

Abstract
Although ischemia-reperfusion (I/R) can initiate apoptosis, the timing and contribution of the mitochondrial/cytochrome c apoptosis death pathway to I/R injury is unclear. We studied the timing of cytochrome c release during I/R and whether subsequent caspase activation contributes to reperfusion injury in confluent chick cardiomyocytes. One-hour simulated ischemia followed by 3-h reperfusion resulted in significant cell death, with most cell death evident during the reperfusion phase and demonstrating mitochondrial cytochrome c release within 5 min after reperfusion. By contrast, cells exposed to prolonged ischemia for 4 h had only marginally increased cell death and no detectable cytochrome c release into the cytosol. Caspase activation could not be detected after ischemia only, but it significantly increased after reperfusion. Caspase inhibitors benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone, Ac-Asp-Gln-Thr-Asp-H, or benzyloxycarbonyl-Leu-Glu (Ome)-His-Asp-(Ome)-fluoromethyl ketone given only at reperfusion significantly attenuated cell death and resulted in return of contraction. Antixoxidants decreased cytochrome c release, nuclear condensation, and cell death. These results suggest that reperfusion oxidants initiate cytochrome c release within minutes, and apoptosis within hours, significant enough to increase cell death and contractile dysfunction.
Keywords: oxidants, ischemia-reperfusion, reperfusion injury, caspase inhibition
apoptosis within the heart can be induced by ischemia-reperfusion (I/R), contributing to the cell injury produced by necrosis (for reviews see Refs. 1 and 16). Studies (10, 17) of I/R injury in animal models have shown significant apoptotic events in infarcted hearts, but there have been few studies examining the molecular pathway of I/R-induced apoptosis in isolated cardiomyocyte models. such models have the advantage of distinguishing between the events of ischemia and early reperfusion in one cell type (32, 34). In I/R injury,mitochondria-initiated apoptosis of cardiac cells may be a significant contributor to cell death (21). This intrinsic pathway of apoptosis is initiated by the release of cytochrome c from the intermembrane space of the mitochondria into the cytoplasm through mechanisms not entirely understood. In the cytoplasm, cytochrome c interacts with apoptotic protease-activating factor-1 (Apaf-1), which recruits pro-caspase-9 forming the macromolecular complex called the apoptosome. During this process caspase-9 is cleaved into active subunits. Active caspase-9, in turn, cleaves downstream caspases, such as caspase-3 and caspase-7. These and perhaps other caspases cleave numerous substrates, including major cell structural proteins, such as α-fodrin, resulting in the terminal biochemical and morphological events of apoptotic death.
We have found in previous work that chick cardiomyocytes exposed to simulated I/R exhibit significant cell death in which the greatest acceleration of death occurs during the reperfusion phase (i.e., reperfusion injury). Indeed, cells exposed to 1-h simulated ischemia/3-h reperfusion exhibit more cell death and membrane damage than cells exposed to prolonged ischemia (4 h) alone (34). Oxidants appear to play a significant role, because a burst of reactive oxygen species (ROS) can be detected after 5 min reperfusion (32), and antioxidants given just at reperfusion significantly decrease cell death and improve functional recovery of synchronous contractions (30).
In addition, this cell system exhibits preconditioning (30, 31), a highly conserved adaptive response found in multiple organs (including heart) and species (including human; see Refs. 18 and 29), which elicits one of the most profound protections known against I/R injury. Thus if apoptosis plays an important role in mediating I/R injury in this system, this model may be useful to study how either oxidant generation or preconditioning may affect the events of apoptosis during I/R. The present study was designed to test what role the intrinsic pathway of apoptosis may play within this relatively acute timeframe of I/R injury and whether apoptosis is initiated during the ischemic or reperfu-sion phases in our model. Finally, we tested whether caspase inhibitors given just at reperfusion could significantly protect chick cardiomyocytes against cell death and contractile dysfunction.
MATERIALS AND METHODS
Materials
The broad-spectrum tripeptide caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk) and the caspase-9 inhibitor benzyloxycarbonyl-Leu-Glu (Ome)-His-Asp-(Ome)-fluoromethyl ketone (zLEHD-fmk) were obtained from Enzyme System Products (Livermore, CA). The caspase-7/3 inhibitor Ac-Asp-Gln-Thr-Asp-H (Ac-DQTD-CHO) was obtained from Peptide Institute (Louisville, KY). An antibody that recognizes the pro and active forms of caspase-9 was used for Western blot analyses and was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-cytochrome c monoclonal antibody was purchased from PharMingen International (San Diego, CA). Buffer reagents were obtained from Sigma (St. Louis, MO). Propidium iodide (PI) for measuring cell viability and 2’,7’-dichlorofluorescin diacetate for measuring oxidant generation were obtained from Molecular Probes (Eugene, OR). Anti-a-fodrin was purchased from ICN Biochemicals (Aurora, OH). N-benzyloxycarbonyl-Phe-Ala-fluoromethyl ketone (ZFA-fmk) is a cysteine protease inhibitor with no known anti-caspase activity (15). As reported by others, this agent can be used as a negative control for zVAD-fmk and was purchased from Enzyme Systems Products (model FK-029; Livermore, CA)(2). The antioxidants 2-mercaptopropionylglycine (MPG) and 1,10-phenanthroline (Phen) were obtained from Sigma.
Cell Preparation and Perfusion System
Cardiomyocyte culture.
Ventricular embryonic chick cardiac myocytes were prepared as previously described (31, 34). Briefly, 10-day-old chicken embryo hearts were resected, and ventricular tissue was minced into 0.5-mm fragments, enzymatically dispersed with 0.025% trypsin (Invitrogen, Grand Island, NY), and centrifuged. Cells (0.7 × 106) were then pipetted onto 25-mm coverslips and incubated (5% CO2, 37°C). Coverslips were checked for nonmuscle cell contamination (33). Experiments were performed with 3–5 day cardiac cell cultures, by which time a synchronously contracting layer of cells could be visualized and viability exceeded 99%.
Perfusion system.
Coverslips with contracting cells were placed in a 1.2-ml Sykes-Moore perfusion chamber (Bellco Glass; Vineland, NJ) as described in previous work (30). Perfusate was pumped through the chamber (0.25 ml/min) by a roller pump via water-jacketed tubing that controlled temperature (37°C). Tubing to the chamber was made of stainless steel to minimize diffusion of ambient O2. Normoxic perfusate used for baseline conditions and for reperfusion subsequent to ischemia consisted of oxygenated balanced salt solution (BSS) with a Po2 of 149 Torr, Pco2 of 40 Torr, pH of 7.4, and a concentration of K of 4.0 meq/l, containing glucose (5.6 mM). There is no standard simulation of ischemia in a cell model. To simulate key aspects of ischemia for cardiomyocytes, we combined hypoxia, hypercarbia with lower pH, hyperkalemia, and glucose deprivation. Specifically, BSS solution with a potassium concentration of 8.0 meq/l is equilibrated with 80% N2 and 20% CO2 to produce a Po2 of ~5 Torr, a Pco2 of 144 Torr, and a final pH of 6.8. As previously reported, we added 2-deoxyglucose (20 mM) to the ischemia solution to prevent these cells from using their large glycogen reserves and thus truly simulate the substrate deprivation of ischemia (30–34).
Video/fluorescent microscopy.
An Olympus IMT-2 inverted phase/epifluorescent microscope was used for cell imaging. Phase contrast Hoffmann modulation optics and a charge-coupled device camera were used to monitor contractions and morphologic membrane changes in the same field of cells (~50 × 70 μm) over time. Fluorescent images were acquired from a cooled slow-scanning PC-controlled camera (Hamamatsu, Hamamatsu City, Japan), and changes in fluorescence intensity over time were quantified with Image-One software (Image-Pro Plus; Fryer, Chicago, IL).
Measures of Viability and Oxidant Generation
Viability assay.
Cell viability was assessed with fluorochrome PI (5 μM). PI has been used previously to predict the transition from reversible to irreversible cell injury in cultured cardiomyocytes (4, 34). It is excluded from viable cells and binds to chromatin after loss of cell membrane integrity, becoming highly fluorescent (an excitation wavelength of 540 nm and a 590 nm band-pass emission filter). The dye was used to quantify cell death throughout the entire time of each experiment, exhibiting no toxicity in control cells even after a 10-h exposure. All cells in the field studied were stained with PI at the end of the experiment by permeabilizing with digitonin (300 μM), and percent cell death was calculated. Cell death was expressed as the PI fluorescence at any given time point relative to the maximal value seen after digitonin exposure (100%).
Measure of intracellular ROS generation.
Intracellular oxidant stress due to ROS was monitored with the intracellular probe 2’,7’-dichlorofluorescin (DCFH) diacetate (DCFH-DA; 5 μM) as previously described (8, 30–32). This dye is cleaved by cellular esterases on entry, trapping the nonfluorescent DCFH inside. ROS, particularly hydrogen peroxide and hydroxyl radical, generate the fluorescent product dichlorofluorescein (DCF) by oxidizing DCFH (8, 32). Thus increases in DCF fluorescence result from DCFH oxidation to DCF and imply hydrogen peroxide or hydroxyl radical generation. DCF fluorescence was measured at an excitation wavelength of 480 nm and a 520-nm band-pass emission filter. All fluorescence measures are expressed in arbitrary units of fluorescence.
Cell contraction.
As part of functional viability, cell contractions were assessed by observing movement within the same field of cells as previously reported (33). A return of contraction after simulated I/R was indicated when contractions could be seen throughout the field of cells after the 3-h period of reperfusion. Video clips of cell contractions before and after experimental conditions are included in the supplement.
Measures of Apoptosis During I/R
Cell preparation for studies of cytochrome c release, caspase-9 activation, and fodrin cleavage.
Cytochrome c release into the cytosol, caspase-9 cleavage to the active p10 subunit, and cleavage of α-fodrin (a major cell cytoskeleton component cleaved by active caspase-3) were also measured during I/R. Cytosolic extracts were prepared for measurement of cytochrome c release at sequential time points during the I/R protocol. Cells (0.7 × 106/sample coverslip; 2 coverslips taken for each time point) were removed from the chamber, and cytosolic extracts were prepared according to the method described by Bossy-Wetzel et al. (5). Briefly, each sample was washed twice with ice-cold PBS solution by centrifugation at 200 g for 5 min, 4°C. The cell pellets were suspended in 5 volumes of extraction buffer, containing (in mM) 220 mannitol, 68 sucrose, 50 PIPES-KOh (pH 7.4), 50 KCl, 5 EGTA, 2 MgCl2, 1 DTT, and protease inhibitors [leupeptin, antipain, chymostatin, and pepstatin A (all from Sigma) were added at a final concentration of 5 mg/ml each]. Cells were left on ice for 30 min, allowed to swell, and homogenized with 40 strokes using a Kontes glass homogenizer and pestle B. Supernatants were collected after centrifugation of homogenates at 14,000 g for 10 min at 4°C and then stored at −70°C for the Western blot analysis.
Western blot analysis.
Three- to five-day-old cultured chick cardiomyocytes were either treated with staurosporine (20 μM, 4 h) or subjected to our I/R protocol. At various time points during the I/R protocol, protein isolates were collected. Briefly, cells were scraped, pelleted, and lysed in 1% Triton X-100, 50 mM Tris (pH 7.5), 40 mM β-glycerophosphate, 100 mM NaCl, 2 mM EGTA, 50 mM NaF, 200 μM sodium vanadate, and 200 μM PMSF. Protein concentration was determined using the Bradford assay (Bio-Rad). Proteins from total cell lysates were resolved on 7.5 and 14% SDS-PAGE gels for detection of α-fodrin and the caspase-9 p10 subunit, respectively. Cytosolic extract proteins were run on 15% SDS-PAGE gel for cytochrome c detection. Separated proteins were electrophoretically transferred to nitrocellulose membranes. Proteins in the membranes were then immuno-blotted with antibodies to cytochrome c, caspase-9 p10 subunit, α-fodrin, or actin according to the vendor’s protocols. Blots were developed with enhanced chemiluminescence (Amersham Pharmacia Biotech; Piscataway, NJ) and auto-visualized by exposure to X-ray films (BioMax; Kodak, Rochester, NY).
Hoechst 33342 staining.
With the use of Hoechst 33342 staining for chromatin condensation and fragmentation of nuclei, nuclear morphology was monitored at intermittent times throughout I/R, and as a positive control in response to known apoptotic stimuli, such as staurosporine exposure. Three- to five-day-old cultured chick cardiomyocytes were either treated with staurosporine (20 μM, 4 h) or subjected to our I/R protocol for 1 h of ischemia alone or 1-h ischemia and 3-h reperfusion. Cells were then washed briefly in ice-cold PBS, fixed in 10% neutral buffered formalin for 10 min at room temperature, and washed again in ice-cold PBS. Coverslips were stored at 4°C until stained. Hoechst 33342 stain was applied to the coverslips for 2 min then washed three times in PBS. Coverslips were mounted on glass slides and stored at —20°C until viewed.
Data Analysis
For each experiment, a field of ~500 cells on one 25-mm coverslip was observed. Treatment and control groups were matched using additional coverslips containing cells isolated and cultured on the same day so as to eliminate variability due to cell batch. Results are reported as means ± SE; and two-tailed unpaired t-tests were performed as post hoc tests of significance, with P < 0.05 considered to be significant.
RESULTS
Induction of Accelerated Cell Death at Reperfusion Versus Ischemia
As seen in Fig. 1, cell death in cells exposed to 1-h simulated ischemia/3-h reperfusion was significantly higher than cells exposed to 4-h prolonged simulated ischemia, similar to results previously published (34). In addition, significant cell membrane blebbing occurred in cells at reperfusion after ischemia.
Fig. 1.
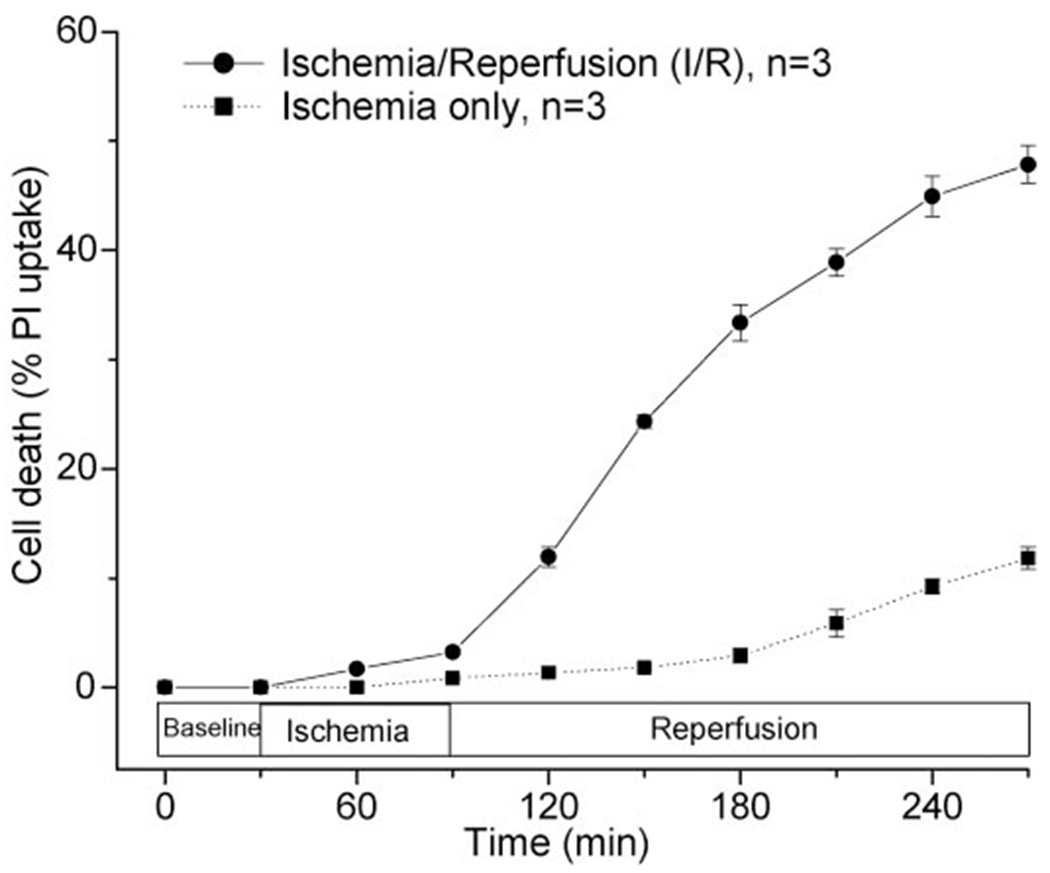
Timing of cell death during ischemia-reperfusion (I/R) vs. prolonged ischemia. Reperfusion of cardiomyocytes after 1 h of simulated ischemia induces rapid cell death to 47.8 ± 1.7% by 3-h reperfusion, with most cell death observed during the reperfusion phase (n = 3). Exposure of cells to ischemia alone resulted in significantly less cell death (P < 0.001) as measured by the exclusion dye propidium iodide (PI), with 11.8 ± 1.0% cell death by 4-h prolonged ischemia (n = 3).
Release of Cytochrome c and Activation of Caspase-9 at Reperfusion
Increased levels of cytochrome c were detected in the cytosolic fractions as early as 5 min into reperfusion and continued to increase ≤30 min reperfusion (see Fig. 2). We tested whether prolonged (4 h) ischemia alone would result in some increase in cytochrome c release. As seen, the level of cytochrome c release was not significantly different from controls or cells exposed to 1-h ischemia without reperfusion. This lack of cytochrome c release corresponded to less cell death as reflected by significantly less PI uptake in nonreperfused cells (see Fig. 1).
Fig. 2.

Cytochrome c (Cyto c) detection and caspase 9 cleavage in the chick cardiomyocyte cytosol during I/R. A: cytochrome c was measured in cardiomyocyte cytosolic extract at 0, 5, 15, and 30 min into reperfusion after 60-min simulated ischemia and compared with actin levels (A). In addition, Cyto c was measured in control cells not exposed to ischemia and after prolonged ischemia ≤4 continuous hours (i.e., 240 min) (B). Cyto c release increased from baseline levels as early as 5 min into reperfusion, increasing significantly within the first 30 min of reperfusion. By contrast, even prolonged ischemia had little impact on Cyto c release. Data represent 7 Western blots. C: total protein extracts of cardiomyocytes were examined using an antibody that detects the caspase 9 (Casp-9) active subunit (p10) during ischemia and reperfusion phases (inset shows selected time points). At 60-min ischemia with 30-min reperfusion (i.e., 60 + 30), no detectable 10 kDa band could be detected. However, within 1 h after the start of reperfusion, after 1-h ischemia (i.e., 60 + 60), significant cleavage of Casp-9 could be observed. This cleavage continued to increase at 120-min and 180-min reperfusion after 60-min simulated ischemia. Data represent 3 Western blots.
To better understand the initiation of apoptosis during I/R injury, we examined caspase-9 activation via Western blot analysis using an antibody that detects the active p10 subunit. As seen in Fig. 2B, this subunit could not be detected at 30-min reperfusion but was detectable by 60-min reperfusion, increasing further at 2- and 3-h reperfusion. Equivalent protein loading was confirmed as before with actin labeling.
Cleavage of Caspase-3 Substrate α-Fodrin and Nuclear Morphology
Cleavage of α-fodrin, a major cell cytoskeleton component and target of caspase-3, has been observed during apoptosis induced by a variety of stimuli (7, 22, 26, 36). Two major fodrin fragments are usually seen in cell lysates. The 150-kDa fragment is often present in control (untreated) samples from cells (26) and represents the cleavage of fodrin at a hypersensitive site by low levels of endogenously active proteases (14). The 120-kDa fodrin cleavage product is specifically associated with caspase-3 activation during apoptosis (19, 24, 26).
As seen in Fig. 3A, compared with chick cardiomyocytes kept in media (lane 1), cells treated with staurospaurine (20 μM; 4 h) (lane 2) as a positive control demonstrated the caspase-3-specific fodrin cleavage product. This 120-kDa cleavage product was also seen in cells exposed to 1-h ischemia/3-h reperfusion (lane 4), but was not seen in cells either exposed to 1-h ischemia without reperfusion (lane 3), or cells exposed to the standard I/R protocol treated with zVAD-fmk (100 μM at first 30-min reperfusion) (lane 5). Figure 3B shows an additional time course of fodrin cleavage at 0, 1, 2, and 3-h reperfusion after 1-h simulated ischemia. After 1-h simulated ischemia, a 120-kDa band could be detected by 2-h reperfusion (see lane 1 + 2), but was not detectable at 0- or 1-h reperfusion (see lanes 1 + 0 and 1 + 1, respectively) or in control cells kept in media (see lane C). Nuclear staining with Hoechst dye showed that inhibition with zVAD-fmk blocked most of the nuclear condensation and fragmentation associated with I/R-induced apoptosis. As seen in Fig. 3C, Hoechst staining of cardiomyocytes exposed to staurospaurine (20 μM, 4 h) (Fig. 3C,2) revealed increased nuclear condensation and fragmentation (examples of normal nuclei highlighted by blank arrows; condensed/fragmented nuclei by solid arrows). Ischemia without reperfusion (Fig. 3C,3) had similar levels of condensed nuclear staining as control cells exposed to media only. However, cells exposed to 1-h ischemia/3-h reperfusion (Fig. 3C,4) had increased nuclear condensation and fragmentation, an outcome attenuated by zVAD-fmk (Fig. 3C,5).
Fig. 3.

Fodrin cleavage and Hoechst 33342 staining after I/R or staurosporine exposure. A: α-fodrin breakdown was studied in cardiomyocytes at baseline (control, lane 1) and exposed to the following conditions: staurosporine (20 μM, 4 h) (lane 2), 1-h ischemia without reperfusion (lane 3), standard 1-h ischemia plus 3-h reperfusion (lane 4), or I/R with benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk; 100 μM) treatment at reperfusion only (lane 5). The 120-kDa fodrin fragment is present in the staurosporine and I/R lanes only. The cleavage fragment seen in I/R cells is blocked by zVAD-fmk treatment, suggesting that caspase inhibition prevents reperfusion-induced cleavage of α-fodrin. The 150-kDa band can be detected even at baseline conditions, a result found by other investigators and reflecting attack at the fodrin protein site prone to cleavage by endogenous proteases. Data represent 3 Western blots. B: α-fodrin breakdown in cardiomyocytes was measured at sequential time points after reperfusion. Compared with cells incubated in media (control, see lane C) after 1-h simulated ischemia, the 120-kDa fodrin fragment could be detected as early as 2-h reperfusion (see lane 1 + 2), but could not be detected at 0 or 1-h reperfusion after simulated ischemia (see lanes 1 + 0 and 1 + 1, respectively). Data represent 4 blots. C: Hoechst 33342 staining of cardiomyocytes exposed to the same conditions listed in A was studied. Cells at baseline (C,1) or exposed to 1-h ischemia only (C,3) had few nuclei demonstrating condensation or fragmentation. However, cells exposed to staurosporine (20 μM; 4 h) (C,2) or I/R (C,4) had increased nuclear condensation and fragmentation. These nuclear apoptotic changes after I/R were abrogated by treatment with zVAD-fmk (100 μM) given at reperfusion only (C,5). Open arrows point out normal nuclear morphology, and closed arrows highlight condensed and fragmented nuclei. Data represent 5 coverslips stained at each time point.
Fig. 4.

Effect of zVAD-fmk on reperfusion injury. A: cardiomyocytes were perfused with normoxia buffer for 30 min and exposed to 60 min of simulated ischemia followed by 180 min of normoxia reperfusion. ZVAD-fmk (100 μM) was given during the first 30 min of reperfusion. zVAD-fmk treatment during reperfusion alone decreased cell death from 49 ± 1.4% (n = 4) to 25 ± 4.4% (n = 4) in treated cells (*P < 01) compared with the untreated control group. B: to test whether the protective effect seen in A was due to an antioxidant effect, intracellular oxidant stress was monitored with the intracellular probe 2’,7’-dichlorofluorescin diacetate (DCFH-DA, 5 μM). A burst of oxidant generation, suggested by the increase in oxidation of DCFH to the fluorescent product dichlorofluorescein (DCF), was seen within minutes after the start of reperfusion in untreated cells (n = 4). This increase in oxidant generation was not affected by zVAD treatment (n = 4), suggesting that reactive oxygen species (ROS) are not downstream of caspase activation in this model. a.u., Arbitrary units.
Fig. 5.

Effect of the negative control N-benzyloxycarbonyl Phe-Ala-fluoromethylketone (ZFA-fmk) peptide on reperfusion injury. ZFA-fmk was used as a negative control for zVAD-fmk, because it is a similar peptide without caspase inhibitory activity. ZFA-fmk (100 μM) was administered to cells during the first 30 min of reperfusion as zVAD-fmk was previously given. Observed cell death was 59.1 ± 10% cell death by 3-h reperfusion in treated cells (n = 5) compared with cell death in untreated cells of 45.0 ± 5.2% (n = 3) (P = 0.61).
Effect of Caspase Inhibitors Given Only at Reperfusion
Because reperfusion seemed to induce the caspase cascade, we tested whether the I/R injury seen in our model could be significantly attenuated with caspase inhibitors given only at the start of reperfusion. For these experiments, we attempted to maximize exposure of cells to caspase inhibitors without wasting inhibitor agents. To do this, we designed a protocol that prevented washing the inhibitor out of the chamber after it entered the Sykes-Moore perfusion chamber. Cells were treated with the caspase inhibitor during the first 15 min of reperfusion and then perfusion of the chamber was stopped for 15 min (allowing an additional 15 min of incubation with the inhibitor). Although flow is stopped in the chamber, the amount of oxygenated media with substrate far exceeds the volume of cells in the chamber and oxygen tension, as measured by an optical phosphorescence quenching technique (Oxyspot; Medical Systems), remains constant at normoxic conditions (results not shown). The perfusion of the chamber was then continued for the remainder of the 3-h reperfusion without added caspase inhibitor, allowing the inhibitor in the chamber to wash out. Matched, untreated cells were perfused in the same way (i.e., perfusion of the chamber was stopped at 15-min reperfusion) but without caspase inhibitor in the reperfusion solution.
As seen in Fig. 4A, the broad-spectrum caspase inhibitor zVAD-fmk (100 μM) decreased cell death from 49 ± 1.4% in untreated cells exposed to I/R (n = 4) to 25 ± 4.4% in treated cells at early reperfusion (n = 4, P < 0.01). This improved survival was associated with return of spontaneous contractions in 4 of 4 treated cell groups versus 0 of 4 untreated cells. As seen in the supplementary material online (25), the first three video segments (“Without Treatment”) show sequentially the same 50 × 70-μm field of untreated cardiomyocytes at baseline physiological conditions (“baseline”), 1 h into simulated ischemia (“1 hour after ischemia”), and 3 h into reperfusion (“3 hours after reperfusion”). In the subsequent three video segments (“Treatment with ZVAD-fmk”), a 50 × 70-μm field of cardiomyocytes treated with zVAD-fmk given at the start of reperfusion is followed through the same simulated I/R protocol. The cells appear similar to untreated cells during baseline conditions and after simulated ischemia before zVAD-fmk treatment, but after being treated with zVAD-fmk at the start of reperfusion, the return of spontaneous contractions are seen that persist into 3-h reperfusion (25).
Given that activated caspases can increase ROS generation (37), it is possible that some of the protection rendered by zVAD-fmk is due to an “antioxidant” effect, i.e., inhibiting caspases upstream of ROS production. In addition, as a peptide, it could conceivably act as an antioxidant scavenger. To test these possibilities, we studied whether zVAD-fmk given at reperfusion attenuates ROS generation. As seen in Fig. 4B, the burst of ROS, as measured by DCFH oxidation to DCF, occurs within 1–2 min and peaks at 5 min of reperfusion. This burst was unaffected by the caspase inhibitor, suggesting that caspase activation in this system is downstream of the reperfusion ROS burst. To further rule out a nonspecific effect, we also tested the effect of an inactive placebo control for zVAD-fmk. N-benzyloxycarbonyl Phe-Ala-fluoromethylketone (ZFA-fmk) is a small peptide cysteine protease inhibitor with no known anti-caspase activity that can be used as a negative control for zVAD-fmk (2). As seen in Fig. 5, a 100 μM dose of ZFA-fmk, the same dose as that used for zVAD-fmk, given at reperfusion did not offer any protection. Thus the protective effects of zVAD-fmk are likely to be due to its specific anti-caspase activity.
We further tested whether more specific caspase inhibitors would be protective. As seen in Fig. 6A, the caspase-7/3 inhibitor Ac-DQTD-CHO (50 μM) given only during the first 30 min of reperfusion (as before with zVAD-fmk) decreased cell death from 42.6 ± 4.2% in untreated cells (n = 3) to 13.0 ± 0.9% in treated cells (n = 3, P < 0.01). Again, this cell death was associated with the return of spontaneous contractions in three of three groups. As seen in Fig. 6B, the caspase-9 inhibitor zLEHD-fmk (20 μM) decreased cell death from 49.7 ± 1.6% in the untreated group (n = 3) to 25.0 ± 2.7% in cells treated with this inhibitor (n = 3, P < 0.01), with return of contraction in all experiments.
Fig. 6.

Effect of the caspase-7/3 inhibitor Ac-Asp-Gln-Thr-Asp-H (Ac-DQTD-CHO) and Casp-9 inhibitor benzyloxycarbonyl-Leu-Glu (Ome)-His-Asp-(Ome)-fluoromethyl ketone (zLEHD-fmk) on I/R-induced cell death. A: Ac-DQTD-CHO (50 μM) was administered into cells during the first 30 min of reperfusion as zVAD-fmk was previously given. Observed cell death was 13.0 ± 0.9% at 3-h reperfusion (n = 3) compared with cell death in untreated cells of 42.6 ± 4.2% (n = 3) (*P < 0.01); B: zLEHD-fmk (20 μM) given at reperfusion resulted in 25.0 ± 2.7% cell death by 3-h reperfusion (n = 3) compared with 49.7 ± 1.6% (n = 3) in corresponding control cells (*P < 0.01).
Effect of Antioxidants on Early and Late Apoptotic Events and Cell Death
Our results showing that DCFH oxidation was upstream of caspase activation (Fig. 4B) suggested that oxidants might play a role in initiating apoptosis during I/R. To test this hypothesis further, we studied the effect of the antioxidants MPG (400 μM, all course) and Phen (10 μM, all course), a thiol agent and metal chelator previously shown in our cardiomyocyte model to attenuate oxidant generation and cell death (30, 32). As seen in Fig. 7A, these antioxidants attenuated cell death from 56.0 ± 5.4% in untreated cells (n = 3) to 15.1 ± 2.5% cell death in cells treated with antioxidants (n = 3) (*P < 0.01). In addition, this decreased cell death corresponded to a significant attenuation of cytochrome c release into the cytosol within the first 30 min of reperfusion (see Fig. 7B) and decreased cardiomyocyte nuclear condensation and fragmentation as detected by Hoechst 33342 staining at 3-h reperfusion (Fig. 7, C–E). Collectively, these results suggest that oxidants generated by the events of I/Rhelp precipitate early and late events of intrinsic apoptosis.
Fig. 7.

Effect of antioxidants on early and late apoptotic events and cell death. A: treatment of cardiomyocytes with the antioxidants 2-mercaptopropionylglycine (MPG, 400 μM, all course) and 1,10-phenanthroline (Phen, 10 μM, all course) reduced cell death from 56.0 ± 5.4% in untreated cells (n = 3) to 15.1 ± 2.5% cell death in cells treated with antioxidants (n = 3) (*P < 0.01). Three of three cell groups treated with antioxidants exhibited return of synchronous contraction vs. 0/3 in untreated cells. B: Cyto c was measured in cytosolic extracts as previously described (Fig. 2) in the absence (−) or presence ( + ) of MPG and Phen at the concentrations indicated in A. Cyto c release was significantly increased by 30 min of reperfusion after 1-h simulated ischemia in the absence of antioxidants but inhibited at all time points in the presence of the antioxidants. Data represent 2 Western blots. C–E: Hoechst 33342 staining of cardiomyocyte nuclei demonstrated normal nuclear morphology (open arrow) before reperfusion (C) and nuclear condensation and fragmentation (D, closed arrows) after 1-h ischemia + 3-h reperfusion, which was blocked significantly by antioxidant (MPG & Phen) treatment (E). Data represent 4 coverslips stained at each time point.
DISCUSSION
Results of the present study suggest that the “intrinsic” (i.e., mitochondrial/cytochrome c) pathway of apoptosis may contribute significantly to the accelerated cell injury and death seen minutes to hours after ischemia in our cardiomyocyte model of simulated I/R. That cytochrome c release and other markers of apoptosis are not seen after simulated ischemia without reperfusion, suggests that the reperfusion phase is an important initiator of this pathway. Given previous work showing that a burst of ROS is generated 5 min after reperfusion in this model, the current work suggests the following sequence of events: after 1-h simulated ischemia, reoxygenation and its associated burst of ROS generation induces cytochrome c release into the cytosol as early as 5 min after the start of reperfusion. This cytochrome c release leads to activation of caspase-9 that is seen by 1-h reperfusion and increases ≤3-h reperfusion. Caspase-3-mediated fodrin cleavage is seen by 2- to 3-h reperfusion. This fodrin cleavage corresponds to increased membrane injury (visualized by PI uptake into cells, indicating cell death and membrane blebbing seen on phase microscopy) and failure of recovery of synchronous contractions. Caspase inhibitors, such as zVAD-fmk (given only at reperfusion) decrease this fodrin cleavage, nuclear condensation and fragmentation, and cell death and restore return of synchronous contractions by 3-h reperfusion. Thus intrinsic apoptosis appears to be initiated by reperfusion within minutes and contributes subsequently within the next 3 h to reperfusion injury in this cardiomyocyte system (Fig. 8).
Fig. 8.

Sequence of apoptotic events during I/R-induced death in cardiomyocytes. Our results suggest that significant apoptosis is initiated within minutes of the start of reperfusion and causes significant functional injury and death within 3-h reperfusion. ROS generation and Cyto c release are detected first (within 1–2 min, 5 min, respectively), followed by Casp-9 activation (detected at 1-h reperfusion), with fodrin cleavage, nuclear DNA condensation, and fragmentation seen by 2- to 3-h reperfusion. Cell death and contractile dysfunction can be inhibited by multiple caspase inhibitors given only at the start of reperfusion. Finally, the antioxidants MPG plus Phen decrease Cyto c(⚫) release, nuclear condensation/fragmentation, and cell death. In addition, these interventions restore spontaneous contractile activity not seen in untreated cells.
Cytochrome c release in cardiac cells has been recognized to be a feature of ischemic injury for over two decades (20). However, the role of cytochrome c-induced apoptosis in mediating ischemic injury in cardiac cells has been demonstrated only in the last few years. Bialik et al. (3) showed that exposure of neonatal rat cardiomyocytes to simulated ischemia using serum-free, glucose-free media containing 2-deoxyglucose resulted in cytochrome c release into the cytosol by 18 h of deprivation, resulting in activation of caspase-9, caspase-3, and internucleosomic cleavage of DNA. In addition to glucose and serum deprivation, hypoxia is another component of ischemia. Tanaka et al. (28) showed that neonatal rat cardiomyocytes exposed to hypoxia displayed DNA fragmentation as early as 12-h exposure, and this effect was associated with an up-regulation of Fas mRNA levels. However, cytochrome c release was not studied. In adult rat cardiomyocytes, Moissac et al. (23) found that just 1-h hypoxia stimulated cytochrome c release into the cytosol and activation of caspase-3. In addition, results from a rabbit heart Langendorff perfusion model of I/R showed that cytochrome c release into the cytosol could be detected in heart tissue as early as 15 min of reperfusion after 30-min global ischemia (10).
Our results are consistent with these studies and extend them in several ways. This is one of the first studies of a confluent cardiomyocyte system (embryonic or neonatal) displaying release of cytochrome c in a time frame similar to that seen in adult cells or perfused organ systems. Rather than occurring after a number of hours, the cytochrome c release was observed minutes after the start of reperfusion after exposure to just 1-h simulated ischemia. This may be due to the combined use of hypoxia (PO2 < 5 Torr) and metabolic inhibition used in our system to more closely simulate in vivo ischemia. This is also one of the first studies to test specifically whether conditions of ischemia alone versus those of reperfusion initiate the cytochrome c release. Gottlieb et al. (13) used in vivo coronary artery occlusion in rabbit hearts to induce infarction, and they detected significant nucleosome DNA laddering after 30 min of ischemia followed by 4 h of reperfusion. However, they could not detect any DNA laddering in hearts exposed to continuous ischemia for 4.5 h without reperfusion. The present study appears to confirm this work in a cellular model by demonstrating earlier events of apoptosis (especially cytochrome c release), only after reperfusion of ischemic cells. In our study, it would have been quite possible that cytochrome c release all occurred during ischemia, and reperfusion simply provided the needed milieu to complete caspase activation and cell death. This study highlights the possible importance of intervening quickly during the first minutes of reperfusion to prevent additional injury from apoptosis.
In addition to studying the timing of initiation of the intrinsic pathway of apoptosis in our cardiomyocyte model, we also attempted to determine the role of subsequent caspase activation in mediating further injury after ischemia. Given that the release of cytochrome c into the cytosol was observed within 15 min after reperfusion, it is likely that subsequent activation of caspases contributes to the cell injury and death during the 3-h reperfusion phase studied after ischemia. Timing of caspase-3 activation as detected by fodrin cleavage on Western blot analysis suggests that by 2-h reperfusion enough caspase-3 activity had occurred for fodrin cleavage to be detected. It is quite possible that caspase-3 activity was increased before this time but was simply not detected by this assay. Our results using three different caspase inhibitors given only at reperfusion, each able to decrease cell death significantly and restore spontaneous synchronous contractions, would suggest that apoptosis is a significant component of “reperfusion injury” and that reperfusion may not be too late to undo some of the injury of I/R.
Our results are similar to those of Holly et al. (17) who showed that the myocardial infarct size in a rabbit model of 30-min coronary occlusion followed by 3-h reperfusion could be decreased by ~31% using caspase inhibition. In their model Holly et al. (17) report evidence of caspase-2, −3, and −7 activation in regions of the heart exposed to I/R. Our results extend this work by examining the temporal relationship of the ischemia and reperfusion phases to both cytochrome c release and evidence of caspase activation. In addition, their in vivo study gave the caspase inhibitor treatment both before coronary occlusion (i.e., pretreatment) and at the time of reperfusion. By contrast, we were able to protect our cardiomyocytes with treatment given only at the start of reperfusion. The prevention of cell death and loss of contractile function in cells treated with caspase inhibitors just at reperfusion suggests that significant apoptotic cell death occurs after reperfusion of ischemic tissue and causes a loss of contractile function that can be blocked by treatment in the first 3 h of reperfusion. Because caspase inhibitors did not block all cell death in our system, it is possible that cell death due to other events, such as oncosis/necrosis also play a role in cell injury and death in our model of I/R. The goal of this present study was not to determine the exact contribution of apoptosis, but to ask simply whether it plays an important role and if so, its timing during simulated ischemia versus reperfusion.
The relationship among the events of I/R, ROS generation, and apoptosis are not well defined. Although oxidative stress can induce cardiac myocyte apoptosis in vitro (11, 35), it is possible that ROS generation is also a later event of apoptosis. Many conditions and agents that induce apoptosis later increase ROS generation (6). In addition, events such as cytochrome c release may increase free radical generation due to dysfunctional electron transfer within the mitochondria. Further work will be needed to clarify this relationship in this model between ROS and cytochrome c release.
The present study has several potential limitations. First, clinical ischemia may be quite different from the simulated ischemia we use. Unfortunately, there is currently no accepted standard that constitutes a clinically relevant “simulated ischemic exposure” for cells. Simulating the ischemic environment of the extracellular fluid that bathes the cells is quite complex due to the fact that there are alterations in many factors (O2, CO2, K+, glucose, lactate, lipid oxidation products, other cellular products, etc.). Simulating all of these events is not currently possible. So, whereas the use of simulated ischemia is not perfect, we believe it recreates a number of the important components of clinical ischemia. Other systems that use different animals or organs also do not have one recipe for ischemia, because the amount of collateral flow among such animals varies considerably, such that ischemic conditions are likely different in these systems. The advantage of the cell culture system is that these factors can be measured and reliably reproduced for one particular cell type.
Another potential limitation is that an embryonic avian system may not model the events of apoptosis during I/R in a mammalian system. However, it is one of the few cardiomyocyte models reported to exhibit three important qualities reported in many intact mammalian heart systems: oxidant generation at reperfusion, preconditioning protection, and synchronous contractile function. The chick cardiomyocyte system exhibits the same level of protection as seen in animal models of preconditioning, that is, an almost 70% reduction in cell death resulting from I/R. In addition, this protection appears to be mediated by protein kinase C activation and mitochondrial ATP sensitive K+ (KATP) channel opening (30), both thought to be central processes for mediation of preconditioning protection in multiple mammalian species. Furthermore, some of the first work describing oncogenes, such as Bcl-2 were first described in the chicken, with expression in both the adult and embryonic heart (12, 27), and high degrees of homology to both human and mouse (27). Because this cellular system exhibits apoptotic injury during I/R and is capable of preconditioning against this injury, it may be a useful tool for further study of how preconditioning alters key mediators of apoptosis.
Acknowledgments
This study was supported by National Heart, Lung, and Blood Institute Grants HL-65558, HL-03779, and HL-68951.
REFERENCES
- 1.Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, and Kajstura J. Apoptosis and myocardial infarction. Basic Res Cardiol 93, Suppl 3: 8–12, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, and Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci 17: 553–562, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bialik S, Vincent CL, Drincic A, Miyata S, Wollowick AL, Srinivasan A, and Kitsis R. The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes. Circ Res 85: 403–414, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Bond JM, Herman B, and Lemasters JJ. Recovery of cultured rat neonatal myocytes from hypercontracture after chemical hypoxia. Res Commun Chem Pathol Pharmacol 71: 195–208, 1991. [PubMed] [Google Scholar]
- 5.Bossy-Wetzel E, Newmeyer DD, and Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 17: 37–49, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredesen D Neural apoptosis. Ann Neurol 38: 839–851, 1995. [DOI] [PubMed] [Google Scholar]
- 7.Brown T, Patil S, Cianci C, Morrow J, and Howe P. Transforming growth factor beta induces caspase-3-independent cleavage of alpha II-spectrin (alpha-fodrin) coincident with apoptosis. J Biol Chem 274: 23256–23262, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Carter WO, Narayanan PK, and Robinson JP. Intracellular hydrogen peroxide and superoxide anion detection in endothelial cells. J Leukoc Biol 55: 253–258, 1994. [DOI] [PubMed] [Google Scholar]
- 9.Cazals-Hatem DL, Louie DC, Tanaka S, and Reed JC. Molecular cloning and DNA sequence analysis of cDNA encoding chicken homologue of the Bcl-2 oncoprotein. Biochim Biophys Acta 1132: 109–113, 1992. [DOI] [PubMed] [Google Scholar]
- 10.Chen M, He H, Zhan S, Krejewski S, Reed JC, and Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem 276: 30724–30728, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Cook SA, Sugden PH, and Clerk A. Regulation of bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: association with changes in mitochondrial membrane potential. Circ Res 85: 940–949, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Eguchi Y, Ewert DL, and Tsujimoto Y. Isolation and characterization of the chicken bcl-2 gene: expression in a variety of tissues including lymphoid and neuronal organs in adult and embryo. Nucleic Acids Res 20: 4187–4192, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottlieb RA, and Burleson KO, Kloner RA, Babior BM, and Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94: 1621–1628, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris A and Morrow J. Proteolytic processing of human brain alpha spectrin (fodrin): identification of a hypersensitive site. J Neurosci 8: 2640–2651, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harth G, Andrews N, Mills AA, Engel JC, Smith R, and McKerrow JH. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. Mol Biochem Parasitol 58: 17–24, 1993. [DOI] [PubMed] [Google Scholar]
- 16.Haunstetter A and Izumo S. Apoptosis: basic mechanisms and implications for cardiovascular disease. Circ Res 82: 1111–1129, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Holly T, Drincic A, Byun Y, Nakamura S, Harris K, Klocke F, and Cryns V. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J Mol Cell Cardiol 31: 1709–1715, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Ikonomidis JS, Tumiati LC, Weisel RD, Mickle DA, and Li RK. Preconditioning human ventricular cardiomyocytes with brief periods of simulated ischeaemia. Cardiovasc Res 28: 12851291, 1994. [DOI] [PubMed] [Google Scholar]
- 19.Janicke R, Ng P, Sprengart M, and Porter A. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem 273: 15540–15545, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Kahles H, Goring GG, Nordbeck H, Preusse CJ, and Spieckermann PG. Functional behaviour of isolated heart muscle mitochondria after in situ ischemia. Polarographic analysis of mitochondrial oxidative phosphorylation. Basic Res Cardiol 72: 563–574, 1977. [DOI] [PubMed] [Google Scholar]
- 21.Kang P, Haunstetter A, Usheva A, and Izumo S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ Res 87: 118–125, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Martin S, O’Brien G, Nishioka W, McGahon A, Mahboubi A, Saido T, and Green D. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem 270: 6425–6428, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Moissac D, Gurevich RM, Zheng H, Singal P, and Kirshenbaum LA. Caspase activation and mitochondrial cytochrome c release during hypoxia-mediated apoptosis of adult ventricular myocytes. J Mol Cell Cardiol 32: 53–63, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Nath R, Huggins M, Glantz S, Morrow J, McGinnis K, Nadimpalli R, and Wanga K. Development and characterization of antibodies specific to caspase-3-produced alpha II-spectrin 120 kDa breakdown product: marker for neuronal apoptosis. Neurochem Int 37: 351–361, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Ohlfs B ZVAD [Online]. University of Chicago Emergency Medicine. http://www.erdoctor.org/zvad.wmv [2002, February 14]. [Google Scholar]
- 26.Shackelford D, Tobaru T, Zhang S, and Zivin J. Changes in expression of the DNA repair protein complex DNA-dependent protein kinase after ischemia and reperfusion. J Neurosci 19: 4727–4728, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takayama S, Cazals-Hatem DL, Kitada S, Tanaka S, Miyashita T, Hovey LR 3rd, Huen D, Rickinson A, Veerapandian P, and Krajewski S. Evolutionary conservation of function among mammalian, avian, and viral homologs of the Bcl-2 oncoprotein. DNA Cell Biol 13: 679–692, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka M, Ito H, Adachi S, Akimoto H, Nishikawa AH, Kasajima T, Marumo F, and Hiroe M. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyoyctes. Circ Res 75: 426–33, 1994. [DOI] [PubMed] [Google Scholar]
- 29.Teoh LK, Grant R, Hulf JA, Pugsley WB, Yellon DM. The effect of preconditioning (ischemic and pharmacological) on myocardial necrosis following coronary artery bypass graft surgery. Cardiovasc Res 53: 175–180, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Vanden Hoek TL, Becker LB, Shao Z, Li C, and Schumacker PT. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circ Res 86: 534–540, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Vanden Hoek TL, Becker LB, Shao Z, Li C, and Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem 273: 18092–18098, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Vanden Hoek TL, Li C, Shao Z, Schumacker PT, and Becker LB. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol 29: 2571–2583, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Vanden Hoek TL, Shao Z, Li C, Schumacker PT, and Becker LB. Mitochondrial electron transport can become a significant source of oxidative injury in cardiomyocytes. J Mol Cell Cardiol 29: 2441–2450, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, and Becker LB. Reperfusion injury in cardiac myocytes after simulated ischemia. Am J Physiol Heart Circ Physiol 270: H1334–H1341, 1996. [DOI] [PubMed] [Google Scholar]
- 35.von Harndorf R, Li P, and Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 99: 2934–2941, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Wang K, Pasmantur R, Nath R, McGinnis K, Whitton M, Talanian R, Glantz S, and Morrow J. Simultaneous degradation of alpha II-and beta II-spectrin by caspase-3 (CPP32) in apoptotic cells. J Biol Chem 273: 22490–22497, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Xia T, Jiang C, Li L, Wu C, Chen Q, and Liu S. A study on permeability transition pore opening and cytochrome c release from mitochondria, induced by caspase-3 in vitro. FEBS Lett 510: 62–66, 2002. [DOI] [PubMed] [Google Scholar]