatkin M. Natural selection and resistance to HIV. Nature 2001; 411: 545–546. [DOI] [PubMed] [Google Scholar]
- 38.Michon P, Woolley I, Wood EM, et al. Duffy-null promoter heterozygosity reduces DARC expression and abrogates adhesion of the P. vivax ligand required for blood-stage infection. FEBS Lett 2001; 495: 111–114. [DOI] [PubMed] [Google Scholar]
- 39.Hill AV. Malaria resistance genes: a natural selection. Trans R Soc Trop Med Hyg 1992; 86: 225–226. [DOI] [PubMed] [Google Scholar]
- 40.Horuk R, Chitnis CE, Darbonne WC, et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science 1993; 261: 1182–1184. [DOI] [PubMed] [Google Scholar]
- 41.Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet 1995; 10: 224–228. [DOI] [PubMed] [Google Scholar]
- 42.Mayr FB, Spiel AO, Leitner JM, et al. Racial differences in endotoxin-induced tissue factor-triggered coagulation. J Thromb Haemost 2009; 7: 634–640. [DOI] [PubMed] [Google Scholar]
- 43.Mayr FB, Spiel AO, Leitner JM, et al. Duffy antigen modifies the chemokine response in human endotoxemia. Crit Care Med 2008; 36: 159–165. [DOI] [PubMed] [Google Scholar]
- 44.He W, Neil S, Kulkarni H, et al. Duffy antigen receptor for chemokines mediates trans-infection of HIV-1 from red blood cells to target cells and affects HIV-AIDS susceptibility. Cell Host Microbe 2008; 4: 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lachgar A, Jaureguiberry G, Le Buenac H, et al. Binding of HIV-1 to RBCs involves the Duffy antigen receptors for chemokines (DARC). Biomed Pharmacother 1998; 52: 436–439. [DOI] [PubMed] [Google Scholar]
- 46.McKeigue PM, Carpenter JR, Parra EJ, Shriver MD. Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann Hum Genet 2000; 64: 171–186. [DOI] [PubMed] [Google Scholar]
- 47.Hoggart CJ, Parra EJ, Shriver MD, et al. Control of confounding of genetic associations in stratified populations. Am J Hum Genet 2003; 72: 1492–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian C, Gregersen PK, Seldin MF. Accounting for ancestry: population substructure and genome-wide association studies. Hum Mol Genet 2008; 17: R143–R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walley NM, Julg B, Dickson SP, et al. The Duffy antigen receptor for chemokines null promoter variant does not influence HIV-1 acquisition or disease progression. Cell Host Microbe 2009; 5: 408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winkler CA, An P, Johnson R, et al. Expression of Duffy antigen receptor for chemokines (DARC) has no effect on HIV-1 acquisition or progression to AIDS in African Americans. Cell Host Microbe 2009; 5: 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Julg B, Reddy S, van der Stok M, et al. Lack of Duffy antigen receptor for chemokines: no influence on HIV disease progression in an African treatment-naive population. Cell Host Microbe 2009; 5: 413–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horne KC, Li X, Jacobson LP, et al. Duffy antigen polymorphisms do not alter progression of HIV in African Americans in the MACS cohort. Cell Host Microbe 2009; 5: 415–417. [DOI] [PubMed] [Google Scholar]
- 53.Nalls MA, Wilson JG, Patterson NJ, et al. Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am J Hum Genet 2008; 82: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reich D, Nalls MA, Kao WH, et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet 2009; 5: e1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forbes WH, Johnson RD, Consolazio F. Leukopenia in Negro workmen. Am J Med Sci 1941; 201: 407–412. [Google Scholar]
- 56.Kiberd BA, Clase CM. Cumulative risk for developing end-stage renal disease in the US population. J Am Soc Nephrol 2002; 13: 1635–1644. [DOI] [PubMed] [Google Scholar]
- 57.Smith MW, Patterson N, Lautenberger JA, et al. A high-density admixture map for disease gene discovery in African Americans. Am J Hum Genet 2004; 74: 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patterson N, Hattangadi N, Lane B, et al. Methods for high-density admixture mapping of disease genes. Am J Hum Genet 2004; 74: 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kao WH, Klag MJ, Meoni LA, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 2008; 40: 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 2008; 40: 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Freedman BI, Hicks PJ, Bostrom MA, et al. Non-muscle myosin heavy chain 9 gene MYH9 associations in African Americans with clinically diagnosed type 2 diabetes mellitus-associated ESRD. Nephrol Dial Transplant 2009; 24: 3366–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Freedman BI, Hicks PJ, Bostrom MA, et al. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int 2009; 75: 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Working Group on Mother-To-Child Transmission of HIV. Rates of mother-to-child transmission of HIV-1 in Africa, America, and Europe: results from 13 perinatal studies. J Acquir Immune Defic Syndr Hum Retrovirol 1995; 8: 506–510. [DOI] [PubMed] [Google Scholar]