

Abstract
Generation of DNA double strand breaks has historically been taught as the mechanism through which radiotherapy (RT) kills cancer cells. Recently, it was recognized that radiation-induced cytosolic DNA release and activation of the cGAS/STING pathway, with ensuing induction of interferon secretion and immune activation, are important mechanisms for RT-mediated anti-tumor efficacy. Here we demonstrate that radiation-induced activation of endogenous retroviruses (ERVs) also plays a major role in regulating the anti-tumor immune response during RT. Radiation-induced ERV-associated dsRNA transcription and subsequent activation of the innate antiviral MDA5/MAVS/TBK1 pathway led to downstream transcription of interferon stimulated genes. Additionally, genetic knockout of KAP1, a chromatin modulator responsible for suppressing ERV transcription sites within the genome, enhanced the effect of radiation-induced anti-tumor response in vivo in two different tumor models. This anti-tumor response was immune-mediated and required an intact host immune system. Our findings indicate that radiation-induced ERV-dsRNA expression and subsequent immune response plays critical roles for clinical radiotherapy, and manipulation of epigenetic regulators and the dsRNA-sensing innate immunity pathway could be promising targets to enhance the efficacy of RT and cancer immunotherapy.
Introduction
Radiotherapy (RT) plays broad roles in cancer treatment and management, with over 50% of cancer patients undergoing RT as a treatment modality(1). Historically, radiation has been utilized to induce local tumor control in patients. According to established radiobiology paradigm, exposure to ionizing radiation results in DNA single- and double-strand breaks, which are mainly responsible for radiation-induced tumor cell death and subsequent tumor control(2–4). Consistent with this paradigm, there is ample evidence for radiation-induced DNA strand breaks causing cell cycle arrest as well as tumor cell death via apoptosis, necrosis, or autophagy(5–8).
Recently it was shown that the host immune response is a key component of the overall tumor response to RT. A role for immune effector cells in mediating tumor response to RT was initially discovered in the late 1970s when it was demonstrated that the efficacy of RT was significantly attenuated in immunocompromised mice(9, 10). More recently, it was demonstrated that radiation could significantly alter the tumor immune microenvironment, and much of the efficacy of RT is dependent on the subsequent immune response(11–13). One of the primary mechanisms by which radiation induces an immune response is by activating the cGAS-STING pathway(14–16). Cytosolic double-stranded DNA (dsDNA) as a result of radiation-induced DNA damage activates cyclic GMP-AMP synthase (cGAS), whose product cyclic GMP-AMP activates the Stimulator of Interferon Genes (STING) pathway and its downstream type I interferon genes(17). Type I interferon activation stimulates anti-tumor T cell-mediated immunity and inhibits tumor growth(18, 19). Furthermore, mammalian cells appeared to possess mechanisms to down-regulate radiation-induced activation of the cGAS/STING pathway(16, 20). Because of the new insights into the molecular mechanisms of radiation-induced immune signaling and robust preclinical studies have demonstrated that RT can induce a potent immune-mediated anti-tumor response, there are quickly growing numbers of clinical trials being initiated focusing on testing combination of RT and immunotherapy(21–23).
While most radiation-induced immune signaling studies have focused on DNA damage and the cGAS/STING dsDNA sensing pathway, it was recently discovered that an interferon response could also be induced through activation of endogenous retroviruses (ERVs)(24). ERVs are ancient deactivated retroviral elements that account for almost 8% of the human genome(25). Under normal circumstances, they are mostly dormant and kept silent by heterochromatin maintenance factors such as DNA methyltransferases and histone methyltransferases, including DNMT1, KAP1/TRIM28, and SETDB1(26, 27). ERVs are traditionally considered to be carcinogenic due to retrotransposon recombination generating genomic mutations, but evidence is accumulating that ERVs may also be a key factor in some forms of cancer therapy(28, 29). Recently, it was shown that DNA methyltransferase inhibitor (DNMTi) anti-cancer agents such as azacytidine or decitabine could induce a type I interferon response through activation of ERV transcription, which led to cytosolic presence of double-stranded RNAs (dsRNA)(30). Cytosolic dsRNA was sensed by the pattern recognition receptor (PRR) MDA5, which subsequently activated the MDA5/MAVS/TBK1 pathway, separate from the cGAS/STING pathway, to promote type I interferon activation and subsequent immune activation and tumor control(31). Other studies involving histone deacetylase inhibitors or RNA-induced silencing complex inhibition also showed a similar effect of ERV accumulation and subsequent interferon activation(32–34).
Though it has been shown previously that radiation can also induce upregulation of ERV transcripts(35), to our knowledge it has not yet been demonstrated if radiation-induced ERVs stimulate subsequent interferon activation, or if radiation-induced ERV activation has any relevance in radiation induced anti-tumor immune response. In this study, we evaluated if ionizing radiation exposure could induce increased activation of ERVs, as well as if manipulating KRAB-associated protein 1 (KAP1/TRIM28) could influence this process. Furthermore, we determined if ERV activation contributes to a radiation-induced type I interferon response, as well as whether the dsRNA-induced type I interferon response was relevant for RT in immunocompetent murine mouse models. Our work demonstrated that KAP1 is a potential therapeutic target to enhance the radiation-induced anti-tumor immune response.
Materials and Methods
Cell Culture and Radiation Treatment
HEK 293T, A549, B16F10, and 4T1 cells were obtained from the Cell Culture Facility at Duke University and routinely cultured at 37°C and 5% CO2 in high-glucose Dulbecco’s modified Eagle medium (Sigma-Aldrich LLC, St. Louis, MO), supplemented with 10% heat-inactivated fetal bovine serum (Corning Inc, Corning, NY) and 1x penicillin/streptomycin (Gibco, Thermo Fisher Scientific, MA). Cells were irradiated in an XRAD 320 irradiator (Precision, North Branford, CT) with a single 8Gy dose of X-ray at a dose rate of 2.31Gy/min.
CRISPR/Cas9 Knockout
CRISPR/Cas9 deletions were carried out using a protocol described previously(36). LentiCRISPR v2 was a gift from Dr. Feng Zhang (Addgene plasmid #52961; http://n2t.net/addgene:52961; RRID:Addgene_52961)(36), and sgRNA sequences were designed using CHOPCHOP (https://chopchop.cbu.uib.no/)(37) (Table 1) and synthesized by IDT (Coralville, IA). Virus generation was performed in HEK 293T cells using psPAX2 and pMD2.G plasmids provided by Didier Trono (Addgene plasmid #12259; http://n2t.net/addgene:12259; RRID:Addgene_12259 and Addgene plasmid12260; http://n2t.net/addgene:12260; RRID:Addgene_12260). Single clones were selected by puromycin for two weeks after infection and subjected to western blotting for detection of pure knockouts.
Table 1:
RNA primers used for CRISPR/Cas9 and qPCR
| Gene Names | Species | Usage | Forward sequence | Reverse sequence (if applicable) |
|---|---|---|---|---|
| TRIM28/KAP1 | Human | sgRNA | GTTCGCATCCTGGGCGTCGG | |
| TRIM28/KAP1 | Human | sgRNA | AATTATTTCATGCGTGATAG | |
| TRIM28/KAP1 | Mouse | sgRNA | CGCCGCAGCGAATAATTCGG | |
| TRIM28/KAP1 | Mouse | sgRNA | TATGCGTGATAGTGGCAGTA | |
| MAVS | Mouse | sgRNA | GCCACCAGACATCCTCGCGA | |
| MAVS | Mouse | sgRNA | GAGGACAAACCTCTTGTCTG | |
| GAPDH | Human | qPCR | AGGGCTGCTTTTAACTCTGGT | CCCCACTTGATTTTGGAGGGA |
| MER21C | Human | qPCR | GGAGCTTCCTGATTGGCAGA | ATGTAGGGTGGCAAGCACTG |
| MER57B1 | Human | qPCR | CCTCCTGAGCCAGAGTAGGT | ACCAGTCTGGCTGTTTCTGT |
| MLT1C49 | Human | qPCR | TATTGCCGTACTGTGGGCTG | TGGAACAGAGCCCTTCCTTG |
| ACTB | Mouse | qPCR | GAAATCGTGCGTGACATCAAA | TGTAGTTTCATGGATGCCACA |
| IFIT1 | Mouse | qPCR | CAAGGCAGGTTTCTGAGGAG | GACCTGGTCACCATCAGCAT |
| CXCL10 | Mouse | qPCR | GCCGTCATTTTCTGCCTCA | CGTCCTTGCGAGAGGGATC |
| ISG15 | Mouse | qPCR | CTAGAGCTAGAGCCTGCAG | AGTTAGTCACGGACACCAG |
| CCL5 | Mouse | qPCR | CAAGTGCTCCAATCTTGCAGTC | TTCTCTGGGTTGGCACACAC |
Immunofluorescence
Cells were cultured in 35mm glass bottom plates (MatTek, Ashland, MA) prior to irradiation and staining. Cells were fixed in 4% PFA, followed by membrane permeabilization with 0.1% Triton X-100 (Sigma-Aldrich LLC, St. Louis, MO). After blocking in 1% BSA and 22.52mg/mL glycine in 1X phosphate buffered saline with Tween-20 (PBST), cells were incubated overnight at 4°C in primary J2 antibody in 1% BSA in PBST. After 3 washes in PBS, cells were incubated in the dark for 1 hour with Alexa Fluor secondary antibodies (Invitrogen, Carlsbad, CA), followed by 3 more washes in PBS. Cellular fluorescence was imaged on a TCS SP5 inverted confocal microscope (Leica, Wetzlar, Germany) and quantified using ImageJ(38). J2 antibody was purchased from Scicons (Szirák, Hungary).
Western Blots
Cells were lysed in RIPA buffer with protease inhibitor cocktail (Sigma-Aldrich LLC, St. Louis, MO). Equal amounts of protein were loaded onto SDS/PAGE gels using a constant voltage of 120V and transferred onto 0.2μm PVDF membranes using a constant voltage of 70V for 2 hours. Membranes were blocked in 5% milk in tris-buffered saline with Tween 20 (TBST) (Sigma-Aldrich LLC, St. Louis, MO), and incubated with primary antibody overnight at 4°C. After 3 washes with 5% milk in TBST, membranes were incubated in HRP-conjugated secondary antibody for 1 hour at room temperature, followed by 3 washes in TBST. Membranes were immersed in chemiluminescent reagent to visualize protein bands (Thermo Fisher Scientific, Waltham, MA). Antibodies and suppliers are listed in Table 2.
Table 2:
Antibodies and reagents used for western blots and flow cytometry
| Protein | Usage | Product code/Company |
|---|---|---|
| KAP1 | Western blot | A300–274A, Bethyl Laboratories Inc. (Montgomery, TX) |
| IFIH1/MDA5 | Western blot | 21775–1-AP, Proteintech (Rosemont, IL) |
| GAPDH | Western blot | 60004–1-Ig, Proteintech (Rosemont, IL) |
| TBK1 | Western blot | 3013S, Cell Signaling Technologies (Danvers, MA) |
| Phospho-TBK1 (Ser172) | Western blot | 5843S, Cell Signaling Technologies (Danvers, MA) |
| MAVS | Western blot | 4983S, Cell Signaling Technologies (Danvers, MA) |
| LIVE-DEAD stain | Flow cytometry | L34965, Thermo Fisher Scientific (Waltham, MA) |
| TruStain FcX™ CD16/32 | Flow cytometry | 101320 Clone 93, Biolegend (Carlsbad, CA) |
| CD45 | Flow cytometry | 103108 Clone 30-F11, Biolegend (Carlsbad, CA) |
| CD8a | Flow cytometry | 100766 Clone 53–6.7, Biolegend (Carlsbad, CA) |
| CD4 | Flow cytometry | 100424 Clone GK1.5, Biolegend (Carlsbad, CA) |
| NK1.1 | Flow cytometry | 108707 Clone PK136, Biolegend (Carlsbad, CA) |
Quantitative Reverse Transcription PCR (qRT-PCR)
Extraction of total RNA from cells was performed using the E.Z.N.A. Total RNA kit (Omega-Biotek, Norcross, GA) following manufacturer’s protocol. Synthesis of complementary DNA library was performed on 1μg of RNA using the SuperScript II Reverse Transcriptase kit with random hexamers (Invitrogen, Carlsbad, CA). Quantitative PCR amplification was performed with 2x qPCRBIO SyGreen Mix Lo-ROX (PCR Biosystems, London, UK) on a ViiA 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). Target genes were normalized to GAPDH in human cells and ACTB in mouse cells. Endogenous retroviral primers were selected as previously described(39), all other primers were generated using the NCBI Primer Design Tool. Primers were synthesized by IDT (Coralville, IA) and are listed in Table 1.
Animals and X-ray Irradiation
All animal experiments were approved by the Duke University Institutional Animal Care and Use Committee. Specific-pathogen-free C57BL/6J and BALB/C female mice were purchased from the Jackson Laboratory, while NOD scid gamma (NSG) female mice were obtained from the Division of Lab Animal Resources at Duke University (Durham, NC). Upon receipt, up to five mice per cage were housed in a specific-pathogen-free facility under constant temperature at 25°C and a 12:12 hour light cycle and fed a standard ad libitum mouse diet with water. After 4 days acclimatization, tumor cells were injected into the right hindlimb of each mouse, and tumor growth rates were tracked every other day. Knockout tumor cell injections were a mixture of two single clones from different sgRNAs. Hindlimb tumors were irradiated using lead shielding in an XRAD 320 (Precision, North Branford, CT) with a single 8Gy dose of X-ray at a dose rate of 3.28Gy/min. Mice were euthanized when tumor size reached endpoint, set at 2000mm3 as defined by Volume=0.5xLengthxWidth2. Lungs were extracted at endpoint and preserved in 10% formalin prior to analysis.
Flow Cytometry analysis
Mouse tumors were excised at 11 days post-injection and dissociated to single cell suspension in cell staining buffer (2% FBS in PBS) using a modified version of a triple enzyme mouse tumor digestion protocol described previously(40). 106 cells were first blocked in CD16/32 antibody for 10 minutes on ice in the dark, prior to primary antibody incubation for 30 minutes on ice, following manufacturer’s recommended dilution. Following wash in cell staining buffer twice, cells were sorted on a BD FACSCantoII flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo version 10.6 software. Antibodies and suppliers are listed in Table 2.
TCGA Analyses of human patient data
Patient survival analyses were conducted using the online GEPIA survival analysis platform (http://gepia.cancer-pku.cn/)(41). mRNA co-enrichments relative to KAP1 mRNA expression levels for lung adenocarcinoma (LUAD) were downloaded from cBioPortal (q-value<0.01) and subjected to pathway enrichment analysis by PANTHER through the Gene Ontology Resource website utilizing Reactome pathway classifications (http://geneontology.org/)(42–48). All p-values were corrected for false discovery rate (FDR). Immune infiltration scores were obtained from the Tracking Tumor Immunophenotype (TIP) online platform pancancer analysis and linked to KAP1 mRNA expression by patient ID (http://biocc.hrbmu.edu.cn/TIP/index.jsp)(49).
Statistical Analyses
Quantitative data are expressed as mean ± the standard error of the mean (SEM). Comparisons between groups were performed using two-tailed unpaired Student’s T test or one-way analysis of variance (ANOVA) as appropriate. P<0.05 was considered statistically significant. All statistical analyses were conducted using JMP Pro version 14.0 software.
Results
Irradiation increased cytosolic dsRNA, including ERV transcripts
We first sought to determine if irradiation upregulated levels of cytosolic dsRNA. Using the J2 antibody that was specific for dsRNA(50), we immunostained A549 human lung cancer cells that had been treated with either sham or 8Gy of X-ray radiation. As shown in Fig. 1A–B, cytosolic dsRNA was significantly upregulated in irradiated cells. Similar findings were also observed in B16F10 mouse melanoma cells treated with radiation (Fig. 1C–D), demonstrating general applicability of our finding in mammalian cells. The upregulation of dsRNA was consistent with activation of ERV transcription, as qRT-PCR results demonstrated significant increases in irradiated A549 cells of transcripts specific to ERVs (Fig. 1E). Thus, X-ray irradiation led to increased cytosolic dsRNA expression, which was likely mediated by the activation of ERV transcription.
Figure 1. Radiation-inducted upregulation of dsRNA, including ERVs in tumor cells.
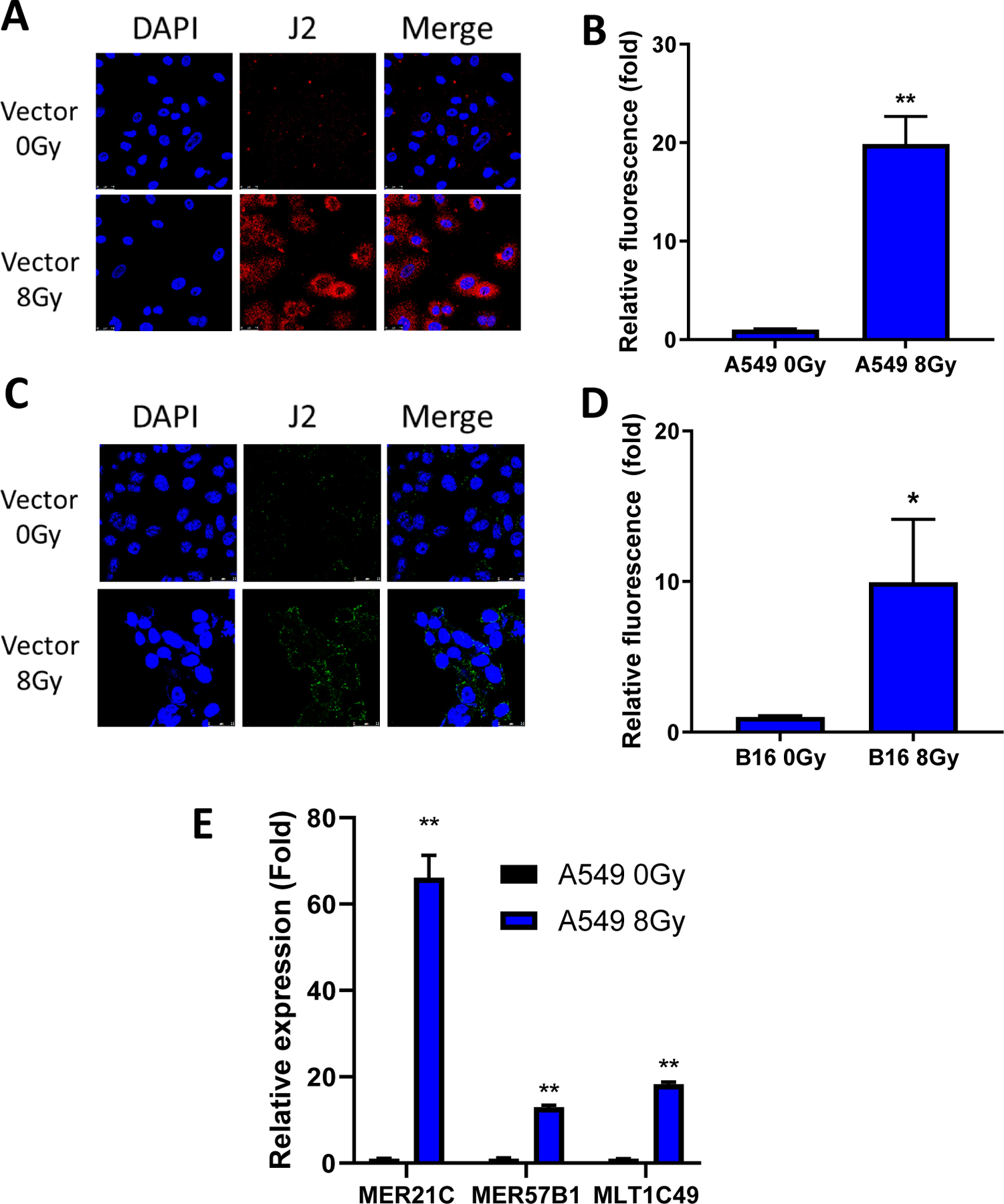
(A, B). Representative immunofluorescence images (A) and quantification (B) of J2 antibody staining of vector control A549 cells 5 days post irradiation with sham or 8Gy X-rays.
(C, D). Representative immunofluorescence images (C) and quantification (D) of J2 antibody staining of vector control B16F10 cells 5 days post irradiation with sham or 8Gy X-rays.
(E) Quantification of qRT-PCR of ERVs in vector control A549 cells 5 days post irradiation with sham or 8Gy X-rays.
Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ns, not significant, as determined by unpaired Student’s T-test (B, D, and E)
KAP1 knockout enhanced radiation-induced dsRNA expression
ERVs make up over 8% of the human genome but are normally repressed by KAP1-mediated heterochromatin formation at ERV sites within the genome(51, 52). However, it was shown previously that KAP1-mediated heterochromatin repression could be temporarily reversed upon detection of DNA damage to facilitate DNA damage repair processes(53). This process was regulated by the Ataxia Telangiectasia Mutated (ATM) protein kinase through phosphorylation of KAP1, which abrogated its repressive effect(54). We hypothesized that knockout of KAP1 would lead to increased basal ERV transcription and enhance radiation-induced ERV reactivation(55, 56). As shown in Fig. 2A–B, knockout of KAP1 by CRISPR-Cas9 in A549 human lung cancer cells did cause an increase in cytosolic dsRNA as determined by J2 staining, and the effect was significantly enhanced by radiation. We also observed similar findings in KAP1 knockout B16F10 mouse melanoma cells treated with irradiation (Fig. 2C–D). Additionally, specific ERV transcripts in KAP1 knockout of human A549 lung cancer cells were further increased after radiation, indicating a combinatorial effect between KAP1 knockout and irradiation in ERV activation (Fig. 2E).
Figure 2. KAP1 knockout stimulates dsRNA and ERV expression.

(A, B). Representative J2 immunofluorescence staining (A) and quantification (B) of KAP1 knockout A549 cells 5 days post irradiation with sham or 8Gy X-rays.
(C, D). Representative J2 immunofluorescence staining (C) and quantification (D) of KAP1 knockout B16F10 cells 5 days post irradiation with sham or 8Gy X-rays.
(E). Quantification of qRT-PCR of ERVs in vector control or KAP1 knockout A549 cells 5 days post irradiation with sham or 8Gy X-rays.
Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ns, not significant, as determined by unpaired Student’s T-test (B, D, and E)
Activation of MDA5/MAVS dsRNA-sensing pathway and interferon stimulated genes (ISGs) in irradiated tumor cells
It has been demonstrated prior that cytosolic dsRNA activates the innate antiviral immune pathway, specifically through dsRNA sensing by MDA5 followed by MAVS activation and phosphorylation of TBK1(30, 57, 58). In order to confirm the relevance of radiation-induced cytosolic dsRNA to immune stimulation, we examined the activation of dsRNA sensing pathways and activated interferons via immunoblotting and qRT-PCR. As shown in Fig. 3A, irradiation and KAP1 knockout significantly upregulated expression of phosphorylated TBK1, indicating activation of the MDA5/MAVS/TBK1 pathway. Additionally, qRT-PCR analysis of mRNA expression of downstream ISGs were also enhanced in irradiated cells, and the effect was amplified by KAP1 knockout (Fig. 3B). To establish the causal relationship between MDA5/MAVS activation and ISG expression, we generated KAP1 and MAVS double knockout cells. Western blot analysis showed that MAVS knockout abrogated radiation-induced TBK phosphorylation (Fig. 3C), suggesting the MDA5/MAVS pathway played an important role in radiation-induced ISG upregulation.
Figure 3. KAP1 knockout amplified radiation induced activation of ISGs.
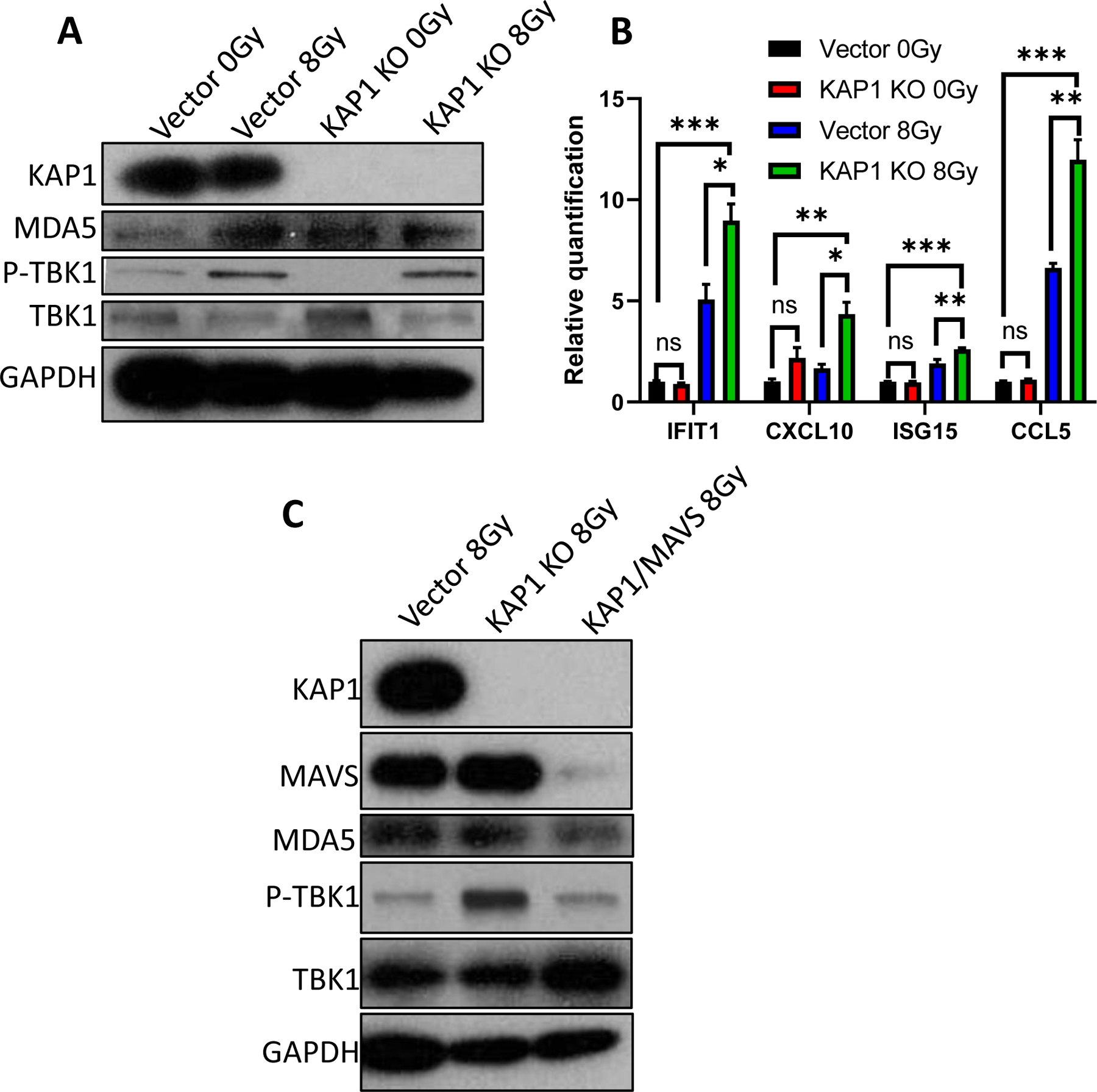
(A). Immunoblot of dsRNA sensing pathway members in vector control or KAP1 knockout B16F10 cells 3 days post irradiation with sham or 8Gy X-rays.
(B) qRT-PCR quantification of interferon-stimulated genes in vector control or KAP1 knockout B16F10 cells 3 days post exposure to sham or 8Gy X-rays.
(C). Immunoblot of dsRNA sensing pathway members from KAP1/MAVS double knockout B16F10 cells 3 days after exposure to 8Gy X-rays.
Error bars represent SEM. *p,<0.05, **p<0.01, ***p<0.001, ns, not significant, as determined by unpaired Student’s T-test (B)
KAP1 inhibition enhanced RT of B16F10 mouse melanomas in vivo
Next, we moved to an in vivo mouse model to investigate the effect of KAP1 knockout on tumor growth. We inoculated 105 vector control or KAP1 knockout B16F10 cells subcutaneously into the right hindlimb of syngeneic C57BL/6J mice and irradiated the tumors with sham or 8Gy X-ray irradiation on the eighth day post-inoculation. As shown in Fig. 4A–B, RT and KAP1 knockout resulted in significant tumor growth delay and prolonged host mice survival, respectively. Furthermore, the combination of both had the greatest effect in suppressing tumor growth. To determine if an intact immune system was required for the observed effect of KAP1 knockout, we inoculated KAP1 knockout B16F10 cells into immunodeficient NSG mice. Strikingly, we failed to see a difference in tumor growth rate or survival in the KAP1 knockout mice when compared to vector control tumors (Fig. 4C–D). These results indicated that the KAP1-knockout-mediated B6F10 melanoma growth inhibition required an intact host immune system.
Figure 4. KAP1 knockout inhibits B16F10 melanoma growth in syngeneic C57BL/6 and immunodeficient NSG mice.
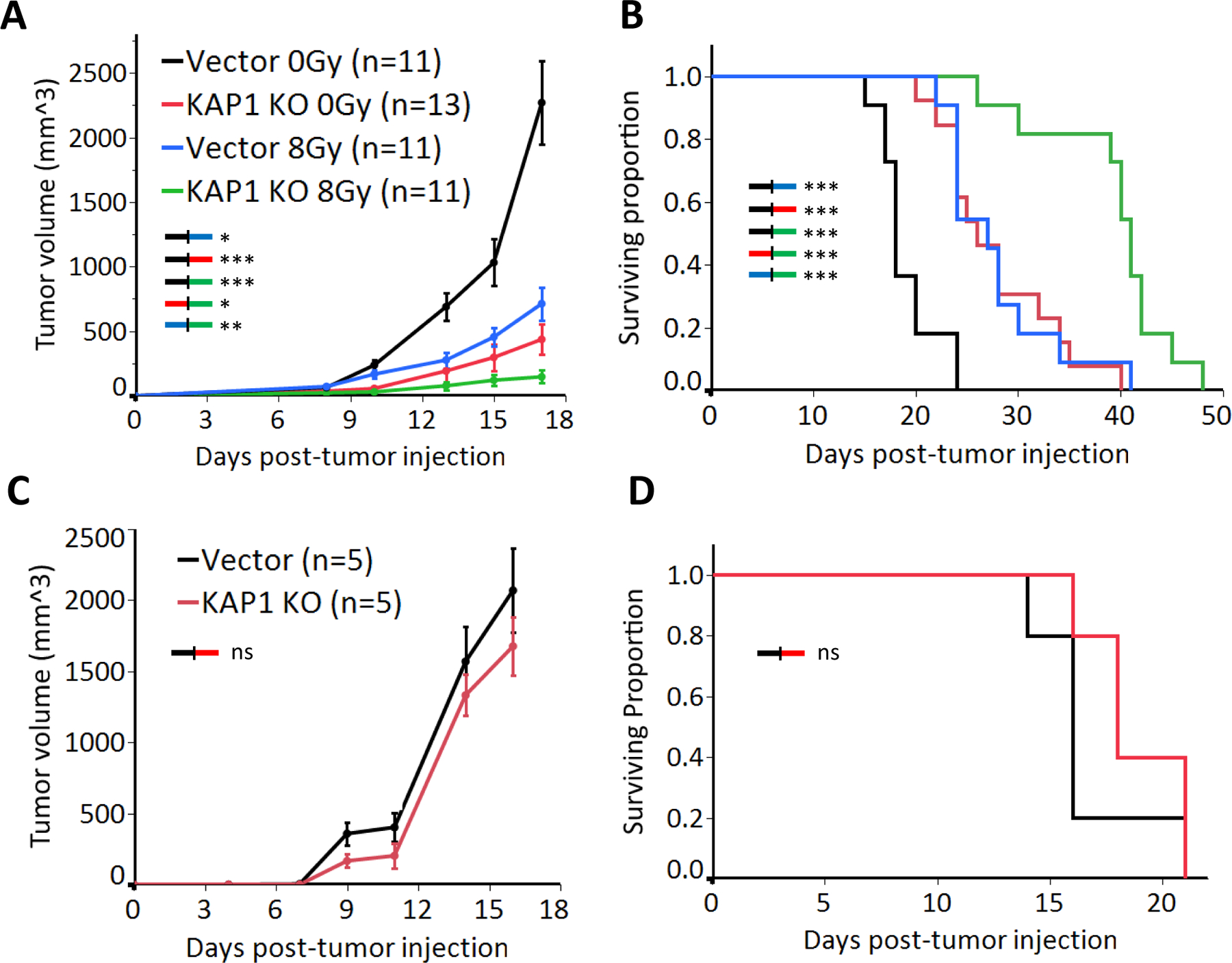
(A, B). Tumor growth (A) and Kaplan-Meier survival curve in immunocompetent C57BL/6 host mice(B) inoculated with 105 vector control or KAP1 knockout B16F10 cells and treated with 0Gy or 8Gy X-rays at day 8 post inoculation.
(C, D). Tumor growth (C) and survival curves (D) in immunodeficient NSG mice inoculated with 105 vector control or KAP1 knockout B16F10 cells.
Data in A&B represent three separate experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ns, not significant, as determined by one-way ANOVA (A and C) or Log-Rank test (B and D)
KAP1 knockout inhibited tumor metastases in the 4T1 murine breast cancer model
To confirm if the effect of KAP1 knockout could be extended to a different tumor model, we inoculated the right hindlimb of syngeneic BALB/c mice with 5×105 vector or KAP1 knockout 4T1 murine breast cancer cells, which were known to be aggressive and metastatic(59), and irradiated tumors with sham or 8Gy X-ray irradiation. As shown in Fig. 5A–B, irradiation or KAP1 knockout alone did not have a significant effect on tumor growth rates or survival, but the combination treatment significantly inhibited tumor growth and extended survival. Furthermore, examination of lungs of mice that had reached end points demonstrated that KAP1 knockout alone, with or without radiation, was sufficient to inhibit metastasis to the lung (Fig. 5C–E), suggesting that immune-stimulatory effects of KAP1 knockout allowed for the immune system to detect and destroy migrating metastatic cells.
Figure 5. KAP1 knockout inhibits metastasis and delays tumor growth conjunction with RT in syngeneic 4T1 breast cancer model.

(A, B). Tumor growth (A) and Kaplan-Meier survival curves (B) of Balb/C mice inoculated with 5×105 vector control or KAP1 knockout 4T1 cells and treated with 0Gy or 8 Gy of X-rays at day 8 post inoculation.
(C). Representative images of lungs of mice inoculated vector control and KAP1 knockout 4T1 cells.
(D, E). Quantification of lung metastases by mass (D) and metastatic surface nodules (E) (n=5 per group).
Data represent two independent experiments (A and B). Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ns, not significant, as determined by two-way ANOVA (A), log-rank (B), or unpaired Student’s T-test (D and E)
KAP1 knockout promoted intratumoral immune infiltration in murine tumor models
Previously it was shown that immune activation of the tumor microenvironment significantly influenced the efficacy of RT and subsequent tumor prognosis(60, 61). In order to determine if KAP1 inhibition was sufficient to influence tumor immune infiltration, we elected to analyze the constitution of immune effector cells in vector control and KAP1 knockout B16F10 tumors grown in syngeneic C57BL/6 mice. We harvested tumors at eleven days post-injection, prior to tumors reaching endpoint size. Flow cytometry analysis of dissociated tumor cells (see Fig. 6A for gating strategy) indicated there was no significant difference in CD45+ cell populations between vector control and KAP1 knockout tumors (Fig. 6B), despite the trend towards increased population in KAP1 knockouts. However, the percentage of CD8+ cells was significantly increased in KAP1 knockout tumors compared to vector controls, suggesting an increased cytotoxic immune cell population in KAP1 knockout tumors (Fig. 6C–D). NK1.1+ populations were unchanged between vector control and knockout tumors (Fig. 6D), suggesting that tumor inhibition in KAP1 knockouts was primarily mediated by recruitment of CD8+ cytotoxic immune cells as opposed to NK cells.
Figure 6. KAP1 knockout increases CD45+CD8+ leukocyte infiltration into B16F10 tumors.
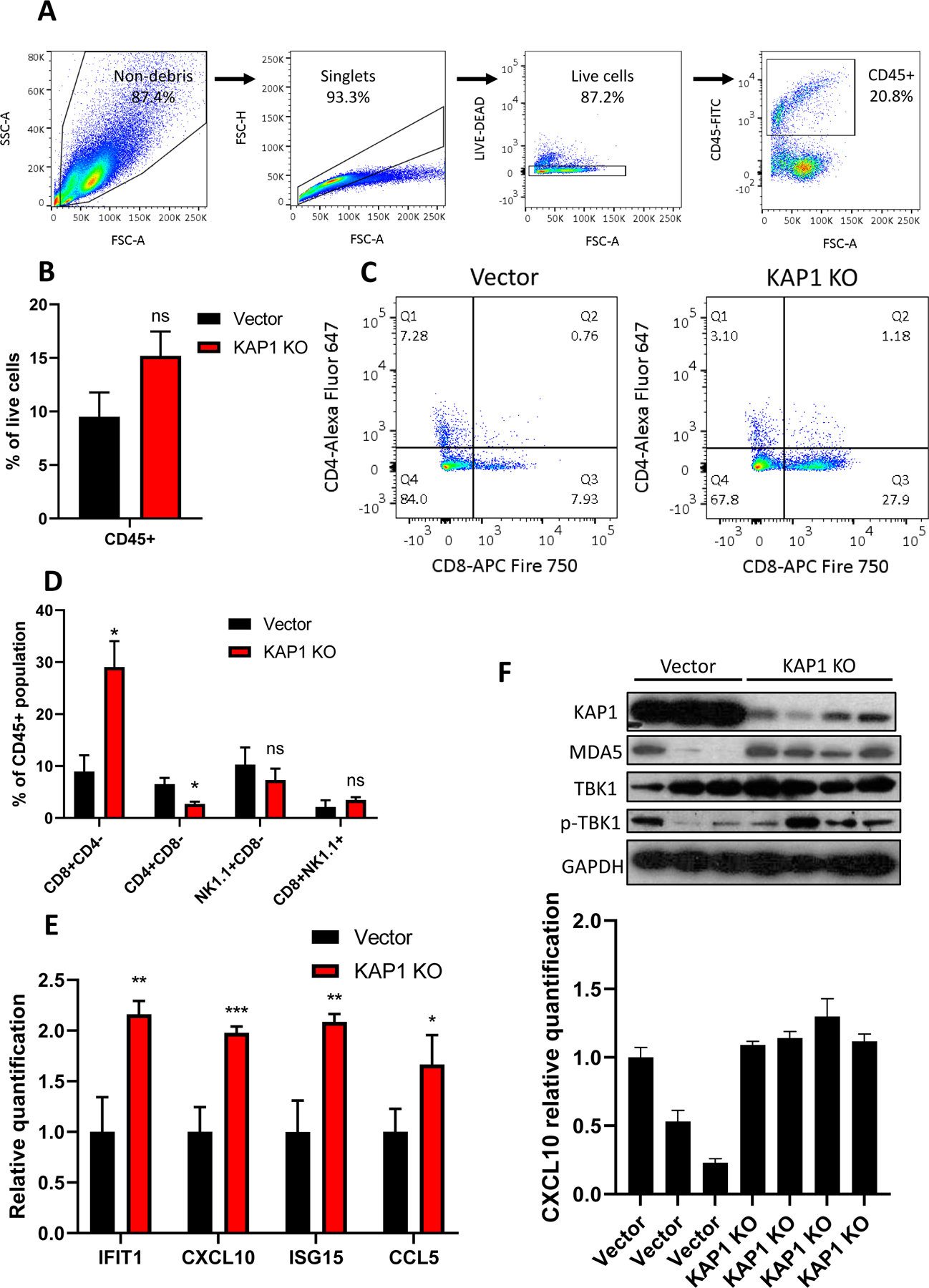
(A). Gating strategy for selection of live CD45+ leukocyte population
(B). Fraction of CD45+ cell population in vector control or KAP1 knockout B16F10 tumors formed in syngeneic C57BL/6 mice.
(C). Flow cytometry analysis of CD8+ vs CD4+ populations among CD45+ cells from vector control or KAP1 knockout tumors grown in C57BL/6 mice.
(D). Fractions of various immune effector cell populations in among CD45+ cells from vector control and KAP1 knockout tumors.
(E). Quantification by qRT-PCR of interferon-stimulated genes in vector control vs KAP1 knockout tumors; samples normalized to GAPDH.
(F). Western blot analysis of vector control and KAP1 knockout tumors (upper) correlated to qRT-PCR quantified mRNA expression of CXCL10 (lower) in individual tumors
Tumors were excised at day 11 post inoculation of tumor cells. Error bars represent SEM. *p<0.05, **p<0.01, ***P<0.001, ns, not significant, as determined by unpaired Student’s T-test (B, D, and E)
We also carried out qRT-PCR analysis of harvested tumor cells for ISGs. Our analysis showed significant upregulation of ISGs in KAP1 knockout tumors when compared to vector controls (Fig. 6E), indicating significant interferon activation within the tumor. Additionally, levels of MDA5 and phosphorylated TBK1 were upregulated in KAP1 knockout tumors compared to control tumors, and correlated with ISG induction (Fig. 6F) on an individual tumor basis, demonstrating a direct link between dsRNA sensing pathway induction (Fig.6F, top panel) and interferon activation (Fig. 6F, lower panel). Taken as a whole, our data indicated that loss of KAP1 activated the MDA5/MAVS/TBK1 antiviral pathway, which in turn stimulated the transcription of downstream ISGs that provoked cytotoxic immune cells and inhibited tumor growth.
KAP1/Trim28 expression levels were correlated with patient survival and intratumoral immune infiltration in patients
To determine if our findings were relevant to patients, we analyzed patient survival rates from the 33 TCGA cancer datasets, comparing patients with high or low Trim28 mRNA expression levels (as defined by 75th and 25th percentile). We discovered that Trim28 expression levels had a significant effect on survival when compared across all 33 cancer types within the TCGA dataset, with a greater effect in specific cancers such as lung adenocarcinoma (LUAD) and skin cutaneous melanoma (SKCM) (Figure 7A–C). Next, we identified the most significantly under-enriched genes in Trim28-high tumor samples from SKCM patients and subjected them to Reactome pathway enrichment analysis. As expected, we found that Trim28 was negatively associated with a wide variety of immune regulatory, cytokine signaling, and antigen presentation pathways (Fig. 7D).
Figure 7. Trim28 mRNA levels correlate with patient survival in TCGA cancer cohorts.

(A, B, and C). Kaplan-Meier survival curves for all 33 TCGA cancers (A), lung adenocarcinoma (B), or skin cutaneous melanoma (C), separated into Trim28 high vs low by 75th vs 25th quartile Trim28 mRNA expression levels.
(D) REACTOME pathway analysis of the most significantly downregulated genes in SKCM Trim28-high patients.
(E). Immune effector cell recruitment scores in Trim28-high vs low SKCM patients.
To determine if Trim28 expression levels affected intratumoral lymphocyte infiltration in patients, we analyzed expression of genes specific to immune and T cell recruitment in the melanoma patient cohort by use of the TIP online database (http://biocc.hrbmu.edu.cn/TIP/index.jsp)(49). Gene expression data were analyzed and different immune effector subsets were identified by use of the CIBERSORT method(62), and each patient was assigned immune infiltration scores, which were then correlated to patient Trim28 mRNA expression levels. Our analysis demonstrated that multiple scores for immune infiltration were significantly increased in Trim28-low SKCM patients, suggesting an inverse relationship between the two (Fig. 7E). Taken together, our results were consistent with the hypothesis that KAP1/Trim28-mediated ERV repression was an important factor in anti-tumor immune signaling in cancer patients.
Discussion
The discovery of the cGAS/STING dsDNA sensing pathway and its role in regulating tumor immunity in response to RT has sparked much research regarding the combination of RT and immunotherapy. However, other pathways potentially acting in parallel to activate interferon signaling have remained understudied. Here, we demonstrate that radiation-induced ERV induction plays a significant role in modulating the tumor immunity response to radiation, through an alternative dsRNA sensing pathway. This has major implications for understanding the various immune-mediated effects of radiation, such as the abscopal effect(63).
Ionizing radiation induces a multitude of effects on a cell, many involving DNA damage and genomic instability(64). Other related effects also include activation of DNA damage repair proteins, heterochromatin decondensation, and upregulation of pro-survival factors(65–67). Previous work has demonstrated direct involvement of KAP1 in linking DNA damage repair to heterochromatin decondensation, and subsequent expression of ERVs(52, 53). Here, we show that irradiation alone could stimulate ERV transcription, and this effect was enhanced through loss of KAP1. Moreover, the ensuing interferon stimulation was mediated by the MDA5/MAVS/TBK1 dsRNA sensing pathway rather than the cGAS/STING dsDNA sensing pathway, as TBK1 phosphorylation and ISG induction was abrogated by MAVS knockout, which is not involved in the cGAS/STING signaling pathway(68). Further studies are needed to determine the relative contributions of the dsDNA, dsRNA, and other possible sensing pathways to overall radiation-induced ISG induction.
Interestingly, although KAP1 knockouts show only minor effects in vitro, the in vivo transplant murine tumor models of KAP1 knockouts had significant effects on tumor growth and metastasis, suggesting the in vivo tumor microenvironment was sufficient to provoke ERV production and interferon response. Consistently, we found KAP1 knockout in B16F10 melanomas was sufficient to promote immune infiltration, correlated with ISG induction. ISGs have been implicated in regulating antigen presentation(69), which is a potential explanation for our observed difference in immune infiltration in both experimental and clinical data.
There are currently no drugs or small molecules that specifically target KAP1. However, the chemotherapeutic doxorubicin (dox) has been previously reported to upregulate ATM-mediated KAP1 phosphorylation, as well as subsequent histone eviction from the chromatin(54, 70). Thus, in the absence of drugs targeting KAP1 specifically, it may be profitable in the meantime to target the heterochromatic control of KAP1, using therapies such as histone methyltransferase inhibitors or histone deacetylase inhibitors to induce greater ERV transcription and render tumors more sensitive to combination therapies(71, 72). Furthermore, our study provides a strong rational to develop KAP1-targeted small molecules to enhance RT and/or immunotherapy.
In conclusion, our study demonstrates that transcriptional control of ERVs by KAP1 is an important factor in tumor immunogenicity, and that KAP1 can be an appealing pharmacological target for enhancing the effects of RT and immunotherapy in cancer treatment.
Acknowledgement:
The study is supported in part by grants ES024015, CA208852, CA216876 from US National Institutes of Health (to C-Y. Li). We thank Michael Cook and colleagues at the Flow Cytometry Shared Resource, and Yasheng Gao and colleagues at the Microscopy Share Resource of Duke University School of Medicine for their expert services.
References
- 1.Barton MB, Jacob S, Shafiq J, Wong K, Thompson SR, Hanna TP, et al. Estimating the demand for radiotherapy from the evidence: a review of changes from 2003 to 2012. Radiother Oncol. 2014;112(1):140–4. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. 2010;31(4):363–72. [DOI] [PubMed] [Google Scholar]
- 3.Vignard J, Mirey G, Salles B. Ionizing-radiation induced DNA double-strand breaks: a direct and indirect lighting up. Radiother Oncol. 2013;108(3):362–9. [DOI] [PubMed] [Google Scholar]
- 4.Moding EJ, Kastan MB, Kirsch DG. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov. 2013;12(7):526–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377(6549):552–7. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300(5622):1155–9. [DOI] [PubMed] [Google Scholar]
- 7.Nehs MA, Lin CI, Kozono DE, Whang EE, Cho NL, Zhu K, et al. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery. 2011;150(6):1032–9. [DOI] [PubMed] [Google Scholar]
- 8.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61(2):439–44. [PubMed] [Google Scholar]
- 9.Jurin M, Suit HD. In vivo and in vitro studies of the influence of the immune status of C3Hf-Bu mice on the effectiveness of local irradiation of a methylcholanthrene-induced fibrosarcoma. Cancer Res. 1972;32(10):2201–11. [PubMed] [Google Scholar]
- 10.Suit HD, Kastelan A. Immunologic status of host and response of a methylcholanthrene-induced sarcoma to local x-irradiation. Cancer. 1970;26(1):232–8. [DOI] [PubMed] [Google Scholar]
- 11.Demaria S, Bhardwaj N, McBride WH, Formenti SC. Combining radiotherapy and immunotherapy: a revived partnership. Int J Radiat Oncol Biol Phys. 2005;63(3):655–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71(7):2488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2 Pt 1):728–34. [PubMed] [Google Scholar]
- 14.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41(5):843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe. 2015;18(3):333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192(12):5993–7. [DOI] [PubMed] [Google Scholar]
- 17.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–14. [DOI] [PubMed] [Google Scholar]
- 19.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanpouille-Box C, Formenti SC, Demaria S. TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology. 2017;6(9):e1339857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front Pharmacol. 2018;9:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cushman TR, Caetano MS, Welsh JW, Verma V. Overview of ongoing clinical trials investigating combined radiotherapy and immunotherapy. Immunotherapy. 2018;10(10):851–0. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimoto Y, Suzuki Y, Mimura K, Ando K, Oike T, Sato H, et al. Radiotherapy-induced anti-tumor immunity contributes to the therapeutic efficacy of irradiation and can be augmented by CTLA-4 blockade in a mouse model. PLoS One. 2014;9(3):e92572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bannert N, Hofmann H, Block A, Hohn O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front Microbiol. 2018;9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bannert N, Kurth R. The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet. 2006;7:149–73. [DOI] [PubMed] [Google Scholar]
- 26.Sharif J, Endo TA, Nakayama M, Karimi MM, Shimada M, Katsuyama K, et al. Activation of Endogenous Retroviruses in Dnmt1(−/−) ESCs Involves Disruption of SETDB1-Mediated Repression by NP95 Binding to Hemimethylated DNA. Cell Stem Cell. 2016;19(1):81–94. [DOI] [PubMed] [Google Scholar]
- 27.Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, et al. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development. 2013;140(3):519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448(7153):595–9. [DOI] [PubMed] [Google Scholar]
- 29.Kassiotis G, Stoye JP. Making a virtue of necessity: the pleiotropic role of human endogenous retroviruses in cancer. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162(5):974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162(5):961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell. 2017;171(6):1284–300 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell. 2018;174(3):549–63 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, Ning S, Nolley R, Scicinski J, Oronsky B, Knox SJ, et al. The immunomodulatory anticancer agent, RRx-001, induces an interferon response through epigenetic induction of viral mimicry. Clin Epigenetics. 2017;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JR, Ahn K, Kim YJ, Jung YD, Kim HS. Radiation-induced human endogenous retrovirus (HERV)-R env gene expression by epigenetic control. Radiat Res. 2012;178(5):379–84. [DOI] [PubMed] [Google Scholar]
- 36.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44(W1):W272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chatterjee A, Rodger EJ, Ahn A, Stockwell PA, Parry M, Motwani J, et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience. 2018;4:312–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National Cancer Institute. Triple Enzyme Mouse Tumor Digestion 2006. [Available from: https://ccr.cancer.gov/sites/default/files/triple_enzyme_mouse_tumor_digestion.pdf.
- 41.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The Gene Ontology C The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(D1):D330–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419–D26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018;46(D1):D649–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fabregat A, Sidiropoulos K, Viteri G, Forner O, Marin-Garcia P, Arnau V, et al. Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics. 2017;18(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu L, Deng C, Pang B, Zhang X, Liu W, Liao G, et al. TIP: A Web Server for Resolving Tumor Immunophenotype Profiling. Cancer Res. 2018;78(23):6575–80. [DOI] [PubMed] [Google Scholar]
- 50.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80(10):5059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf D, Goff SP. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell. 2007;131(1):46–57. [DOI] [PubMed] [Google Scholar]
- 52.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463(7278):237–40. [DOI] [PubMed] [Google Scholar]
- 53.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8(8):870–6. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, et al. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem. 2007;282(50):36177–89. [DOI] [PubMed] [Google Scholar]
- 55.White D, Rafalska-Metcalf IU, Ivanov AV, Corsinotti A, Peng H, Lee SC, et al. The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol Cancer Res. 2012;10(3):401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem. 2011;286(30):26267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dias Junior AG, Sampaio NG, Rehwinkel J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019;27(1):75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, et al. MAVS activates TBK1 and IKKepsilon through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017;13(11):e1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2(5):331–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farooque A, Singh N, Adhikari JS, Afrin F, Dwarakanath BS. Enhanced antitumor immunity contributes to the radio-sensitization of ehrlich ascites tumor by the glycolytic inhibitor 2-deoxy-D-glucose in mice. PLoS One. 2014;9(9):e108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strom T, Harrison LB, Giuliano AR, Schell MJ, Eschrich SA, Berglund A, et al. Tumour radiosensitivity is associated with immune activation in solid tumours. Eur J Cancer. 2017;84:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vanpouille-Box C, Formenti SC, Demaria S. Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin Cancer Res. 2018;24(2):259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morgan WF, Day JP, Kaplan MI, McGhee EM, Limoli CL. Genomic instability induced by ionizing radiation. Radiat Res. 1996;146(3):247–58. [PubMed] [Google Scholar]
- 65.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. [DOI] [PubMed] [Google Scholar]
- 66.Falk M, Lukasova E, Kozubek S. Higher-order chromatin structure in DSB induction, repair and misrepair. Mutat Res. 2010;704(1–3):88–100. [DOI] [PubMed] [Google Scholar]
- 67.Hein AL, Ouellette MM, Yan Y. Radiation-induced signaling pathways that promote cancer cell survival (review). Int J Oncol. 2014;45(5):1813–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jacobs JL, Coyne CB. Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol. 2013;425(24):5009–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One. 2012;7(3):e32542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun. 2013;4:1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49(7):1052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. 2014;3(1):e27414. [DOI] [PMC free article] [PubMed] [Google Scholar]