

Abstract
Mutations in the GBA1 gene encoding the lysosomal enzyme β-glucocerebrosidase (GCase) represent the most common risk factor for Parkinson’s disease (PD). GCase has been identified as a potential therapeutic target for PD and current efforts are focused on using chemical chaperones to translocate mutant GCase into lysosomes. However, for several GBA1-linked forms of PD and PD associated with mutations in LRRK2, DJ-1, and PARKIN, activating wild-type GCase represents an alternative approach. We developed a new small-molecule modulator of GCase called S-181 that increased wild-type GCase activity in iPSC-derived dopaminergic neurons from sporadic PD patients, as well as patients carrying the 84GG mutation in GBA1, or mutations in LRRK2, DJ-1, or PARKIN who had decreased GCase activity. S-181 treatment of these PD iPSC-derived dopaminergic neurons partially restored lysosomal function and lowered accumulation of oxidized dopamine, glucosylceramide and α-synuclein. Moreover, S-181 treatment of mice heterozygous for the D409V GBA1 mutation (Gba1D409V/+) resulted in activation of wild-type GCase and consequent reduction of GCase lipid substrates and α-synuclein in mouse brain tissue. Our findings point to activation of wild-type GCase by small-molecule modulators as a potential therapeutic approach for treating familial and sporadic forms of PD that exhibit decreased GCase activity.
Overline: Parkinson’s Disease
One sentence summary:
New small-molecule modulators of wild-type glucocerebrosidase improve pathogenic phenotypes in different cellular models of Parkinson’s disease.
Editor’s Summary
Boosting GCase activity in PD
Mutations in the GBA1 gene encoding the lysosomal enzyme β-glucocerebrosidase (GCase) represent the most common risk factor for Parkinson’s disease (PD). Because decreased wild-type GCase activity may be contributing to PD pathogenesis, activating the wild-type enzyme is a potential therapeutic approach. Burbulla et al. developed a new small molecule modulator of wild-type GCase called S-181. This compound increased wild-type GCase activity and improved pathogenic phenotypes in iPSC-derived dopaminergic neurons generated from fibroblasts from PD patients with both genetic and idiopathic forms of the disease. These findings point to activation of wild-type GCase as a potential therapeutic approach for various forms of PD that exhibit decreased GCase activity.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease affecting 2–3% of the population ≥65 years of age (1). It is characterized by loss of dopaminergic neurons in the substantia nigra and by Lewy bodies containing α-synuclein (2, 3). GBA1 encodes the lysosomal enzyme β-glucocerebrosidase (GCase), which converts its substrate glucosylceramide into ceramide and glucose within the lysosome. Mutations in GCase are linked to the lysosomal storage disorder Gaucher’s disease and represent the most common genetic risk factor for PD (4). Ultimately, loss of function GBA1 mutations lead to the accumulation of its substrate glucosylceramide and α-synuclein oligomers contributing to a toxic pathogenic cascade (5), thus making GCase an important potential therapeutic target for PD.
Most of the translational efforts have focused on improving trafficking of mutant GCase into lysosomes (6, 7). However, this is not a viable strategy for heterozygous carriers of the 84GG GBA1 mutation, where the mutant protein is not expressed and patients must rely on residual wild-type GCase activity. Moreover, we recently showed that wild-type GCase enzyme activity was reduced in induced pluripotent stem cell (iPSC)-derived dopaminergic neurons from patients with genetic or idiopathic PD who did not harbor GBA1 mutations (non-GBA-PD) (8, 9). This suggested that decreased wild-type GCase activity may contribute to pathogenesis in other forms of PD and that activating wild-type GCase may represent a potential therapeutic strategy.
In this study, we developed a small-molecule modulator that was capable of stabilizing wild-type GCase protein and improving its activity. We further investigated whether activating wild-type GCase as an early intervention was sufficient to improve lysosomal function in iPSC-derived dopaminergic neurons from patients with PD with decreased wild-type GCase activity. Activation of wild-type GCase improved lysosomal function and other pathogenic phenotypes such as dopamine oxidation and α-synuclein accumulation in multiple cellular models of PD. These included iPSC-derived dopaminergic neurons from sporadic PD patients, as well as patients with heterozygous 84GG GBA1 mutations or mutations in genes encoding LRRK2, Parkin, or DJ-1. Administration of the GCase modulator S-181 to Gba1+/+ and Gba1D409V/+ mice led to activation of GCase in the central nervous system (CNS) and reduced accumulation of GCase lipid substrates and α-synuclein.
Results
Lysosomal dysfunction and oxidized dopamine accumulation in iPSC-derived dopaminergic neurons with a heterozygous 84GG GBA1 mutation
We obtained fibroblasts from patients with PD harboring the heterozygous 84GG mutation (c.84dupG frame-shift mutation) in GBA1 (GBA-PD), which results in complete loss of mutant GCase and reduced wild-type protein from a single copy of the wild-type GCase allele (fig. S1A and B). These fibroblasts were reprogrammed into induced pluripotent stem cells (iPSCs) (fig. S1C and D), and multiple GBA-PD and control iPSC lines were obtained (table S1). These lines were differentiated into midbrain dopaminergic neurons using previously established protocols (fig. S1, E and F) (5, 8, 10). We found that the 84GG mutation did not affect the reprogramming of fibroblasts into iPSCs. In addition, differentiation efficiencies and expression of midbrain dopaminergic neuronal markers were comparable in all lines examined. Dopaminergic neurons differentiated from iPSCs derived from fibroblasts from two GBA1 mutation carriers (GBA-PD1 and GBA-PD2) showed reduced amounts of GCase and decreased GCase enzymatic activity (P < 0.01) (Fig. 1, A to D). CRISPR-Cas9 correction of the heterozygous 84GG GBA1 mutation in iPSCs (figs. S1 and S2) increased the amount of wild-type GCase and enzymatic activity in corrected iPSC-derived dopaminergic neurons (P < 0.01) (Fig. 1, E and F). iPSC-derived dopaminergic neurons from patients with GBA-PD also demonstrated increased accumulation of the GCase substrate glucosylceramide (GluCer) (Fig. 1G; P < 0.05 for GBA-PD1 and P < 0.01 for GBA-PD2) and decreased proteolysis in lysosomes (Fig. 1H). Lysosomal function was rescued in isogenic CRISPR-corrected iPSC-derived dopaminergic neurons (Fig. 1H; P < 0.01). We recently found that iPSC-derived dopaminergic neurons from patients with familial or sporadic PD exhibited accumulation of oxidized dopamine that contributed to lysosomal dysfunction in neurons (8). Here, using our recently developed near-infrared fluorescence assay (11), we found that GBA-PD iPSC-derived dopaminergic neurons also demonstrated accumulation of oxidized dopamine (Fig. 1, I and J), which was reversed in CRISPR-corrected isogenic iPSC-derived dopaminergic neurons (Fig. 1K; P < 0.01).
Figure 1. Lysosomal dysfunction and oxidized dopamine accumulation in PD patient iPSC-derived dopaminergic neurons.
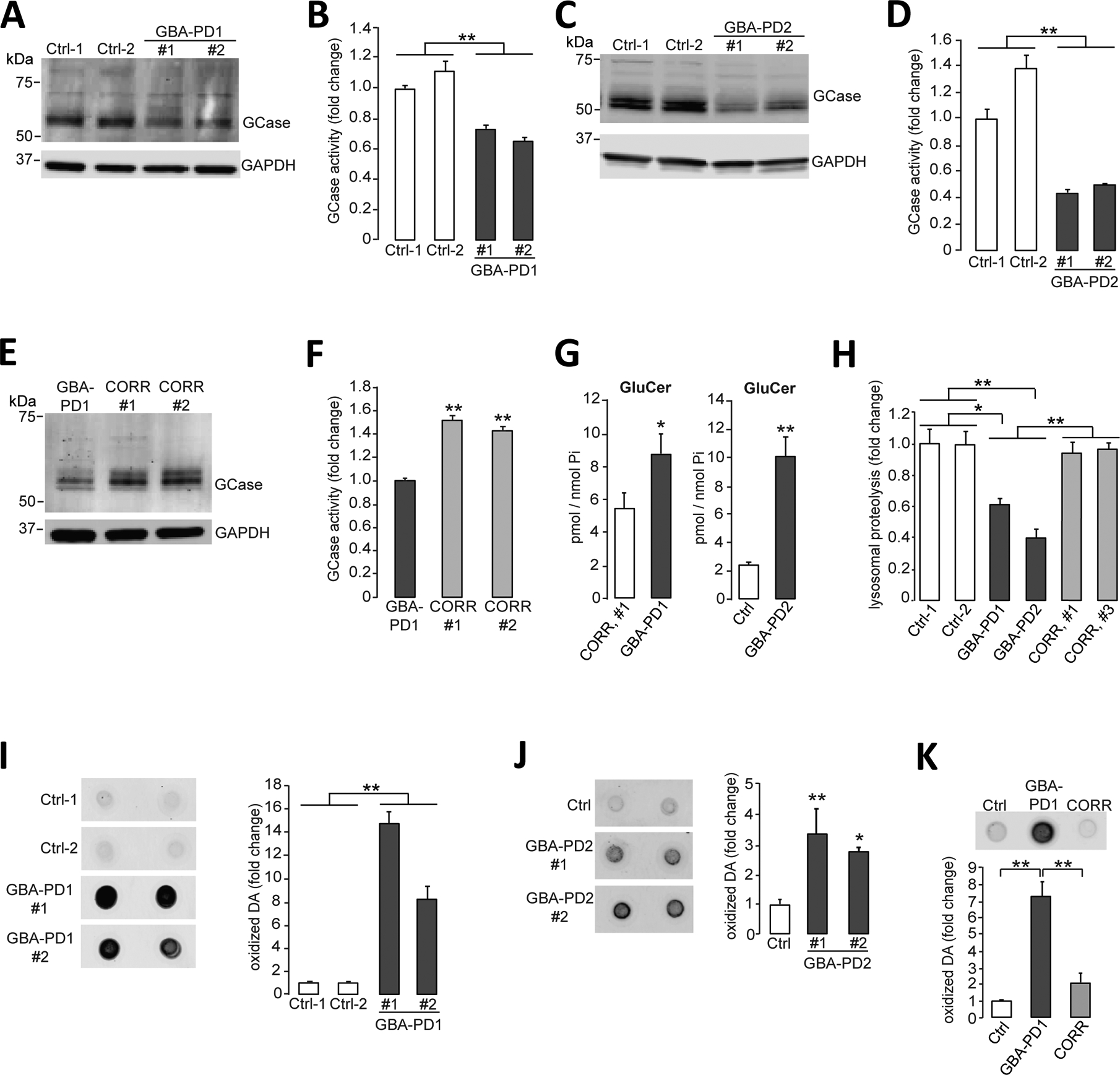
(A and B) Triton-soluble lysates of iPSC-derived dopaminergic neurons obtained from fibroblasts from healthy control individuals or patients with PD carrying the heterozygous 84GG GBA1 mutation (GBA-PD1, clone #1 and clone #2) were prepared at day 80 of differentiation. Neuronal lysates were analyzed for (A) GCase protein amounts by immunoblotting and (B) GCase activity by in vitro enzyme activity assay (N=3 independent experiments). (C and D) Triton-soluble lysates of iPSC-derived dopaminergic neurons obtained from fibroblasts from healthy control individuals or patients with PD carrying the heterozygous 84GG GBA1 mutation (GBA-PD2, clone #1 and #2) were prepared at day 60 of differentiation. Neuronal lysates were analyzed for (C) GCase protein amounts by immunoblotting and (D) GCase activity by in vitro enzyme activity assay (N=3 independent experiments). (E and F) Triton-soluble lysates from GBA-PD1 iPSC-derived dopaminergic neurons and isogenic control iPSC-derived dopaminergic neurons with the GBA1 mutation corrected by CRISPR-Cas9 gene editing (CORR, clone #1 and #2) were prepared at day 60 of differentiation. Neuronal lysates were analyzed for (E) GCase protein amounts by immunoblotting (representative blot shown) and (F) GCase activity by in vitro enzyme activity assay (N=3 independent experiments). (G) Quantification of intracellular glucosylceramide (GluCer) by mass spectrometry normalized to internal phosphate (Pi) was performed in GBA-PD1 mutant iPSC-derived dopaminergic neurons and CRISPR-Cas9-corrected GBA1 isogenic control iPSC-derived dopaminergic neurons (CORR, clone #1) (left, N=3 to 4 independent experiments). Quantification of intracellular glucosylceramide (GluCer) by mass spectrometry normalized to internal phosphate (Pi) was performed in GBA-PD2 mutant and healthy control iPSC-derived dopaminergic neurons (Ctrl) (right, N=3 independent experiments) at day 70 of differentiation. (H) Quantification of lysosomal proteolysis of long-lived proteins was measured by radioactive pulse-chase at day 80 of differentiation in healthy control (Ctrl), in GBA-PD1 and GBA-PD2 iPSC-derived dopaminergic neurons, and in CRISPR-Cas9 gene-edited GBA1 isogenic control iPSC-derived dopaminergic neurons (CORR, clone #1 and #3) (N=4 independent experiments). (I to K) Detection and quantification of oxidized dopamine (DA) by near-infrared fluorescence assay was performed (I) in healthy control iPSC-derived dopaminergic neurons (Ctrl) and GBA-PD1 mutant iPSC-derived dopaminergic neurons (clone #1 and #2) at day 80 of differentiation (N=3 independent experiments), (J) in healthy control iPSC-derived dopaminergic neurons (Ctrl) and GBA-PD2 mutant iPSC-derived dopaminergic neurons (clone #1 and #2) at day 60 of differentiation (N=5 to 6 independent experiments), and (K) in healthy control (Ctrl) and GBA-PD1 mutant iPSC-derived dopaminergic neurons and CRISPR-Cas9 gene-edited GBA1 isogenic control iPSC-derived dopaminergic neurons (CORR) at day 50 to130 of differentiation (N=3 independent experiments). Error bars, mean ± SEM. *P<0.05 and **P<0.01, Student’s t test (G) or one-way ANOVA with Tukey post hoc test (B, D, F, and H to K).
A small-molecule S-181 activates wild-type glucocerebrosidase in GBA-PD patient iPSC-derived dopaminergic neurons
We generated a series of new noninhibitory modulators of wild-type GCase by further examining the structure activity relationship of previously described quinazoline inhibitors (12). On the basis of our recent GCase crystallography data (13), the hydrogen bond interaction between quinazoline inhibitors and GCase Ser345 is critical for their inhibitory activity. We recently found that introduction of an N-methyl group was sufficient to disrupt this hydrogen bond interaction and convert these inhibitors into effective GCase activators. Here, we identified S-181 (Fig. 2A), which showed the most promising effect in in vitro GCase enzyme activity assays with a concentration of half-maximal activity (AC50) value of 1.49 μM and a maximum activation of 780% (Fig. 2B). In contrast, there was no effect of S-181 on in vitro enzyme activity of the lysosomal enzymes acid α-glucosidase and α-galactosidase A (fig. S3). To determine whether S-181 could further modulate wild-type GCase activity in human neurons, we treated iPSC-derived dopaminergic neurons from two healthy control subjects with increasing concentrations of S-181 for 10 consecutive days and found that concentrations of 5 to 25μM were sufficient to increase wild-type GCase activity (Fig. 2C).
Figure 2. Activation of wild-type GCase by the small molecule S-181.
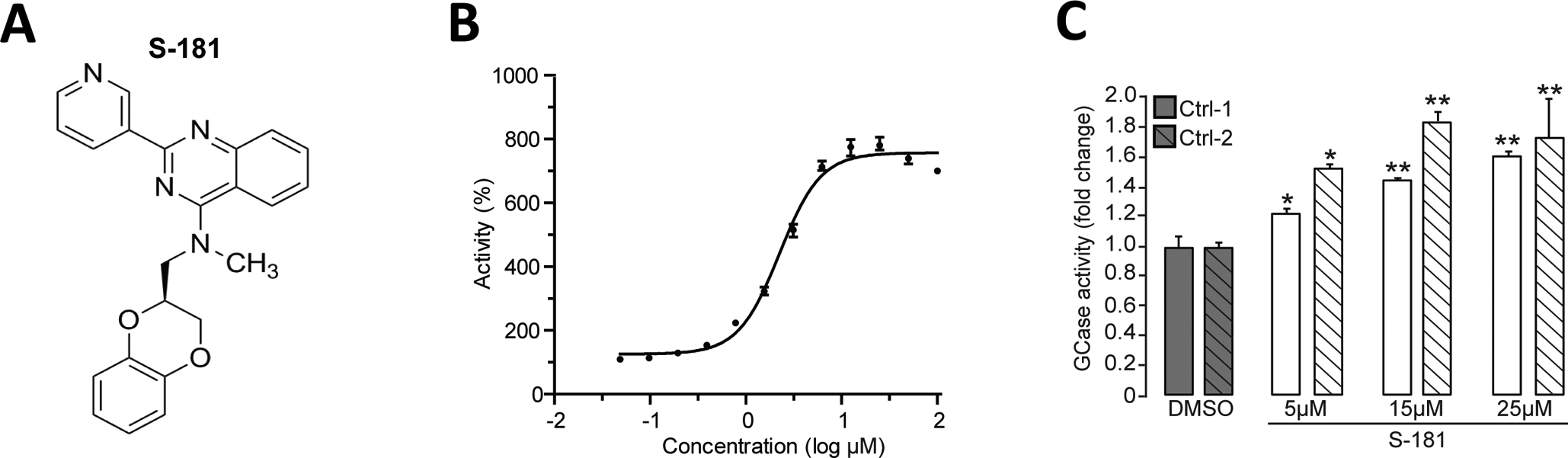
(A) Structure of the GCase modulator S-181. (B) Shown is the cell-free in vitro enzyme activity of GCase with S-181 with a concentration of half-maximal activity (AC50) of 1.49μM. (C) Measurement of GCase activity in Triton-soluble lysates of iPSC-derived dopaminergic neurons in vitro (derived from two healthy control subjects) treated with either dimethyl sulfoxide (DMSO) (vehicle) or S-181 (5, 15, and 25μM) for 10 days (N=3–5 independent experiments). Error bars, mean ± SEM. *P<0.05 and **P<0.01, one-way ANOVA with Tukey post hoc test (C).
Wild-type glucocerebrosidase activation partially rescues pathogenic phenotypes in GBA-PD iPSC-derived dopaminergic neurons
Next, we examined whether increasing wild-type GCase activity was sufficient to rescue defective GCase activity and other pathogenic phenotypes (Fig. 1) in GBA-PD iPSC-derived dopaminergic neurons. Treatment of GBA-PD iPSC-derived dopaminergic neurons with S-181 increased overall amounts of lysosomal GCase (Fig. 3, A and C). Consistent with increased GCase protein amounts, treatment with S-181 enhanced wild-type GCase enzymatic activity in GBA-PD iPSC-derived dopaminergic neurons (Fig. 3, B and D) with no effect on expression of the wild-type GBA1 allele (fig. S4). In contrast, a control compound that was structurally similar to S-181 (fig. S5A) but with no detectable in vitro GCase enzyme activation effect (fig. S5B) did not alter GCase enzyme activity in GBA-PD iPSC-derived dopaminergic neurons (Fig. 3E). Moreover, activation of wild-type GCase with S-181 reduced the accumulation of the GCase substrate GluCer (Fig. 3F), as well as partially corrected deficient lysosomal proteolysis in GBA-PD iPSC-derived dopaminergic neurons (Fig. 3G; P < 0.05). In addition, we found that S-181 treatment decreased dopamine oxidation in these neurons (Fig. 3, H and I; P < 0.05 and P < 0.01).
Figure 3. Activation of wild-type GCase with S-181 partially rescues pathogenic phenotypes in GBA-PD iPSC-derived dopaminergic neurons.
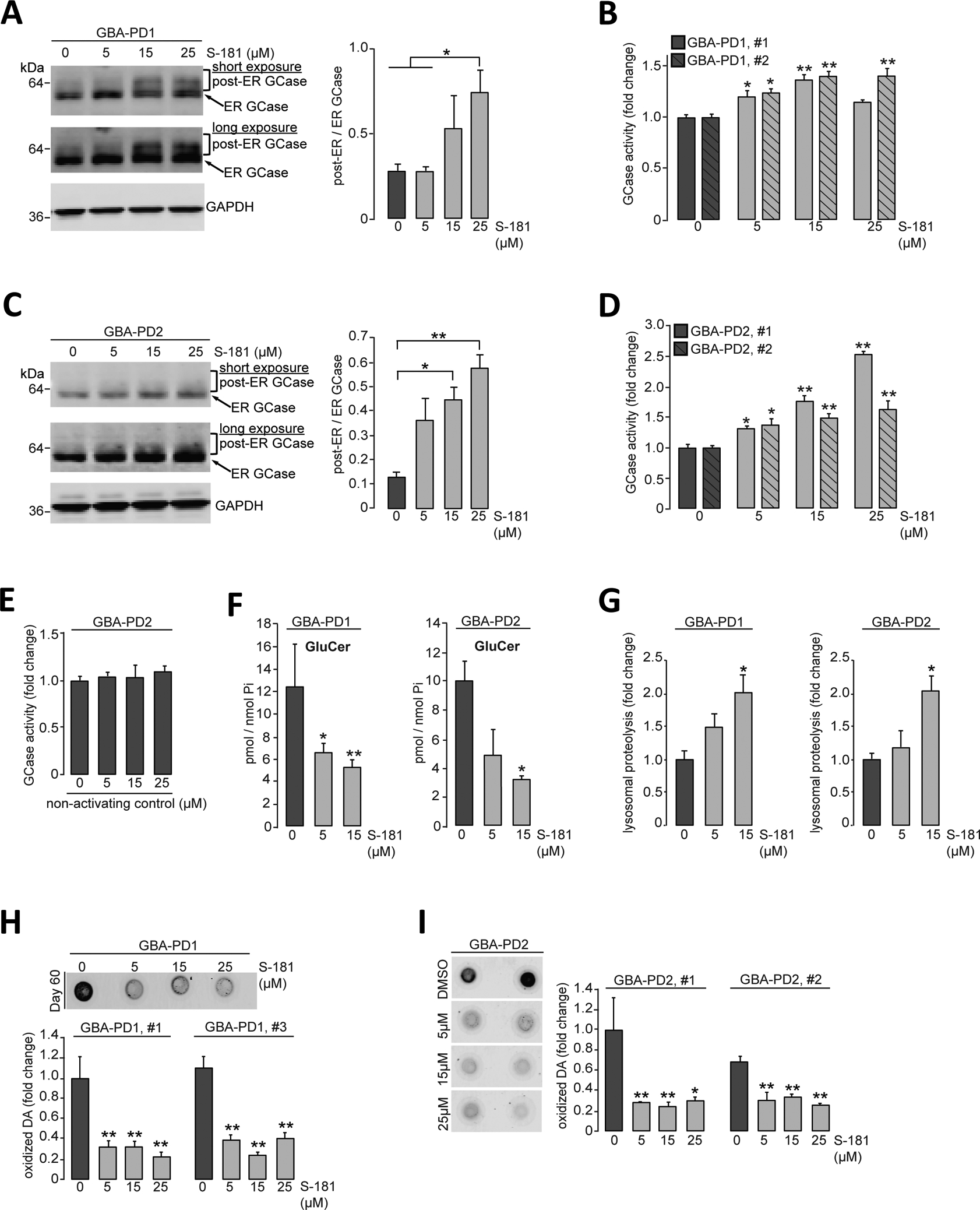
(A and B) Heterozygous 84GG GBA1 mutant iPSC-derived dopaminergic neurons (GBA-PD1 clone #1 and #2) were treated with either DMSO (vehicle) or S-181 (5, 15, and 25μM) for 10 days. Triton-soluble lysates were obtained at day 60 of differentiation and analyzed for (A) GCase protein amount by immunoblotting with quantification of post-ER/ER GCase (N=3 independent experiments) and (B) GCase activity assessed by in vitro enzyme activity assay (N=3 independent experiments). (C and D) Heterozygous 84GG GBA1 mutant iPSC-derived dopaminergic neurons (GBA-PD2, clone #1 and #2) were treated with either DMSO (vehicle) or S-181 (5, 15, and 25μM) for 10 days. Triton-soluble lysates were analyzed at day 60 of differentiation for (C) GCase protein amounts by immunoblotting with quantification of post-ER/ER GCase (N=3 independent experiments) and (D) GCase activity assessed by in vitro enzyme activity assay (N=3 to 6 independent experiments). (E) Heterozygous 84GG GBA1 mutant iPSC-derived dopaminergic neurons (GBA-PD2) were treated with either DMSO (vehicle) or a nonactivating control compound (5, 15, and 25μM) for 10 days. GCase activity was analyzed in Triton-soluble lysates at day 60 of differentiation (N=3 independent experiments). (F) Quantification of intracellular glucosylceramide (GluCer) by mass spectrometry normalized to internal phosphate (Pi) was performed at day 70 of differentiation in heterozygous 84GG GBA1 mutant iPSC-derived dopaminergic neurons [GBA-PD1 (left), N=3 to 4 independent experiments and GBA-PD2 (right), N=3 independent experiments] treated with either DMSO (vehicle) or S-181 (5 and 15μM) for 10 days. (G) Lysosomal proteolysis of long-lived proteins was quantified at day 80 of differentiation by radioactive pulse-chase of heterozygous 84GG GBA1 mutant iPSC-derived dopaminergic neurons [GBA-PD1 (left), N=3 independent experiments and GBA-PD2, N=3 to 5 independent experiments] treated with either DMSO (vehicle) or S-181 (5 and 15μM) for 10 days. (H and I) Detection and quantification of oxidized dopamine (DA) was performed by near-infrared fluorescence assay at day 60 of differentiation of heterozygous 84GG GBA1 mutant iPSC-derived dopaminergic neurons (H) GBA-PD1 clone #1 and #3 (N=3 to 4 independent experiments), (I) GBA-PD2 clone #1 and #2 (N=4 to 6 independent experiments) treated with either DMSO (vehicle) or S-181 (5, 15, and 25μM) for 10 days. Error bars, mean ± SEM. *P<0.05 and **P<0.01, one-way ANOVA with Tukey post hoc test.
Wild-type glucocerebrosidase activation partially rescues pathogenic phenotypes in non-GBA-PD iPSC-derived dopaminergic neurons
Our recent studies showed that wild-type GCase enzyme activity was decreased in mutant LRRK2, Parkin, and DJ-1 iPSC-derived dopaminergic neurons (8, 9) and in iPSC-derived dopaminergic neurons as well as brain tissue from patients with sporadic PD without GBA1 mutations (non-GBA-PD) (8, 14, 15). This suggested that decreased activity of wild-type GCase may play a role in PD pathogenesis in different forms of the disease. We found accumulation of oxidized dopamine in iPSC-derived dopaminergic neurons from patients with various forms of non-GBA-PD (8, 9). Therefore, we examined whether treating non-GBA-PD iPSC-derived dopaminergic neurons with the S-181 compound could increase GCase enzymatic activity and rescue the accumulation of oxidized dopamine and other pathogenic phenotypes. S-181 treatment increased wild-type GCase activity in iPSC-derived dopaminergic neurons from patients with PD carrying LRRK2 mutations (LRRK2-PD) (Fig. 4A; P < 0.001 for LRRK2-PD1 and P < 0.05 for LRRK2-PD2) or Parkin mutations (Parkin-PD) (Fig. 4C; P < 0.01). In addition, S-181 treatment increased wild-type GCase activity in iPSC-derived dopaminergic neurons lacking DJ-1- through CRISPR-Cas9 genetic engineering (Fig. 4E; P < 0.05) and in iPSC-derived dopaminergic neurons from two patients with idiopathic PD (iPD) without any known PD mutations (Fig. 4G; P < 0.01 at 25μM S-181 in iPD-1 and P < 0.05 at 15 and 25μM S-181 in iPD-2). Moreover, we found that S-181 treatment decreased dopamine oxidation in iPSC-derived dopaminergic neurons from patients with non-GBA1-linked forms of PD (Fig. 4B, D, F, and H). S-181 treatment also reduced the amounts of total and oxidized α-synuclein in LRRK2-PD and iPD iPSC-derived dopaminergic neurons (Fig. 4, I and J, P < 0.05).
Figure 4. Activation of wild-type GCase by S-181 lowers oxidized dopamine and α-synuclein in non-GBA1-linked PD patient iPSC-derived dopaminergic neurons.
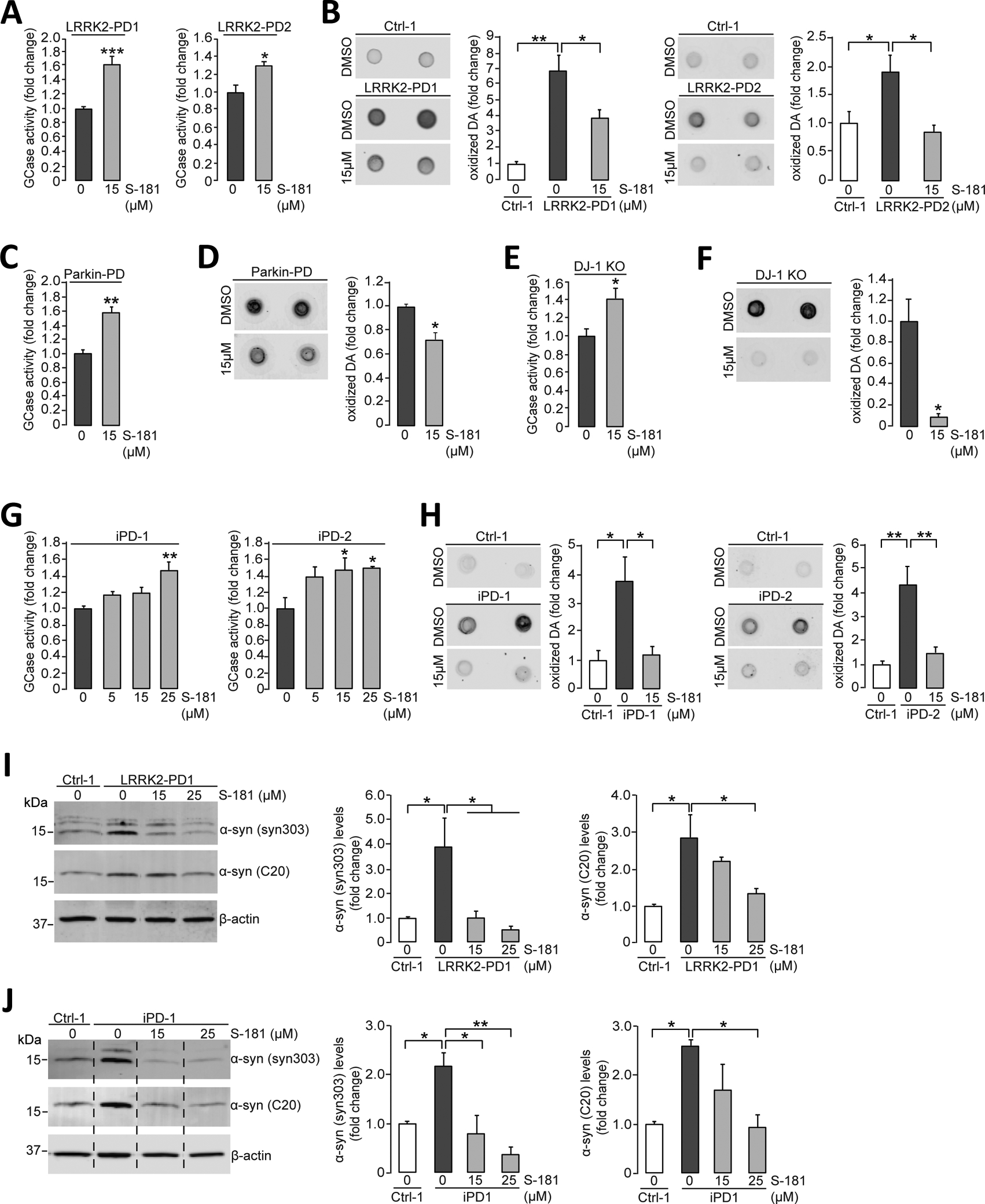
(A) Mutant iPSC-derived dopaminergic neurons carrying the LRRK2 R1441C mutation (LRRK2-PD1) or G2019S mutation (LRRK2-PD2) were treated with either DMSO (vehicle) or S-181 (15μM) for 10 days. Cell lysates were then analyzed at day 100 of differentiation for GCase activity assessed by in vitro enzyme activity assay (N=6 to 9 independent experiments for LRRK2-PD1; N=3 independent experiments for LRRK2-PD2). (B) Mutant iPSC-derived dopaminergic neurons (LRRK2-PD1 and LRRK2-PD2) and healthy control iPSC-derived dopaminergic neurons were treated with either DMSO (vehicle) or S-181 (15μM) for 10 days. Cell lysates were analyzed at day 100 of differentiation for oxidized dopamine (DA) by near-infrared fluorescence assay (N=7 independent experiments for LRRK2-PD1; N= 4–5 independent experiments for LRRK2-PD2). (C and D) Mutant iPSC-derived dopaminergic neurons generated from fibroblasts of a patient with a compound heterozygous PARKIN mutation were treated with either DMSO (vehicle) or S-181 (15μM) for 10 days. Cell lysates were analyzed at day 150 of differentiation for (C) GCase activity assessed by in vitro enzyme activity assay (N=3 independent experiments) and (D) oxidized DA measured by near-infrared fluorescence assay (N=3 independent experiments). (E and F) Mutant iPSC-derived dopaminergic neurons in which DJ-1 was deleted by CRISPR-Cas9 gene editing (DJ-1 KO) were treated with either DMSO (vehicle) or S-181 (15μM) for 10 days. Cell lysates were analyzed at day 160 of differentiation for (E) GCase activity by in vitro enzyme activity assay (N=3 independent experiments) and (F) oxidized DA measured by near-infrared fluorescence assay (N=3 independent experiments). (G) iPSC-derived dopaminergic neurons generated from fibroblasts from two patients with idiopathic PD (iPD-1 and iPD-2) were treated with either DMSO (vehicle) or S-181 (5, 15, and 25μM) for 10 days. Cell lysates were analyzed at day 130 of differentiation for GCase activity (N=4 to 5 independent experiments for iPD1; N=3 to 5 independent experiments for iPD2). (H) iPSC-derived dopaminergic neurons generated from fibroblasts from two patients with idiopathic PD (iPD-1 and iPD-2) and one healthy control (Ctrl-1) were treated with either DMSO (vehicle) or S-181 (15μM) for 10 days. Cell lysates were analyzed at day 130 of differentiation for oxidized DA measured by near-infrared fluorescence assay (N=3 to 4 independent experiments for iPD1; N= 3 to 6 independent experiments for iPD2). (I) Mutant iPSC-derived dopaminergic neurons carrying the LRRK2 G2019S mutation (LRRK2-PD1) and healthy control iPSC-derived dopaminergic neurons (Ctrl-1) were treated with either DMSO (vehicle) or S-181 (15 and 25μM) for 10 days. Triton-soluble lysates were analyzed at day 100 of differentiation for the amount of α-synuclein by immunoblotting with C20 and syn303 anti-α-synuclein antibodies. β-Actin was used as a loading control (N=3 to 4 independent experiments). (J) iPSC-derived dopaminergic neurons generated from fibroblasts from a patient with idiopathic PD (iPD-1) and a healthy control (Ctrl-1) were treated with either DMSO (vehicle) or S-181 (15 and 25μM) for 10 days. Triton-soluble lysates were analyzed at day 100 of differentiation for the amount of α-synuclein by immunoblotting with C20 and syn303 anti-α-synuclein antibodies. β-Actin was used as a loading control (N=3 to 4 independent experiments). Error bars, mean ± SEM. *P<0.05, **P<0.01, and ***P<0.001, Student’s t test (A and C to F) or one-way ANOVA with Tukey post hoc test (B and G to J).
Wild-type glucocerebrosidase activation in mice lowers lipid substrate and α-synuclein accumulation
Our pharmacokinetic analysis showed that an intraperitoneal single administration of S-181 (50 mg/kg) into wild-type mice resulted in a Cmax of 2.9μM in brain and a half-life of 20.7 hours, with higher concentrations in brain than in plasma after 2 hours (Fig. 5A and table S2). In addition, we performed plasma protein and brain homogenate binding assays to determine the amount of unbound compound that was available to reach GCase in the mouse brain. S-181 exhibited binding to brain tissue in mouse brain homogenates (table S3) and to proteins in plasma (table S4) with a minimum stability of 6 hours. Treatment with S-181 compound had no impact on brain weight of the treated mice (fig. S6). The ability of S-181 to cross the brood-brain barrier led to a significant increase in GCase activity in brain tissue from wild-type (Gba1+/+) and Gba1D409V/+ mutant mice treated with S-181 (50 mg/kg, twice daily) for 15 days compared to vehicle-treated mice (Fig. 5B; P < 0.05). Activation of GCase with S-181 reduced the accumulation of the GCase substrates GluCer (Fig. 5C; P < 0.05) and glucosylsphingosine (Fig. 5D; P < 0.05) in brain tissue from S-181-treated Gba1D409V/+ mutant mice. Moreover, treatment of Gba1D409V/+ mutant mice with S-181 reduced the amount of insoluble α-synuclein (16) in brain tissue compared to brain tissue from vehicle-treated Gba1D409V/+ mutant mice (Fig. 5E; P < 0.001). These results demonstrated that the GCase modulator S-181 could modify GCase activity in vivo.
Figure 5. Activation of wild-type GCase by S-181 reduces lipid substrates and α-synuclein in mice.
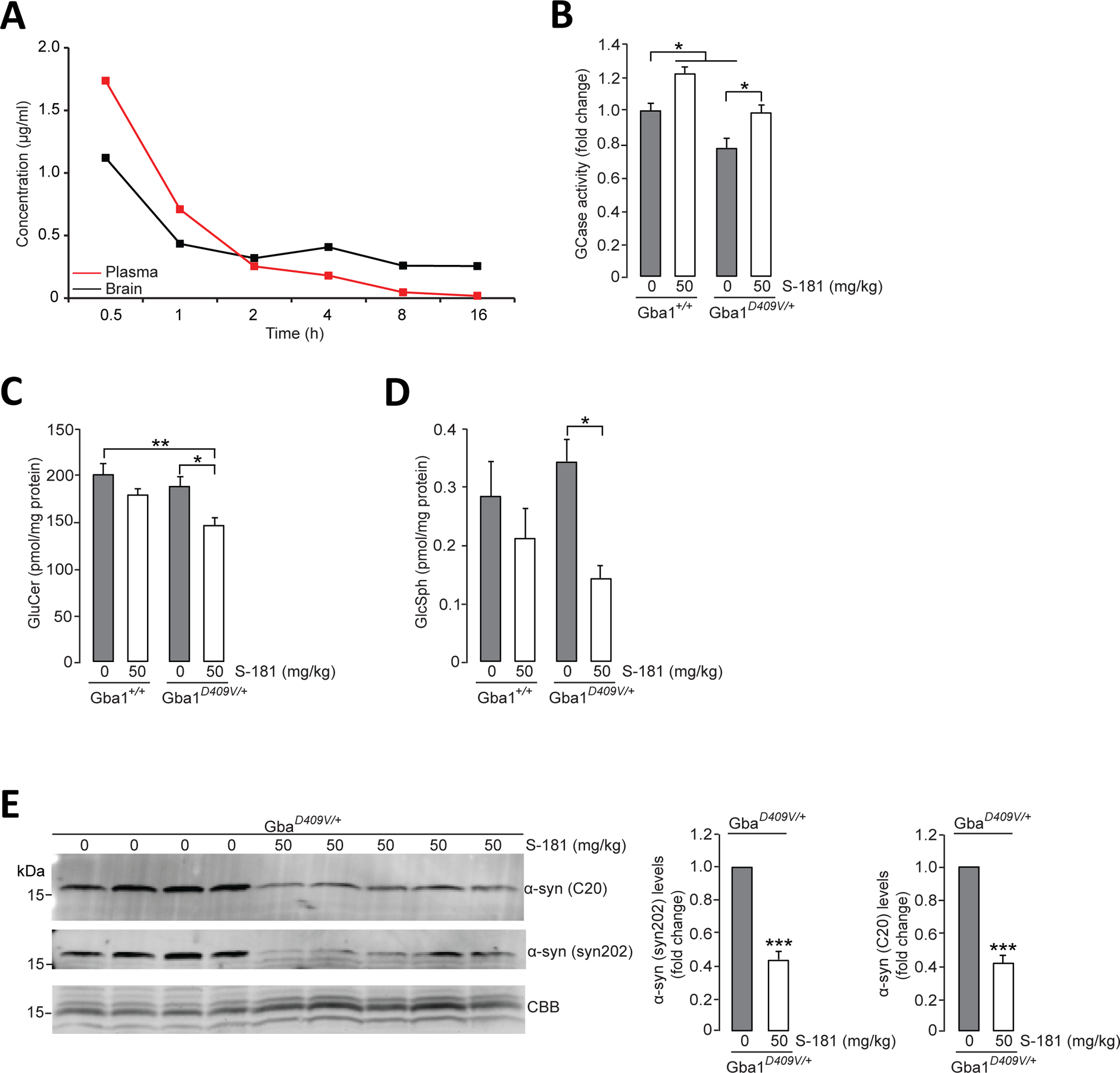
(A) Mean concentration-time profiles for S-181 in plasma and brain tissue after administration of intraperitoneal single dose (50 mg/kg) to C57BL/6 wild-type mice (n=3 mice per time point). (B) GCase activity was measured by in vitro enzyme activity assay in lysates of hippocampal tissue from wild-type Gba1+/+ mice and from Gba1D409V/+ heterozygous mutant mice that had been treated with 5% dextrose (vehicle) (n=8 mice per group) or S-181 (50mg/kg; n=5 mice per group) intraperitoneally twice daily for 15 days. (C and D) Quantification of intracellular (C) glucosylceramide (GluCer) and (D) glucosylsphingosine (GluSph) by mass spectrometry in hippocampal tissue from wild-type Gba1+/+ and Gba1D409V/+ heterozygous mutant mice treated with 5% dextrose (vehicle) (n=8 mice per group) or S-181 (50mg/kg; n=5 to 6 mice per group) intraperitoneally twice daily for 15 days. (E) Immunoblot analysis of α-synuclein (using C20 and syn202 anti-α-synuclein antibodies) in Triton-insoluble lysates of hippocampal tissue from Gba1D409V/+ heterozygous mutant mice treated with 5% dextrose (vehicle) (n=8 mice per group) or S-181 (50mg/kg; n=5 mice per group) intraperitoneally twice daily for 15 days. Coomassie Brilliant Blue (CBB) was used as a loading control. Mice (mixed group of males and females) were between 13 and 15 months of age. Error bars, mean ± SEM. *P<0.05, **P<0.01, and ***P<0.001, Student’s t test (E) or one-way ANOVA with Tukey post hoc test (B to D).
Discussion
There is an urgent need to identify new effective therapies for PD, a debilitating neurodegenerative disease for which there still remains no neuroprotective treatment. The identification of genetic forms of PD caused by mutations in, for example, the LRRK2 and GBA1 genes has facilitated development of targeted therapies for PD. A well-studied potential target is the lysosomal enzyme GCase in patients who have PD linked to GBA1 mutations. Recent evidence suggests that these patients with GBA-PD exhibit loss of GCase activity in lysosomes, which, in turn, results in accumulation of oxidized dopamine and α-synuclein. Therefore, direct activation of GCase in lysosomes and using chemical chaperones to promote translocation of mutant GCase from the endoplasmic reticulum into lysosomes (7, 17, 18) are potential therapeutic strategies.
Our work and studies by others have shown that wild-type GCase activity is also reduced in brain tissue and neurons from patients with idiopathic PD and patients with other genetic forms of PD that are not linked to GBA1 mutations (7–9, 14, 15, 19, 20), suggesting that wild-type GCase could also be a potential therapeutic target. To this end, our study explored whether activation of wild-type GCase with a small molecule could enhance lysosomal function and rescue pathological phenotypes in iPSC-derived dopaminergic neurons from patients with sporadic or familial forms of PD. We identified the GCase modulator S-181 as a small molecule that increased the activity of wild-type GCase and partially ameliorated lipid substrate accumulation, lysosomal dysfunction, α-synuclein accumulation, and dopamine oxidation in both GBA1-linked and non-GBA1-linked PD iPSC-derived dopaminergic neurons. Our work thus suggests that rescuing GCase activity is sufficient to improve lysosomal function and to reduce accumulation of toxic oxidized dopamine in iPSC-derived dopaminergic neurons in vitro. In turn, decreased accumulation of oxidized dopamine results in diminished oxidation-mediated modifications to GCase that disrupts its enzymatic activity (8). We found that this deleterious feedback cycle could be interrupted by targeting wild-type GCase with the small-molecule modulator S-181 in iPSC-derived dopaminergic neurons from patients with PD. Moreover, our in vivo analysis in mice revealed that S-181 could penetrate the CNS, enhance wild-type GCase enzyme activity in brain tissue, and reduce accumulation of GCase lipid substrates and α-synuclein. This is in agreement with previous work demonstrating that viral delivery of GBA1 increased wild-type GCase enzyme activity and reduced pathological phenotypes in mice (21, 22). These findings point to the relevance of therapeutically targeting GCase in different forms of PD, including familial PD caused by mutations in LRRK2, DJ-1-, or Parkin and possibly also sporadic PD.
This study primarily examined PD patient iPSC-derived human midbrain dopaminergic neurons because we have previously observed that certain key pathogenic phenotypes, such as accumulation of oxidized dopamine and neuromelanin, were only observed in human midbrain dopaminergic neurons in vitro but not in mice (8). Consistent with this notion is prior work showing that positive therapeutic effects in mouse studies did not translate to human clinical trials. Whereas studies of human midbrain dopaminergic neurons clearly provide a unique advantage for drug development in PD, they also pose some limitations. For example, cultured human midbrain dopaminergic neurons primarily address the cell-autonomous dysfunction of PD in the absence of other cell types and neuronal circuits that likely contribute to PD disease pathogenesis. Although we tested our GCase modulator in a mouse model to demonstrate CNS penetrance and GCase activity in vivo, we were unable to examine the most relevant pathogenic phenotypes in mouse midbrain dopaminergic neurons because these neurons remain intact in this and other genetic models of PD. Therefore, it will be important to utilize iPSC-derived dopaminergic neurons from patients with PD to test candidate therapeutic interventions that target midbrain dopaminergic neurons.
Development of targeted therapies that are based on the genetics of PD is likely to have a major impact on this neurodegenerative disease. Our study established that modulation of GCase activity may be a promising therapeutic approach for GBA1-linked PD and other forms of PD that exhibit decreased wild-type GCase activity. Future studies will be needed to fully define the pharmacokinetic and dynamic properties of our GCase modulator S-181 in normal and affected tissues and to develop the most effective analogs for testing in human clinical trials.
MATERIALS AND METHODS
Study design
The objective of this study was to examine the effects of a new chemical series of glucocerebrosidase (GCase) modulators that increase wild-type GCase activity in iPSC-derived dopaminergic neurons in vitro. These iPSC-derived dopaminergic neurons were obtained from fibroblasts from patients with PD carrying GBA1-linked or non-GBA1-linked mutations in which mutant GCase was not expressed and wild-type GCase enzyme activity was decreased. In these cases, activating wild-type GCase could represent an alternative therapeutic approach for decreasing pathogenic phenotypes such as lysosomal dysfunction and accumulation of α-synuclein, oxidized dopamine and GluCer. Skin fibroblasts from five patients with PD having mutations in GBA1 (84GG), LRRK2 or PARKIN and from two patients with idiopathic PD with no known PD mutations were used to generate iPSCs in vitro. These iPSC lines together with CRISPR-Cas9-generated iPSCs lacking DJ-1 were differentiated into midbrain dopaminergic neurons in vitro. In addition, isogenic control iPSC lines with the 84GG GBA1 mutation corrected by CRISPR-Cas9 and two iPSC lines from healthy control subjects were differentiated into midbrain dopaminergic neurons in vitro. GCase activity and protein amounts were measured in iPSC-derived dopaminergic neurons in vitro followed by measurements of lysosomal proteolysis, oxidized dopamine, α-synuclein, and lipids. These parameters were evaluated after treatment of iPSC-derived dopaminergic neurons with the small-molecule GCase modulator S-181 or vehicle control in a randomized fashion. A nonactivating control compound structurally similar to S-181 was used to confirm the specificity of S-181. To determine whether S-181 would be effective in increasing wild-type GCase enzyme activity and reducing PD pathology in vivo, a mixed group of male and female Gba1D409V/+ mice were treated with S-181 and compared to vehicle-treated mice in a randomized fashion. Brain penetrance of S-181 was confirmed, and GCase activity in mouse brain tissue was confirmed followed by measurements of α-synuclein and lipids. The number of independent experiments or number of mice per group, respectively, is included in the figure legends.
Generation of human iPSCs and differentiation into midbrain dopaminergic neurons
Skin fibroblasts from two patients with PD carrying the 84GG (c.84dupG) mutation in GBA1 (GBA-PD1, GBA-PD2) were reprogrammed into iPSCs by the Northwestern University Stem Cell Core Facility. Reprogramming was done using Sendai Virus (SeV) based delivery of key reprogramming factors including the four Yamanaka transcription factors (Oct3/4, Sox2, Klf4 and c-Myc). Reprogrammed iPSC lines were characterized for expression of pluripotency markers and genomic integrity as described previously (8). Briefly, all iPSC lines were characterized for expression of pluripotency markers by immunocytochemistry (NANOG, OCT4, SSEA4, TRA1–81) and by quantitative RT-PCR (SOX2, cMYC, OCT4, NANOG). Total RNA was prepared using the RNeasy kit (Qiagen) and then reverse transcribed into cDNA with the SuperScript III First-Strand Synthesis System (Invitrogen). Quantitative RT-PCR was performed with SYBR GreenER (Invitrogen) on the 7500 Fast Real-Time PCR system (Applied Biosystems). Karyotype analysis was performed by Cell Line Genetics using standard protocols for high-resolution G-banding.
iPSCs were cultured in mTeSR media (Stem Cell Technologies) and passaged every 5 to 7days on Matrigel (Corning) coated plates. All iPSC lines were routinely tested for mycoplasma contamination.
We used iPSC lines already generated from fibroblasts from three patients with familial PD: One patient had a PARKIN mutation (23), and two patients carried LRRK2 mutations [R1441C LRRK2-PD1 (9); G2019S LRRK2-PD2, obtained from NINDS Repository at Coriell Institute for Medical Research; catalog number, ND29423). We also used iPSC lines derived from fibroblasts from two patients with idiopathic PD [iPD-1 (8) and iPD-2 obtained from NINDS Repository at Coriell Institute for Medical Research; catalog number, ND39896) and from two healthy control subjects (8, 24, 25). CRISPR-Cas-edited iPSCs lacking DJ-1 were described and characterized previously (8). Differentiation of iPSC lines into midbrain dopaminergic neurons was done according to published protocols (10) and as previously described (8). Briefly, cells were passaged mechanically at day 12 into 2-mm2 blocks and plated into poly-D-lysine/laminin-coated dishes. At day 25 to day 30, these blocks were passaged by Accutase into single cells and plated into poly-D-lysine/laminin-coated plates. Neural growth factors were removed at day 40, and cells were maintained in Neurobasal Media (Thermo Fisher Scientific no. 21103–049) with NeuroCult SM1 supplement (STEMCELL Technologies, no. 5711). Neurons were extensively characterized by immunostaining and Western blotting for the midbrain markers FOXA2, LMX1a, and tyrosine hydroxylase (TH) at different time points in the differentiation protocol, and all iPSC lines were found to differentiate at similar efficiencies.
Sequencing Analysis
DNA samples were extracted from cell pellets using Mini prep kits (QIAGEN). Eluted gDNA was used as a template for PCR amplification using Phusion Master Mix (NEB). PCR product was purified using PCR Purification Kit (QIAGEN) and sent for sequencing through the NUSeq Core or University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility. Primers used for PCR amplification and sequencing are GBA 84GG Seq F (gtgggccttgtcctaatgaa) and GBA 84GG Seq R (caccccaaagttggtctcag).
Generation of isogenic lines from heterozygous 84GG (c.84dupG) GBA mutant iPSCs
GBA-001 transcript sequence (ENST0000368373.7) on Ensembl genome browser was used to design the isogenic line. Single-guide RNAs (sgRNA) were designed using Zhang Lab CRISPR Design Tool (crispr.mit.edu). Guides were selected on the basis of high predicted activity and low off-targets. Furthermore, guides were chosen so that double stranded nicks occur within 20 base pairs (bp) of the homologous recombination site. These guides were cloned into pSpCas9n(BB)-2A-GFP (green fluorescent protein) (pX461, Addgene, no. 48140) using an established protocol (24). Single stranded oligonucleotides (ssODN) were designed with 80 bp, surrounding the left and right side of the mutation correction site for efficient homologous recombination. GBA1 mutant iPSC lines were grown to 70% confluency on Matrigel (Corning)-coated 10cm plates with mTeSR media (STEMCELL Technologies). One hour before electroporation, cells were replaced with 1:1 media (MEF conditioned iPS media: mTeSR media). Next, cells were washed with 1x phosphate-buffered saline (PBS) and dissociated with Accutase for 5 min at 37⁰C. Dissociated cells were centrifuged at 200xg for 5min. Supernatant was removed, and cell pellet was resuspended in 1x PBS to count the cells. Subsequently, cells were centrifuged as described before, and the pellet was resuspended in Buffer E2 at 50million/ml dilution. A mixture composed of the following was prepared: 100ul of resuspended cells, 1ul of 1mg/ul of each px461 with inserted guides, and 3ul of 100uM ssODN. Electroporation was done using Neon Transfection System (Invitrogen) using two 850V, 30ms pulses. Transfected cells were plated in mTeSR media containing Y-27632. Cells were sorted 36 hours after transfection using BD FACS Aria 4-Laser for GFP-positive cells. Next, cells were expanded and screened by PCR genotyping and sequencing analysis.
Preparation of compounds
Preparation of (S)-N-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)-N-methyl-2-(pyridin-3-yl) quinazolin-4-amine (S-181) and of N-(2-phenoxyethyl)-N-phenyl-2-(pyridin-3-yl)quinazolin-4-amine (non-activating control) is provided in the Supplementary materials.
Wild-type and Gba1D409V/+ mice
Wild-type (C57BL/6) and Gba1D409V/+ (C57BL/6N-GBAtm1.1Mjff) mice (the Jackson laboratory) were bred following Institutional Animal Care and Use Committee at Northwestern University guides and handled in accordance with the US National Institutes of Health Guide to the Care and Use of Laboratory Animals and Society for Neuroscience guidelines. Mice either received dextrose or S-181 (50mg/kg) through intraperitoneal injection, twice a day, for 15 days at 13 to 15 months of age. All animals were chosen randomly (blinded) for S-181 treatment.
Protein binding assay and cell-free enzyme activity assay
Brain tissue binding measurements have been performed according to published protocols (26) and conducted by Pharmaron. The cell-free in vitro enzyme activity assay was performed as described previously (12).
Pharmacokinetics and pharmacodynamics of S-181
The intraperitoneal dosing solution of S-181 hydrochloride was prepared in 5% dextrose. Brain samples were homogenized with three volumes of PBS (pH 7.4) before sample extraction. Liquid chromatography-tandem mass spectroscopy (LC−MS/MS) analysis of samples was done with a Shimadzu Nexera X2 UHPLC Binary System, C18 column, with a flow rate of 0.5 mL/min and a mobile phase consisting of solvent A (H2O−0.1% FA), solvent B (ACN−0.1% FA). For pharmacokinetics study, wild-type mice were treated with one dose of S-181 hydrochloride (50mg/kg; in 5% dextrose) or vehicle. After 30min and 1, 2, 4, 8, and 16 hours, blood and brain samples were collected. For pharmacodynamics study, wild-type (Gba1+/+) and Gba1D409V/+ mice were treated with S-181 hydrochloride (50mg/kg; in 5% dextrose) or vehicle twice per day for 15 days. Brain and other organs were collected, weighted, and analyzed. All animals were chosen randomly (blinded) for S-181 treatment.
Sequential biochemical extraction
Sequential biochemical extraction of proteins from iPSC-derived neurons and mouse tissue has been previously described (8). Neurons were harvested in ice-cold PBS and centrifuged at 300g for 5min. Cell pellets were homogenized for extraction in 1% Triton X-100 lysis buffer (containing 10% glycerol, 150mM NaCl, 25mM Hepes pH7.4, 1mM EDTA, 1.5mM MgCl2, and proteinase inhibitor cocktail) for 30 min on ice and were centrifuged for 100,000g for 30 min at 4⁰C. Supernatant was analyzed as Triton-soluble fraction. Subsequently, insoluble pellets were extracted in 2% SDS/50mM tris (pH 7.4) buffer by boiling and sonication. SDS buffer volume was normalized to total protein per Triton-soluble fraction. Samples were centrifuged at 150,000g for 30 min at 4⁰C, and the supernatant was used for analysis as SDS-soluble fraction. SDS-insoluble pellets were further used for near-infrared fluorescence assay. For biochemical extraction of brain tissue from Gba1D409V/+ mice, freshly dissected hippocampal brain tissue was homogenized in 1% Triton X-100 lysis buffer according to tissue weight. Insoluble pellets from a 100,000g spin were further extracted in 2% SDS/50mM Tris (pH 7.4) by boiling and sonication. Samples were centrifuged at 150,000g for 30 min at 4⁰C, and the supernatant was used for analysis as SDS-soluble fraction.
Near infrared fluorescence (nIRF) detection of oxidized dopamine
nIRF assay was performed as described previously (8, 11). Leftover pellets from SDS extraction were extracted in 1M NaOH by incubating at 55⁰C overnight. The volume of NaOH for extraction was calculated depending on the protein concentration of the soluble fraction. Next day, the samples were left to dry. The leftover pellet was washed once with Nanopure H2O for removal of hydroxides and to lower pH levels and then lyophilized in Speed Vac Concentrator before the dried pellet was taken up in Nanopure H2O and lastly analyzed. Ten microliters of samples were dropped on a Biodyne Nylon Transfer Membrane (Pall). In addition to samples, standards made from a 10mM oxidized dopamine stock were loaded at different concentrations. For preparation of oxidized DA, 10mM DA (in D-PBS) was mixed with 20mM NaIO4 (in D-PBS), vortexed briefly, and incubated for 5min at room temperature. The solution was then handled in the same way as the cell samples including centrifugation, sonication, and lyophilization steps. The final pellet was taken up in Nanopure H2O and standard dilution prepared. Membranes were scanned using an Odyssey infrared imaging system (LI-COR) with the 700 channel. Samples were quantified by obtaining integrated spot intensities using Odyssey infrared imaging software, version 3.1 (LI-COR).
Assay for in vitro activity of GCase in neurons and mouse brain tissue
Neurons were washed in PBS and harvested in 1% Triton lysis buffer, and activity was measured from whole-cell lysates as described previously (7). Hippocampal brain tissue from mice was homogenized in 1% Triton X-100 lysis buffer according to tissue weight.
Enzyme reaction was prepared as a mixture containing the following: 5ug of protein lysate, 10ul of 10% BSA in activity assay buffer [0.25% w/v taurocholic acid and 1mM EDTA in citrate/phosphate buffer (pH 5.4)], 20ul of 5mM 4-methylumbelliferyl-β-D-glucopyranoside substrate (4MU-Gluc, Sigma-Aldrich) in activity assay buffer, and activity assay buffer to bring the final volume to 100ul. The mixture was incubated at 37⁰C for 30 min (±8ul of 25mM conduritol-b-epoxide, CBE) and stopped by addition of 100ul of 1M glycine (pH 12.5). 4MU fluorescence was determined using a SpectraMax i3 plate reader (Molecular Devices) (excitation, 365nm; emission, 445nm; top read). Enzyme activity was quantified by subtracting CBE-treated lysates from non-CBE-treated lysates to obtain activity derived from GBA1.
Immunofluorescence analysis
Immunofluorescence analysis was performed as described previously (8). Neurons were fixed in 4% paraformaldehyde and permeabilized with 0.3% Triton X-100 in PBS. Neurons were blocked for 30min in blocking buffer (1% BSA and 5% normal goat serum in PBS-Triton) and then incubated with the following primary antibodies overnight at 4⁰C: anti-β-III-tubulin (Covance, no. MRB-435P; 1:1000), tyrosine hydroxylase (Millipore, no. 657012; 1:1000), HNF-3β (FOXA2) (Santa Cruz Biotechnology, no. sc-101060; 1:100), LMX1A (EMD Millipore, no. AB10533; 1:1000), NANOG (Abcam, no. ab80892; 1:1000), OCT4 (Abcam, no. ab19857; 1:300), SSEA4 (Merck, no. MAB4304; 1:100), and TRA-1–81 (Merck, no. MAB4381; 1:50). Cells were washed in PBS and incubated with Alexa-conjugated anti-rabbit or anti-mouse antibodies for 1 hour at room temperature and lastly mounted in ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). Neurons were imaged using Leica Confocal Microscope (Leica).
Assay for lysosomal proteolysis
Long-lived protein degradation assay was performed by radioactive pulse-chase using tritium-labeled leucine (Perkin-Elmer, no. NET460A001MC) as previously described (8, 27). Proteins were labeled with radioactive leucine for 36 hours (pulse period), followed by a chasing period of 28 hours. Short-lived proteins were excluded from the analysis by replacing the media after 1 hour of chasing period with fresh chasing media. For lysosomal inhibition, 100 mM of leupeptin was added to the initial medium (pulse period) and 100 mM of leupeptin and 5 mM of NH4Cl were added to the chasing medium. Aliquots of culture media were sampled after 8, 20 and 28 hours during chasing period and precipitated with 20% (v/v) trichloroacetic acid with BSA (0.5mg/ml) for a minimum of 8 hours at 4°C followed by centrifugation at 20,000g for 20 min at 4°C. Pellets were resuspended in 0.1 M NaOH / 0.1% NaDexoycholate. After the last time point, cells were scrapped and harvested in 0.1 M NaOH / 0.1% NaDexoycholate. Radioactive counts of cell lysates and secreted proteins were measured using a liquid scintillation analyzer (TriCarb 2800TR, Perkin Elmer). Percentage of secreted proteins was determined by dividing the radioactive signal obtained from the media by the total radioactive counts obtained from the cell lysate. Lysosomal proteolysis was calculated as the difference between control and inhibited conditions.
Quantification of lipids by SFC/MS/MS analysis
Neurons were harvested in cold PBS, centrifuged at 200g for 5min, and cell pellets were rapidly stored at −80⁰C until analysis. Brain hippocampal tissue from mice was homogenized in homogenization buffer according to tissue weight and samples were rapidly stored at −80⁰C until analysis. Quantification of glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph) was performed as a service provided by the Lipidomics Core facility at the Medical University of South Carolina (http://hcc.musc.edu/research/sharedresources/lipidomics/lipidomicsanalytics.htm). The samples of iPSC-derived neurons were normalized to total cellular phosphate levels (Pi) and expressed as picomole/nanomole Pi. The samples of hippocampal mouse tissue were normalized to total protein and expressed as picomole/milligram protein.
Statistical Analysis
Comparison of multiple groups was performed using analysis of variance (ANOVA) followed by Tukey’s post-hoc test; comparisons between two groups were performed using t test. p values less than 0.05 were considered significant. Statistical calculations were performed using Microsoft Excel or GraphPad Prism Software, version 5.0 (http://www.graphpad.com/scientific-software/prism/). All data shown are representative of experiments from n≥3 biologically independent samples (see figure legends). All errors bars shown in the figures are standard error of the mean (SEM).
Supplementary Material
Fig. S1. Characterization of heterozygous 84GG (c.84dupG) GBA1 mutant iPSCs, CRISPR/Cas9 corrected isogenic iPSCs and control iPSCs, and the midbrain dopaminergic neurons differentiated from them.
Fig. S2. CRISPR-Cas9 editing of heterozygous 84GG (c.84dupG) GBA1 mutant iPSCs.
Fig. S3. Cell-free in vitro enzyme activity assays for acid α-glucosidase and α-galactosidase A after treatment with S-181.
Fig. S4. Measurement of GBA1 expression by qPCR in GBA1 mutant iPSC-derived dopaminergic neurons treated with S-181 or vehicle for 10 days.
Fig. S5. Structure and in vitro enzyme activity of a nonactivating control compound.
Fig. S6. Brain weights of wild-type (Gba1+/+) and Gba1D409V/+ mice treated with a dose of S-181 (50mg/kg) or 5% dextrose (vehicle) intraperitoneally twice daily (n=5 to 8 per group).
Table S1. List of human iPSC lines used in the study including summary of clinical and genotype information for patients with PD.
Table S2. Pharmacokinetics (PK) for S-181 determined in mouse plasma and brain tissue after a single intraperitoneal dose (50mg/kg) in wild-type mice.
Table S3. Protein binding of S-181 and control compound in mouse brain homogenate.
Table S4. Protein binding of S-181 and control compound in mouse plasma.
Data File S1. Individual level data for all figures
Acknowledgments
We thank K. Merchant and Y.C. Wong for helpful discussions. We thank the Northwestern Stem Cell Core Facility for the generation of iPSC lines from GBA1 mutation carriers. We thank K. Hunt for help with the protein binding assays. Lipid analyses were performed by the Lipidomics Shared Resource, Hollings Cancer Center, and Medical University of South Carolina. Pharmacokinetics were performed by the Northwestern Developmental Therapeutics Core and Integrated Molecular Structure Education and Research Center (IMSERC) at Northwestern University. We are grateful to N. Marotta for help with quantitative PCR.
Funding: This work was supported by NIH grants no. R01NS076054 and R01NS096240 (to D.K.); WildKat, LLC; Buckeye Research LLC (to D.K.), The Michael J. Fox Foundation for Parkinson’s Research (to J.Z.), and, in part by NIH core support grant no. P30 NS081774.
Footnotes
Competing interests: D.K is Founder and Scientific Advisory Board Chair of Lysosomal Therapeutics Inc. D.K. serves on the scientific advisory boards of The Silverstein Foundation, Intellia Therapeutics, and Prevail Therapeutics and is a Venture Partner at OrbiMed. D.K., R.B.S. and J.Z. are coinventors on patent no. PCT/US2018/043703 entitled “Substituted fused pyrimidine compounds and uses thereof”. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or Supplementary Materials. S-181 and iPSC lines used in this study are available from D.K. under a materials transfer agreement (MTA) with Northwestern University.
REFERENCES
- 1.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE, Parkinson disease. Nature reviews. Disease primers 3, 17013 (2017); published online EpubMar 23 ( 10.1038/nrdp.2017.13). [DOI] [PubMed] [Google Scholar]
- 2.Surmeier DJ, Obeso JA, Halliday GM, Parkinson’s Disease Is Not Simply a Prion Disorder. The Journal of neuroscience : the official journal of the Society for Neuroscience 37, 9799–9807 (2017); published online EpubOct 11 ( 10.1523/JNEUROSCI.1787-16.2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997); published online EpubAug 28 ( 10.1038/42166). [DOI] [PubMed] [Google Scholar]
- 4.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG, Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361, 1651–1661 (2009); published online EpubOct 22 ( 10.1056/NEJMoa0901281). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D, Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 (2011); published online EpubJul 8 ( 10.1016/j.cell.2011.06.001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, Schoenen FJ, Westbroek W, Zheng W, Sullivan P, Fujiwara H, Sidhu R, Khaliq ZM, Lopez GJ, Goldstein DS, Ory DS, Marugan J, Sidransky E, A New Glucocerebrosidase Chaperone Reduces alpha-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 7441–7452 (2016); published online EpubJul 13 ( 10.1523/JNEUROSCI.0636-16.2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, Patnaik S, Sidransky E, Marugan JJ, Sue CM, Krainc D, Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 7693–7706 (2016); published online EpubJul 20 ( 10.1523/JNEUROSCI.0628-16.2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Kruger R, Surmeier DJ, Krainc D, Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261 (2017); published online EpubSep 22 ( 10.1126/science.aam9080). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen M, Krainc D, LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, (2018); published online EpubMay 7 ( 10.1073/pnas.1717590115). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L, Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551 (2011); published online EpubNov 6 ( 10.1038/nature10648). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzulli JR, Burbulla LF, Krainc D, Ischiropoulos H, Detection of Free and Protein-Bound ortho-Quinones by Near-Infrared Fluorescence. Analytical chemistry 88, 2399–2405 (2016); published online EpubFeb 16 ( 10.1021/acs.analchem.5b04420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng J, Chen L, Schwake M, Silverman RB, Krainc D, Design and Synthesis of Potent Quinazolines as Selective beta-Glucocerebrosidase Modulators. Journal of medicinal chemistry 59, 8508–8520 (2016); published online EpubSep 22 ( 10.1021/acs.jmedchem.6b00930). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng J, Chen L, Skinner OS, Ysselstein D, Remis J, Lansbury P, Skerlj R, Mrosek M, Heunisch U, Krapp S, Charrow J, Schwake M, Kelleher NL, Silverman RB, Krainc D, beta-Glucocerebrosidase Modulators Promote Dimerization of beta-Glucocerebrosidase and Reveal an Allosteric Binding Site. Journal of the American Chemical Society 140, 5914–5924 (2018); published online EpubMay 9 ( 10.1021/jacs.7b13003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH, Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Annals of neurology 72, 455–463 (2012); published online EpubSep ( 10.1002/ana.23614). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM, Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain : a journal of neurology 137, 834–848 (2014); published online EpubMar ( 10.1093/brain/awt367). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL, Cheng SH, Shihabuddin LS, CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proceedings of the National Academy of Sciences of the United States of America 108, 12101–12106 (2011); published online EpubJul 19 ( 10.1073/pnas.1108197108). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khanna R, Benjamin ER, Pellegrino L, Schilling A, Rigat BA, Soska R, Nafar H, Ranes BE, Feng J, Lun Y, Powe AC, Palling DJ, Wustman BA, Schiffmann R, Mahuran DJ, Lockhart DJ, Valenzano KJ, The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. The FEBS journal 277, 1618–1638 (2010); published online EpubApr ( 10.1111/j.1742-4658.2010.07588.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aflaki E, Stubblefield BK, Maniwang E, Lopez G, Moaven N, Goldin E, Marugan J, Patnaik S, Dutra A, Southall N, Zheng W, Tayebi N, Sidransky E, Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Science translational medicine 6, 240ra273 (2014); published online EpubJun 11 ( 10.1126/scitranslmed.3008659). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M, Chinnery PF, Morris CM, Calabresi P, Parnetti L, Beccari T, Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Molecular neurodegeneration 10, 15 (2015); published online EpubMar 27 ( 10.1186/s13024-015-0010-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, Isacson O, Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Annals of clinical and translational neurology 2, 433–438 (2015); published online EpubApr ( 10.1002/acn3.177). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, Beagan J, McLean JR, Izen SC, Perez-Torres E, Hallett PJ, Isacson O, Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiology of disease 82, 495–503 (2015); published online EpubOct ( 10.1016/j.nbd.2015.09.009). [DOI] [PubMed] [Google Scholar]
- 22.Morabito G, Giannelli SG, Ordazzo G, Bido S, Castoldi V, Indrigo M, Cabassi T, Cattaneo S, Luoni M, Cancellieri C, Sessa A, Bacigaluppi M, Taverna S, Leocani L, Lanciego JL, Broccoli V, AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Molecular therapy : the journal of the American Society of Gene Therapy 25, 2727–2742 (2017); published online EpubDec 6 ( 10.1016/j.ymthe.2017.08.004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grunewald A, Voges L, Rakovic A, Kasten M, Vandebona H, Hemmelmann C, Lohmann K, Orolicki S, Ramirez A, Schapira AH, Pramstaller PP, Sue CM, Klein C, Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PloS one 5, e12962 (2010); published online EpubSep 27 ( 10.1371/journal.pone.0012962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D, Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 5970–5976 (2011); published online EpubApr 20 ( 10.1523/JNEUROSCI.4441-10.2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D, alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proceedings of the National Academy of Sciences of the United States of America 113, 1931–1936 (2016); published online EpubFeb 16 ( 10.1073/pnas.1520335113). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maurer TS, Debartolo DB, Tess DA, Scott DO, Relationship between exposure and nonspecific binding of thirty-three central nervous system drugs in mice. Drug metabolism and disposition: the biological fate of chemicals 33, 175–181 (2005); published online EpubJan ( 10.1124/dmd.104.001222). [DOI] [PubMed] [Google Scholar]
- 27.Kaushik S, Cuervo AM, Methods to monitor chaperone-mediated autophagy. Methods in enzymology 452, 297–324 (2009) 10.1016/S0076-6879(08)03619-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of heterozygous 84GG (c.84dupG) GBA1 mutant iPSCs, CRISPR/Cas9 corrected isogenic iPSCs and control iPSCs, and the midbrain dopaminergic neurons differentiated from them.
Fig. S2. CRISPR-Cas9 editing of heterozygous 84GG (c.84dupG) GBA1 mutant iPSCs.
Fig. S3. Cell-free in vitro enzyme activity assays for acid α-glucosidase and α-galactosidase A after treatment with S-181.
Fig. S4. Measurement of GBA1 expression by qPCR in GBA1 mutant iPSC-derived dopaminergic neurons treated with S-181 or vehicle for 10 days.
Fig. S5. Structure and in vitro enzyme activity of a nonactivating control compound.
Fig. S6. Brain weights of wild-type (Gba1+/+) and Gba1D409V/+ mice treated with a dose of S-181 (50mg/kg) or 5% dextrose (vehicle) intraperitoneally twice daily (n=5 to 8 per group).
Table S1. List of human iPSC lines used in the study including summary of clinical and genotype information for patients with PD.
Table S2. Pharmacokinetics (PK) for S-181 determined in mouse plasma and brain tissue after a single intraperitoneal dose (50mg/kg) in wild-type mice.
Table S3. Protein binding of S-181 and control compound in mouse brain homogenate.
Table S4. Protein binding of S-181 and control compound in mouse plasma.
Data File S1. Individual level data for all figures