

Abstract
BACKGROUND:
Nicotine intake induces addiction through neuroplasticity of the reward circuitry, altering the activity of dopaminergic neurons of the ventral tegmental area. Prior work demonstrated that altered circuit activity can change neurotransmitter expression in the developing and adult brain. Here we investigated the effects of neonatal nicotine exposure on the dopaminergic system and nicotine consumption in adulthood.
METHODS:
Male and female mice were used for two-bottle-choice test, progressive ratio breakpoint test, immunohistochemistry, RNAscope, quantitative polymerase chain reaction, calcium imaging, and DREADD (designer receptor exclusively activated by designer drugs)-mediated chemogenic activation/inhibition experiments.
RESULTS:
Neonatal nicotine exposure potentiates drug preference in adult mice, induces alterations in calcium spike activity of midbrain neurons, and increases the number of dopamine-expressing neurons in the ventral tegmental area. Specifically, glutamatergic neurons are first primed to express transcription factor Nurr1, then acquire the dopaminergic phenotype following nicotine re-exposure in adulthood. Enhanced neuronal activity combined with Nurr1 expression is both necessary and sufficient for the nicotine-mediated neurotransmitter plasticity to occur.
CONCLUSIONS:
Our findings illuminate a new mechanism of neuroplasticity by which early nicotine exposure primes the reward system to display increased susceptibility to drug consumption in adulthood.
Keywords: Dopamine, Neurotransmitter-switching, Nicotine, Plasticity, Tyrosine hydroxylase, VTA
Tobacco is one of the most abused drugs. Its main neuroactive component, nicotine, acts on the brain through nicotinic acetylcholine receptors (nAChRs) (1). Chronic nicotine exposure alters dopaminergic (DAergic) neuron activity in the ventral tegmental area (VTA) and dopamine (DA) release in nucleus accumbens (NAc), a major reward center of the brain (2–5) that is involved in addiction (6). Maternal smoking and early postnatal exposure to nicotine have been associated with altered children’s behaviors (7) and increased propensity for drug abuse (8) in humans. In rodents, maternal nicotine exposure during gestation and lactation elicits long-term plasticity of the DA-mediated reward system, promoting drug-induced reinforcement (9–13). Several nAChR subtypes, including α4β2 and homomeric α7 configurations expressed in the VTA, are permeable to Ca2+ (14) and can enhance neuronal excitability by triggering voltage-gated Ca2+ channels (15–17). Chronic nicotine exposure upregulates voltage-gated calcium channels (18), leading to long-term amplification of intracellular Ca2+ signaling through Ca2+-induced Ca2+ release mechanisms (19). Such nicotine-induced perturbation of neuronal calcium homeostasis has been shown to elicit changes in gene transcription and neuronal activity (20–22).
Because changes in calcium signaling (23–25) and sustained circuit activation (26,27) have been shown to induce a novel form of DA plasticity referred to as a neurotransmitter (NT) switching, we hypothesized that neonatal nicotine (NN) exposure enhances drug preference in the adult through a mechanism related to DA switching. Consistent with this, recent studies have shown that the number of neurons expressing tyrosine hydroxylase (TH), the DA-synthesizing enzyme, increases in the adult mouse substantia nigra compacta in response to photoperiod (28) and in the VTA following deep brain stimulation (29), social defeat (30), and chronic exposure to methamphetamine(31). Given that the transcription factor Nurr1 (32) is involved in the acquisition (33) and maintenance (34) of the DAergic phenotype (35), and that its expression is activity dependent (36,37), we investigated whether Nurr1 and TH expression are affected by nicotine-mediated circuit activation during development.
METHODS AND MATERIALS
Mice were housed in standard conditions in accordance with the guidelines of University of California San Diego Institutional Animal Care and Use Committee. Neonatal mice were exposed to nicotine from postnatal day 2 (P2) to P16 through lactation. RNAscope (Advanced Cell Diagnostics, USA, Newark, CA) was performed following the manufacturer’s instructions. Quantitative reverse transcriptase polymerase chain reaction, calcium imaging, two-bottle-choice test, DAB staining, and fluorescent immunocytochemistry were performed as previously described (38). Brain injections were performed in a stereotaxic apparatus with a continuous flow of 1% isoflurane. Stereological counts of DAB-stained neurons were obtained with Stereologer2000 software (Stereology Resource Center, Tampa, FL). Data were analyzed with SPSS (version 24.0; IBM Corp., Armonk, NY).
Detailed methods are reported in the Supplement.
RESULTS
NN-Treated Mice Display Enhanced Drug Preference as Adults
Minipumps (delivering 2 mg/kg/day nicotine for 15 days) were implanted into wild-type dams with P2 litters (Figure 1A). This achieved a concentration of 25 ng/mL nicotine in the dam’s blood, which is within the range (5–50 ng/mL) found in the blood of smokers and nicotine replacement therapy users. High blood concentrations of nicotine and cotinine were also detectable by high-pressure liquid chromatography (Supplemental Figure S1A) in pups. Litter mortality (Supplemental Figure S1B) and pup weight (Supplemental Figure S1C) were not affected by NN exposure.
Figure 1.
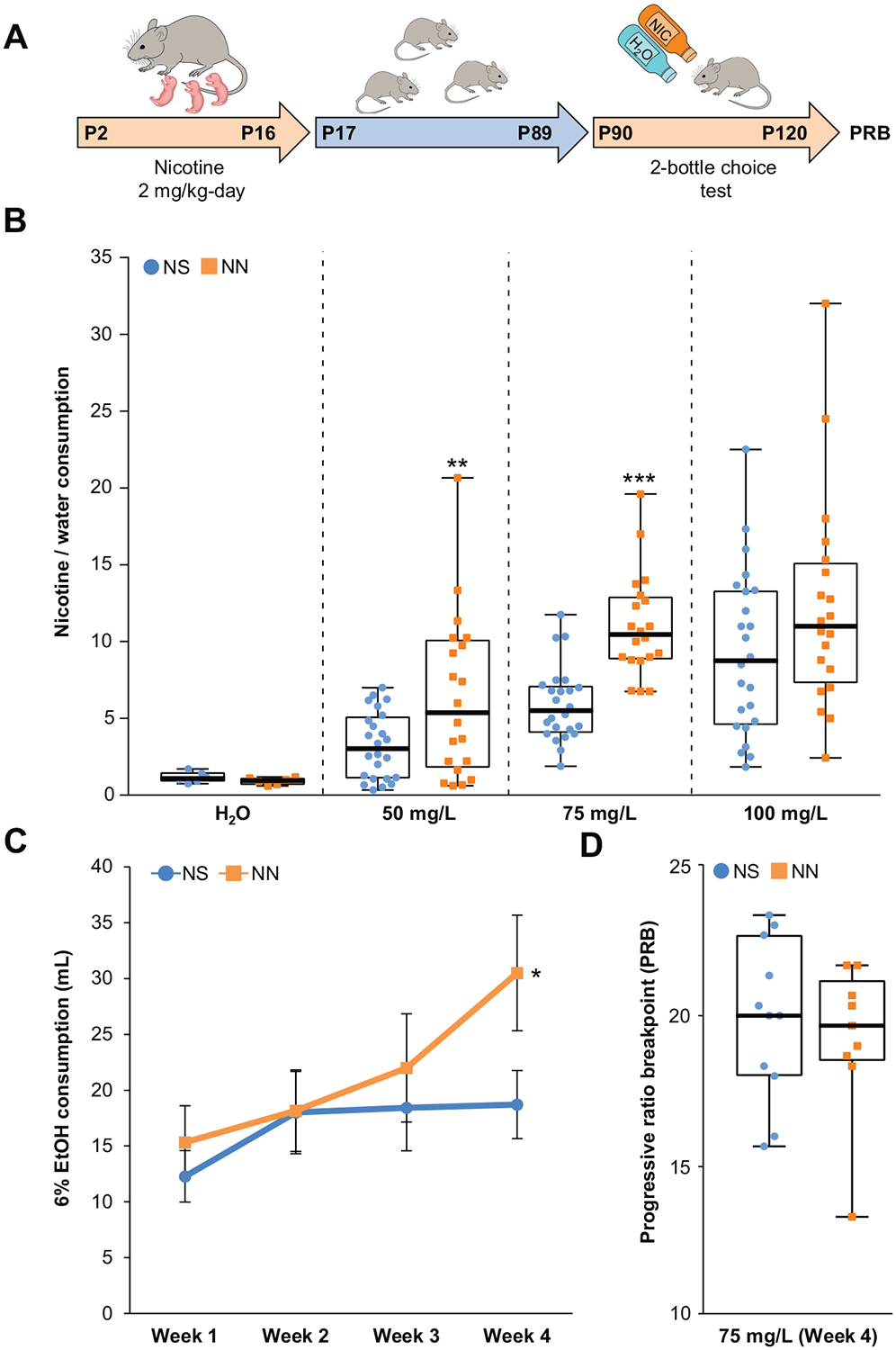
Neonatal nicotine (NN) exposure increases drug preference in adult mice. (A) Experimental timeline: mice were neonatally exposed to nicotine (or saline [NS]) from postnatal day 2 (P2) to P16 through lactation, then raised in standard conditions until adults (P17–P89). At P90, they underwent 4 weeks of two-bottle-choice test for (50, 75, or 100 mg/L) nicotine or 6% ethanol (EtOH). At P120, mice performed the progressive ratio breakpoint (PRB) test. (B) NN treatment significantly increases nicotine (50–75 mg/L) preference over water (nNS = 24, nNN = 20; 50 mg/L, t42 = 2.64, p, .01; 75 mg/L, t42 = 5.86, p, .001). Graph shows all data points (fourth week) with medians and interquartile range. **p, .01, ***p, .001. (C) Quantification of 6% ethanol consumption over 4 weeks (n = 6 per group, repeated measures analysis of variance F3,8 = 0.7, p, .05). Graph shows mean ± SE. *p, .05. (D) NN exposure does not alter motivation to drug seeking measured by PRB test to compare NS- and NN-treated mice. Graph shows all data points with medians and interquartile ranges.
To investigate whether NN treatment promotes drug preference in these mice as adults, we administered nicotine with the two-bottle-choice test to NN- and neonatal saline (NS)- treated mice at P90 (Figure 1A). NN exposure enhanced preference for nicotine over water (Figure 1B) at both 50 mg/L and 75 mg/L nicotine concentrations, without altering total fluid intake (mean ± SD: NS = 35 ± 8 mL/week, NN = 34.5 ± 3.9 mL/week; nNS = 24, nNN = 20). Weekly measurements of nicotine consumption revealed that mice displayed a significant increase in nicotine intake by the fourth week of the experiment (Supplemental Figure S1D). When a separate cohort of NN-treated mice (P90) was tested for ethanol preference with the two-bottle-choice test, NN-treated mice, compared with NS-treated control mice, displayed an increased consumption for 6% ethanol over time (Figure 1C). Increased consumption for a drug to which the animals have not been neonatally exposed and lack of preference for a bitter substance, such as quinine (t10 = 21.72, p = .12; unpaired t test) (Supplemental Figure S1E), excludes the possibility that the increased adult nicotine preference observed in NN-treated mice is due to decreased sensitivity to aversive effects or taste of nicotine.
To determine whether a general motivational component may have contributed to the enhanced drug consumption observed in adult NN-treated mice, we performed the progressive ratio breakpoint test following the two-bottle choice test (P120). We found that the breakpoint at which NN-treated mice stopped pressing the lever to obtain a food pellet reward did not differ from that of control mice (Figure 1D). This indicates that NN exposure does not affect the overall motivation to obtain a reward, but rather increases drug preference in the adult.
NN-Treated Mice Display an Increased Number of DAergic Neurons in Response to Adult Nicotine Consumption
Because the VTA is key in mediating reward-seeking behaviors (39–41) and its neurons can be induced to acquire a DAergic phenotype (23–25), we tested the hypothesis that NN treatment induces an increase in the number of TH-expressing VTA neurons. We found that NN-exposed mice (P120) showed a significant increase in the total number of TH+ neurons in response to adult nicotine (AN) consumption (Figure 2A), in both the paranigral and parabrachial pigmented subnuclei of the VTA. The increased number of DAergic cells was not present before AN exposure (P90) and was not paralleled by an increase in the total number of cells in the VTA (Supplemental Figure S2A, B). This indicates that newly TH-expressing neurons result from de novo acquisition of the DAergic phenotype in preexisting non-DAergic neurons. Such TH plasticity appears to be VTA specific, given that we observed no changes in the number of TH+ neurons in other DAergic nuclei such as the substantia nigra compacta (Figure 2B) and paraventricular nucleus (Figure 2C). To determine whether the increase in the number of TH1 neurons in the VTA is paralleled by an increase in DA content in the VTA-target region NAc, we measured DA and its metabolites in the NAc following AN exposure (Supplemental Figure S2E). Multivariate analysis revealed an overall increase in the concentration of DA and its metabolites in NAc tissue of NN+AN–treated mice, suggesting an increased DA release by VTA terminals in the NAc. Highly significant (p = .001, post hoc pairwise comparison) was the increase of 3,4-dihydroxyphenylacetic acid metabolite, a direct product of DA degradation.
Figure 2.
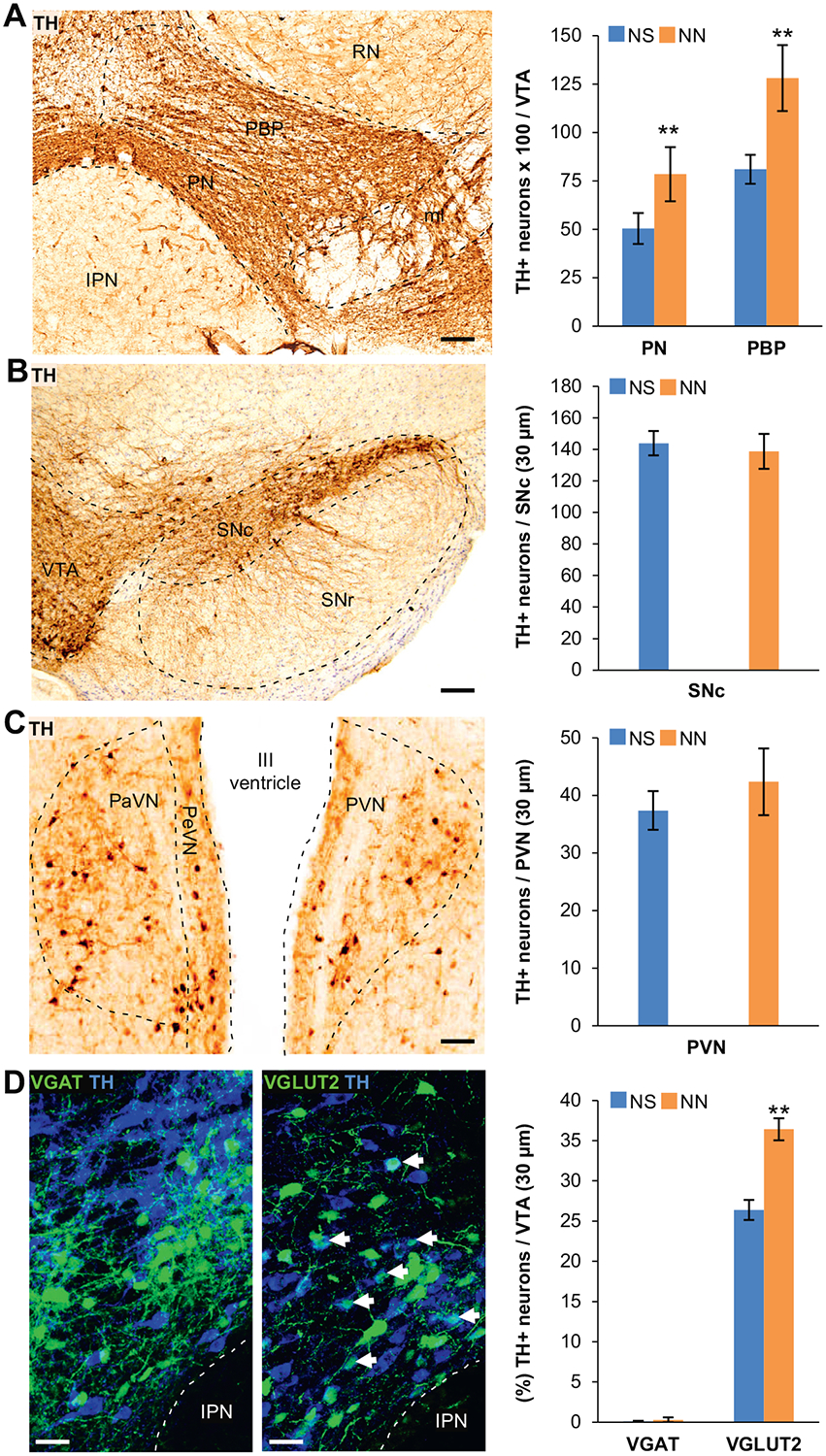
Neonatal nicotine (NN)-treated mice display an increased number of tyrosine hydroxylase (TH)1 neurons in the ventral tegmental area (VTA) after adult nicotine (AN) consumption. (A) Representative image of DAB immunoreactivity of the VTA labeled with TH marker after AN exposure. DAB stereological quantification shows that after AN exposure, the number of TH+ neurons per VTA section (30 μm) in paranigral (PN) (n = 6 mice per group, t10 = 3.71, p, .01) and parabrachial pigmented (PBP) (n = 6 mice per group, t10 = 3.39, p, .01) subnuclei of the VTA is significantly increased in NN-treated mice compared with control mice (neonatal saline [NS]). Graph shows mean 6 SE. **p, .01. Scale bar = 100 mm. (B, C) Representative images of TH-DAB immunostaining performed on the substantia nigra compacta (SNc) (B) and paraventricular nuclei (PaVN) (C) after AN exposure. Stereological quantification of DAB TH-immunoreactivity (graphs) shows that NN treatment does not affect the number of TH1 neurons in other dopaminergic regions of the brain. Graph shows mean 6 SE. Scale bars = 100 mm. (D) Confocal images showing TH and green fluorescent protein (GFP) immunofluorescence in the VTA of NN/AN-treated VGAT-Cre (left) and VGLUT2-Cre (right) mice injected with Cre-dependent green fluorescent protein–adeno-associated virus reporter. Epifluorescence quantification (%) of TH1/GFP1 neurons following AN exposure shows a significant increase in TH expression within glutamatergic neurons (arrows) in NN-treated mice (n = 6 mice per group, t10 = 25.43, p, .01). Graph shows means 6 SE. **p, .05. Scale bars = 50 mm. IPN, interpeduncular nucleus; PVN, periventricular nucleus; RN, red nucleus; SNr, substantia nigra reticulata.
To determine the NT identity of VTA non-DAergic neurons before their nicotine-induced DA phenotype acquisition, we quantified TH expression in VGAT+ (gamma-aminobutyric acidergic [GABAergic]) and VGLUT2+ (glutamatergic) neurons in NS- and NN-treated VGAT-Cre and VGLUT2-Cre mice, respectively. A floxed green fluorescent protein (GFP) viral vector was injected at P70 to visualize these cells following AN exposure (Figure 2D). We found that the total number of VGAT+ and VGLUT2+ neurons did not change across conditions (Supplemental Figure S2D), but a fraction of the VGLUT2+ VTA neurons acquired the DAergic phenotype, as shown by their enhanced rates of TH coexpression (Figure 2D, arrows). Almost no GABAergic neurons coexpressed TH, indicating that the acquisition of the DA phenotype was confined to VGLUT2+ neurons.
NN Exposure Enhances Activity of Adult VTA Neurons
Increased neuronal activity can induce NT switching (23–27). Accordingly, we investigated whether NN treatment induces increased activity levels in the VTA. NN induction of cFos was observed in brain areas such as VTA, substantia nigra, nucleus accumbens, dorsal raphe, lateral septum, cingulate, and auditory cortex at P90 (Supplemental Figure S2C). Because Nurr1 is a transcription factor that precedes and regulates the DAergic phenotype in the midbrain (32–34), we focused on Nurr1-expressing neurons in the VTA (Figure 3A). Quantification of the number of cFos1 neurons in NS- and NN-exposed mice (P90) showed that NN-exposed mice, compared with NS-treated control mice, displayed a significant increase in cFos expression within the Nurr1+ neuronal pool of the VTA (Figure 3B). The increase in cFos activity observed in NN- treated mice could be mediated by nAChRs upregulation (42). Coherently, we found that NN significantly increased the level of nAChR transcripts of α4, α7, and b2 subunits in the VTA (Figure 3C) and the number of VGLUT2+ and VGAT+ neurons expressing a4 messenger RNA (Supplemental Figure S4D).
Figure 3.
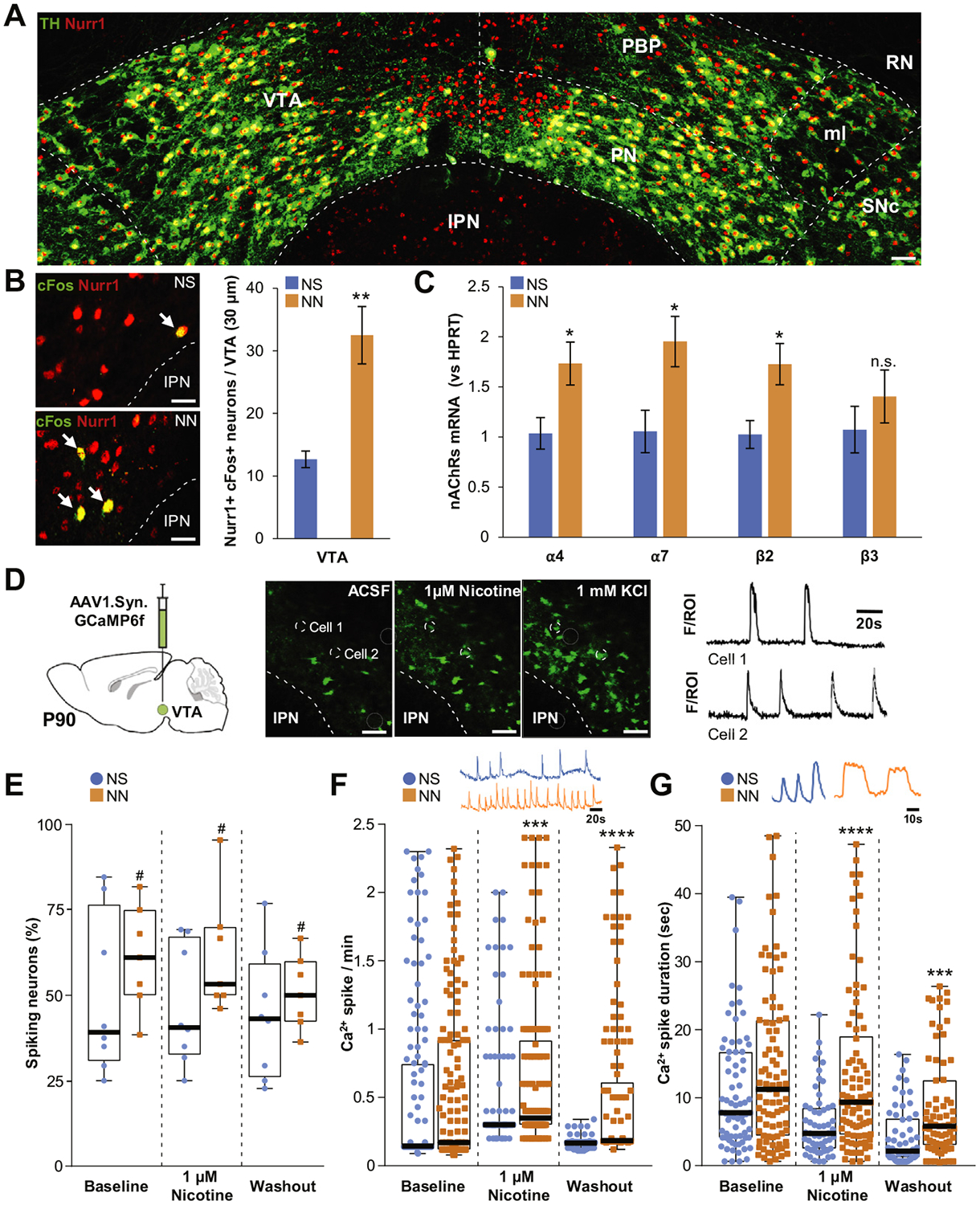
Neonatal nicotine (NN) exposure alters activity of adult ventral tegmental area (VTA) neurons. (A) Representative image of tyrosine hydroxylase (TH)/Nurr1 immunoreactivity in the VTA of adult mouse (postnatal day 90 [P90]). Scale bar = 60 μm. (B) Nurr1 and cFos epifluorescence immunoreactivity staining (left panels) in neonatal saline (NS)- exposed (top) and NN-exposed (bottom) mouse VTA. NN increases cFos immunoreactivity (arrows) in Nurr11 VTA neurons (graph, n = 6 mice per group, t10 = 3.59, p, .01). Graph shows mean ± SE. **p, .01. (C) NN exposure upregulates the messenger RNAs (mRNAs) of specific nicotinic acetylcholine receptor (nAChR) subunits in the VTA. Quantification shows a significant relative increase of a4 (t10 = 2.7, p, .05), a7 (t10 = 2.74, p, .05), and b2 transcripts (t10 = 2.94, p, .05) in NN-exposed mice (n = 6 mice per group). Graph shows mean 6 SE. *p, .05. (D) Calcium indicator GCaMP6f was expressed in VTA neurons via adeno-associated virus (AAV) injections (left). Ca21 activity (middle panels) was recorded during artificial cerebrospinal fluid (ACSF), 1 mM nicotine, or 1 mM KCl perfusion. Changes in fluorescence (F) were analyzed for individual neurons (region of interest [ROI]) to generate calcium spike traces (cell 1, cell 2, right). Dashed circles show two representative VTA neurons responding to 1 mM nicotine perfusion. Scale bar = 100 mm. (E) NN-treated mice display an overall increase of active (spiking) neurons (%) (F1,39 = 4.35, p, .05). #Main effect: pNN, .05. (F) NN-treated mice display enhanced Ca2+spike frequency during nicotine perfusion (U = 6364, p, .001, Mann-Whitney U test) that persists during washout (U = 1185, p, .0001, Mann-Whitney U test). (G) NN exposure increases average Ca21 spike duration during (U = 1462, p, .0001, Mann-Whitney U test) and after (U = 1257, p, .001, Mann-Whitney U test) nicotine perfusion. Graphs (E–G) show all data points with medians and interquartile ranges. ***p, .001, ****p, .0001. Additional statistics are presented in Supplemental Table S1. IPN, interpeduncular nucleus; ml, medial lemniscus; n.s., not significant; PBP, parabrachial pigmented area; PN, paranigral nucleus; RN, red nucleus; SNc, substantia nigra compacta.
Because of the causal role of calcium spike activity in NT switching (23–25), we next tested for NN-induced increases in calcium-mediated activity in VTA. We performed ex vivo calcium imaging on VTA slices of adult mice (P90) previously injected with an adeno-associated virus (AAV) vector carrying the calcium indicator GCaMP6f (Figure 3D, Supplemental Video). The spontaneous calcium spike activity and nicotine-evoked responses were measured in a total of 159 and 148 VTA neurons from 8 NS- and 6 NN-exposed mice, respectively. Two-way analysis of variance revealed a major effect of NN exposure on the fraction of active neurons (Figure 3E), which was not altered by acute nicotine. While baseline calcium-spiking frequency was not significantly altered in NN-treated animals, VTA neurons did show significantly greater responses to nicotine perfusion (Figure 3F, Supplemental Table S1). This enhancement persisted at washout, indicating a prolonged nicotine-mediated activation of VTA neurons. We observed a similar effect on the average duration of calcium spikes (Figure 3G, Supplemental Table S1). NN exposure did not alter spontaneous calcium spike duration, though the duration did significantly increase during nicotine stimulation, and this effect too persisted during washout. These findings indicate that following NN exposure, adult VTA neurons display a significant boost in the fraction of overall active neurons and in the nicotine-evoked calcium spiking activity. To investigate potential changes in nAChRs desensitization properties in NN-exposed mice, we measured the effect of a longer nicotine bath application (15 minutes) on calcium spiking. Again, NN-treated mice showed an increase in calcium spike frequency and longer calcium spike duration (Supplemental Figure S6F). However, a longer nicotine bath application seemed to increase the fraction of spiking neurons in control animals to a level comparable to that of the NN-exposed mice.
To further characterize the priming mechanism of enhanced calcium signaling in non-DAergic neurons of NN-exposed mice, we performed calcium imaging in both VGLUT2-Cre and VGAT-Cre mice injected with a floxed GCaMP6f vector. We found that NN treatment affects calcium spike dynamics of VTA glutamatergic and GABAergic neurons in a cell-type–specific manner. Although both primed glutamatergic and GABAergic neurons display increased baseline calcium spike frequency (Supplemental Figure S3B, E), they showed opposite changes in terms of spike duration in NN-treated mice compared with control mice (Supplemental Figure S3C, F). Moreover, in NN-exposed mice, acute nicotine application increased the number of spiking glutamatergic neurons and reduced the number of spiking GABAergic neurons (Supplemental Figure S3A, D).
Another difference was their baseline calcium spike frequency (Supplemental Figure S3B, E), with GABAergic neurons displaying higher frequency (mean ± SD: 0.19 ± 0.2 spike/min, n = 97 cells) of spontaneous calcium spikes than glutamatergic neurons (mean ± SD: 0.12 ± 0.02 spike/min, n = 104 cells). Such difference (p = .0011, Mann-Whitney U test) might contribute to the activity-dependent threshold required for TH induction within specific subpopulations of VTA neurons.
NN Exposure Induces De Novo Nurr1 Expression in Non-DAergic Neurons
To investigate the mechanism underlying the acquisition of the DAergic phenotype in non-DAergic neurons, we monitored the expression of transcription factor Nurr1 in the VTA. Adult NN-exposed mice (P90), compared with control mice (NS), showed a significantly higher number of Nurr1+ TH− neurons (Figure 4A, arrows) in both paranigral and parabrachial pigmented subnuclei of the VTA (Figure 4B), but this was not paralleled by an increase in total TH+ neurons (Figure 4C). We confirmed the transcriptional regulation of Nurr1 by quantitative RNAscope in situ hybridization (ISH) (Figure 4D). We found a 60% increase in the number of Nurr1-ISH+ neurons in NN-exposed mice (P90) (Figure 4E) and no difference in the number of TH-ISH+ neurons across groups. Quantification of Nurr1+ TH− neurons and total TH expression at P16 revealed that enhanced Nurr1 expression in non-DAergic neurons was already detectable shortly after completion of NN exposure (Supplemental Figure S4A, B) without affecting TH expression (Supplemental Figure S4C).
Figure 4.
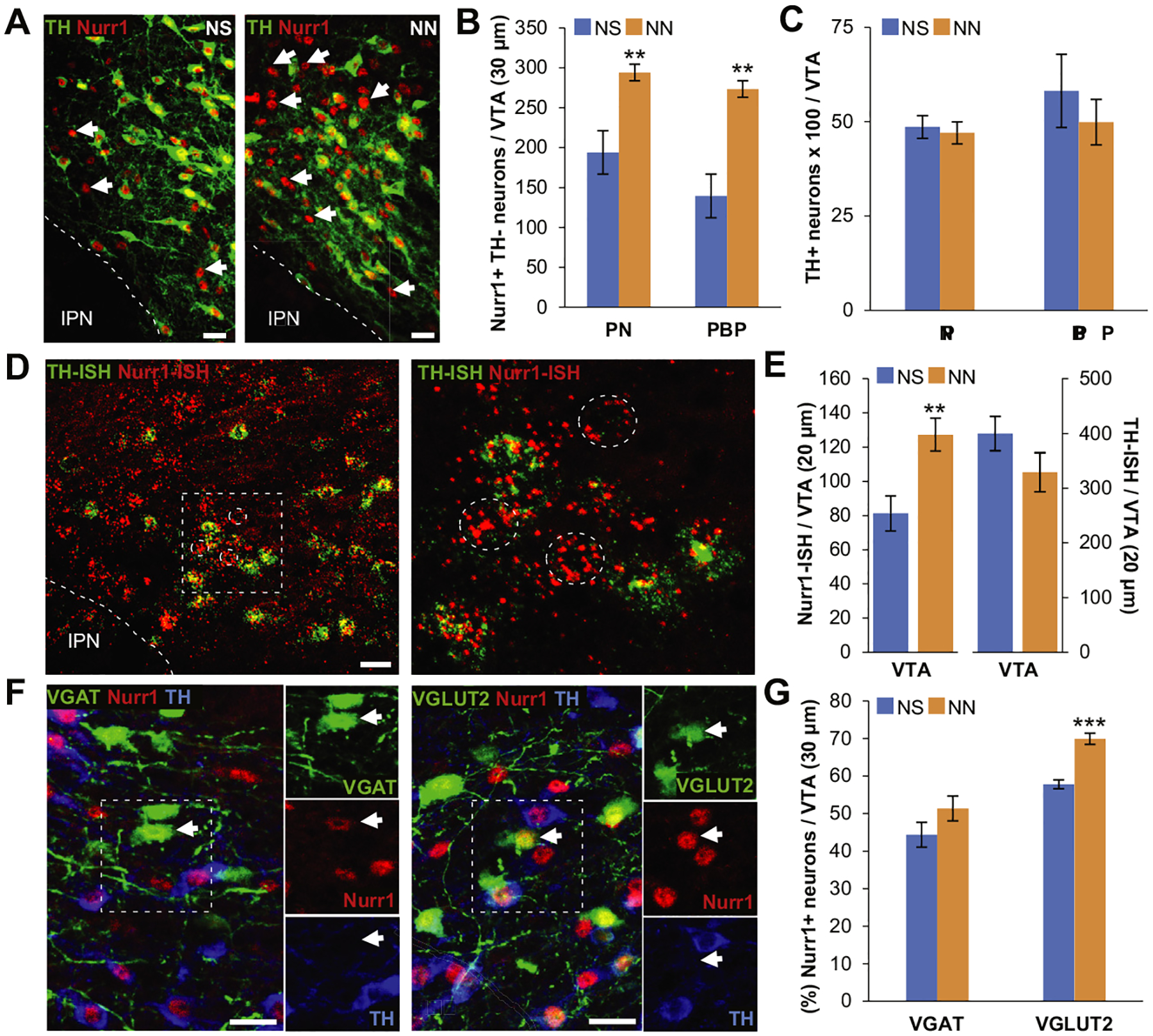
Neonatal nicotine (NN) exposure increases Nurr1 expression in nondopaminergic ventral tegmental area (VTA) neurons. (A) Nurr1 and tyrosine hydroxylase (TH) immmunoreactivity of the VTA in neonatal saline (NS)-treated (left) and NN-treated (right) adult (postnatal day 90) mice. Arrows indicate Nurr11 TH2 neurons. Scale bars = 50 mm. (B, C) Epifluorescence quantification indicates that NN exposure increases the number of Nurr11 neurons in both paranigral nucleus (PN) (n = 6 mice per group, t10 = 4.41, p, .001) and parabrachial pigmented area (PBP) (n = 6 mice per group, t10 = 4.37, p, .01) subnuclei of the VTA (B), without affecting the total number of TH1 neurons (C). Graph shows mean ± SE. **p, .01. (D) Nurr1/TH in situ hybridization (ISH) in the VTA of NN-treated adult (postnatal day 90) mice (left). Inset is shown at a higher magnification (right) to indicate nondopaminergic (TH-ISH2) Nurr1-ISH1 neurons (dashed circles). Scale bar = 50 mm. (E) Epifluorescence quantification shows that NN exposure induces an increase in the number of Nurr1-ISH1 neurons (n = 8 sections per group, t14 = 23.27, p, .01), while the number of TH-ISH1 neurons remains constant. Graph shows mean 6 SE. *p, .05. (F) Nurr1, TH, and green fluorescent protein immune reactivity in the VTA of NN-treated VGAT-Cre (left) and VGLUT2-Cre (right) mice injected with a Cre-dependent adeno-associated virus–green fluorescent protein reporter. Arrows indicate nondopaminergic (TH2) gamma-aminobutyric acidergic or glutamatergic neurons expressing Nurr1. Scale bars = 50 mm. (G) Epifluorescence quantification (%) of Nurr11 neurons coexpressing VGAT1 or VGLUT21 in the VTA. NN treatment induces a significant increase in the percentage of Nurr1-expressing glutamatergic neurons (n = 6 mice per group, t10 = 25.8, p, .001). Graph shows mean 6 SE. ***p, .001. IPN, interpeduncular nucleus.
In addition to the DAergic population, VTA contains both GABAergic and glutamatergic NAc-projecting neurons (43). To determine the NT identity of the newly expressing Nurr1 neurons in response to NN, VTA Nurr1+ neurons were colabeled with TH by immunohistochemistry (IHC) in either VGAT-Cre or VGLUT2-Cre mice injected with a reporter viral vector carrying a floxed GFP construct (Figure 4F). We found that Nurr1 is expressed in a fraction of both VGAT+ TH− and VGLUT2+ TH− neurons, but NN exposure induced a selective increase of Nurr1 expression in the glutamatergic subpopulation (Figure 4G).
To determine whether non-DAergic Nurr1+ neurons share the same target area with VTA DAergic neurons, fluorescent retrobeads were injected into the NAc and 10 days later were visualized in the cell body of VTA-to-NAc projection neurons (Supplemental Figure S5A). Retrobeads were localized in Nurr1+ TH− VTA somata (Supplemental Figure S5C, circles) in addition to expected Nurr1+ TH+ projection neurons (Supplemental Figure S5C, arrows). This indicates that a fraction of non-DAergic Nurr1-expressing neurons exhibits direct connectivity with the NAc. Retrograde bead labeling performed in either VGAT-zsGreen or VGLUT2-Cre mice injected with a GFP reporter virus revealed both VGAT+ TH− and VGLUT2+ TH− VTA-to-NAc projecting neurons (Supplemental Figure S5D, circles), with a higher Nurr1 colabeling (Supplemental Figure S5E, arrows) in the VGLUT2+ pool (Supplemental Figure S5F).
Increased Neuronal Activity Induces Nurr1 Expression in the VTA
Previous studies have reported that chronic alteration of neuronal activity is sufficient to elicit the acquisition of a different NT phenotype (23,24). We used a chemogenetic approach (Figure 5) to test whether a chronic increase of neuronal activity in selected classes of NN-primed non-DAergic neurons is sufficient to induce DAergic switching. We injected a viral vector carrying a floxed excitatory mCherry-DREADD (designer receptor exclusively activated by designer drugs) (Gq) construct in the VTA of NS- and NN-exposed VGLUT2-Cre and VGAT-Cre mice (P70). At P90, mice were treated twice a day with either 0.01 mg/kg clozapine (CLZ) or vehicle (saline) for 2 weeks. We validated the excitatory effect of CLZ-mediated DREADD activation in the VTA by ex vivo calcium imaging of mCherry-labeled neurons (Supplemental Figure S6A–C). Following CLZ treatment, mice were first tested with the open-field paradigm (Supplemental Figure S6D), then processed for IHC analysis with Nurr1 and TH markers. Both control and CLZ-treated NS- and NN-exposed VGLUT2-Cre mice showed a major increase in the time (%) spent in the center of the arena (Supplemental Figure S6D) in the second half (10–20 minutes), when challenged with a single CLZ injection. This supports an effect of chemogenetic excitation of glutamatergic VTA cells on increasing exploratory behavior.
Figure 5.
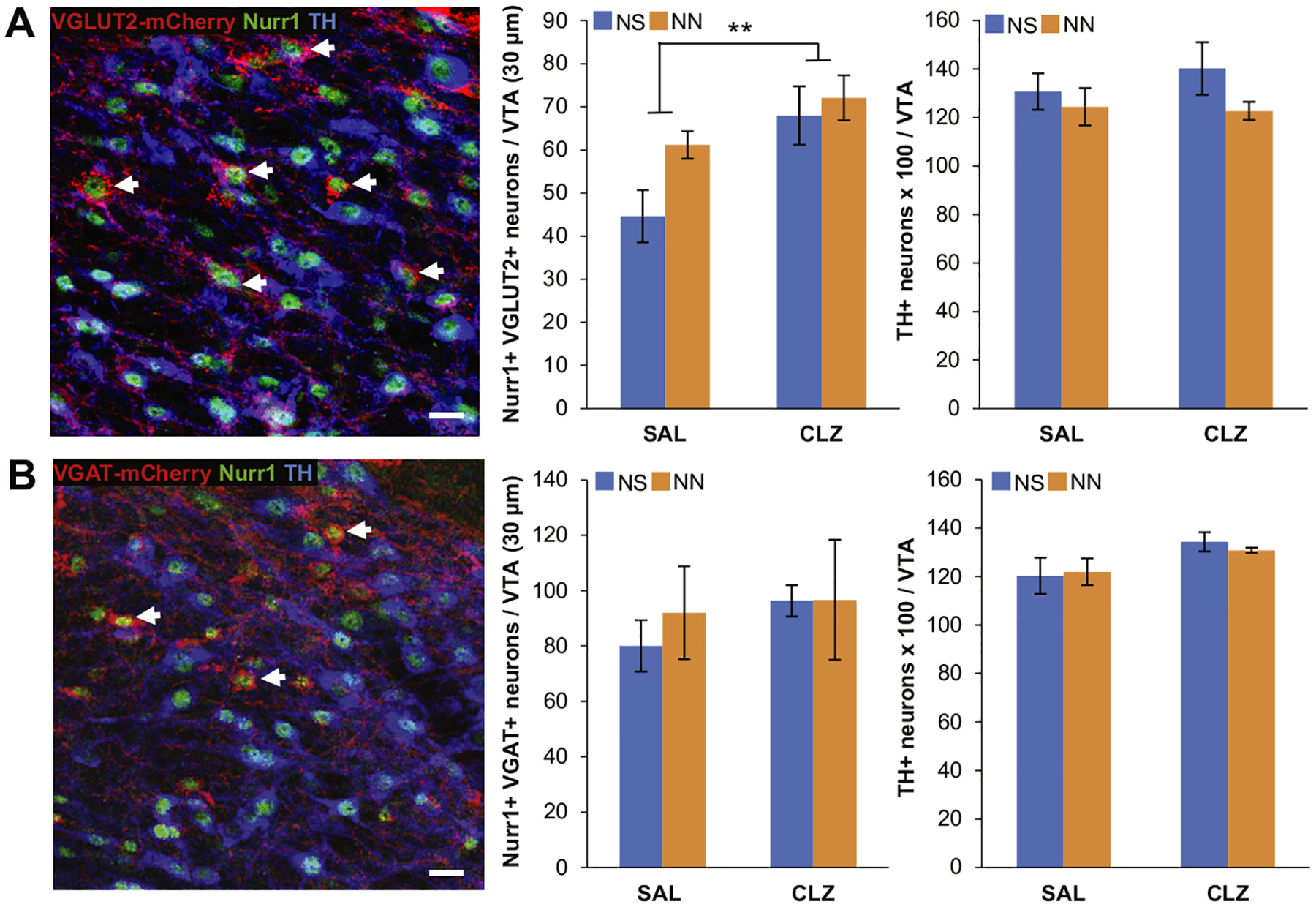
Increased neuronal activity is not sufficient to induce tyrosine hydroxylase (TH) expression in neonatal nicotine (NN)-exposed mice. (A) Colocalization of TH and Nurr1 immunoreactivity in the ventral tegmental area (VTA) of NN-exposed VGLUT2-Cre mice injected with a Cre-dependent DREADD (designer receptor exclusively activated by designer drugs) (Gq)-mCherry adeno-associated virus (arrows, left). Chronic depolarization of VGLUT2-expressing neurons induces them to acquire Nurr1 (middle) (n = 8 mice per group, F1,16 = 8.97, p, .01), without affecting the total number of TH1 neurons (right) in the VTA. Graph shows mean 6 SE. **p, .01. Scale bar = 50 mm. (B) Colocalization of TH and Nurr1 immunoreactivity in the VTA of NN-exposed VGAT-Cre mice injected with a Cre-dependent DREADD (Gq)-mCherry adeno-associated virus (arrows, left). Chronic depolarization of VGAT-expressing neurons does not affect Nurr1 (middle) or TH (right) expression in gamma-aminobutyric acidergic neurons of the VTA. Graph shows mean 6 SE. Scale bar = 50 mm. CLZ, clozapine; NS, neonatal saline; SAL, saline vehicle.
Chronic CLZ treatment had a major effect on the number of Nurr1-expressing VGLUT2-mCherry+ neurons (Figure 5A), indicating that DREADD-mediated activation of VGLUT2 neurons further induces Nurr1 expression. This suggests an activity-dependent multistep priming mechanism of glutamatergic neurons. Two-way analysis of variance showed no effects of adult DREADD-mediated activation on the total number of TH+ neurons (Figure 5A). In summary, chronic activation of VTA glutamatergic neurons boosts Nurr1 expression but is not sufficient to induce the DAergic phenotype observed in NN1AN-treated mice. DREADD-mediated activation of VGAT-expressing neurons did not elicit enhancement of either Nurr1 or TH expression within VGAT-mCherry neurons in the VTA (Figure 5B). Similarly, CLZ treatment performed in mice injected with a control virus carrying GFP did not affect the number of Nurr1- and TH- expressing neurons (Supplemental Figure S6E).
Concomitant Enhancement of Nurr1 Expression and Activity Are Sufficient Priming Steps for DA Acquisition in Response to AN Exposure
Our results suggest that NN exposure primes non-DAergic neurons of the VTA, resulting in acquisition of DAergic phenotype after AN challenge. We tested whether Nurr1 expression alone, increased neuronal activity alone, or both are sufficient priming steps for non-DAergic neurons to acquire the DAergic phenotype in response to AN consumption. We injected a Nurr1-overexpressing viral vector (AAV.Nurr1) alone or in combination with a pan-neuronal excitatory DREADD (Gq) into the VTA of adult (P70) mice that did not receive NN treatment. Control mice were injected with a viral vector expressing GFP (AAV.ctl) in lieu of AAV.Nurr1 (Figure 6A). Animals were then given the nicotine two-bottle choice test for 4 weeks while being treated daily with CLZ (0.01 mg/kg) or vehicle. Efficacy of transduction and Nurr1 overexpression in the VTA were confirmed by IHC with Nurr1 and TH markers at P90 (Figure 6B, Supplemental Figure S7A). As expected, AAV.Nurr1-injected mice displayed an increase in the number of Nurr1+ neurons in the VTA (Supplemental Figure S7A). Nurr1 overexpression alone did not affect the total number of TH+ neurons either before (P90) (Supplemental Figure S7A) or after AN exposure (P120) (Supplemental Figure S7B). This shows that ectopic Nurr1 expression alone in non-DAergic neurons is not sufficient to induce a TH phenotype even in combination with AN induction.
Figure 6.
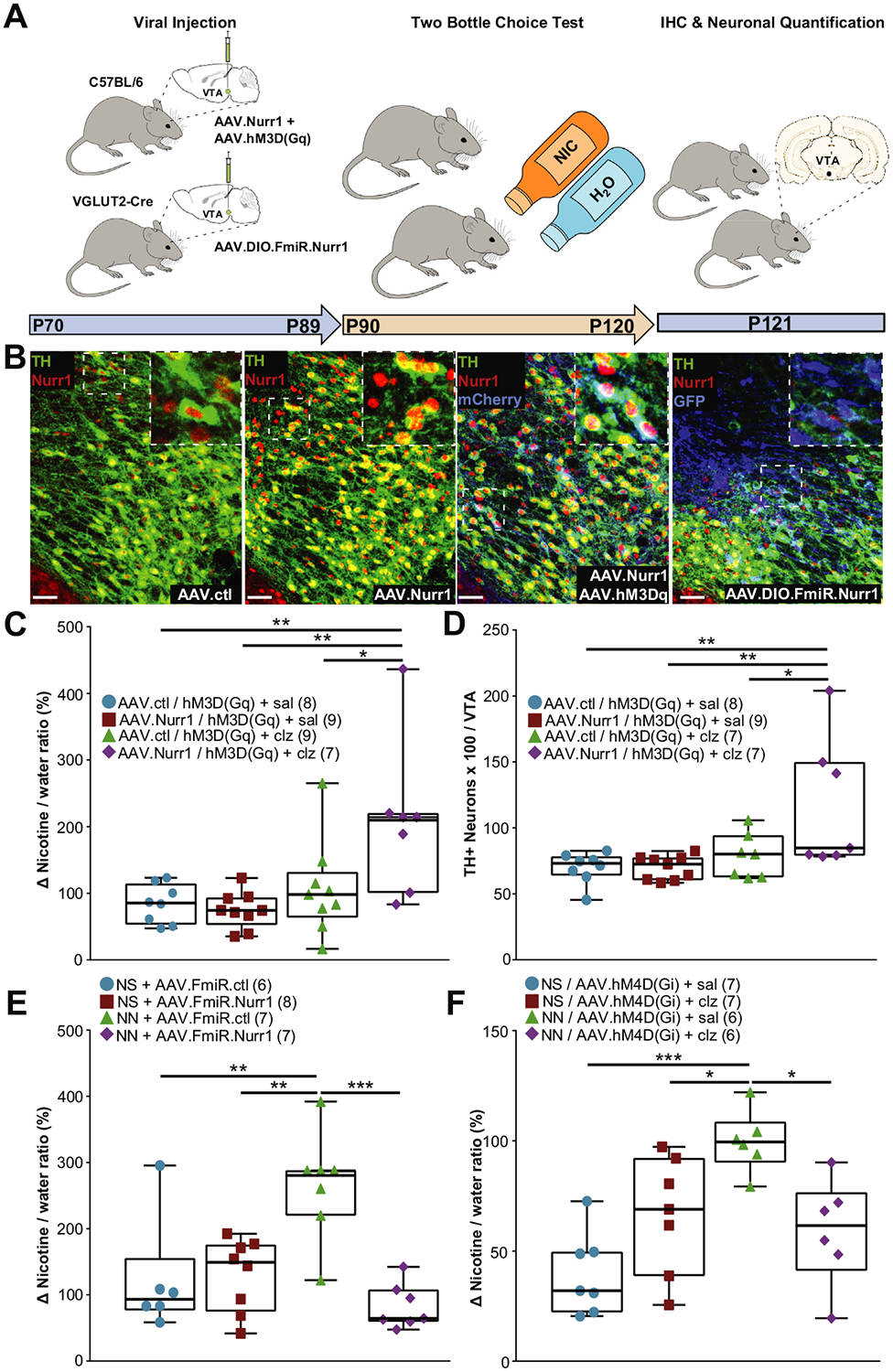
Concomitant Nurr1 overexpression and increased neuronal activity are sufficient to induce tyrosine hydroxylase (TH) expression affecting nicotine (NIC) preference. (A) Diagram of experimental design. The ventral tegmental area (VTA) of postnatal day 70 (P70) C57BL/6 mice (top, left) or neonatal nicotine (NN)-treated VGLUT2-Cre mice (bottom, left) was injected with a combination of viruses to either overexpress Nurr1 and DREADD (designer receptor exclusively activated by designer drugs) or selectively downregulate Nurr1 in glutamatergic neurons. At P90, animals underwent nicotine two-bottle choice test (middle). At P121, brains were processed for immunohistochemistry (IHC) and neuronal quantification (right). (B) Representative confocal images of the VTA showing Nurr1 and TH immunoreactivity in mice injected with AAV.ctl (adeno-associated virus control), AAV.Nurr1, AAV.Nurr11AAV.hM3Dq, or Cre-dependent AAV.Nurr1 microRNA (AAV.FmiR. - Nurr1), or a scrambled FmiR control. Scale bars = 60 mm. Insets (dashed box) are shown at higher magnification on the right top corner of each image. (C, D) Concomitant Nurr1 overexpression and DREADD-mediated depolarization in the VTA increases nicotine preference measured as the change (D) in preference between the first and last weeks of the experiment (one-way analysis of variance [ANOVA]: F32 = 6.18, p, .01; Tukey’s multiple comparison: Nurr1/clozapine [clz] vs. ctl/saline [sal], p, .01; Nurr1/clz vs. Nurr1/sal p, .01; Nurr1/clz vs. ctl/clz, p, .05) (C) and induces a significant increase in the number of 3,3- diaminobenzidine-TH1 neurons in the VTA (one-way ANOVA: F30 = 5.53, p, .01; Tukey’s multiple comparison: Nurr1/clz vs. ctl/sal, p, .01; Nurr1/clz vs. Nurr1/sal, p, .01; Nurr1/clz vs. ctl/clz, p, .05)(D). Graphs show all data points with medians and interquartile ranges. The number of animals is annotated in parenthesis for each condition. *p, .05, **p, .01. (E) Selective Nurr1 downregulation in VTA glutamatergic neurons abolishes the increase in adult nicotine preference displayed by NN-treated mice (one-way ANOVA: F28 = 8.05, p, .01; Tukey’s multiple comparison: NN/FmiR.ctl vs. NS/FmiR.ctl, p, .05; NN/FmiR.ctl vs. NS/FmiR.- Nurr1, p, .01; NN/FmiR.ctl vs. NN/FmiR.Nurr1, p, .0001). Graph shows all data points with medians and interquartile ranges. The number of animals is annotated in parenthesis for each condition. **p, .01, ***p, .0001. (F) NN-treated mice no longer display enhanced adult nicotine preference when VTA neurons are chronically hyperpolarized during the nicotine two-bottle choice test (one-way ANOVA: F25 = 8.62, p, .01; Tukey’s multiple comparison: NN/sal vs. neonatal saline [NS]/sal, p, .0001, NN/sal vs. NS/clz, p, .05; NN/sal vs. NN/clz, p, .05). Graph shows all data points with medians and interquartile ranges. The number of animals is annotated in parenthesis for each condition. *p, .05, ***p, .0001. FmiR, Nurr1 microRNA; hM3Dq, human M3 muscarinic excitatory Gq-coupled DREADD receptor.
Our data showed that neither Nurr1 overexpression alone nor enhanced VTA activity alone was sufficient to increase AN preference (Figure 6C) or TH expression (Figure 6D). Concomitant Nurr1 overexpression and DREADD-enhanced VTA activity elicited a significant increase of both AN preference and the number of TH+ neurons in the VTA (Figure 6C, D). These findings indicate that concomitant Nurr1 expression and enhanced neuronal activity are sufficient to mimic the NN-mediated priming mechanism through which VTA neurons become responsive to AN and are recruited to a DAergic phenotype.
Recruitment of VTA Neurons to the DAergic Phenotype Affects Nicotine Preference
To investigate the contribution of newly expressing DAergic neurons on enhanced nicotine preference displayed by adult NN-treated mice, we downregulated Nurr1 expression in VGLUT2+ neurons to selectively abolish their ability to acquire a TH phenotype during AN exposure. NN-treated mice were injected with a viral vector (AAV.FLEX.FmiR.GFP) carrying a floxed version of a microRNA targeting Nurr1 messenger RNA (or a scrambled control microRNA) into the VTA of NS- and NN-exposed VGLUT2-Cre mice (P70). Knockdown of Nurr1 was validated using IHC against the transduction marker GFP and Nurr1 (Figure 6B, Supplemental Figure S7C). Mice were then given the two-bottle-choice test (P90–P120) to measure AN consumption (Figure 6A). We found that Nurr1 downregulation in VGLUT2+ VTA neurons abolished the increase in AN preference (Figure 6E) as well as the acquisition of the TH phenotype (Supplemental Figure S7D) that is typically observed in NN+AN-exposed mice.
To inhibit circuit activity in the VTA during AN consumption, we injected a pan-neuronal viral vector carrying an inhibitory DREADD-mCherry (Gi) construct into the VTA of P70 NN- or NS-treated mice (Supplemental Figure S7E, arrows) and then treated them twice a day with 0.01 mg/kg CLZ (or vehicle) from P90 to P120 while testing for AN or ethanol preference. We found that silencing VTA neurons during AN consumption abolished the increase in nicotine (Figure 6F) as well as ethanol (Supplemental Figure S1F) preference and reduced the number of TH+ neurons in the VTA (Supplemental Figure S7E), indicating that increased acquisition of the TH phenotype by VTA neurons no longer occurred. The reduction in TH+ neurons in NN-treated mice indicates that NN priming made VTA neurons more responsive to both excitatory and inhibitory changes in neuronal activity.
DISCUSSION
Our results demonstrate that early NN exposure primes the reward neurocircuit through a novel form of neuroplasticity that leads to an increase in the number of VTA DAergic neurons following nicotine re-exposure in adult mice. Newly converted DAergic neurons are recruited from a non-DAergic pool that begins to express the DAergic determinant Nurr1 and display enhanced calcium spike activity and nicotine-mediated responses (Figure 7) at P16 throughout adulthood (P90). Remarkably, adult NN-primed mice display a higher preference for nicotine and other drugs of abuse (such as alcohol). During adult drug exposure, NN-primed glutamatergic neurons of the VTA are recruited to acquire a DAergic phenotype, thus increasing the total number of DAergic neurons via activity-dependent NT switching (24–27).
Figure 7.
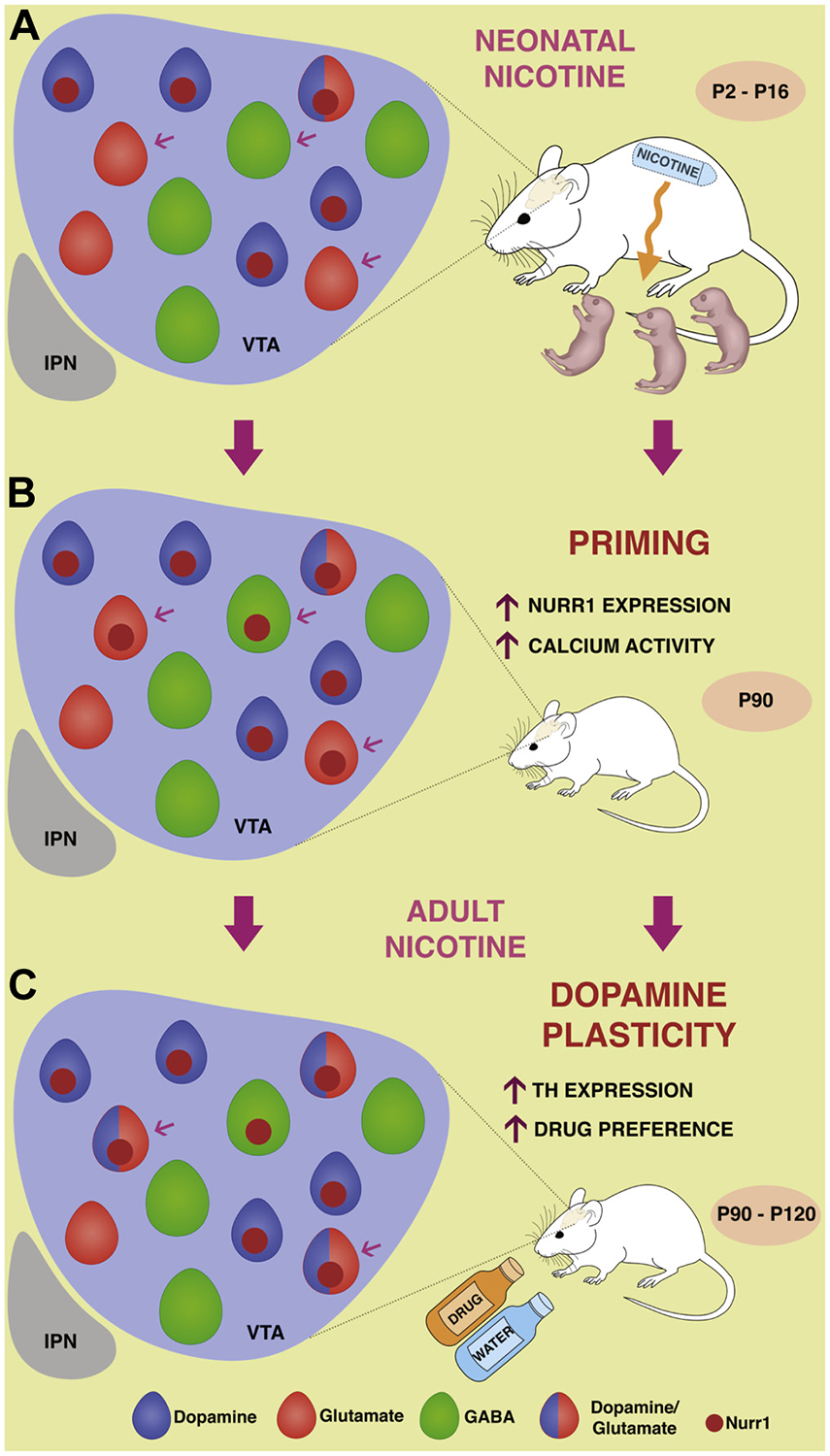
Neonatal nicotine primes ventral tegmental area (VTA) glutamatergic neurons to express tyrosine hydroxylase (TH) following nicotine exposure in adulthood. Illustration showing neonatal nicotine priming and subsequent dopamine induction affecting nicotine preference in adulthood. (A) Pups are exposed to nicotine through lactation during a critical window (postnatal days 2–16 [P2–P16]) for neurotransmitter specification. The VTA is a heterogeneous nucleus composed of different neuronal classes (dopaminergic [DAergic], gamma-aminobutyric acidergic [GABAergic], and glutamatergic). (B) Neonatal exposure to nicotine “primes” VTA neurons by inducing an increase in calcium spike activity and in the expression of DAergic transcription factor Nurr1 in non-DAergic neurons (arrows). (C) Adult exposure (P90–P120) to nicotine recruits Nurr1-expressing glutamatergic neurons to acquire the DAergic phenotype affecting drug preference. IPN, interpeduncular nucleus.
Previous work (26) has shown that prolonged changes in circuitry activation in the mature brain can induce NT plasticity affecting behavior. Similarly, we identified a neuronal population of glutamatergic Nurr1-expressing neurons of the VTA that project to the NAc and affect drug preference. The developmental priming of the “switching” neuronal pool does not implicate TH acquisition at P16 or P90 but rather induces expression of the transcription factor Nurr1. Thus, NN exposure appears to have primed a reserve pool of resident neurons to a fate (44) that they would not normally have taken on, namely the ability to express a DAergic phenotype when properly induced with AN exposure. The priming effects observed following NN exposure, which include Nurr1 over-expression, enhanced calcium signaling, and increased nAChR expression, are also developmentally regulated to finely tune DAergic phenotype (37,45,46). Calcium-mediated activity has been shown to affect Nurr1 expression (37) and membrane depolarization to promote DA neuron differentiation. This allows transcriptional activators, such as Nurr1, to access promoter regions of relevant genes associated with DA differentiation (45). DAergic transcription factors, such as Nurr1 and Pitx3, regulate the expression of specific nAChR subunits in the developing midbrain (47), therefore affecting basal responsiveness of VTA neurons to nicotine. NN-induced priming recruits these developmental mechanisms to alter nicotine preference via DA plasticity.
Nurr1 expression was also observed in VTA GABAergic neurons, but the glutamatergic population is the reserve pool undergoing DA plasticity after AN exposure. This is consistent with the established role of glutamatergic transmission on drug reward and addiction (48). Despite nAChRs being expressed in both GABA (49) and glutamate (45) neurons of the VTA, differences in subunit expression, subcellular localization, and sensitization to nicotine might explain the differential NN effect on calcium dynamics and the selective NN-induced recruitment of glutamatergic versus GABAergic neurons.
Because control mice display a cell-type–specific baseline of spontaneous calcium spike frequency, VTA GABAergic and glutamatergic neurons might be differentially prone to respond to activity manipulations. This is in agreement with previous developmental studies showing that spontaneous calcium signatures determine the signaling threshold that different cell types need to reach to be recruited to the DA phenotype (46) or other homeostatic forms of NT plasticity (23).
The functional potential of DA acquisition by VTA glutamatergic neurons is corroborated by previous studies showing that a fraction of them express TH and corelease dopamine in relevant target areas (41) and that their activity is regulated by cholinergic signaling through nAChRs (45). Recent findings showing that DA injury provokes an upregulation of a glutamatergic coidentity in substantia nigra compacta DA neurons(50) suggest that NT plasticity could go both ways in these classes of neurons. Downregulation of Nurr1 expression in VTA glutamatergic neurons of NN-exposed mice abolished both DA switching and enhancement in AN preference, indicating that this reserve pool of neurons plays a critical role in the acquired NN-induced susceptibility to drug consumption in adulthood.
Nicotine can cause prolonged increase of DA release by direct activation of nAChRs onto VTA DAergic neurons (51), long-term potentiation of excitatory inputs to brain reward areas (5), enhanced burst DA release from synaptic terminals (52), increased expression of TH messenger RNA in the VTA (53), and increased DA receptors in the NAc (54). Here we show how nicotine consumption can also modulate the number of DA-expressing neurons in the adult VTA of NN-exposed animals. Given the involvement of DA release in the reinforcing effect of nicotine and other drugs (55), the appearance of new DAergic neurons is a novel form of plasticity that may contribute to the life-long propensity toward nicotine dependence by increasing the magnitude of the DA response to a rewarding stimulus.
DA neurons of the VTA display a heightened affinity for nicotine binding in the early postnatal period (56) in rodents, which corresponds to the third trimester of human pregnancy(57). Pregnant smokers are often offered nicotine replacement therapy in the form of nicotine patches, electronic cigarettes, or mouth sprays (58). Maternal smoking is linked to increased rates of adolescent smoking (59). Behavioral alterations induced by gestational exposure to nicotine have been recapitulated in animal models. Rodents perinatally exposed to nicotine show altered locomotor activity (9), spatial learning and memory deficits (10), anxiety (11), and nicotine sensitivity (12). In addition to alterations in neuronal proliferation and differentiation (13), the effects of early exposure to nicotine on behavior have been linked to long-term effects on cholinergic and catecholaminergic NT systems. Examples include abundance of nAChRs and upregulation of DA receptors (60).
The effects of developmental NN-induced plasticity on other drug preferences may be pertinent to the increase in prevalence of substance abuse such as alcohol abuse (61) observed in human subjects gestationally exposed to nicotine.
Supplementary Material
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by the National Institute of Drug Addiction (Grant No. R21-DA047455 [to DD]), the Kavli Institute for Brain and Mind (Grant No. 2012-008 [to DD]), and the Tobacco-Related Disease Research Program (Grant No. 27IR-0020 [to DD]). Construction and evaluation of Nurr1 vectors were supported by the National Institute of Neurological Disorders and Stroke (Grant No. 5R21NS098079-02 [to FPM]).
DD and BR planned the project. BR, DD, and AFL designed and carried out the experiments and performed data analysis. IMS, FPM, and TSH provided experimental tools. BR and DD wrote the manuscript. TSH, DKB, and FPM reviewed the manuscript.
We thank N. Prakash for critical comments on the manuscript and G. Lippi, S. Barnes, and R. Pritchard for technical support.
Footnotes
All authors report no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2019.04.019.
REFERENCES
- 1.Dani JA (2001): Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry 49:166–174. [DOI] [PubMed] [Google Scholar]
- 2.Heath CJ, Picciotto MR (2009): Nicotine-induced plasticity during development: Modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacology 56(suppl 1):254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luscher C (2013): Drug-evoked synaptic plasticity causing addictive behavior. J Neurosci 33:17641–17646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ikemoto S, Qin M, Liu ZH (2006): Primary reinforcing effects of nicotine are triggered from multiple regions both inside and outside the ventral tegmental area. J Neurosci 26:723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansvelder HD, McGehee DS (2000): Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349–357. [DOI] [PubMed] [Google Scholar]
- 6.Laviolette SR, Van der Kooy D (2004): The neurobiology of nicotine addiction: Bridging the gap from molecules to behaviour. Nat Rev Neurosci 5:55–65. [DOI] [PubMed] [Google Scholar]
- 7.Weitzman M, Gortmaker S, Sobol A (1992): Maternal smoking and behavior problems of children. Pediatrics 90:342–349. [PubMed] [Google Scholar]
- 8.Porath AJ, Fried PA (2005): Effects of prenatal cigarette and marijuana exposure on drug use among offspring. Neurotoxicol Teratol 27:267–277. [DOI] [PubMed] [Google Scholar]
- 9.Nordberg A, Zhang XA, Fredriksson A, Eriksson P (1991): Neonatal nicotine exposure induces permanent changes in brain nicotinic receptors and behaviour in adult mice. Brain Res Dev Brain Res 63:201–207. [DOI] [PubMed] [Google Scholar]
- 10.Eppolito AK, Smith RF (2006): Long-term behavioral and developmental consequences of pre-and perinatal nicotine. Pharmacol Biochem Behav 85:835–841. [DOI] [PubMed] [Google Scholar]
- 11.Eppolito AK, Bachus SE, McDonald CG, Meador-Woodruff JH, Smith RF (2010): Late emerging effects of prenatal and early postnatal nicotine exposure on the cholinergic system and anxiety-like behavior. Neurotoxicol Teratol 32:336–345. [DOI] [PubMed] [Google Scholar]
- 12.Levin ED, Lawrence S, Petro A, Horton K, Seidler FJ, Slotkin TA (2006): Increased nicotine self-administration following prenatal exposure in female rats. Pharmacol Biochem Behav 85:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst M, Moolchan ET, Robinson ML (2001): Behavioral and neural consequences of prenatal exposure to nicotine. J Am Acad Child Adolesc Psychiatry 40:630–641. [DOI] [PubMed] [Google Scholar]
- 14.Shen JX, Yakel JL (2009): Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol Sin 30:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, et al. (2009): Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol 78:703–711. [DOI] [PubMed] [Google Scholar]
- 16.Tredway TL, Guo JZ, Chiappinelli VA (1999): N-type voltage-dependent calcium channels mediate the nicotinic enhancement of GABA release in chick brain. J Neurophysiol 81:447–454. [DOI] [PubMed] [Google Scholar]
- 17.Dani JA, Bertrand D (2007): Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 47:699–729. [DOI] [PubMed] [Google Scholar]
- 18.Katsura M, Shibasaki M, Kurokawa K, Tsujimura A, Ohkuma S (2007): Up-regulation of L-type high voltage-gated calcium channel subunits by sustained exposure to 1,4-and 1,5-benzodiazepines in cerebrocortical neurons. J Neurochem 103:2518–2528. [DOI] [PubMed] [Google Scholar]
- 19.Ziviani E, Lippi G, Bano D, Munarriz E, Guiducci S, Zoli M, et al. (2011): Ryanodine receptor-2 upregulation and nicotine-mediated plasticity. EMBO J 30:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang KT, Berg DK (2001): Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron 32:855–865. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Gutala R, Hwang YY, Kim JM, Konu O, Ma JZ, Li MD (2008): Strain-and region-specific gene expression profiles in mouse brain in response to chronic nicotine treatment. Genes Brain Behav 7:78–87. [DOI] [PubMed] [Google Scholar]
- 22.West AE, Griffith EC, Greenberg ME (2002): Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3:921–931. [DOI] [PubMed] [Google Scholar]
- 23.Borodinsky LN, Root CM, Cronin JA, Sann SB, Gu X, Spitzer NC (2004): Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 429:523–530. [DOI] [PubMed] [Google Scholar]
- 24.Dulcis D, Spitzer NC (2012): Reserve pool neuron transmitter respe-cification: Novel neuroplasticity. Dev Neurobiol 72:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dulcis D, Spitzer NC (2008): Illumination controls differentiation of dopamine neurons regulating behaviour. Nature 456:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dulcis D, Jamshidi P, Leutgeb S, Spitzer NC (2013): Neurotransmitter switching in the adult brain regulates behavior. Science 340:449–453. [DOI] [PubMed] [Google Scholar]
- 27.Dulcis D, Lippi G, Stark CJ, Do LH, Berg DK, Spitzer NC (2017): Neurotransmitter switching regulated by miRNAs controls changes in social preference. Neuron 95:1319–1333.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aumann TD (2016): Environment-and activity-dependent dopamine neurotransmitter plasticity in the adult substantia nigra. J Chem Neuroanat 73:21–32. [DOI] [PubMed] [Google Scholar]
- 29.Dela Cruz JAD, Hescham S, Adriaanse B, Campos FL, Steinbusch HWM, Rutten BPF, et al. (2015): Increased number of TH-immunoreactive cells in the ventral tegmental area after deep brain stimulation of the anterior nucleus of the thalamus. Brain Struct Funct 220:3061–3066. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg GD, Steinman MQ, Doig IE, Hao R, Trainor BC (2015): Effects of social defeat on dopamine neurons in the ventral tegmental area in male and female California mice. Eur J Neurosci 42:3081–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kesby JP, Najera JA, Romoli B, Fang Y, Basova L, Birmingham A, et al. (2017): HIV-1 TAT protein enhances sensitization to methamphetamine by affecting dopaminergic function. Brain Behav Immun 65:210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Y (2012): The function and mechanisms of Nurr1 action in midbrain dopaminergic neurons, from development and maintenance to survival. Int Rev Neurobiol 102:1–22. [DOI] [PubMed] [Google Scholar]
- 33.Smits SM, Ponnio T, Conneely OM, Burbach JPH, Smidt MP (2003): Involvement of Nurr1 in specifying the neurotransmitter identity of ventral midbrain dopaminergic neurons. Eur J Neurosci 18:1731–1738. [DOI] [PubMed] [Google Scholar]
- 34.Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, et al. (2009): Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci 9:15923–15932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paliga D, Raudzus F, Leppla SH, Heumann R (2019): Lethal factor domain-mediated delivery of Nurr1 transcription factor enhances tyrosine hydroxylase activity and protects from neurotoxin-induced degeneration of dopaminergic cells. Mol Neurobiol 56:3393–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu N, Baker H (1999): Activity-dependent Nurr1 and NGFI-B gene expression in adult mouse olfactory bulb. Neuroreport 10:747–751. [DOI] [PubMed] [Google Scholar]
- 37.Tokuoka H, Hatanaka T, Metzger D, Ichinose H (2014): Nurr1 expression is regulated by voltage-dependent calcium channels and calcineurin in cultured hippocampal neurons. Neurosci Lett 559:50–55. [DOI] [PubMed] [Google Scholar]
- 38.Lippi G, Fernandes CC, Ewell LA, John D, Romoli B, Curia G, et al. (2016): MicroRNA-101 regulates multiple developmental programs to constrain excitation in adult neural networks. Neuron 92:1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, et al. (2012): Input-specific control of reward and aversion in the ventral tegmental area. Nature 491:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tritsch NX, Ding JB, Sabatini BL (2012): Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 490:262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, et al. (2010): Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron 65:643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narayanan U, Birru S, Vaglenova J, Breese CR (2002): Nicotinic receptor expression following nicotine exposure via maternal milk. Neuroreport 13:961–963. [DOI] [PubMed] [Google Scholar]
- 43.Taylor SR, Badurek S, Dileone RJ, Nashmi R, Minichiello L, Picciotto MR (2014): GABAergic and glutamatergic efferents of the mouse ventral tegmental area. J Comp Neurol 522:3308–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson BL, Levitt P, Stanwood GD (2009): Prenatal exposure to drugs: Effects on brain development and implications for policy and education. Nat Rev Neurosci 10:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan Y, Peng C, Arvin MC, Jin XT, Kim VJ, Ramsey MD, et al. (2018): Nicotinic cholinergic receptors in VTA glutamate neurons modulate excitatory transmission. Cell Rep 23:2236–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velazquez-Ulloa NA, Spitzer NC, Dulcis D (2011): Contexts for dopamine specification by calcium spike activity in the CNS. J Neurosci 31:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakrabarty K, Von Oerthel L, Hellemons A, Clotman F, Espana A, Groot Koerkamp M, et al. (2012): Genome wide expression profiling of the mesodiencephalic region identifies novel factors involved in early and late dopaminergic development. Biol Open 1:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’Souza MS (2015): Glutamatergic transmission in drug reward: Implications for drug addiction. Front Neurosci 9:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Semenova S, D’Souza MS, Stoker AK, Markou A (2014): Involvement of glutamatergic and GABAergic systems in nicotine dependence: Implications for novel pharmacotherapies for smoking cessation. Neuropharmacology 76:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinkellner T, Zell V, Farino ZJ, Sonders MS, Villeneuve M, Freyberg RJ, et al. (2018): Role for VGLUT2 in selective vulnerability of midbrain dopamine neurons. J Clin Invest 128:774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nisell M, Nomikos GG, Svensson TH (1994): Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse 16:36–44. [DOI] [PubMed] [Google Scholar]
- 52.Wang L, Shang S, Kang X, Teng S, Zhu F, Liu B, et al. (2014): Modulation of dopamine release in the striatum by physiologically relevant levels of nicotine. Nat Commun 5:3925. [DOI] [PubMed] [Google Scholar]
- 53.Serova L, Sabban EL (2002): Involvement of alpha 7 nicotinic acetylcholine receptors in gene expression of dopamine biosynthetic enzymes in rat brain. J Pharmacol Exp Ther 303:896–903. [DOI] [PubMed] [Google Scholar]
- 54.Pinheiro CR, Oliveira E, Manhães AC, Fraga MC, Claudio-Neto S, Younes-Rapozo V, et al. (2015): Exposure to nicotine increases dopamine receptor content in the mesocorticolimbic pathway of rat dams and offspring during lactation. Pharmacol Biochem Behav 136:87–101. [DOI] [PubMed] [Google Scholar]
- 55.Stolerman IP, Jarvis MJ (1995): The scientific case that nicotine is addictive. Psychopharmacology (Berl) 117:2–10. [DOI] [PubMed] [Google Scholar]
- 56.Azam L, Chen Y, Leslie FM (2007): Developmental regulation of nicotinic acetylcholine receptors within midbrain dopamine neurons. Neuroscience 144:1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobbing J, Sands J (1979): Comparative aspects of the brain growth spurt. Early Hum Dev 3:79–83. [DOI] [PubMed] [Google Scholar]
- 58.Kapaya M, Tong V, Ding H (2015): Nicotine replacement therapy and other interventions for pregnant smokers: Pregnancy Risk Assessment Monitoring System, 2009–2010. Prev Med 78:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DiFranza JR, Aligne CA, Weitzman M (2004): Prenatal and postnatal environmental tobacco smoke exposure and children’s health. Pediatrics 113(suppl 4):1007–1015. [PubMed] [Google Scholar]
- 60.Dwyer JB, McQuown SC, Leslie FM (2009): The dynamic effects of nicotine on the developing brain. Pharmacol Ther 122:125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fergusson DM, Woodward LJ, Horwood LJ (1998): Maternal smoking during pregnancy and psychiatric adjustment in late adolescence. Arch Gen Psychiatry 55:721–727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.