

Abstract
In industrial and military settings, individuals who suffer from one episode of acoustic trauma are likely to sustain another episode of acoustic stress, creating an opportunity for a potential interaction between the two stress conditions. We previously demonstrated that acoustic overstimulation perturbs the cochlear immune environment. However, how the cochlear immune system responds to repeated acoustic overstimulation is unknown. Here, we used a mouse model to investigate the cochlear immune response to repeated stress. We reveal that exposure to an intense noise at 120 dB SPL for 1 hour activates the cochlear immune response in a time-dependent fashion with substantial expansion and activation of the macrophage population in the cochlea at 2-days post-exposure. At 20-days post-exposure, the number and pro-inflammatory phenotypes of cochlear macrophages have significantly subsided, but have yet to return to homeostatic levels. Monocytes with anti-inflammatory phenotypes are recruited into the cochlea. With the presence of this residual immune activation, a second exposure to the same noise provokes an exaggerated inflammatory response as evidenced by exacerbated maturation of macrophages. Furthermore, the second noise causes greater sensory cell pathogenesis. Unlike the first noise-induced damage that occurs mainly between 0 and 2 days post-exposure, the second noise-induced damage occurs more frequently between 2 and 20 days post-exposure, the period when secondary damage takes place. These observations suggest that repeated acoustic overstimulation exacerbates cochlear inflammation and secondary sensory cell pathogenesis. Together, our results suggest that the cochlear immune system plays an important role in modulating cochlear responses to repeated acoustic stress.
Keywords: Noise-induced hearing loss, Acoustic overstimulation, Immune cells, Macrophages, Inflammation, Cochlea
Introduction
According to the National Institute for Occupational Safety and Health (NIOSH), noise-induced hearing loss (NIHL) is one of the most common occupational injuries in the United States (Basner et al., 2014; Dobie, 2008). The World Health Organization (WHO) estimates that 10% of the world’s population is at risk of NIHL, and one-third of all cases of hearing loss can be attributed to noise exposure (Le et al., 2017; Oishi et al., 2011). NIHL can be caused by a single, intense exposure or by prolonged exposure to moderate-intensity noise (Henderson et al., 1995; Plontke et al., 2004; Saunders et al., 1991). Acoustic overstimulation damages cochlear sensory hair cells (Harris et al., 2006; Hu et al., 2002; Hu et al., 2000; Saunders et al., 1985; Tanaka et al., 2009), spiral ganglion neurons (Kujawa et al., 2006; Puel et al., 1998) and cells in the cochlear lateral wall (Gratton et al., 1992; Hirose et al., 2003; Li et al., 2017; Ulehlova, 1983), leading to hearing loss and impaired auditory processing (Frisina et al., 2001; Khullar et al., 2011; Ohlemiller et al., 2008).
Repeated acoustic exposure is a common human health hazard. Individuals who receive one episode of acoustic trauma are likely to have future encounters. Prior acoustic injury likely affects the cochlear response to subsequent stressors; however, the interaction between the initial and subsequent exposures is complex. Subjects with pre-existing permanent threshold shift (PTS) display potentiated PTS upon subsequent noise exposure (Perez et al., 2004). However, in subjects with a recent history of temporary threshold shift (TTS), subsequent noise exposure causes less PTS than in subjects without prior noise-induced TTS (Mills, 1973). This phenomenon is known as conditioning-related protection (Campo et al., 1991; Henselman et al., 1994; Niu et al., 2002). Presently, the underlying mechanisms responsible for these interactions are unclear.
Inflammation has been implicated in the pathophysiology of acute cochlear stress including acoustic trauma (Abdallahi et al., 1999; Chistiakov et al., 2017; Frye et al., 2017; Fujioka et al., 2006; Kirkegaard et al., 2006; Nakamoto et al., 2012; Vethanayagam et al., 2016; Wakabayashi et al., 2010; Yang et al., 2016). Inflammatory responses involve both immune molecules and immune cells. Cochlear tissues constitutively express a large group of immune molecules (Patel et al., 2013b; Yang et al., 2016), many of which are expressed in the supporting cells and cochlear lateral wall cells (Cai et al., 2014a; Fujioka et al., 2006). Acoustic injury augments the expression of immune molecules linked to the JAK-STAT signaling pathways (Wilson et al., 2014) as well as Toll-like (Vethanayagam et al., 2016) and NOD-like receptors (Leichtle et al., 2015). The cochlea also contains immune cells involved in homeostasis and acute inflammation (Hirose et al., 2005; Tornabene et al., 2006). Under physiological conditions, macrophages are present in multiple cochlear locations, including the lateral wall, basilar membrane, and neural regions (Hirose et al., 2005; Lang et al., 2006; Okano et al., 2008). After acoustic injury, this immune cell pool markedly expands as blood monocytes, precursors of tissue macrophages, migrate from the circulation into the cochlea (Frye et al., 2017; Hirose et al., 2005; Sautter et al., 2006; Tornabene et al., 2006; Wakabayashi et al., 2010), possibly attracted by cytokines such as CCL2 (MCP-1), IL-6, IL-1β, and TNF (Fujioka et al., 2006; Ichimiya et al., 2000). Upon reaching the inflamed cochlea, macrophages acquire an activated morphology and participate in local inflammation (Fredelius et al., 1990; Fujioka et al., 2006; Tornabene et al., 2006), phagocytosis (Fredelius et al., 1990; Hirose et al., 2017), and antigen presentation (Yang et al., 2015).
Previous studies have revealed that activation of the systemic immune system affects cochlear responses to subsequent stress (Hirose et al., 2014; Koo et al., 2015), suggesting that systemic immune activation can affect the cochlear immune state by potentially enhancing the cochlea’s susceptibility to subsequent pathological insults. Although noise-induced inflammation is sterile inflammation, it shares many cellular and molecular processes with pathogen-mediated inflammation (Medzhitov, 2008). Therefore, we hypothesized that a prior acoustic injury could prime the cochlear immune system and sensitize the cochlea to subsequent acoustic trauma. To test this hypothesis, we developed a mouse model of repeated stress and examined the cochlear inflammatory state and sensory cell pathogenesis to determine how prior immune activation affects cochlear responses to subsequent acoustic stress. We found that acoustic stress exacerbated the inflammatory response and sensory cell pathogenesis in cochleae with prior noise stress. This suggests that the cochlear immune state could play an important role in cochlear susceptibility to subsequent injury.
Methods
Subjects
C57BL/6J and B6.129P2(Cg)-Cx3Cr1tm1Litt/J mice (Stock number: 005582) (The Jackson Laboratory, Bar Harbor, ME, USA) were used in the study. B6.129P2(Cg)-Cx3cr1tm1Litt/J mice, also known as Cx3cr1GFP/GFP mice, express a green fluorescent protein (GFP) under the control of the endogenous Cx3cr1 locus in monocytes, macrophages, and a subset of dendritic and natural killer cells. The replacement of Cx3cr1 with a GFP-coding gene renders these cells endogenously fluorescent (Jung et al., 2000). These mice were utilized to track the infiltration of monocytes to cochleae during inflammation. Heterozygous mice (Cx3cr1GFP/+) and wild-type littermates were created by cross-breeding B6.129P2(Cg)-Cx3cr1tm1Litt/J with C57BL/6J mice. An equal number of male and female animals were used in the study. All mice were 4–6 weeks old and were housed at the University at Buffalo Lab Animal Facility in a 12-hour light on and 12-hour light off (8 a.m.–8 p.m.) light cycle room. We limited the age range of the mice to 4–6 weeks because C57BL/6J mice develop age-related high-frequency hearing loss starting at 6 weeks of age (Someya et al., 2009). All procedures involving the use and care of the animals were approved by the Institutional Animal Care and Use Committee of the State University of New York at Buffalo.
Experimental Procedures
The basic experiment paradigm included the following steps: a baseline hearing evaluation, acoustic overstimulation, hearing function re-evaluation, sample collection and analyses of sensory cell lesions and cochlear immune status. Mice were randomly divided into 5 groups: control, N1–2d, N1–20d, N2–2d, and N2–20d group (Figure 1). Animals in the control group did not receive any noise treatment. Animals that received only the initial noise exposure were labeled as the N1 group and animals that received both the initial and the second noise exposure were labeled as the N2 group. The cochlear samples were collected 2- or 20-days after the initial or second noise exposure. N1–2d represents 2-days after the initial noise exposure, N1–20d represents 20-days after the initial noise exposure, N2–2d represents 2-days after the second noise exposure and N2–20d represents 20-days after the second noise exposure.
Figure 1. Schematic of group assignments, noise exposures, and sample collection times.
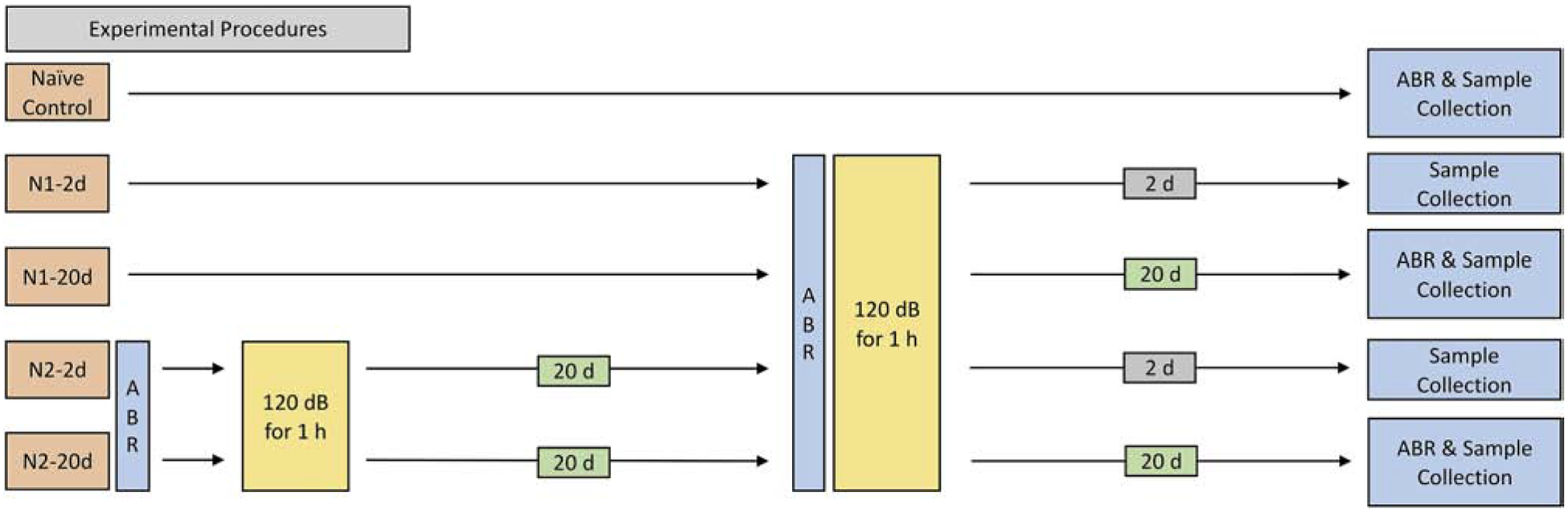
The experimental paradigm included two noise groups: first noise group (N1) and second noise group (N2). Noise injury in all groups was induced by a continuous noise (1–7 kHz) at 120 dB SPL for 1 hour. Samples were collected before and 2- or 20-days after noise exposure for subsequent analyses.
Auditory brainstem response (ABR)
ABRs were measured in both ears of each animal before, 2-days, and 20-days after noise exposure to assess auditory function. All ABR measurements were performed in a soundproof booth. Animals were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). Stainless steel needle electrodes were inserted sub-dermally over the vertex (active), posterior to the stimulated (reference) and non-stimulated (ground) ear. The animal body temperature was maintained at 37.5 °C using a heating system (Homeothermic Blanket Control Unit, Harvard Apparatus, Holliston, MA, USA) during testing.
The acoustic signals were generated and responses were processed using Tucker-Davis Technologies (TDT, Alachua, FL, USA) hardware and software. The ABRs were elicited with tone bursts of 4, 8, 16, and 32 kHz (0.5 ms rise/fall Blackman ramp, 1 ms duration, alternating phase) at the rate of 21/s. These signals were calibrated using a sound level meter (824, Larson Davis, 1/2” microphone). The evoked potentials were collected and sent to a preamplifier (RA16LA, TDT) via a flexible, low-noise cable. The output of the preamplifier was sent to a digital signal processing module (RX5–2, Pentusa Base Station, TDT) controlled by TDT software (BioSigRP, TDT). The average response to 1000 stimuli was obtained over a range of intensities at each frequency. The stimulus was presented at high intensities to elicit a clear response and then decreased first in 10-dB steps and then in 5-dB steps near the threshold. The threshold was defined as the lowest intensity at which an ABR wave I was reliably detectable.
Noise exposure
Animals were exposed to a continuous noise (1–7 kHz) at 120 dB SPL for 1 hour. For the repeated noise group, the time interval between the first and the second exposure was 20 days. This noise condition was selected because it causes permanent hearing loss and sensory cell death (Cai et al., 2014b), allowing us to investigate the cochlear immune response in the context of hair cell pathogenesis. The noise signal was generated using a Real-Time signal processor (RP2.1, Tucker Davis Technologies, TDT, Alachua, FL, USA). The signal was routed through an attenuator (PA5 TDT, Alachua, FL, USA), a power amplifier (Crown XLS 202, Harman International Company, Elkhart, IN, USA) and then to a loudspeaker (NSD2005–8, Eminence, Eminence, KY, USA) positioned 30 cm above the animal’s head. The noise level at the position of the animal’s head in the sound field was calibrated with a sound level meter (LD-PCB, model 800 B, APCB Piezotronics Div., Larson Davis, Depew, NY, USA), preamplifier (LD-PCB, model 825, Larson Davis, Depew, NY, USA), and condenser microphone (LDL 2559, Larson and Davis, Depew, NY, USA). The mice were individually exposed to the noise in a cylindrical holding cage with an approximate diameter of 5.5 cm and a height of 6.5 cm.
Cochlear tissue collection
Cochleae were harvested before, 2-days or 20-days after the first or second noise exposure. The animals were sacrificed by CO2 asphyxiation and subsequently decapitated. The cochleae were quickly removed from the skull. The collected cochleae were processed for either ex vivo staining, PCR analysis, or fixation for subsequent analyses of sensory cell pathology or cochlear immune status.
Assessment of sensory cell damage
Our procedures for assessing sensory cell damage have been reported previously (Yang et al., 2015). Cochleae were fixed overnight with 10% buffered formalin at 4 °C. The sensory epithelium was carefully dissected, and the organs of Corti were collected. Actin, which is abundantly expressed in the stereocilia and cuticular plate, was stained with Alexa Fluor 488- or 594-labeled phalloidin (1:75, Applied Biosystems, Foster City, CA, USA) in 10 mM PBS for 30 minutes at room temperature.
For assessment of hair cell nuclei, the tissues were stained with TO-PRO®−3 (diluted 1:1,000 in 1 μM PBS) for 30 minutes or 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI) (1 μg/ml in PBS) for 10 minutes. Afterward, the tissues were rinsed in PBS and then imaged using a fluorescence microscope (Leica Z6 APO Manual MacroFluo, 10× objective lens) equipped with a Leica DFC digital camera.
Real-time quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to determine the transcriptional expression of the following genes: Ccl2, Tnf-α, Il1ß, and Ccl7. Gene expression assays were performed for Ccl2 (Mm00441242_m1), Tnf-α (Mm00443258_m1), Il1ß (Mm00434228_m1), and Ccl7 (Mm00443113_m1). Animals were sacrificed, and the cochleae were quickly collected. The cochleae were placed in an RNA stabilization reagent (RNAlater, Qiagen, Valencia, CA, USA) following the dissection to collect the sensory epithelium and lateral wall. The collected tissues were transferred to RNase-free PCR tubes and stored at −80 °C before RNA extraction. Tissue from one cochlea was used to generate one sample.
Total RNA was extracted using the RNeasy Micro Kit (Qiagen) as per manufacturer’s instruction. The concentration and quality of isolated total RNAs were assessed using the NanoDrop 1000 (ThermoFisher Scientific). The isolated total RNAs were reverse transcribed into cDNA using a High Capacity cDNA Reverse Transcription Kit (SuperScript™ VILO™ MasterMix, Invitrogen, Carlsbad, CA, USA). The transcriptional expression levels of the target genes were examined using pre-developed TaqMan gene expression primer/probe assays (Applied Biosystems, Foster City, CA, USA). Synthesized cDNA was mixed with a target gene probe and a TaqMan Universal PCR Master Mix (ThermoFisher Scientific) and transferred to a 96-well plate. RT-qPCR was performed on a CFX Connect Real-Time PCR detection system (Bio-Rad, Hercules, CA, USA). Pre-developed GAPDH and RPL13A gene expression assays (Applied Biosystems, Foster City, CA, USA) were used as endogenous controls. Analysis of relative gene expression data was completed with a previously reported standard 2^ΔΔCt method (Livak et al., 2001).
Immunolabeling for immune cell identification and immune molecule expression
Cochleae were fixed with 10% buffered formalin. After dissection, whole-mount preparations were treated with 0.5% Triton X-100 to permeabilize the cells for 30 minutes at room temperature, and then a blocking buffer (pH 7.4) for 1 h at room temperature. The tissues were subsequently incubated overnight at 4 °C with one or two selected primary antibodies: goat anti-CD45 polyclonal antibody (1:100, AF114, RD Inc., Minneapolis, MN, USA), rabbit anti-Iba1 monoclonal antibody (1:200, ab178846, Abcam Inc., Cambridge, MA, USA), anti-F4/80 monoclonal antibody (1:100, ab6640, Abcam Inc., USA), Anti-B220 monoclonal antibody (1:100, 14-0452-82, ThermoFisher Scientific), rat anti-Ly6C monoclonal antibody (1:100, 128001, BioLegend, San Diego, CA, USA), rat anti-mouse CD45R monoclonal antibody (1:100, 14-0452-82, ThermoFisher Scientific, Waltham, MA, USA), rat anti-mouse monoclonal antibody NIMP-R14 (1:100, sc-59338, Santa Cruz Biotechnology, Inc., Dallas, TX, USA), rabbit anti-CD4 polyclonal antibody (1:100, NBP1–19371, Novus Inc., Littleton, CO, USA), rat anti-CD8 monoclonal antibody (1:100, ab112159, Abcam Inc., Cambridge, MA, USA), rabbit anti-MCP1/CCL2 polyclonal antibody (1:100, ab25124, Abcam Inc., Cambridge, MA, USA), and rat anti-mouse CD68 monoclonal antibody (1:100, MCA1957GA, Bio-Rad Laboratories, Inc., Hercules, CA, USA).
After incubation with primary antibodies, the tissues were rinsed 3 times in PBS and incubated in the dark with one or two secondary antibodies for 2 hours at room temperature. The secondary antibodies included Alexa Fluor® 488, 568 or 594 donkey anti-goat, anti-rat, anti-mouse or anti-rabbit (Life Technologies, Grand Island, NY, USA).
The specificity of the primary antibodies used in this study was confirmed in our previous publications (Ding et al., 2018; Frye et al., 2018; Frye et al., 2017). To prevent false-positive identifications from non-specific labeling of secondary antibodies, some samples were only incubated with secondary antibodies. Under these conditions, no clear fluorescence was observed.
Immunolabeling of ganglion neurons and their fibers
Ganglion cells and their fibers were observed using an antibody against neurofilaments (Ding et al., 2011; Ding et al., 2013; Dong et al., 2018). Whole-mount preparations were incubated overnight at 4 °C with an anti-neurofilament antibody (rabbit anti-Neurofilament 200 antibody, N4142, Sigma-Aldrich, St. Louis, MO, USA) diluted in 1% Triton X-100 and 5% donkey serum in 0.1 M PBS (1:100). After incubation with the primary antibody, the tissues were rinsed 3 times with 10 mM PBS and incubated for 2 hours at room temperature with a secondary antibody (1:100 Alexa Fluor® donkey anti-rabbit IgG, Life Technologies, Grand Island, NY, USA) in 10 mM PBS. The specificity of the primary antibody was confirmed in our previous publications (Ding et al., 2011; Ding et al., 2013; Dong et al., 2018).
In vivo and Ex vivo tracking of circulating monocytes
Latex beads were used to track circulating anti-inflammatory monocytes (Olingy et al., 2017; Tacke et al., 2007) into the cochlea. Fluoresbrite® Polychromatic Red Microspheres (0.5 μm) were purchased from Polysciences, Inc. (Warrington, PA, USA). Using the paradigm illustrated in Figure 2, we administrated the latex beads 3 hours prior to noise exposure to track monocytes at the acute phase of noise injury or at 17–20 days after noise exposure to track monocytes at the recovery phase of noise injury. The latex beads were administered via retro-orbital injection. Prior to injection, the stock suspension (2.5% solids) was diluted (1:25) with sterile saline to generate the working suspension. A total of 250 μl suspension was injected with 125 μl per eye. The mice were anesthetized with continuous inhalation of isoflurane mixed with oxygen (2–3%, flow rate 1.5 L/min for induction and 0.8 L/min for maintenance). 0.5% Proparacaine Hydrochloride Ophthalmic Solution (Bausch & Lomb, Bridgewater, NJ, USA) was applied to the eyes as an analgesic before injecting. After the injection, a sterile petrolatum ophthalmic lubricant (Paralube) was applied to the eyes, and the mouse was subsequently placed back into its cage for recovery.
Figure 2. Schematic of latex bead administration, noise exposure, and sample collection times.
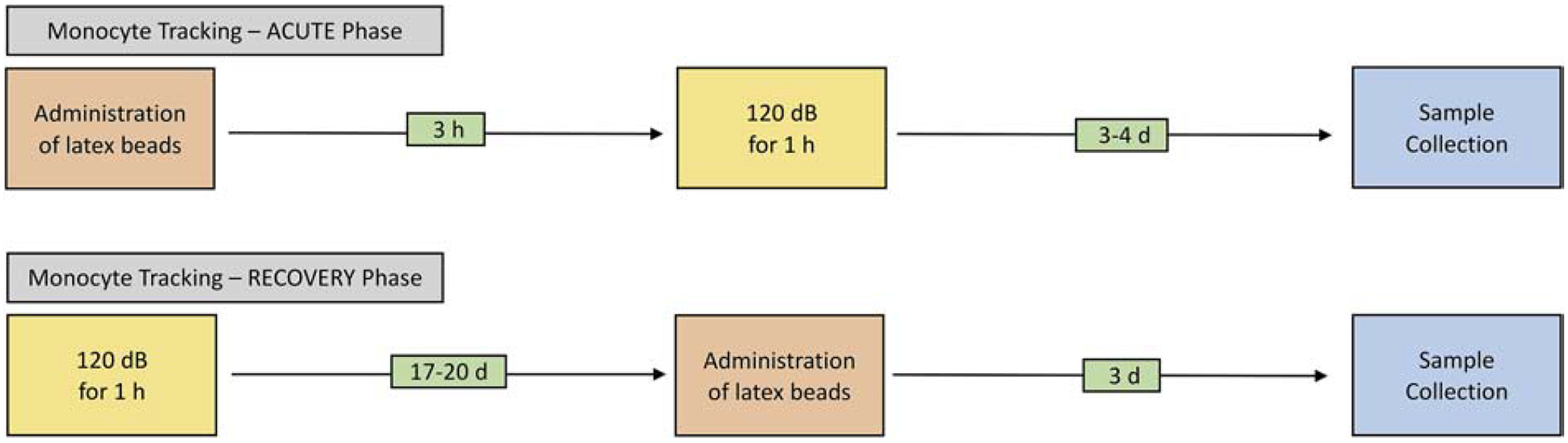
The experimental paradigm included two groups: monocyte tracking in the acute phase and recovery phase. Noise exposures for all groups was a continuous noise (1–7 kHz) at 120 dB SPL for 1 hour. Latex beads were administrated via retro-orbital injection either 3 hours before or 17–20 days after noise exposure. Samples were collected 3–4 days after noise exposure or 3 days after the latex bead administration.
The animals were sacrificed 3- or 4-days after noise exposure or 3 days after latex bead injection. The collected cochleae were fixed with 10% formalin overnight, and were dissected in 10 mM PBS to collect the sensory epithelia and lateral walls. The tissues were stained with an antibody against CD45 using the procedure described above. The stained cochleae were mounted on slides with antifade medium and examined with confocal microscopy.
Ex vivo macrophage tracking was performed in CX3CR1GFP/GFP mice to determine whether GFPlow macrophages observed in the cochleae during the acute phase of acoustic injury are able to engulf latex bead. The animals were decapitated at 1 day after noise exposure under CO2 gas anesthesia. The cochleae were quickly removed, and both round and oval windows were opened. Through the round window, we slowly perfused the cochlea with the latex bead suspension that was freshly prepared by diluting the stock solution (1:30) with RPMI 1640 medium (Life Technologies, Grand Island, NY, USA), a macrophage culture medium, supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA), 2 mM L-glutamine and 1% penicillin-streptomycin-neomycin solution (Life Technologies, Grand Island, NY, USA). The cochleae were placed in an incubator at 37 °C for 20 minutes. After incubation, the cochleae were perfused with cold PBS twice and fixed with 10% formalin overnight. The cochleae were dissected in 10 mM PBS to collect the sensory epithelia and lateral wall. The collected tissues were mounted on slides with antifade medium and examined with confocal microscopy.
Tissue observation and image acquisition
Cochlear tissues were examined first using an epifluorescence illumination microscope (Z6 APO apochromatic zoom system) equipped with a digital camera (DFC3000 G microscope camera) controlled by Leica Application Suite V4 PC-based software (Leica Microsystems, Buffalo Grove, IL, USA). The entire length of the sensory epithelium, lateral wall, and osseous spiral lamina were photographed. For detailed observations, the samples were examined and photographed using a confocal microscope (LSM510 multichannel laser scanning confocal image system) and processed with ZEN 2012 (blue edition) image processing software (Zeiss, Thornwood, NY, USA) as previously reported (Cai et al., 2014b). For macrophage quantification and morphological analysis, Adobe Photoshop CS6 (version 13.0.1, Adobe Systems, San Jose, CA, USA) was used to enhance image contrast.
Quantitative analyses of macrophage morphology and distribution
Macrophages were identified based on their expression of CD45 (pan-leukocyte marker), Iba1 (macrophage-specific marker) and Ly6C (monocyte marker) combined with their unique morphologies. Using both bright-field illumination and epifluorescence illumination, we distinguished immune cells in regions of the osseous spiral lamina, basilar membrane and luminal surface of the scala tympani, as well as within and outside blood vessels.
Analyses of basilar membrane macrophages
Macrophage quantification:
Basilar membrane macrophages are defined as the macrophages on the surface of the scala tympani side of the basilar membrane. Macrophage-grams, generated using techniques described in our previous publications (Frye et al., 2018; Frye et al., 2017), were used to show the distribution of immune cells along the basilar membrane. The number of macrophages was quantified with consecutive 450 μm intervals along the entire length of the basilar membrane (approximately 6000 μm). The values were averaged to produce the group means per unit length from the apex to the base. Group means were acquired by averaging cell counts per unit length across the samples for each experimental group.
Morphological analysis of macrophages:
Our preliminary observation revealed an amoeboid transformation of macrophages on the surface of the basilar membrane in the middle and basal portion of the cochlea, leading to an increase in the soma size of the cells. This morphological change is a sign of macrophage activation (Buttini et al., 1996; McWhorter et al., 2013; Streit et al., 1988). To quantify the change, we selected the ten largest macrophages, which represent the mature state of the cell, from the middle region of the basilar membrane for analyses of cell circularity and length. These morphological indices were utilized to assess the level of morphological changes induced by noise exposure compared to the resting condition.
Cell Circularity:
This parameter was used to define how close an immune cell resembles a perfect circle. The calculation is derived from 4π (area/perimeter2), which has been integrated into a measure provided by Adobe Photoshop (Agley et al., 2012). Upon tracing the cell boundaries, the Record Measurements function of Adobe Photoshop provided the value of circularity with 1.0 indicating a perfect circle.
Cell Length:
Upon tracing the cell boundaries, the Record Measurements function also provided the length of each cell measured.
Quantification of osseous spiral lamina macrophages
Osseous spiral lamina macrophages are those residing in the neural region of the osseous spiral lamina. Quantification of these macrophages was performed by quantifying the number of cells present per length of 350 μm along the longitudinal axis of the osseous spiral lamina. The cells that we included for quantification ran radially toward the edge of the osseous spiral lamina and presented with a branched morphology. The mean for these counts was then computed to produce an average value per unit area in each cochlea. Group means were acquired by averaging cell counts per unit area across samples for each group.
Quantification of macrophages on the luminal surface of the scala tympani
Quantification of macrophages residing on the luminal surface of the scala tympani was performed by identifying the number of cells present in a 100 mm2 area. The mean for these counts was then computed to produce an average value per unit area for each cochlea. Group means were acquired by averaging cell counts per unit area across samples for each group.
Scanning electron microscopy (SEM)
Assessment of macrophage morphology using SEM has been previously reported by our laboratory (Dong et al., 2018). Animals were sacrificed, the cochleae were first fixed with 2% glutaraldehyde in 0.1 M phosphate buffer at room temperature for 1 h and then incubated overnight in the same fixative at 4 °C. The cochleae were decalcified with 10% ethylenediaminetetraacetic acid at 4 °C for 5 days and dissected to expose the scala tympani side of the basilar membrane. The tissues were dehydrated through a graded series of ethanol (30%, 50%, 70%, 85%, 95%, and 100%) and finally air-dried with 100% hexamethyldisilazane. Samples were coated with evaporated carbon and gold in a high vacuum (Denton 502 Evaporator, Denton Vacuum, LLC, Moorestown, NJ, USA). Images were acquired on a Hitachi SU-70 SEM (Hitachi, Tokyo, Japan) at 2.0 KeV using an in-lens secondary electron detector at zero tilt, or a lower detector at 70° tilt. We examined several hundred macrophages in three conditions: control, 2- and 20-days post noise exposure (n=5 cochleae for each condition). Macrophages are identified based on their location, shape, size and surface textures.
Genotyping
To determine the genotype of F2 mice obtained from the intercross of F1 hybrids (B6.129P-Cx3cr1tm1Litt/J and C57BL/6J mice), a small piece of tissue (approximately 2 mm) from the tail was collected. Total DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) as per manufacturer’s instruction. The concentration of isolated DNA was measured using NanoDrop 1000 (ThermoFisher Scientific, Waltham, MA, USA). The following primers were used for polymerase chain reaction (PCR): 5’- GTC TTC ACG TTC GGT CTG GT-3’ (wild-type forward); 5’ CCC AGA CAC TCG TTG TCC TT-3’ (common); 5’-CTC CCC CTG AAC CTG AAA C-3’ (mutant forward). Extracted DNA was mixed with the primers and Sigma JumpStart TM Taq Ready Mix (Sigma Aldrich, St. Louis, MO, USA). PCR temperature-cycling parameters were as follows: initial denaturation at 94°C for 3 minutes followed by 35 cycles of amplification of the target genes. Each amplification cycle consisted of denaturation of the target genes at 94 °C for 30 seconds, primer annealing at 60°C for 30 seconds and primer extension at 72 °C for 2 minutes. PCR was performed on a MyIQ Two-Color Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA).
PCR products were used for standard electrophoresis. Equivalent protein (15 μg protein/lane) and a pre-stained molecular weight marker (SeeBlue Plus2 Pre-Stained Protein Ladder, ThermoFisher Scientific, Waltham, MA, USA) were loaded into the wells of 2% agarose gels in a mini-gel apparatus (Mini-PROTEAN II, Bio-Rad, Hercules, CA, USA). Electrophoresis was conducted at 100 V for 45 minutes, and then the bands were examined with ultraviolet transillumination. The presence of a single band at 410 bp indicated homozygous mice, a single band at 500 bp indicated wild-type mice and both bands indicated heterozygous mice.
Data analyses
All data were presented as mean ± 1 standard deviation. Statistical analyses were performed using SigmaStat (version 10.0.1.25, San Jose, CA, USA). Group means were statistically compared with either a one- or two-tailed Student’s t-test or a one-way, or two-way ANOVA. An α-level of 0.05 was chosen to denote significance for all statistical tests. Normative data and equal variance tests were performed for all statistical analyses. If these two criteria were not satisfied, non-parametric tests were performed. Multiple comparisons were performed using either the Bonferroni t-test or the Mann Whitney U-test.
Results
Auditory dysfunction and cochlear damage following the first and second acoustic overstimulation
To generate acoustic injury, we exposed mice to an intense noise at 120 dB SPL for 1 hour (N1) and examined the functional and structural impact of this noise exposure. ABR thresholds were measured before and 20-days after the noise exposure. Before the noise exposure, the thresholds ranged from 18.8 ± 3.9 dB to 31.6 ± 4.5 dB among the four tested frequencies. At 20-days post-exposure, these thresholds were elevated and ranged from 48.8 ± 4.7 dB to 65.3 ± 9.0 dB among the tested frequencies (Fig. 3A, two-way ANOVA, F (3, 120) = 1.242, P < 0.001; Tukey test, P < 0.001 for all pairwise comparisons). Subsequently, sensory cells were quantified at 20-days post-exposure. In comparison to control cochleae where only a few missing OHCs were scattered along the sensory epithelium, a significant number of missing OHCs were found in the basal portion of the cochlea 20-days post-exposure (Fig. 3B). The increase was statistically significant (Student’s t-test, t (8) = −18.156; P < 0.001; Fig. 3C).
Figure 3. Comparison of ABR thresholds and sensory cell loss before and after the initial and the second noise exposure.
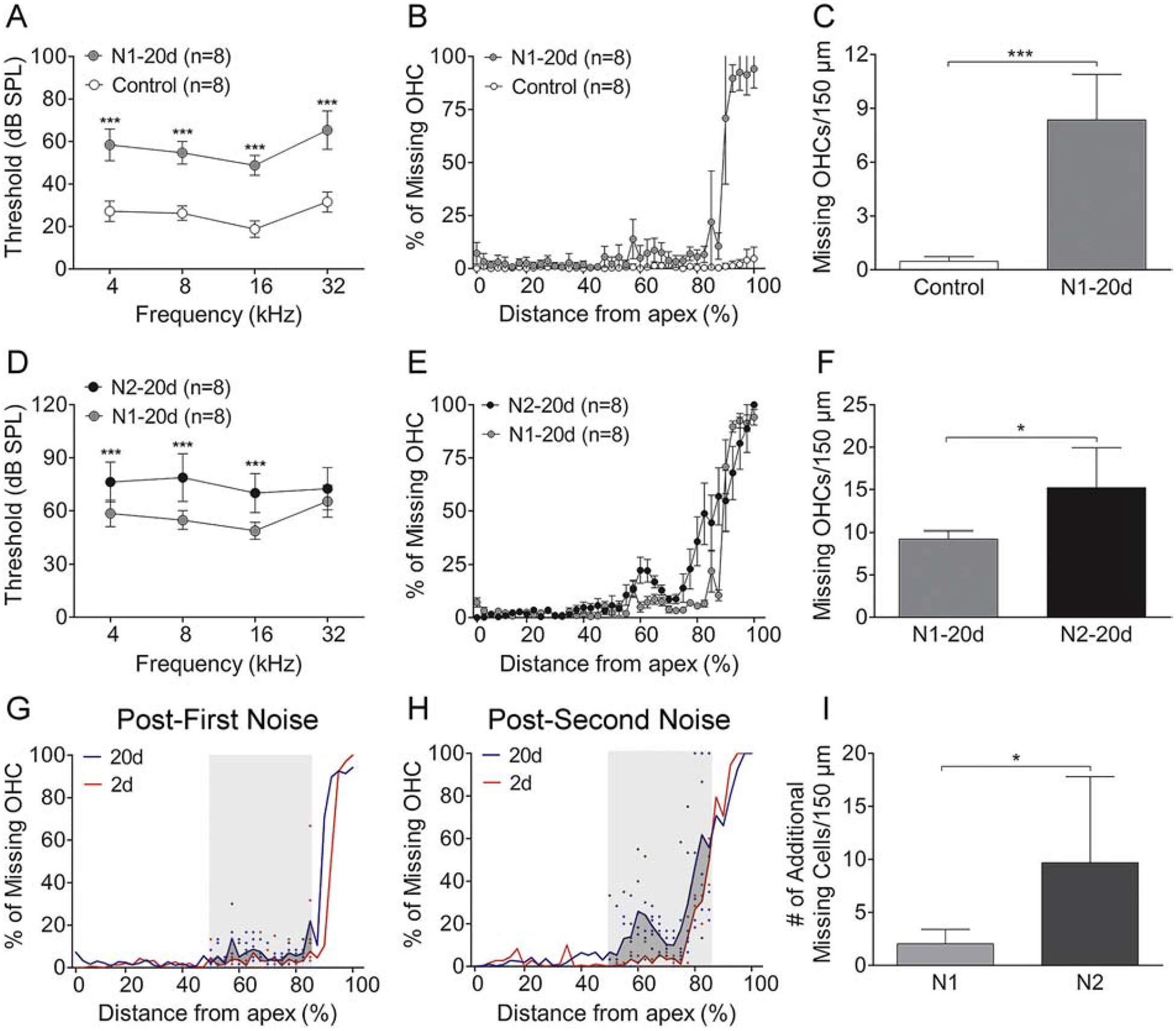
A. Comparison of mean ABR thresholds at the four tested frequencies before and 20 days after first noise exposure. Relative to the baseline thresholds, the thresholds are significantly elevated at 20 days after noise exposure (*** indicates P < 0.001). B. Cochleogram showing the distribution of OHC lesions along the basilar membrane. Notice that OHC lesions are confined in the basal end of the cochlea. C. Comparison of the average number of missing OHCs between the control and the noise group. There is a statistically significant increase in the number of missing OHCs per 150 μm at 20 days after noise exposure (* indicates P < 0.05). D. Comparison of ABR thresholds at the four tested frequencies before and 20 days after the second noise. As compared with the thresholds measured before the second noise exposure, i.e., 20 days after the first noise, the thresholds are significantly elevated at 20 days after the second noise exposure. E. Cochleogram showing the further expansion of OHC lesions into the middle region of the cochlea after the second noise exposure. F. Comparison of the average number of missing OHCs per 150 μm before (i.e., 20-days after the first noise exposure) and 20 days after the second noise exposure. A significant increase in the number of missing OHCs is present after the second noise exposure (* indicates P < 0.05). G and H. Cochleogram showing the level and the distribution of OHC loss along the basilar membrane at 2 and 20 days after the first noise exposure (G) or the second noise exposure (H). Notice that the second noise exposure results in a marked expansion of OHC lesions in the middle region of the cochlea, approximately 50–85% distance from the apex (marked by the gray area in G and H). I. Comparison of the increase in the number of missing cells between the N1 and the N2 group quantified in the region of the delayed damage marked by the gray areas in G and H. The level of the increase, which reflects the number of additional missing cells generated between 2- and 20-days post-noise, is greater after the second noise exposure compared to that observed after the first noise exposure (* indicates P < 0.05). n = 8 cochleae for each group for all graphs in this figure.
To determine how the second noise exposure (N2) affected auditory function, we measured ABR thresholds before and 20-days after the second exposure. We found a further elevation of ABR thresholds 20-days after the second exposure (N2–20d). Using the thresholds measured immediately before the N2 exposure (N1–20d), we calculated the threshold shifts and found significant threshold shifts ranging from 10.0 ± 14.9 dB to 21.3 ± 14.6 dB (two-way ANOVA, F (3, 88) = 84.896, P < 0.001; Tukey test, P < 0.001 for 4 kHz, 8 kHz and 16 kHz; P = 0.063 for 32 kHz; Fig. 3D). The second exposure also caused further expansion of sensory cell lesions as compared with that observed 20-days after the first exposure. Again, the lesions were distributed mainly in the basal portion of the cochlea (Fig. 3E), and the increase was statistically significant (Fig. 3F, Student’s t-test, t (9) = −2.753; P = 0.022).
To further analyze the expansion of sensory cell lesions, we divided the progression of sensory cell lesions into two stages: the early expansion that occurred during the first two days after the noise exposure and the delayed expansion that occurred between 2- and 20-days post-exposure. The numbers of missing OHCs were quantified for each stage. For the N1 group, we found an average of 6.9 ± 0.7 missing cells per 150 μm length of the sensory epithelium at 2-days post-exposure, and additional 2.5 ± 0.2 missing cells at 20-days post-exposure. The increase in the percentage of missing cells between 2-days and 20-days post-exposure represented only 26.3% of all missing cells. This result suggests that the majority of cell loss (73.6%) from the first noise exposure had already occurred by 2-day post-exposure. The second noise exposure resulted in 1.1 ± 0.2 additional missing cells per 150 μm of cochlear length 2-days after the second noise. This value was obtained by comparing the numbers of missing OHCs examined before the second noise (N1–20d) and 2-days after the second noise (N2–2d). Twenty days after the second exposure, there were 4.7 ± 1.2 additional missing cells per 150 μm of cochlear length, which accounts for 68.6% of total missing cells as the result of the second noise exposure. This result suggests that the majority of sensory cell damage as a result of the second noise exposure occurred between 2-days and 20-days post-exposure.
To further quantify the differences in the pattern of lesion growth, we compared the expansion pattern of secondary damage between the N1 and the N2 group. For this analysis, we quantified the number of missing OHCs in the cochlear region ranging from 50 to 85% of the distance from the apex because this region displayed a significant lesion growth during the period of 2- to 20-days after noise exposure (Fig. 3G–H). This analysis revealed that the number of missing cells generated during secondary damage was greater after the second noise exposure than that observed after the first noise exposure (Fig. 3I, Student’s t-test, t (10) = −2.277; P = 0.046). These observations suggest that repeated acoustic overstimulation causes greater delayed sensory cell damage.
Persistent retention of the macrophage population at 20-days post-exposure
Our previous study revealed a time-dependent expansion of the macrophage population in the cochlea following noise exposure (Yang et al., 2015). This change starts at 1-day post-exposure, peaks at 4-days, and then partially recovers at 10-days post-exposure. To examine whether the number of cochlear macrophages can further recover toward the pre-noise level with an extended resting time, we quantified the number of macrophages at 20-days after the first noise exposure. Macrophages revealed by CD45 immunolabeling were verified in a subset of samples by immunostaining for Iba1 and F4/80, two markers used for macrophage identification (Frye et al., 2018; Frye et al., 2017).
We quantified the macrophage number in three cochlear regions: basilar membrane (BM) osseous spiral lamina (OSL), and the luminal surface of the scala tympani (ST) (Fig. 4A–D). Basilar membrane macrophages reside on the tympanic surface of the basilar membrane (Fig. 4B). These cells are in close proximity to outer hair cells, one of the most vulnerable cell types to acoustic trauma. Osseous spiral lamina macrophages reside within the bony tunnels of the osseous spiral lamina (Fig. 4C), where the peripheral fibers of spiral ganglion neurons are located. The neural distribution of macrophages was confirmed by immunoreactivity with an anti-neurofilament antibody that labels nerve fibers (Arslan et al., 2011; Ding et al., 2013; Dong et al., 2018). We quantified the number of BM and OSL macrophages per 450 μm length along the longitudinal axis of the cochlea. For the luminal surface of the scala tympani (Fig. 4D), we quantified the macrophages per 100 mm2 of the surface area in the middle and basal portion of the cochlea. In our current study, we did not quantify immune cells within microvessels because the destiny of these cells was unknown in our study. We used the differential interference contrast (DIC) function offered by our LSM510 Zeiss confocal microscope to illustrate microvessels (Fig. 4E–F). In the three cochlear regions that we examined, we identified microvessels on the luminal surface of the scala tympani. Immune cells within microvessels were not included in our analysis.
Figure 4. Acoustic overstimulation provokes a time-dependent expansion of the macrophage population.
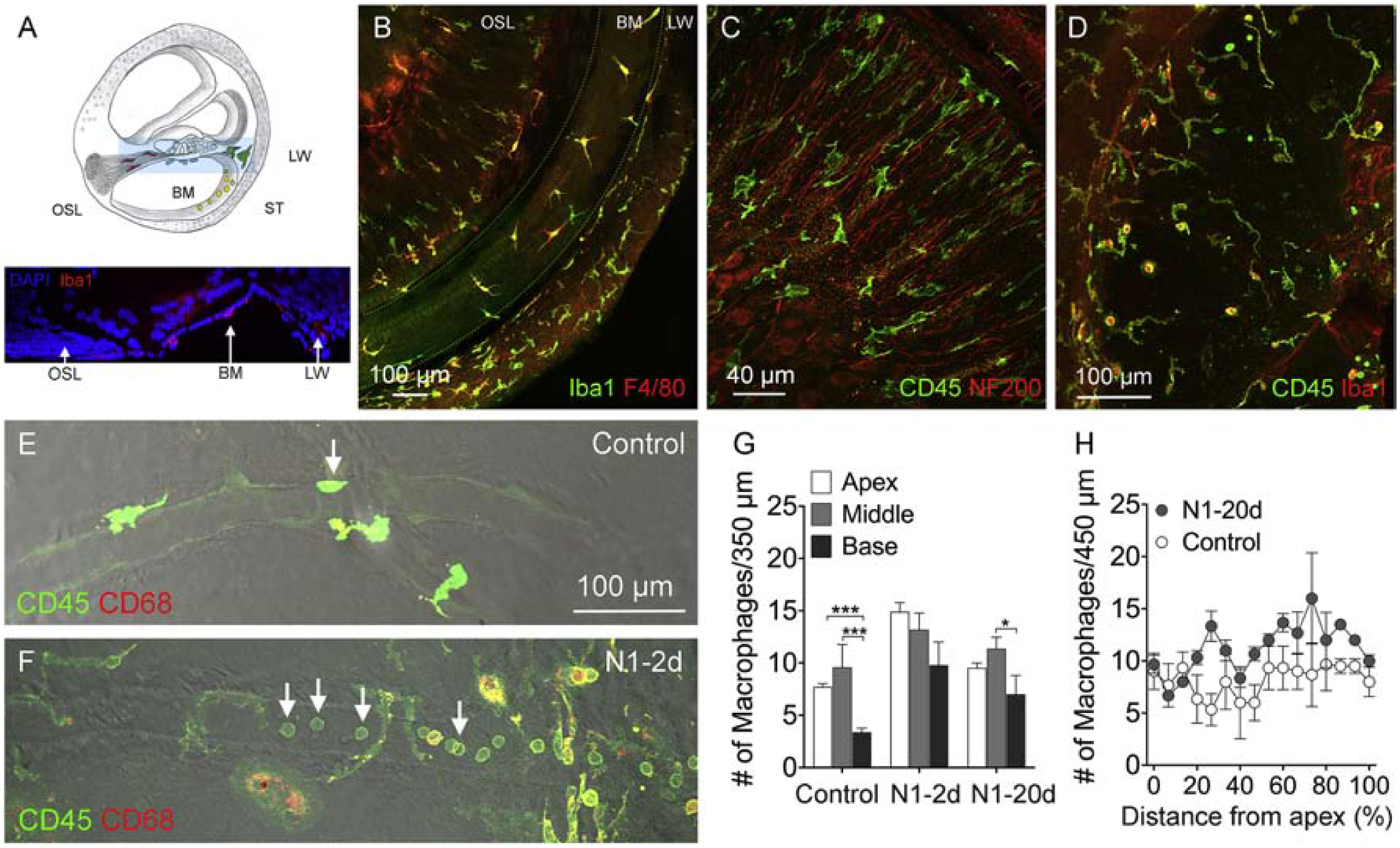
A. Schematic of a side view of the cochlea illustrating the locations of cochlear macrophages examined in the current study. The osseous spiral lamina (OSL) macrophages reside among the peripheral fibers of the spiral ganglia. Basilar membrane (BM) macrophages reside on the scala tympani side of the basilar membrane. Scala tympani (ST) macrophages reside on the luminal surface of the scala tympani cavity. Inset shows a side viewof a cochlea labeled with DAPI (blue) and Iba1 (red). B. BM-macrophages enclosed by the dotted lines. This tissue was stained with Iba1 and F4/80. C. OSL-macrophages among the peripheral bundles of ganglion neurons. The location of the neural fibers is confirmed using neurofilament staining (red). Macrophages in this region have a branched shape and are oriented in parallel to the peripheral fibers of ganglion neurons. D. Macrophages on the luminal surface of the scala tympani illustrated by CD45 and Iba1 immunoreactivities. E and F. Microvessels on the luminal surface of the scala tympani in a control cochlea (E) and a noise-damaged cochlea from the N1–2d group (F) illustrated using the differential inference contrast (DIC) function on the confocal microscope. Immune cells are illustrated by CD45 (green) and CD68 (red) immunostaining. Notice that the microvessel in the control cochlea lacks immune cells. In contrast, the microvessel in the noise-damaged cochlea contains many small, round immune cells. G. Comparison of the number of macrophages between the control, N1–2d, and N1–20d group from the three cochlear partitions, basilar membrane, osseous spiral lamina, and luminal surface of the scala tympani. When combining macrophage numbers for all examined cochlear partitions, the number of total macrophages is significantly increased at 2-days after the noise exposure and partially recovers at 20-days after noise exposure. (** indicates P < 0.01 and *** indicates P < 0.001). n = 6 biological replicates for each group. H. Macrophage-gram showing the distribution of basilar membrane macrophages along the cochlear spiral. The increase is more pronounced in the middle and basal regions of the cochlea. n = 4 biological replicates for each group.
We examined the changes in the macrophage number among the three groups (control, N1–2d, and N1–20d). Since the pattern of noise-induced changes was similar across the three cochlear regions, we combined the data from the three regions to generate the total number of macrophages for each cochlea. As compared with control cochleae, noise-damaged cochleae exhibited a marked increase in the number of macrophages 2-days post-exposure (9.1 ± 0.7 cells for the control group vs. 20.6 ± 0.9 cells for the 2-day group). This increase reduced to 13.8 ± 0.8 cells 20-days post-exposure, but the number is still higher than that observed in the control group (one-way ANOVA, F (2, 8) = 159.119, P < 0.001; Tukey test, P < 0.001 for control vs. N1–2d and N1–2d vs. N1–20d and P = 0.001 for control vs. N1–20d; Fig. 4G).
Further analysis of macrophage distribution along the basilar membrane revealed that the increase was confined largely to the middle and basal regions of the cochlea (approximately 30–90% from the apex, Fig. 4H), consistent with the major site of sensory cell lesions. Collectively, this multi-site analysis revealed that while the cochlear macrophage population has decreased from its peak value, the recovery was still incomplete at 20-days post-exposure.
Persistent activation of cochlear macrophages
Cochlear macrophages acquire an activated phenotype after acoustic trauma (Yang et al., 2015). To determine whether activated macrophages could regain their resting phenotype 20-days after the exposure, we examined both the morphology and the proinflammatory molecule expression of the basilar membrane macrophages. Morphology is an indicator of inflammatory activation (Davis et al., 1979; Neumann et al., 2009; Polliack, 1977; Streit et al., 1988) and changes in macrophage morphology occur during the acute phase of cochlear inflammation (Frye et al., 2018; Frye et al., 2017; Yang et al., 2015). CD45 immunostaining was used to illustrate macrophage morphology, and macrophage identity was confirmed with Iba1 immunoreactivity.
We evaluated the macrophages in the middle region of the cochlea because this region had significant secondary sensory cell pathogenesis as illustrated in the above section describing cochlear damage. In the control ears, tissue macrophages on the surface of the basilar membrane displayed tree-trunk morphologies with thick cell bodies and fine processes (Fig. 5A). Two days after noise exposure, macrophages with monocyte-like morphology (small in size and round in shape) appeared on the basilar membrane (Fig. 5B). At 20-days post-exposure, many macrophages acquired a flat, spread-out morphology or an amoeboid shape with multiple protrusions or fine and short dendritic projections (Fig. 5C). These morphological features are more evident in SEM images (Fig. 5D–F).
Figure 5. Acquisition of the activated morphology of basilar membrane macrophages after acoustic overstimulation.
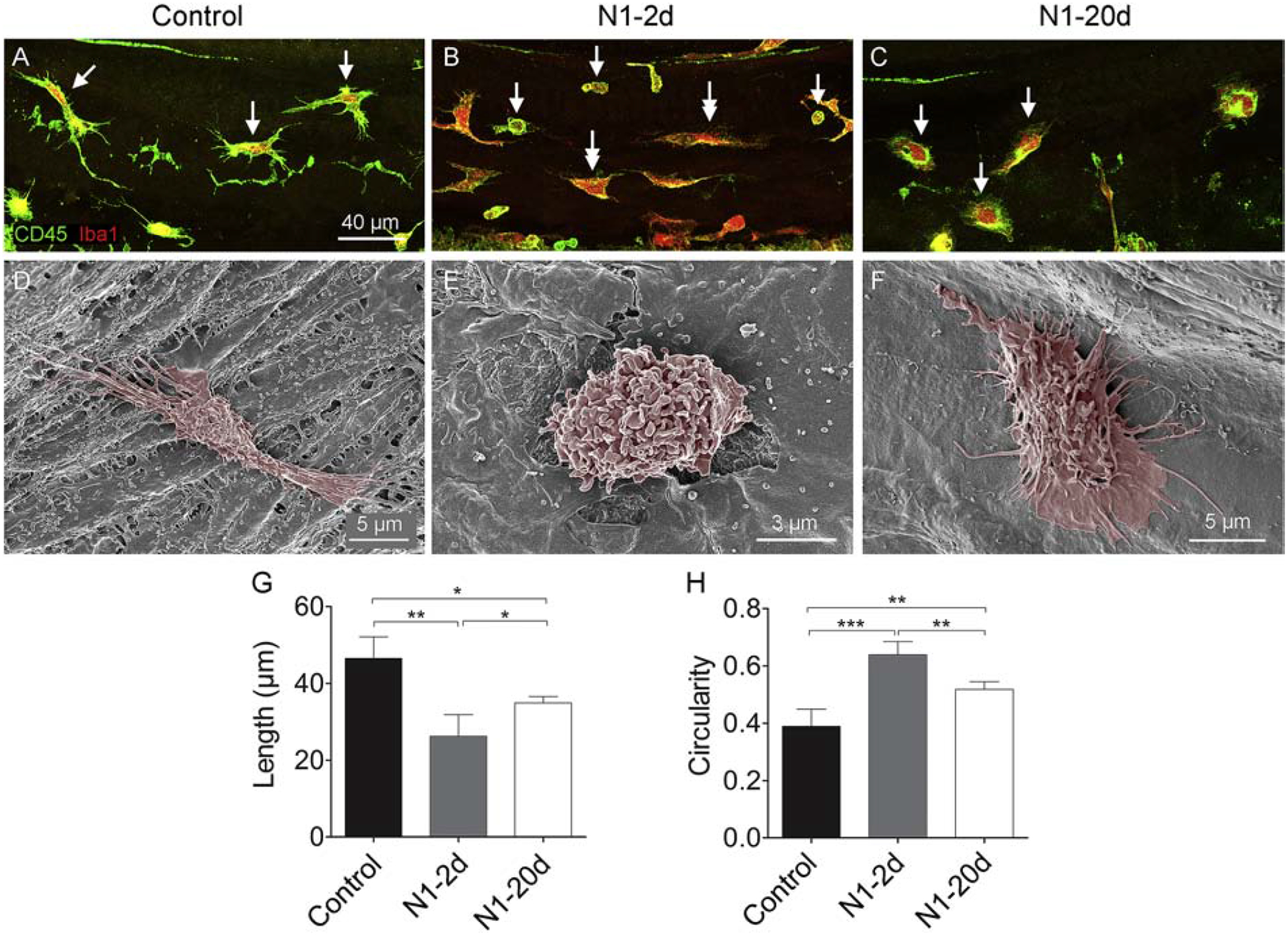
A. Macrophages in the basilar membrane in the middle portion of the cochlea present with a tree-trunk morphology with fine, thin processes (arrows) in a control cochlea. B. Macrophages in a cochlea examined at 2-days after noise exposure. Single-arrows point to the cells with monocyte-like phenotypes: small size with a round shape. These cells are likely newly infiltrated monocytes. Double-arrows indicate cells with the mature macrophage morphology. These cells display a reduced length and have less thin processes as compared with the macrophages observed in the control ears. C. Macrophages in a cochlea examined at 20-days after noise exposure. Notice that macrophages present with a flat, spread morphology or an amoeboid shape with multiple protrusions or fine and short dendritic projections (arrows). D-F. Typical SEM images of basilar membrane macrophages from the middle region of cochleae from the control and the two noise groups. G. Comparison of the macrophage length between the control, N1–2d, and N1–20d groups. There is a significant reduction in the average length of macrophages at 2-days after noise exposure and a partial recovery by 20-days (* indicates P < 0.05 and ** indicates P < 0.01). The reduction of the cell length indicates the retraction of macrophage processes. H. Comparison of macrophage circularity between the control, N1–2d, and N1–20d groups. Cell circularity displays a significant increase at 2-days after noise exposure and a partial recovery at 20-days after noise exposure. The increase in the value of circularity indicates the amoeboid transformation of macrophages (** indicates P < 0.01 and *** indicates P < 0.001). n = 5 biological replicates for each group.
To quantify these morphological changes, we measured the length and circularity of basilar membrane macrophages. We found a reduction in the average length of macrophages at 2-days post-exposure. This change partially recovered 20-days post-exposure (46.5 ± 5.6 μm, 26.6 ± 8.1 μm, and 35 ± 5.9 μm for the control, N1–2d, and N1–20d group, respectively; one-way ANOVA, F (2,12) = 18.917; P < 0.001; Tukey test, P < 0.001 for control vs. N1–2d; P = 0.041 for control vs. N1–20d; P = 0.015 for N1–20d vs. N1–20d; Fig. 5G). The reduction in cell length reflects the retraction of macrophage processes. We also measured the circularity of macrophages. The base-value for macrophage circularity was 0.4 ± 0.1 in control ears. The value increased to 0.6 ± 0.1 at 2-days after noise exposure. At 20-days post-exposure, the value decreased (0.5 ± 0.1), but was still higher than the base-level (one-way ANOVA, F (2, 12) = 36.529; P < 0.001; Tukey test, P < 0.001 for control vs. N1–20d; P = 0.004 for control vs. N1–20d; P = 0.002 for N1–2d vs. N1–20d; Fig. 5H). The increase in circularity indicates the amoeboid transformation of macrophages, a sign of inflammatory activation (Heinrich et al., 2017; Stence et al., 2001). Together, these observations reveal that basilar membrane macrophages have not regained their resting morphology at 20-days post-exposure, indicative of the persistence of a low-level of activation.
To assess the functional state of macrophages, we examined the expression level of CC motif chemokine ligand 2 (CCL2; also known as monocyte chemoattractant protein-1) using immunohistochemistry (Sautter et al., 2006). We selected this molecule as an inflammatory marker because CCL2/CCR signaling has been implicated in cochlear inflammation after acoustic injury (Frye et al., 2018; Sautter et al., 2006; Tornabene et al., 2006). We first examined the level of CCL2 immunoreactivity in macrophages. As shown in Figure 6A–D, CCL2 immunoreactivity was relatively weak in control cochleae, but became significantly stronger 2-days post-exposure (average gray level of 3.2 ± 0.8 for the control group vs. 44.2 ± 15.6 for the N1–2d group). CCL2 immunoreactivity decreased 20-days post-exposure (average gray level of 29.2 ± 10.1; Fig. 6E–F), but the value had not returned to the pre-noise level (one-way ANOVA, F (2, 8) = 119.31; P <0.001; Tukey test, P < 0.001 for control vs. N1–2d; P = 0.003 for N1–2d vs. N1–20d; P < 0.001 for control vs. N1–20d; Fig. 6G). We also quantified the number of CCL2-positive cells on the surface of the basilar membrane and found a dramatic increase from 1.7 ± 0.6 cells per 350 μm under the resting condition to 48.7 ± 2.1 cells per 350 μm 2-days post-noise. At 20-days post-exposure, the number of positive cells decreased to 38 ± 4.6 cells per 350 μm, but the value had not returned to the pre-noise level (one-way ANOVA, F (2,8) = 212.896; P < 0.001; Tukey test, P < 0.001 for control vs. N1–2d; P = 0.01 for N1–2d vs N1–20d; P < 0.001 for control vs. N1 20d; Fig. 6H). CCL2 positive cells accounted for 5.9%, 47.2%, and 21.3% of CD45 positive cells in the control, N1–2d, and N1–20d group, respectively. These positive cells were distributed in the middle portion of the cochlea (Fig. 6I). The finding of persistent CCL2 expression provides evidence for the presence of residual inflammation 20-days post-exposure.
Figure 6. Upregulation of CCL2 in basilar membrane macrophages after acoustic overstimulation.
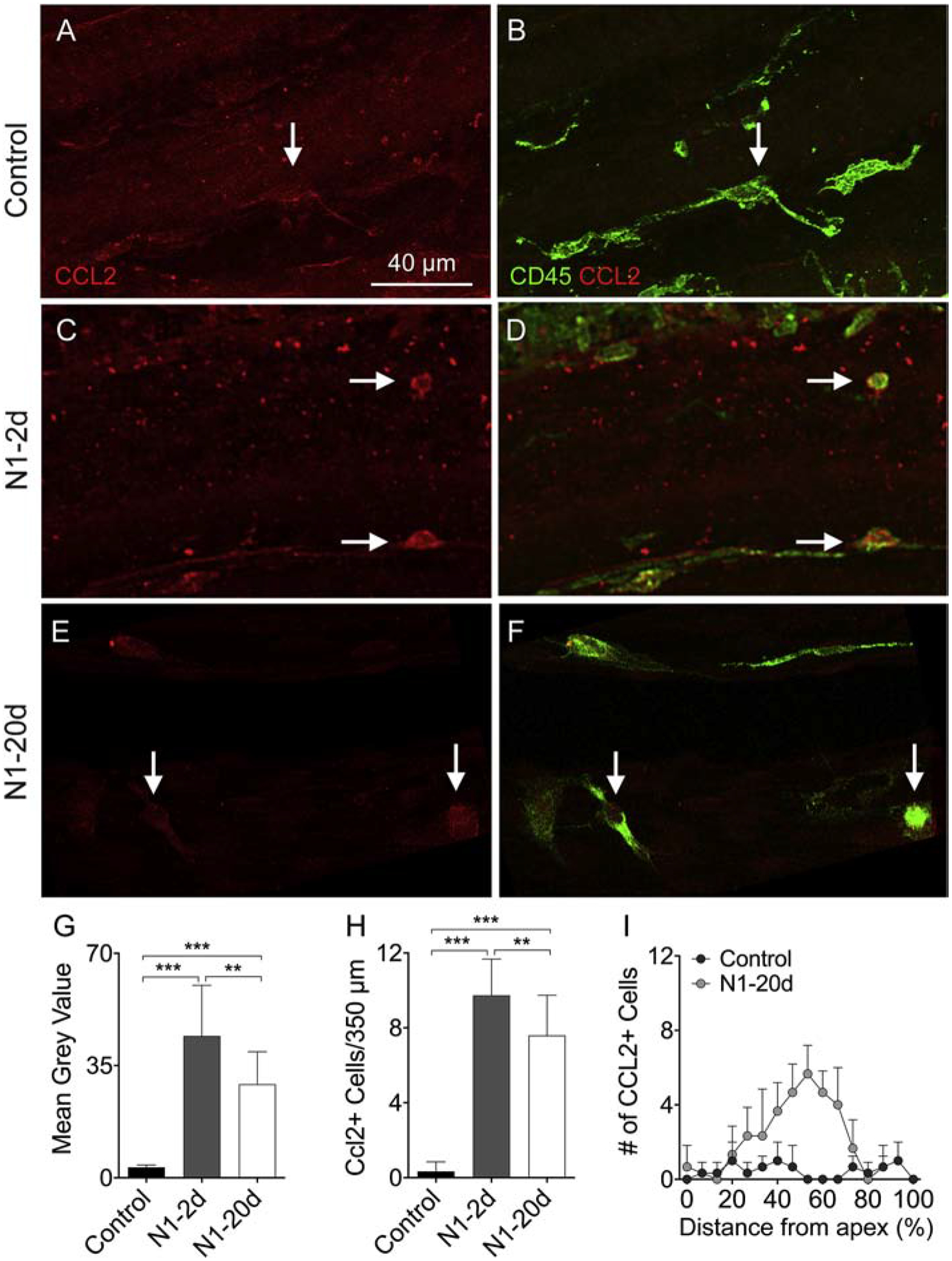
A-B. CCL2 immunoreactivity in basilar membrane macrophages of a control ear. Panel A shows CCL2 immunoreactivity and panel B shows the merged view of CCL2 and CD45. The arrow points a macrophage with weak CCL2 immunoreactivity that is barely visible. C-D. CCL2 immunoreactivity in macrophages examined at 2-days after noise exposure. Arrows indicate small, round macrophages that display strong CCL2 immunoreactivity. E-F. CCL2 immunoreactivity in macrophages examined at 20-days after noise exposure. CCL2 immunoreactivity is still visible in macrophages. G. Comparison of the level of CCL2 immunoreactivity in macrophages among the control, N1–2d, and N1–20d groups. The N1–2d group displays a marked increase in CCL2 immunoreactivity as compared with the control group. The increase is partially decreased in the N1–20d group (** indicates P < 0.01 and *** indicates P < 0.001). H. Comparison of the number of CCL2-positive macrophages between the control, N1–2d, and N1–20d groups. The number of CCL2-positive cells is increased at 2-days after noise exposure, and this increase partially recovers at 20-days after noise exposure (** indicates P < 0.01 and *** indicates P < 0.001). I. Macrophage-gram showing the distribution of CCL2 positive cells along the basilar membrane. Notice that these positive cells are mainly distributed in the middle region (30–70% distance from the apex). n = 5 biological replicates for each group.
Persistence of monocytic macrophages at 20-days after noise exposure
The accumulation of monocyte-like macrophages has been found at the early stage of acoustic injury (Yang et al., 2015). We wanted to know whether this type of cell is still present 20-days post-exposure. As described above, small, round immune cells were present in the cochlea at 2-day post-exposure. We found that cells with similar morphology were also presentat 20-days post-exposure. To determine the immuno-phenotypes of these cells, we stained the tissues with antibodies against CD45, Iba1, F4/80, and Ly6C. As shown in Figure 7, small, round cells displayed stronger CD45 immunoreactivity than mature macrophages that had branched or amoeboid morphologies (Fig. 7A, D, G). Many of these small CD45high cells were F4/80l°w (Fig. 7B–C) and Iba1l°w (Fig. 7E–F). Moreover, these cells were Ly6C-positive (Fig. 7H–I). These immune phenotypes (CD45high, F4/80l°w, Iba1l°w) resemble the phenotypes of infiltrating macrophage precursors (Greter et al., 2015) reported in the brain (Geissmann et al., 2003). Further SEM images revealed that small round cells observed at 20-days post-noise manifested similar macrophage surface texture as those observed 2-days post-exposure (Fig. 7J–K). To determine whether these small round cells were neutrophils, B cells or T cells, we stained the cochleae with the antibodies against Ly6G (neutrophil marker), B220 (B cell marker), CD4 (T cell marker) and CD8 (T cell marker). We found no neutrophils or B cells in the cochleae. A few CD4 and CD8 positive cells were found, but their number was minimal (<4 cells per cochlea). These observations suggest that small round immune cells are primarily macrophages. We quantified the number of these cells and found that their number was significantly higher than in control cochleae. Noticeably, the increase was confined in the basal region (Fig. 7L: two-way ANOVA, F (1, 20) = 6.308, P = 0.021; Tukey test for apex vs. base, control: P = 0.552, P1–20d: P < 0.001). These observations reveal the continuous presence of monocyte-like macrophages 20-days post-exposure.
Figure 7. Phenotyping of immune cells with the monocyte morphology at 20-days after noise exposure.
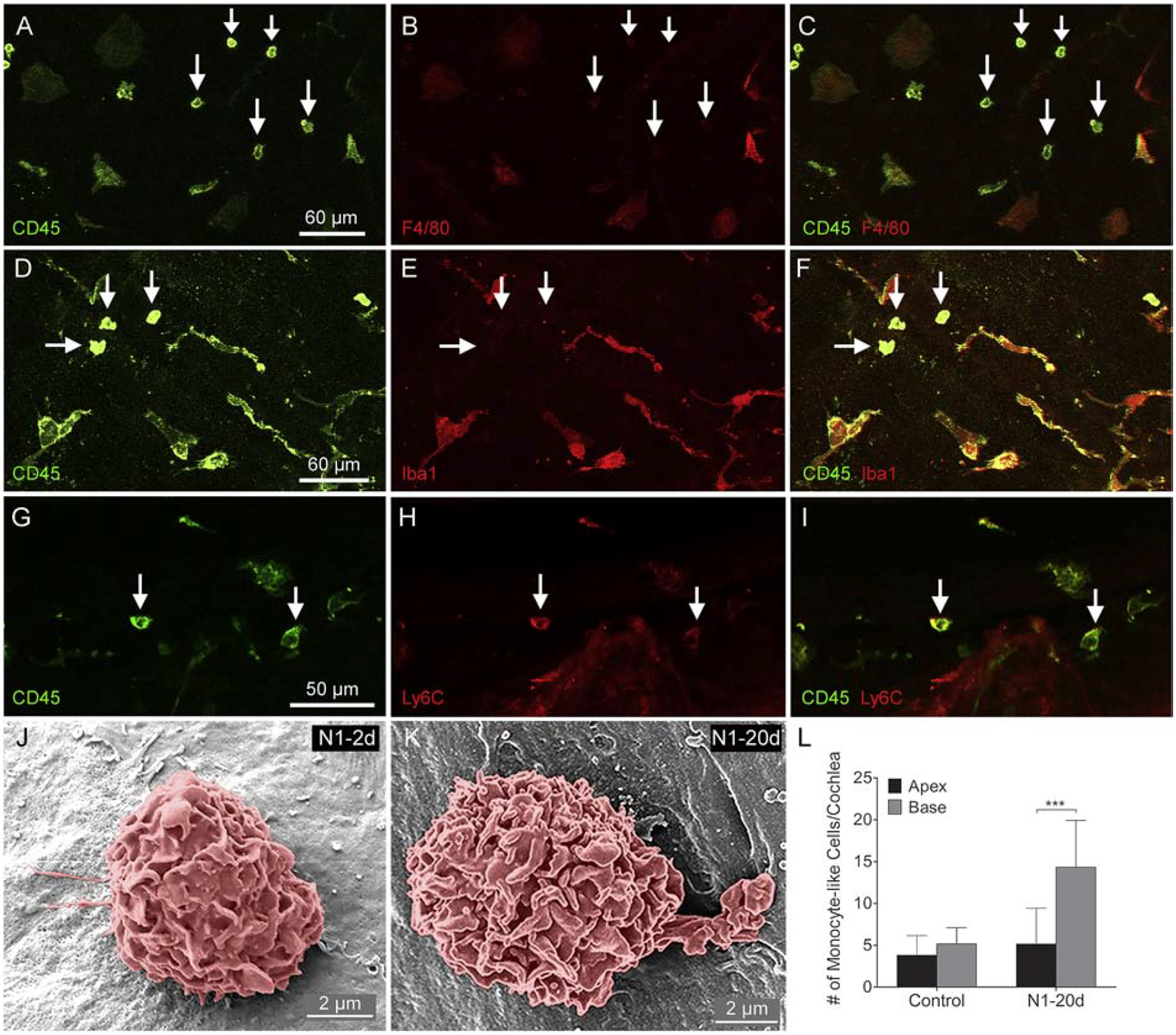
A-C. Double-staining of CD45 and F4/80. Small round CD45high cells are F4/80l°w. D-F. Double-staining of CD45 and Iba1. Small round CD45high cells are Iba1l°w. G-I. Double-staining of CD45 and Ly6C. Small round cells display both CD45 and Ly6C immunoreactivity. J-K. Scanning electron micrographs of monocyte-like cells from cochleae in the N1–2d and N1–20d group. These cells have the surface contour of the monocyte. L. Comparison of the number of small, round monocyte-like cells between the apical and the basal region of the cochleae. There is no difference between the number of monocyte-like cells between the apex and the base of the control cochleae. In contrast, the number of small, round cells is significantly higher in the base compared to that in the apex in the N1–20d (*** indicates P < 0.001). n = 4 biological replicates for each group.
Previous studies have shown that blood monocytes are comprised of two subtypes: proand anti-inflammatory (Geissmann et al., 2003; Ziegler-Heitbrock et al., 2010). Distinguishing these two subpopulations can be achieved by surface markers (Ly6Chigh/CCR2high for pro-inflammatory monocytes and Ly6Cl°w/CCR2l°w for anti-inflammatory). The distinction can also be made based on the GFP fluorescence intensity of monocytes in B6.129P-Cx3cr1tm1Litt/J mice where the Cx3cr1 is replaced by the GFP reporter gene (Jung et al., 2000). Specifically, GFPl°w cells are Ly6Chigh, whereas GFPhigh cells are Ly6Cl°w(Geissmann et al., 2003; Poupel et al., 2013; Rodero et al., 2015; Williams et al., 2017). Here, we wanted to know whether the cells present at 20-days post-exposure are GFPl°w or GFPhigh because the two subsets of monocytes have different roles in inflamed tissues. Because the Cx3cr1 gene is known to have important roles in regulating adhesion and migration of leukocytes (Kaur et al., 2018), we used heterozygous mice (Cx3cr1GFP/+) in which one allele of the Cx3cr1 gene is preserved.
We first examined the cochleae of control mice and found that tissue macrophages in the cochlea were GFPhigh (Fig. 8A). One day after acoustic overstimulation, which represents the beginning of monocyte extravasation into the cochlea, a large number of small, round GFPl°w cells appeared on the surface of the basilar membrane as well as in the lateral wall and osseous spiral lamina (Fig. 8B). In contrast, we found no GFPl°w cells in cochleae 20-days post-exposure. Instead, all small, round GFP-positive cells were GFPhigh (Fig. 8C). Quantitative analysis revealed that the number of GFPl°w cells in cochleae during the acute phase of noise damage was significantly higher than that observed in the control cochleae (Fig. 8D, Student’s t-test, t (6) = −7.961; P < 0.001). Image analysis confirmed the difference in GFP intensity between the GFPhigh cells in the control cochleae, the GFPl°w cells in the cochleae examined at 1-day after noise exposure, and the GFPhigh cells in the cochleae examined at 20-days after noise exposure (Fig. 8E, one-way ANOVA, F (2, 13) = 51.483, P < 0.001; Tukey test, P < 0.001 for control vs. N1–20d and control vs. N1–1d, P = 0.422 for control vs. N1–20d). These analyses revealed that the monocyte-like macrophages observed at 20-days post-exposure display the phenotype previously linked to anti-inflammatory monocytes (Geissmann et al., 2003; Poupel et al., 2013).
Figure 8. Shift from GFPl°w to GFPhigh monocyte-like cells in Cx3cr1GFP/+ cochleae after noise exposure.
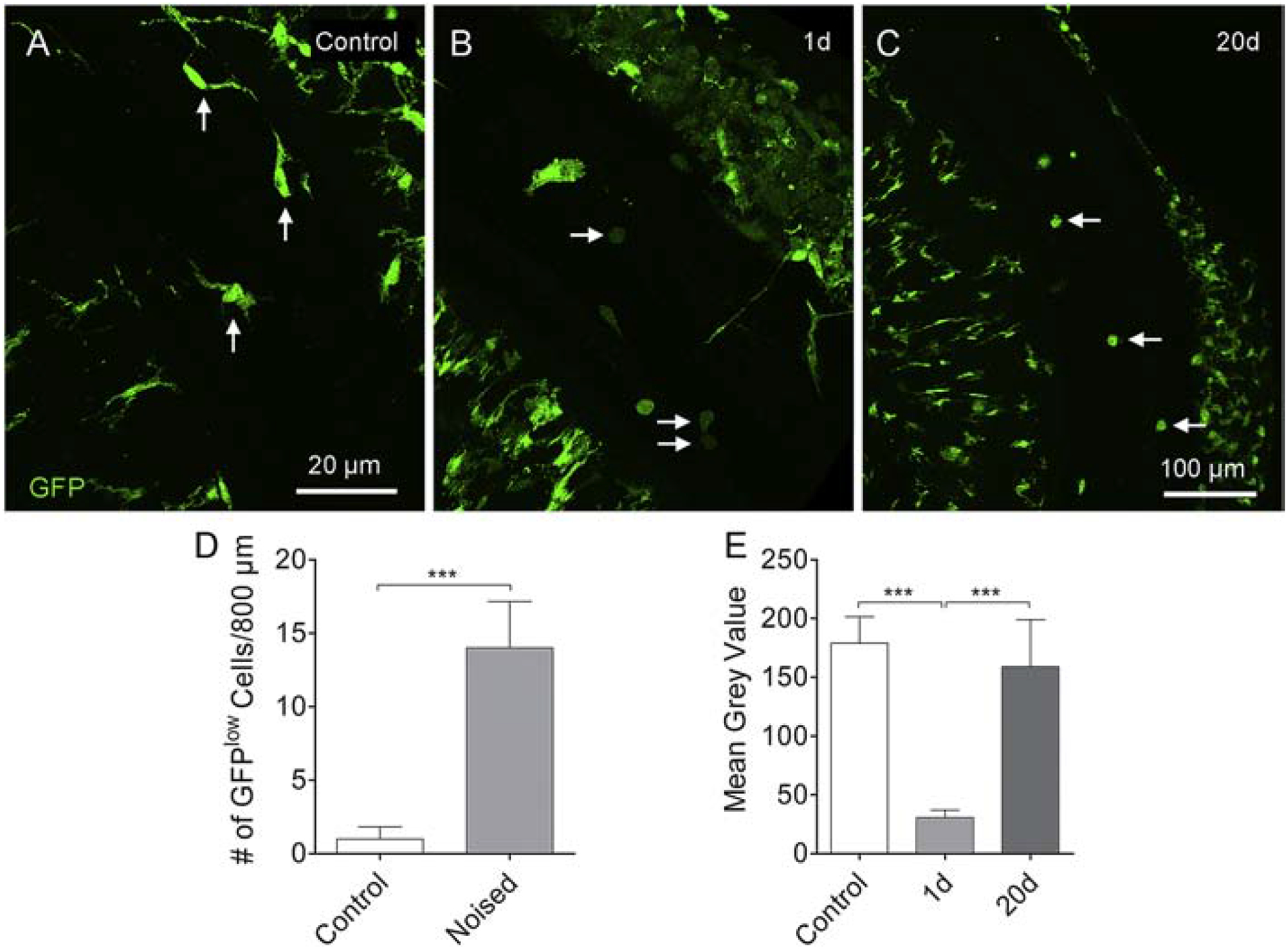
A. Tissue macrophages on the basilar membrane are GFPhigh under resting conditions, and the intensity of the fluorescence is relatively homogenous across GFP-positive cells (arrows). B. One day after noise exposure, GFPl°w cells emerge on the surface of the basilar membrane as well as in the lateral wall and osseous spiral lamina. These cells have a monocyte shape and size (arrows). C. Small, round monocyte-like GFPhigh cells are present in the cochlea examined at 20-days after noise exposure (arrows). D. Comparison of the number of GFPl°w cells between control cochleae and cochleae examined at 1-day after noise exposure. The number of GFPl°w cells in the noise-damaged cochleae is significantly higher than that in the control cochleae. n = 4 cochleae biological replicates for each group. E. Comparison in the GFP fluorescence intensity of basilar membrane macrophages between the control, 1-day and 20-day groups (*** indicates P < 0.001).
To determine whether anti-inflammatory monocytes enter the cochlea at the resolution phase of acute inflammation, we tracked the infiltration of blood monocytes using Fluoresbrite® Polychromatic Microspheres. Previous studies have shown that, when intravenously injected, these latex beads are engulfed exclusively by Ly6Cl°w anti-inflammatory monocytes (Olingy et al., 2017; Tacke et al., 2007) allowing these cells to be tracked into tissues. We administered the latex beads at either the acute (3 hours post-exposure) or the recovery phase (17–20 days post-exposure) of acoustic injury and examined the cochleae at 3-days after latex bead administration. As expected, we did not find latex bead-positive cells at the early phase of inflammation even though a large number of monocytes were identified (Fig. 9A). To further confirm that pro-inflammatory monocytes lack the ability to engulf latex beads, we incubated cochleae from Cx3cr1GFP/+ mice with the latex beads 1-day after post-exposure. This analysis revealed that infiltrated GFPl°w cells were latex-bead negative (Fig. 9B, arrows). Only GFPhigh tissue macrophages contained latex beads (Fig. 9B, inset). In contrast, when the latex beads were injected at the later stage of the inflammation, we identified latex bead-positive cells in the cochleae (Fig. 9C, inset). The number of these positive cells varied from 4 to 10 per cochlea among the examined cochleae. Together, these analyses suggest that monocyte-like cells that were previously defined as anti-inflammatory in the circulation (Varga et al., 2018) enter the cochlea at the recovery phase of acute damage.
Figure 9. Latex bead tracking of monocytes in cochleae.
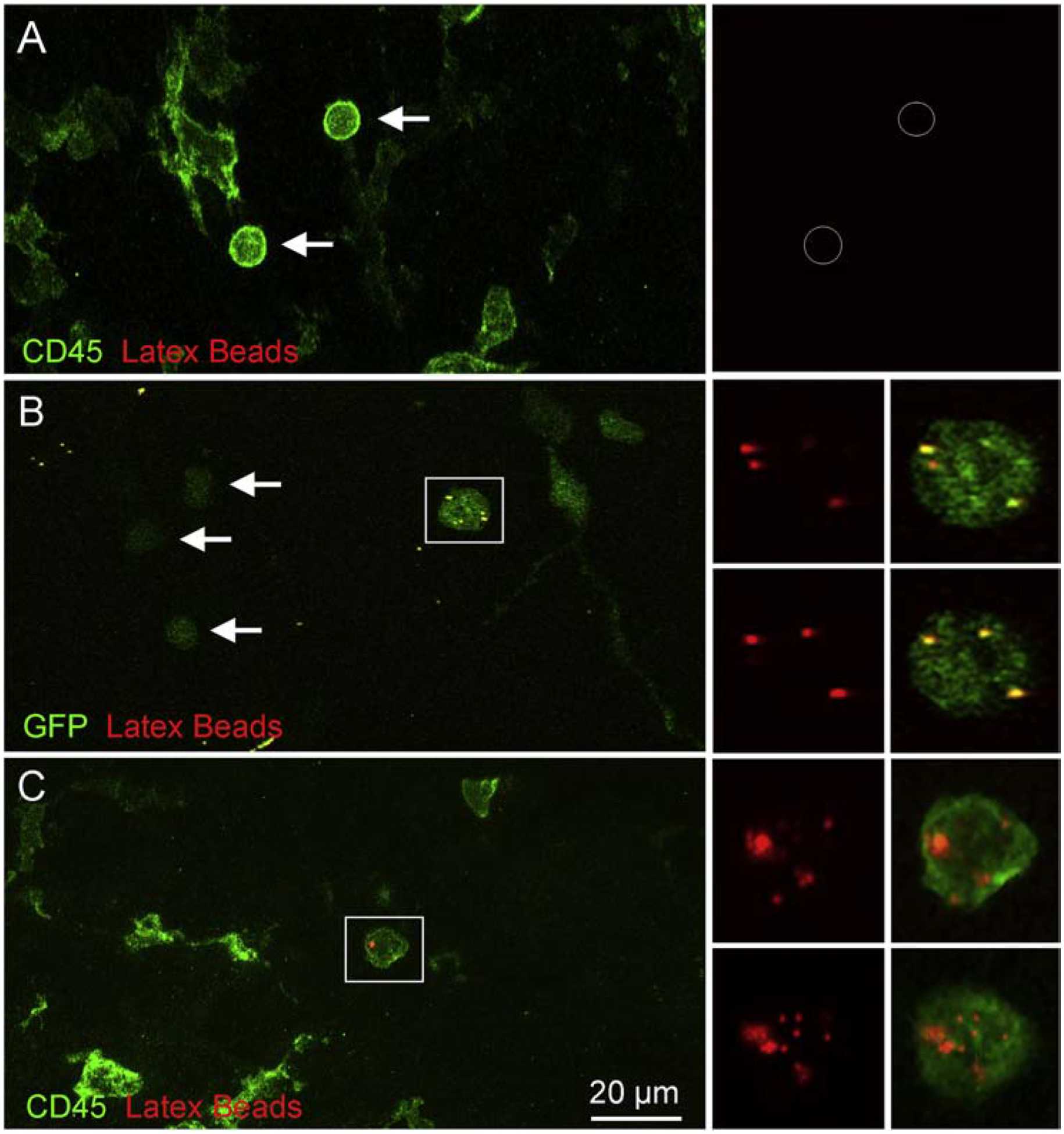
A. Image showing a cochlea examined at 3-days after noise exposure. The animal was injected with latex beads and its cochleae were stained with CD45 to illustrate immune cells. Monocyte-like CD45-positive cells lack latex beads (arrows). B. Image showing a cochlea from a Cx3cr1GFP/+ mouse examined at 1-day after acoustic overstimulation. The cochlea was incubated with latex beads. Arrows point to GFPl°w cells that are latex bead-negative. Notice that GFPhigh cells are latex bead-positive (inset). C. Image showing a cochlea examined at 20-days after noise exposure. The animal was injected with latex beads, and the cochlea was stained with CD45 to illustrate immune cells. The inset illustrates a latex bead-positive cell. n = 4 cochleae biological replicates for each group.
Transcriptional analysis reveals marked variation in the recovery of the expression of inflammatory molecules in the cochlea 20-days after acoustic trauma
The finding of persistent low-level immune cell activation led us to investigate whether cochlear tissues remain inflamed at 20-days post-exposure. We previously observed a marked increase in the expression level of inflammatory molecules in the early phases of acoustic injury (Patel et al., 2013a; Vethanayagam et al., 2016; Yang et al., 2016). To determine whether the increased expression had returned to the base level at 20-days post-exposure, we examined the transcriptional expression of four immune-related genes that play roles in monocyte extravasation (Ccl2 and Ccl7) and inflammation (Tnf and Il-1β). We collected cochlear tissues containing the sensory epithelium and lateral wall before and at two time points after noise-exposure (N1–2d and N1–20d). Using the expression levels examined before noise exposure as the base levels, we calculated the noise-induced changes and found a significant increase in the expression of all four examined genes 2-days post-exposure (Fig. 10A; one-way ANOVA, Ccl2: F (4,15) = 7.982; P = 0.006, Tukey test, P = 0.008 for N1–20d vs. N1–2d; P = 0.024 for control vs. N1–2d; Tnf: F = 18.958; P < 0.001; Tukey test, P < 0.001 for N1–20d vs. N1–2d; P = 0.002 for control vs. N1–2d; Il-1β: F = 7.856; P = 0.007; Tukey test, P = 0.007 for N1–20d vs. N1–2d; P = 0.03 for control vs. N1–2d; Ccl7: F = 23.242, P < 0.001; Tukey test, P < 0.001 for N1–20d vs. N1–2d and control vs. N1–2d). While the group average gene expression levels had returned to pre-noise levels at 20-days post-exposure, the recovery displayed significant individual variation. Figure 10B compares the variation in the expression of the four examined genes between the control and noise groups. The variation in the noise group is much larger than that observed in the control group. This analysis suggests that recovery of immune status is not homogenous across individual ears.
Figure 10. Analysis of transcriptional expression of immune-associated genes in cochlear tissues.
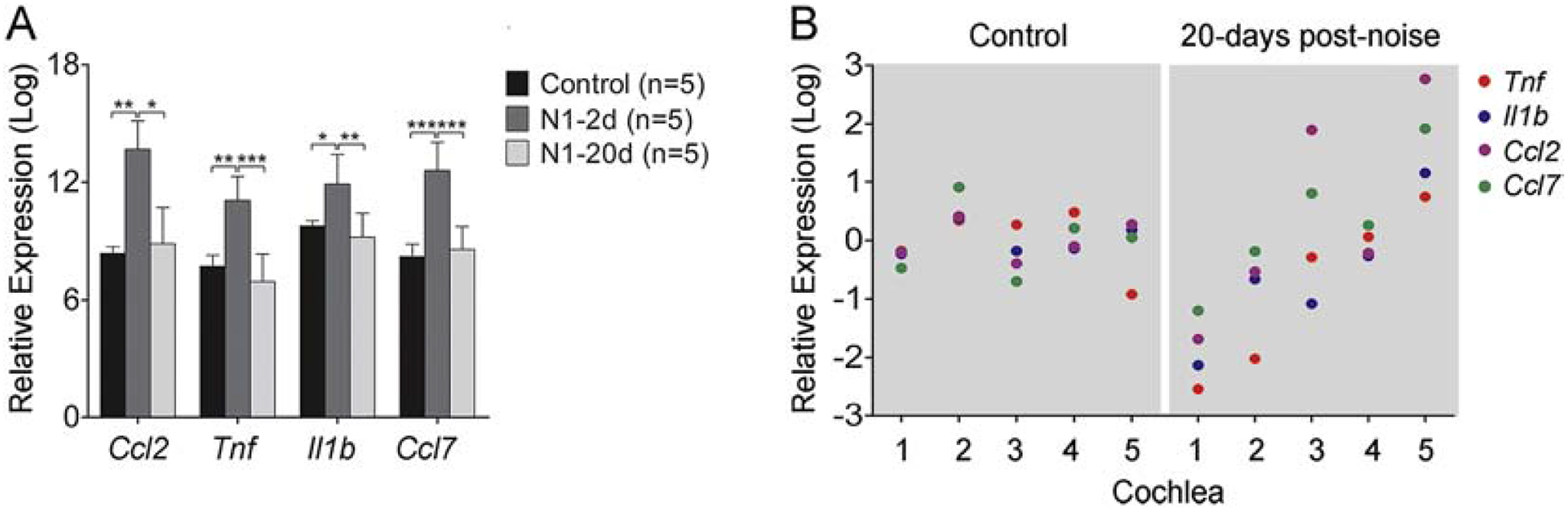
A. Comparison of the transcriptional expression of four immune-related genes in the control, 2-day, and 20-day post-noise groups. There is a significant upregulation in all examined genes at 2-days after noise exposure and the recovery of the expression levels at 20-days after noise exposure. n = 5 biological replicates for each condition (* indicates P < 0.05, ** indicates P < 0.01 and *** indicates P < 0.001). B. Expression variation of the four examined genes across the five examined samples in the control and 20-day noise group (N1–20d). The five samples came from five individual mice. The value of each dot represents the difference between the expression level of a gene in a particular sample and the group mean of the expression of that gene. Notice that the expression variation is relatively homogenous across the five samples in the control group. In contrast, the expression variation increases in the noise group. All four genes in sample 1 display lower expression levels in the noise group than those of the control group. Conversely, all the four genes in sample 5 display higher expression levels in the noise group than those of the control group. This observation suggests that mice display a large individual variation in the recovery of the expression levels of the pro-inflammatory mediators.
Cochleae with prior noise injury display an accelerated immune activation to repeated noise exposure
To determine whether prior acoustic overstimulation can modulate the cochlear immune response to subsequent acoustic trauma, we examined macrophage responses to a second noise that was administered 20-days after the first noise. First, the magnitude of the macrophage population expansion in the regions of the osseous spiral lamina, basilar membrane, and luminal surface of the scala tympani was examined. We evaluated the acute macrophage response by calculating the increases in the macrophage number at 2-days post-exposure, which represents the level of the early expansion of the macrophage population. We found that the increase in number of basilar membrane macrophages was significantly higher in the N2 group when compared to the N1 group (5.5 ± 1.7 cells for N1 group vs. 9.4 ± 3.5 cells per 450 μm for N2 group, Student’s t-test, t (10) = −3.892; P = 0.035, Fig. 11A). We then examined the increase in macrophages along the cochlear spiral and found a relatively even distribution along the cochlear spiral except for the extreme base where the increase was slightly less than that in the apical and middle portions of the cochlea (Fig. 11B). This result suggests that the second noise exposure provoked greater expansion of the macrophage pool.
Figure 11. Expansion of the macrophage population in the acute phase of acoustic injury.
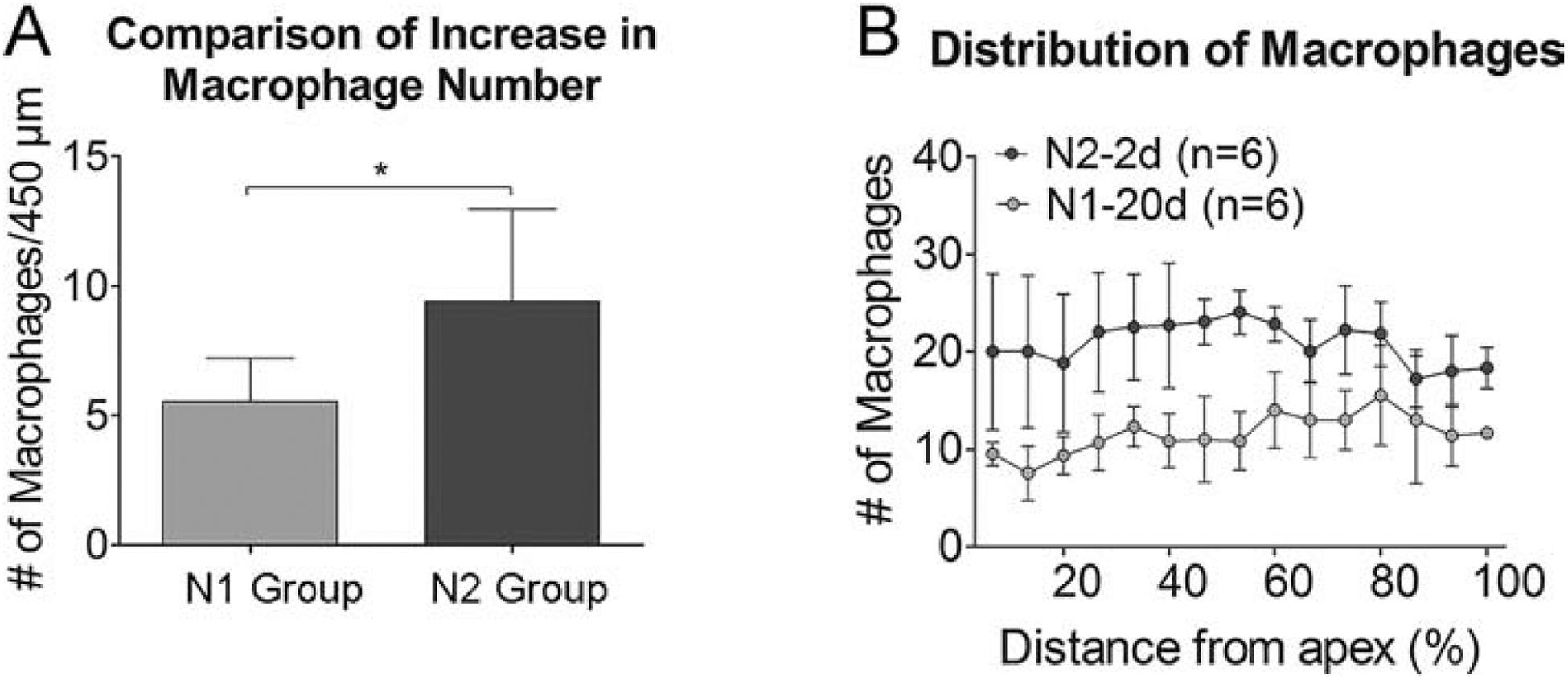
A. Comparison of the increase in the number of basilar membrane macrophages between the N1 and the N2 group examined at 2 days after noise exposure. The magnitude of the increase is significantly higher following the second noise exposure as compared with that observed afterthe initial noise exposure (5.5 ± 1.7 cells per 450 μm vs. 9.4 ± 3.5 cells per 450 μm for the first and the second group, respectively; * indicates P < 0.05). B. Macrophage-gram illustrating the distribution of macrophages along the cochlear spiral before (N1–20d) and 2 days (N2–2d) after exposure to the second noise. The increase of macrophages is present relatively homogeneously across 0–80% distance from the apex. The level of the increase appears slightly smaller in the basal end of the cochlea (marked by the gray area). n = 6 cochleae biological replicates for each group.
Next, we compared the macrophage morphology and found a noticeable difference between the first-noise and the second-noise group. In the N1 group, small round cells were commonly seen on the basilar membrane at 2-days after noise exposure (Fig. 12A). In contrast, we found that many small, round cells displayed short, fine processes after the second noise exposure, indicating that these cells already underwent an active transformation into mature macrophages (Fig. 12B). This morphological difference was more evident in SEM images (Fig. 12C–D). To quantify the changes, we counted the number of small, round monocyte-like cells and mature macrophages on the surface of the basilar membrane. We found that there were more mature macrophages, but less monocyte-like cells, after the second noise exposure compared to the first (mature macrophages: Student’s t-test, t (10) = 3.914, P = 0.003; monocyte-like cells: Student’s t-test, t (10) = 2.393, P = 0.038; Fig. 12E). To quantify this difference, we calculated the percentage of monocyte-like cells over mature macrophages. Monocyte-like cells constitute 11.3 ± 2.6% of the total number of macrophages after the second noise exposure, which is significantly lower than 21.5 ± 5.9% observed after the first noise exposure (Fig. 12F, Student’s t-test, t (10) = 3.914, P = 0.003). Given that the N2 group had a greater number of macrophages, the small percentage of monocyte-like macrophages suggests that more macrophages had acquired the mature morphology, which in turn suggests that macrophages are more advanced in their transition to an activated state following the second noise exposure.
Figure 12. Comparison of the maturation levels of monocyte-like macrophages between the N1 and the N2 group.
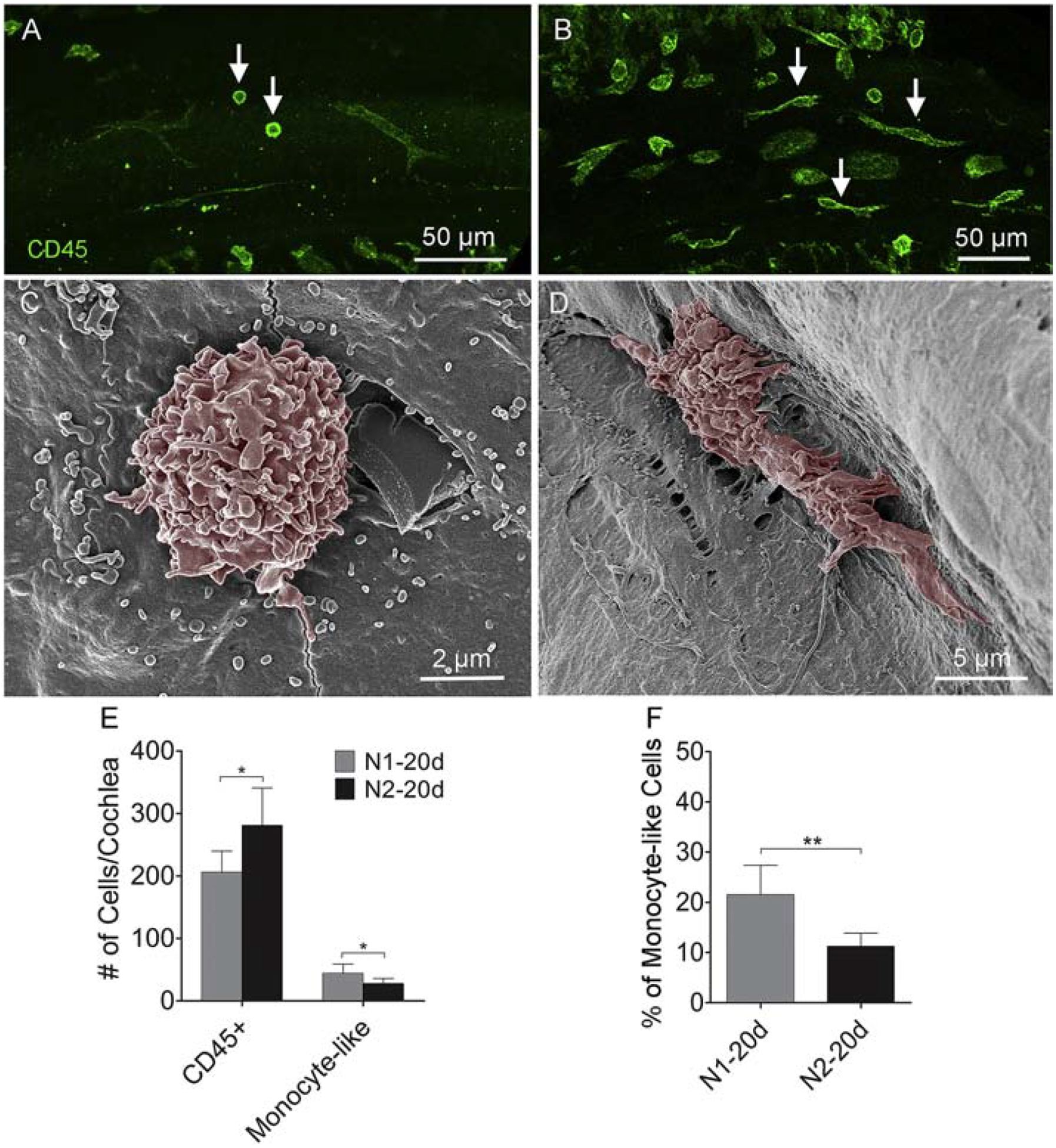
A. Two days after the initial noise exposure, small, round CD45-positive cells are identified on the basilar membrane (arrows). B. Two days after the second noise exposure, small, CD45-positive cells display short, fine processes (arrows), suggesting that these cells are actively transforming into mature macrophages. C-D. Scanning electron micrographs showing the difference in the morphology of typical monocyte-like cells in cochleae from the N1–2d and N2–2d group. E. Comparison of the number of small, round monocyte-like cells and CD45-positive cells on the surface of the basilar membrane between the N1–20d and N2–20d groups. On average, there are more CD45-positive cells, but less monocyte-like cells, after the second noise exposure than those observed after the first noise exposure (* indicates P < 0.05). F. Comparison of the percentage of monocyte-like cells over the total number of basilar membrane macrophages per cochlea between the N1–2d and N2–2d group. Monocyte-like cells constitute approximately 21% of the total number of macrophages in the N1–2d group and 12% in the N2–2d group (** indicates P < 0.01). n = 6 cochleae biological replicates for each group.
Finally, we examined the expression of CD68, a marker for macrophage activation (Chistiakov et al., 2017). Under resting conditions, CD68 immunoreactivity was relatively weak in macrophages (Fig. 13A–C). This immunoreactivity was increased 20-days after the first noise exposure (Fig. 13D–F) and was further increased 20-days after the second noise exposure (Fig. 13G–I). Image analysis of immunoreactivity confirmed these increases (Fig. 13J, the average gray level was 25.8 ± 7.7 for the control group, 41.33 ± 3.80 for the N1–20d and 63.6 ± 14.9 for the N1–20d group; one-way ANOVA, F (2, 8) = 66.396, P < 0.001; Tukey test, P < 0.001 for control vs. N2–20d and N1–20d vs. N2–20d; P = 0.006 for control vs. N1–20d). We also quantified the number of CD68-positive cells on the surface of the basilar membrane. As shown in Figure 13K, the number of CD68-positive cells increased from the base level of 53.0 ± 4.3 per cochlea to 76.0 ±7.21 per cochlea 20-days after the first noise to 97.0 ± 12.7 per cochlea 20-days after the second noise exposure (one-way ANOVA, F (2, 8) = 23.989; P < 0.001; Tukey test, P < 0.001 for control vs. N2–20d, P = 0.037 for N1–20d vs. N2–20d; P = 0.024 for control vs. N1–20d). The finding of greater CD68 expression in the N2 group provides further evidence for enhanced immune activation in cochlea with repeated acoustic stress.
Figure 13. CD68 immunoreactivity in basilar membrane macrophages in control and noise-damaged cochleae.
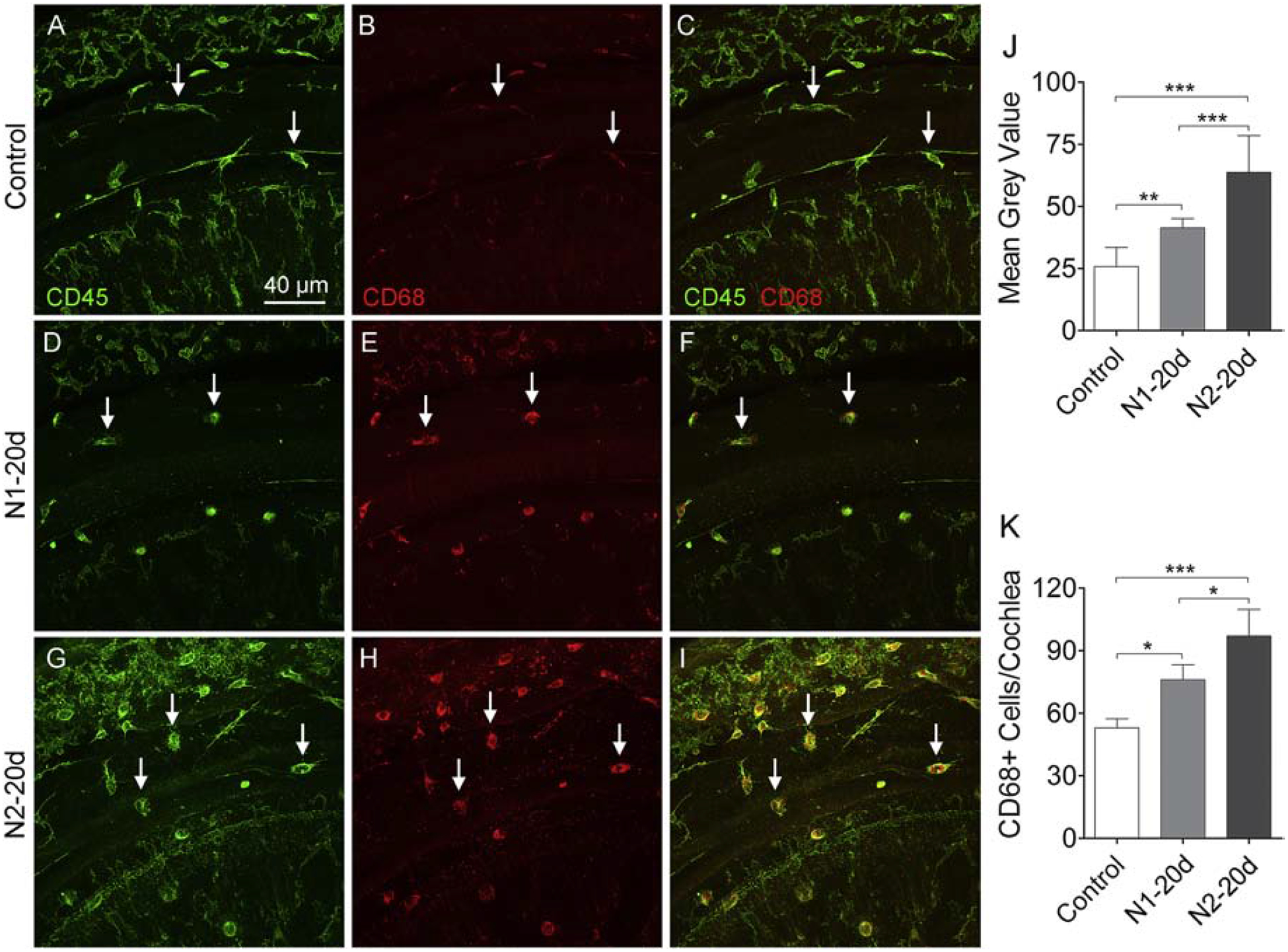
A-C. Macrophages in a control cochlea display weak CD68 immunoreactivity. D-F. 20 days after the initial noise exposure, the number of CD68-positive cells increases along with their immunoreactivity. G-I. 20 days after the second noise exposure, there is a further increase in the number of CD68-positive cells as well as their immunoreactivity. J. Comparison of the immunoreactivity of CD68-positive cells in the control, N1–20d, and N2–20d group. CD68 immunoreactivity is elevated at 20 days after the first noise exposure and further increases after the second noise exposure. K. Comparison of the number of CD68-positive cells on the basilar membrane per cochlea in the control, N1–20d, and N2–20d group. The number of CD68-positive cells increases at 20 days after the initial noise exposure and further increases after the second noise exposure. n = 4 cochleae biological replicates for each group. (*, ** and *** indicate P < 0.05, 0.01 and 0.001, respectively).
Discussion
The goal of the current investigation was to determine how a prior acoustic trauma affects cochlear responses to subsequent stress in order to understand how residual immune activation could affect the cochlear immune response to repeated cochlear damage. Using macrophages as a sensor, we examined the cochlear inflammatory status. The study revealed that enhanced macrophage activities persist after the completion of sensory cell pathogenesis. This macrophage engagement consists of both pro-inflammatory and anti-inflammatory components and is orchestrated by macrophages at multiple cochlear regions. In the presence of prior noise injury, the cochlea becomes prone to subsequent acoustic overstimulation as evidenced by an accelerated immune response and exacerbated secondary sensory cell pathogenesis. Together, these observations implicate the immune reaction in the cochlear response to repeated acoustic overstimulation, emphasizing the possibility of reducing cochlear vulnerability to acoustic injury via immunomodulation.
Recovering cochleae are susceptible to secondary damage from repeated acoustic overstimulation
Acoustic injury is known to consist of two components: the initial mechanical stress and the subsequent secondary damage (Saunders et al., 1985; Wong et al., 2015). The initial mechanical stress occurs during noise exposure and thus can be detected shortly after noise exposure, whereas the secondary damage is caused by biological changes of the cochlea as the consequence of the initial mechanic disruption, and gradually emerges as time elapses. To determine the differential impact of the first and the second noise exposure, we developed a novel strategy to divide the growth of sensory cell lesions into two stages: the early damage that occurs within the first two days after noise exposure and the delayed damage that follows. The early damage is presumably caused by the initial mechanical stress of acoustic overstimulation, whereas the delayed secondary damage is presumably caused by cochlear responses to the initial mechanical disruption.
Our findings reveal that after the initial noise exposure, significant early damage occurs during the first two days. In contrast, delayed cell death accounts for only a small portion of total cell death, indicating that the majority of cell death occurs at the early phase of damage following the initial noise exposure. In contrast to the dominant early hair cell damage following the initial noise exposure, repeated noise exposure caused only a minor increase in the number of early cell death. This reduced early damage can be explained by the overlap of the primary lesion site from the first and the second noise exposure as both the first and the second noise have the same frequency profile and, therefore, are expected to stress the same hair cell region along the tonotopic axis of the cochlea. Because sensory cell loss has already taken place in the basal end of the cochlea after the first noise exposure, no further loss could occur in this region after the second noise exposure. Despite this reduced initial damage, cochleae that sustained the second noise displayed greater later-stage pathogenesis compared to those that sustained only the first noise. Specifically, the late-onset cell death accounts for 68.6% of the total missing hair cells after exposure to the second noise, which is significantly higher than 26.3% observed after the initial noise exposure. Moreover, we found a marked expansion of sensory cell lesions toward the middle region of the cochlea after the second noise exposure, forming a second locus of sensory cell damage. These observations suggest that the cochlea, when recovering from a prior episode of acoustic injury, is highly susceptible to repeated stress.
Low-level immune activity is present prior to the second acoustic overstimulation
We previously reported a partial recovery of cochlear immune activation 10-days after exposure to an intense noise at 120 dB SPL (Yang et al., 2015). Here, we wanted to know the prolonged effect of acoustic injury on the immune system in the cochlea. Our study provides several pieces of evidence for the presence of low-level immune activity 20 days after acoustic injury. First, the number of cochlear macrophages, while reduced from its peak value, has not returned to the pre-noise level. Second, many macrophages retained their activated morphology. Third, the expression level of CCL2, a pro-inflammatory mediator, remained higher than the base level, although a significant reduction from its peak level had occurred. We noticed that the accumulation of CCL2 positive cells is concentrated in the middle region, rather than the basal region where the maximal hair cell loss is present. This discrepancy could be explained by the state of hair cell pathology. At 20 days after acoustic injury, the pathogenesis of sensory cells is complete with no hair cells left in the basal region. Therefore, immune activities shift to the middle region where damaged hair cells may still in the stage of recovering. We also noticed an increase in Iba1 immunoreactivity after noise exposure, which could be a sign of macrophage activation (Ito et al., 2001; Sasaki et al., 2001); however, we did not quantify this phenotype because Iba1 was used only as a marker of macrophages. The persistence of activated phenotypes of macrophages suggests the presence of low-level immune activity at the time of the second noise exposure.
We also found the accumulation of monocyte-like macrophages at 20-days post-exposure, which could be a sign of the continual infiltration of monocytes into the cochlea during the resolution phase of inflammation. Our monocyte tracking experiment supports this speculation. Although multiple immune cell markers were used in the present study to define infiltrating cells, more definitive approaches are needed in future studies to identify these cells as newly infiltrated monocytes.
Additionally, we found evidence of anti-inflammatory activity 20-days post-exposure. Using the GFP fluorescence intensity to distinguish macrophage properties in Cx3cr1GFP/+ mice, we found a large number of GFPl°w monocyte-like macrophages in the cochlea at the early stage of acoustic injury. As demonstrated in previous studies (Geissmann et al., 2003; Poupel et al., 2013; Rodero et al., 2015), blood monocytes that are GFPl°w and Ly6Chigh are pro-inflammatory, whereas blood monocytes that are GFPhigh and Ly6Cl°w are anti-inflammatory. Thus, our result could suggest that pro-inflammatory macrophages are present in the cochlea at the induction phase of acute inflammation after acoustic injury. In contrast, 20-days after noise exposure, macrophages with a monocyte-like morphology were GFPhigh, suggesting that anti-inflammatory macrophages are present in the cochlea at the resolution phase of acute inflammation. To determine whether anti-inflammatory monocytes enter the cochlea at the recovery phase of acoustic trauma, we tracked anti-inflammatory monocytes by injecting latex beads that are engulfed exclusively by Ly6Cl°w anti-inflammatory monocytes (Weinstock et al., 2019). We found that at the early stage of acute inflammation, macrophages with the monocyte morphology were latex bead-negative. In contrast, latex bead-positive cells were present 20-days post-exposure. Although the number of these cells in the cochlea was limited, possibly due to the fact that not all anti-inflammatory blood monocytes are able to engulf latex beads, the presence of these cells is clear evidence for the recruitment of anti-inflammatory monocytes into the cochlea during the recovery process of acute inflammation.
The engagement of anti-inflammatory macrophages in the cochlear response to acoustic overstimulation could suggest beneficial impacts on the cochlear immune environment. These cells may participate in the recovery process, including phagocytosis of damaged cells, promoting differentiation and regeneration (Atri et al., 2018). They can also release proteases that assist in the removal of damaged tissue and secrete growth factors that can enhance cell survival, stimulate cells that have functions in healing and generation of new extracellular matrixes (Kotwal et al., 2017). Further studies are warranted for defining the functional transition of cochlear macrophages and their roles in the activation of pro-resolving mediators for inflammation resolution.
Repeated acoustic overstimulation provokes an exaggerated immune reaction in the cochlea
Our current study provides evidence that cochleae recovering from a prior acoustic injury are prone to an enhanced inflammatory response when encountering further stress. We observed greater expansion of the macrophage population after the second noise exposure compared to the first noise exposure. We also found a marked difference in macrophage morphologies at the induction stage of acute inflammation. Two days after the first noise exposure, small round macrophages were commonly seen. In contrast, in cochleae subjected to the second noise injury, small round macrophages were less common and the number of more mature macrophages was significantly increased. While it is not known whether this increased maturation reflects an early onset of monocyte infiltration or exacerbated macrophage maturation, our findings suggest that macrophages after the second noise are in a more advanced stage in acquiring mature macrophage phenotypes, possibly pro-inflammatory function as well.
The functional impact of exacerbated immune reaction is not clear. Previous studies have shown that the upregulation of pro-inflammatory molecules peaks 1–2 days after acoustic injury (Tan et al., 2016; Vethanayagam et al., 2016), whereas the expansion of the macrophage population peaks 3–7 days after acoustic injury (Fredelius et al., 1990; Hirose et al., 2005; Tornabene et al., 2006; Wakabayashi et al., 2010). This delayed response could allow macrophages to participate in the repair process of the cochlea, instead of the early inflammatory attack. As a second noise exposure leads to early maturation of macrophages, their involvement can be brought forward, which could enhance the early inflammatory attack. While this speculation is in line with our finding of potentiated sensory cell death after the second noise exposure, its verification requires future investigation.
Clinical implications
Our study is clinically relevant because repeated acoustic overstimulation is a common human health hazard. Several laboratories, including ours, have revealed complex interactions between initial and subsequent noise exposures. Exposure to low or moderate levels of acoustic stimuli renders the ear more resistant to traumatic noise (Campo et al., 1991; Henselman et al., 1994; Mills, 1973; Niu et al., 2002). This protective effect has been linked to the augmentation of heat shock proteins and antioxidants (Fairfield et al., 2005; Harris et al., 2006; Hu et al., 1997; Jacono et al., 1998; Yoshida et al., 1999). Unlike these previous studies, our current study used high-level acoustic overstimulation. Intense noise is known to increase the susceptibility of the ear to subsequent noise exposure (Perez et al., 2004), and our pathological analysis reveals potentiation of sensory cell pathogenesis. At present, it is not clear how immune reaction contributes to this increased susceptibility. Our study reveals that macrophage activation remains elevated in the cochlea for a prolonged period after completion of sensory cell death following acoustic overstimulation and that cochleae with residual immune activity are susceptible to subsequent stress. Macrophages have diverse functions in tissue homeostasis and disease formation. The phagocytotic function of macrophages facilitates the removal of damaged and dead cells and, therefore, is beneficial for inflammation resolution (Bang et al., 2018; Oishi et al., 2011). However, excessive and persistent inflammation is detrimental and could be contributory to the secondary damage observed in our model. This speculation is in line with the previous finding that activation of the systemic immune system by application of bacteria-derived molecules potentiates cochlear damage induced by ototoxic drugs (Hirose et al., 2014; Koo et al., 2015). While noise-induced inflammation is sterile inflammation, it shares many similarities with infection-induced inflammation, as both feature infiltration of circulating monocytes and overproduction of inflammatory molecules. Future investigations into the therapeutic effects of pro-resolution mediators for speedy inflammation resolution hold clinical promise for reducing cochlear susceptibility to subsequent acoustic injury.
Highlights.
Low-level immune activation persists for an extended period in the cochlea after acute acoustic injury.
Persistent immune activation involves both pro- and anti-inflammatory macrophages.
Cochleae recovering from a prior noise injury are susceptible to subsequent acoustic stress.
Repeated exposure in recovering cochleae provokes an exaggerated immune reaction.
Acknowledgments:
The authors would like to thank Peter J. Bush for assisting SEM sample preparation and image acquisition, and Carley Cuzzacrea for creating the anatomic schematic of the cochlea.
Funding: This work was supported by the National Institute on Deafness and Other Communication Disorders of the National Institutes of Health R01DC010154 (BHH) and the Mark Diamond Research Fund from the University at Buffalo FA-18-21 (CZ).
Abbreviations
- ABR
Auditory brainstem response
- BM
Basilar membrane
- DAPI
4’, 6-diamidino-2’-phenylindole, dihydrochloride
- NIHL
Noise-induced hearing loss
- OHC
Outer hair cell
- OSL
Osseous spiral lamina
- PBS
Phosphate buffered saline
- PTS
Permanent threshold shift
- ROS
Reactive oxygen species
- RT-qPCR
Quantitative reverse transcriptase-polymerase chain reaction
- SEM
Scanning electron microscopy
- TTS
Temporary threshold shift
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests: The authors declare that they have no competing interests
References
- Abdallahi OM, Hanna S, De Reggi M, Gharib B 1999. Visualization of oxygen radical production in mouse liver in response to infection with Schistosoma mansoni. Liver 19, 495–500. [DOI] [PubMed] [Google Scholar]
- Agley CC, Velloso CP, Lazarus NR, Harridge SD 2012. An image analysis method for the precise selection and quantitation of fluorescently labeled cellular constituents: application to the measurement of human muscle cells in culture. J Histochem Cytochem 60, 428–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan N, Oguz H, Demirci M, Safak MA, Islam A, Kaytez SK, Samim E 2011. Combined intratympanic and systemic use of steroids for idiopathic sudden sensorineural hearing loss. Otol Neurotol 32, 393–7. [DOI] [PubMed] [Google Scholar]
- Atri C, Guerfali FZ, Laouini D 2018. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang S, Xie YK, Zhang ZJ, Wang Z, Xu ZZ, Ji RR 2018. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J Clin Invest 128, 3568–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basner M, Babisch W, Davis A, Brink M, Clark C, Janssen S, Stansfeld S 2014. Auditory and non-auditory effects of noise on health. Lancet 383, 1325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Limonta S, Boddeke HW 1996. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochemistry international 29, 25–35. [DOI] [PubMed] [Google Scholar]
- Cai Q, Vethanayagam RR, Yang S, Bard J, Jamison J, Cartwright D, Dong Y, Hu BH 2014a. Molecular profile of cochlear immunity in the resident cells of the organ of Corti. J Neuroinflammation 11, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Wang B, Coling D, Zuo J, Fang J, Yang S, Vera K, Hu BH 2014b. Reduction in noise-induced functional loss of the cochleae in mice with pre-existing cochlear dysfunction due to genetic interference of prestin. PloS one 9, e113990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo P, Subramaniam M, Henderson D 1991. The effect of ‘conditioning’ exposures on hearing loss from traumatic exposure. Hear Res 55, 195–200. [DOI] [PubMed] [Google Scholar]
- Chistiakov DA, Killingsworth MC, Myasoedova VA, Orekhov AN, Bobryshev YV 2017. CD68/macrosialin: not just a histochemical marker. Lab Invest 97, 4–13. [DOI] [PubMed] [Google Scholar]
- Davis GS, Brody AR, Adler KB 1979. Functional and physiologic correlates of human alveolar macrophage cell shape and surface morphology. Chest 75, 280–2. [DOI] [PubMed] [Google Scholar]
- Ding D, He J, Allman BL, Yu D, Jiang H, Seigel GM, Salvi RJ 2011. Cisplatin ototoxicity in rat cochlear organotypic cultures. Hear Res 282, 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Qi W, Yu D, Jiang H, Han C, Kim MJ, Katsuno K, Hsieh YH, Miyakawa T, Salvi R, Tanokura M, Someya S 2013. Addition of exogenous NAD+ prevents mefloquine-induced neuroaxonal and hair cell degeneration through reduction of caspase-3-mediated apoptosis in cochlear organotypic cultures. PLoS One 8, e79817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ren J, Zhang D, Li Y, Huang X, Hu Q, Wang H, Song Y, Ni Y, Hou Y 2018. A novel stromal lncRNA signature reprograms fibroblasts to promote the growth of oral squamous cell carcinoma via LncRNA-CAF/interleukin-33. Carcinogenesis 39, 397–406. [DOI] [PubMed] [Google Scholar]
- Dobie RA 2008. The burdens of age-related and occupational noise-induced hearing loss in the United States. Ear Hear 29, 565–77. [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhang C, Frye M, Yang W, Ding D, Sharma A, Guo W, Hu BH 2018. Differential fates of tissue macrophages in the cochlea during postnatal development. Hear Res 365, 110–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfield DA, Lomax MI, Dootz GA, Chen S, Galecki AT, Benjamin IJ, Dolan DF, Altschuler RA 2005. Heat shock factor 1-deficient mice exhibit decreased recovery of hearing following noise overstimulation. J Neurosci Res 81, 589–96. [DOI] [PubMed] [Google Scholar]
- Fredelius L, Rask-Andersen H 1990. The role of macrophages in the disposal of degeneration products within the organ of corti after acoustic overstimulation. Acta Otolaryngol 109, 76–82. [DOI] [PubMed] [Google Scholar]
- Frisina DR, Frisina RD, Snell KB, Burkard R, Walton JP, Ison JR 2001. Auditory temporal processing during aging., Functional neurobiology of aging. pp. 565–579. [Google Scholar]
- Frye MD, Zhang C, Hu BH 2018. Lower level noise exposure that produces only TTS modulates the immune homeostasis of cochlear macrophages. J Neuroimmunol 323, 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye MD, Yang W, Zhang C, Xiong B, Hu BH 2017. Dynamic activation of basilar membrane macrophages in response to chronic sensory cell degeneration in aging mouse cochleae. Hear Res 344, 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka M, Kanzaki S, Okano HJ, Masuda M, Ogawa K, Okano H 2006. Proinflammatory cytokines expression in noise-induced damaged cochlea. J Neurosci Res 83, 575–83. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Gratton MA, Wright CG 1992. Hyperpigmentation of chinchilla stria vascularis following acoustic trauma. Pigment Cell Res 5, 30–7. [DOI] [PubMed] [Google Scholar]
- Greter M, Lelios I, Croxford AL 2015. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front Immunol 6, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KC, Bielefeld E, Hu BH, Henderson D 2006. Increased resistance to free radical damage induced by low-level sound conditioning. Hear Res 213, 118–29. [DOI] [PubMed] [Google Scholar]
- Heinrich F, Lehmbecker A, Raddatz BB, Kegler K, Tipold A, Stein VM, Kalkuhl A, Deschl U, Baumgartner W, Ulrich R, Spitzbarth I 2017. Morphologic, phenotypic, and transcriptomic characterization of classically and alternatively activated canine blood-derived macrophages in vitro. PLoS One 12, e0183572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson D, Hamernik RP 1995. Biologic bases of noise-induced hearing loss. Occup Med 10, 513–34. [PubMed] [Google Scholar]
- Henselman LW, Henderson D, Subramaniam M, Sallustio V 1994. The effect of ‘conditioning’ exposures on hearing loss from impulse noise. Hear Res 78, 1–10. [DOI] [PubMed] [Google Scholar]
- Hirose K, Liberman MC 2003. Lateral wall histopathology and endocochlear potential in the noise-damaged mouse cochlea. J Assoc Res Otolaryngol 4, 339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Rutherford MA, Warchol ME 2017. Two cell populations participate in clearance of damaged hair cells from the sensory epithelia of the inner ear. Hear Res 352, 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Discolo CM, Keasler JR, Ransohoff R 2005. Mononuclear phagocytes migrate into the murine cochlea after acoustic trauma. J Comp Neurol 489, 180–94. [DOI] [PubMed] [Google Scholar]
- Hirose K, Li SZ, Ohlemiller KK, Ransohoff RM 2014. Systemic lipopolysaccharide induces cochlear inflammation and exacerbates the synergistic ototoxicity of kanamycin and furosemide. J Assoc Res Otolaryngol 15, 555–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BH, Henderson D 1997. Changes in F-actin labeling in the outer hair cell and the Deiters cell in the chinchilla cochlea following noise exposure. Hear Res 110, 209–18. [DOI] [PubMed] [Google Scholar]
- Hu BH, Henderson D, Nicotera TM 2002. Involvement of apoptosis in progression of cochlear lesion following exposure to intense noise. Hear Res 166, 62–71. [DOI] [PubMed] [Google Scholar]
- Hu BH, Guo W, Wang PY, Henderson D, Jiang SC 2000. Intense noise-induced apoptosis in hair cells of guinea pig cochleae. Acta Otolaryngol 120, 19–24. [PubMed] [Google Scholar]
- Ichimiya I, Yoshida K, Hirano T, Suzuki M, Mogi G 2000. Significance of spiral ligament fibrocytes with cochlear inflammation. Int J Pediatr Otorhinolaryngol 56, 45–51. [DOI] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y 2001. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 32, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Jacono AA, Hu B, Kopke RD, Henderson D, Van De Water TR, Steinman HM 1998. Changes in cochlear antioxidant enzyme activity after sound conditioning and noise exposure in the chinchilla. Hear Res 117, 31–8. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR 2000. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur T, Ohlemiller KK, Warchol ME 2018. Genetic disruption of fractalkine signaling leads to enhanced loss of cochlear afferents following ototoxic or acoustic injury. J Comp Neurol 526, 824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khullar S, Babbar R 2011. Presbycusis and auditory brainstem responses: a review. Asian Pacific Journal of Tropical Disease. pp. 150–157. [Google Scholar]
- Kirkegaard M, Murai N, Risling M, Suneson A, Jarlebark L, Ulfendahl M 2006. Differential gene expression in the rat cochlea after exposure to impulse noise. Neuroscience 142, 425–35. [DOI] [PubMed] [Google Scholar]
- Koo JW, Quintanilla-Dieck L, Jiang M, Liu J, Urdang ZD, Allensworth JJ, Cross CP, Li H, Steyger PS 2015. Endotoxemia-mediated inflammation potentiates aminoglycoside-induced ototoxicity. Sci Transl Med 7, 298ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotwal GJ, Chien S 2017. Macrophage Differentiation in Normal and Accelerated Wound Healing. Results Probl Cell Differ 62, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC 2006. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci 26, 2115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang H, Ebihara Y, Schmiedt RA, Minamiguchi H, Zhou D, Smythe N, Liu L, Ogawa M, Schulte BA 2006. Contribution of bone marrow hematopoietic stem cells to adult mouse inner ear: mesenchymal cells and fibrocytes. J Comp Neurol 496, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TN, Straatman LV, Lea J, Westerberg B 2017. Current insights in noise-induced hearing loss: a literature review of the underlying mechanism, pathophysiology, asymmetry, and management options. J Otolaryngol Head Neck Surg 46, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichtle A, Klenke C, Ebmeyer J, Daerr M, Bruchhage KL, Hoffmann AS, Ryan AF, Wollenberg B, Sudhoff H 2015. NOD-Like Receptor Signaling in Cholesteatoma. Biomed Res Int 2015, 408169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Watanabe K, Fujioka M, Ogawa K 2017. Characterization of slow-cycling cells in the mouse cochlear lateral wall. PLoS One 12, e0179293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–8. [DOI] [PubMed] [Google Scholar]
- McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF 2013. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A 110, 17253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R 2008. Origin and physiological roles of inflammation. Nature 454, 428–35. [DOI] [PubMed] [Google Scholar]
- Mills JH 1973. Threshold shifts produced by exposure to noise in chinchillas with noise-induced hearing losses. J Speech Hear Res 16, 700–8. [DOI] [PubMed] [Google Scholar]
- Nakamoto T, Mikuriya T, Sugahara K, Hirose Y, Hashimoto T, Shimogori H, Takii R, Nakai A, Yamashita H 2012. Geranylgeranylacetone suppresses noise-induced expression of proinflammatory cytokines in the cochlea. Auris Nasus Larynx 39, 270–4. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ 2009. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Canlon B 2002. Protective mechanisms of sound conditioning. Adv Otorhinolaryngol 59, 96–105. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Frisina RD 2008. Age-related hearing loss and its cellular and molecular bases. Auditory trauma, protection, and repair,, 145–194. [Google Scholar]
- Oishi N, Schacht J 2011. Emerging treatments for noise-induced hearing loss. Expert Opin Emerg Drugs 16, 235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano T, Nakagawa T, Kita T, Kada S, Yoshimoto M, Nakahata T, Ito J 2008. Bone marrow-derived cells expressing Iba1 are constitutively present as resident tissue macrophages in the mouse cochlea. J Neurosci Res 86, 1758–67. [DOI] [PubMed] [Google Scholar]
- Olingy CE, San Emeterio CL, Ogle ME, Krieger JR, Bruce AC, Pfau DD, Jordan BT, Peirce SM, Botchwey EA 2017. Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci Rep 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Hu Z, Bard J, Jamison J, Cai Q, Hu BH 2013a. Transcriptome characterization by RNA-Seq reveals the involvement of the complement components in noise-traumatized rat cochleae. Neuroscience 248C, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Hu Z, Bard J, Jamison J, Cai Q, Hu BH 2013b. Transcriptome characterization by RNA-Seq reveals the involvement of the complement components in noise-traumatized rat cochleae. Neuroscience 248, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez R, Freeman S, Sohmer H 2004. Effect of an initial noise induced hearing loss on subsequent noise induced hearing loss. Hear Res 192, 101–6. [DOI] [PubMed] [Google Scholar]
- Plontke S, Zenner HP 2004. Current aspects of hearing loss from occupational and leisure noise. GMS Curr Top Otorhinolaryngol Head Neck Surg 3, Doc06. [PMC free article] [PubMed] [Google Scholar]
- Polliack A 1977. Normal Leukocytes Normal, transformed and leukemic leukocytes: A scanning electron microscopy atlas. pp. 8–52. [Google Scholar]
- Poupel L, Boissonnas A, Hermand P, Dorgham K, Guyon E, Auvynet C, Charles FS, Lesnik P, Deterre P, Combadiere C 2013. Pharmacological inhibition of the chemokine receptor, CX3CR1, reduces atherosclerosis in mice. Arterioscler Thromb Vasc Biol 33, 2297–305. [DOI] [PubMed] [Google Scholar]
- Puel JL, Ruel J, Gervais d’Aldin C, Pujol R 1998. Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport 9, 2109–14. [DOI] [PubMed] [Google Scholar]
- Rodero MP, Poupel L, Loyher PL, Hamon P, Licata F, Pessel C, Hume DA, Combadiere C, Boissonnas A 2015. Immune surveillance of the lung by migrating tissue monocytes. Elife 4, e07847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S, Imai Y 2001. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochemical and biophysical research communications 286, 292–297. [DOI] [PubMed] [Google Scholar]
- Saunders JC, Dear SP, Schneider ME 1985. The anatomical consequences of acoustic injury: A review and tutorial. J Acoust Soc Am 78, 833–60. [DOI] [PubMed] [Google Scholar]
- Saunders JC, Cohen YE, Szymko YM 1991. The structural and functional consequences of acoustic injury in the cochlea and peripheral auditory system: a five year update. J Acoust Soc Am 90, 136–46. [DOI] [PubMed] [Google Scholar]
- Sautter NB, Shick EH, Ransohoff RM, Charo IF, Hirose K 2006. CC chemokine receptor 2 is protective against noise-induced hair cell death: studies in CX3CR1(+/GFP) mice. J Assoc Res Otolaryngol 7, 361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Xu J, Kondo K, Ding D, Salvi RJ, Yamasoba T, Rabinovitch PS, Weindruch R, Leeuwenburgh C, Tanokura M, Prolla TA 2009. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc Natl Acad Sci U S A 106, 19432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stence N, Waite M, Dailey ME 2001. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–66. [PubMed] [Google Scholar]
- Streit WJ, Graeber MB, Kreutzberg GW 1988. Functional plasticity of microglia: a review. Glia 1, 301–7. [DOI] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ 2007. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117, 185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WJ, Thorne PR, Vlajkovic SM 2016. Characterisation of cochlear inflammation in mice following acute and chronic noise exposure. Histochem Cell Biol 146, 219–30. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Chen GD, Hu BH, Chi LH, Li M, Zheng G, Bielefeld EC, Jamesdaniel S, Coling D, Henderson D 2009. The effects of acoustic environment after traumatic noise exposure on hearing and outer hair cells. Hear Res 250, 10–8. [DOI] [PubMed] [Google Scholar]
- Tornabene SV, Sato K, Pham L, Billings P, Keithley EM 2006. Immune cell recruitment following acoustic trauma. Hear Res 222, 115–24. [DOI] [PubMed] [Google Scholar]
- Ulehlova L 1983. Stria vascularis in acoustic trauma. Arch Otorhinolaryngol 237, 133–8. [PubMed] [Google Scholar]
- Varga G, Foell D 2018. Anti-inflammatory monocytes-interplay of innate and adaptive immunity. Mol Cell Pediatr 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vethanayagam RR, Yang W, Dong Y, Hu BH 2016. Toll-like receptor 4 modulates the cochlear immune response to acoustic injury. Cell Death Dis 7, e2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Fujioka M, Kanzaki S, Okano HJ, Shibata S, Yamashita D, Masuda M, Mihara M, Ohsugi Y, Ogawa K, Okano H 2010. Blockade of interleukin-6 signaling suppressed cochlear inflammatory response and improved hearing impairment in noise-damaged mice cochlea. Neurosci Res 66, 345–52. [DOI] [PubMed] [Google Scholar]
- Weinstock A, Fisher EA 2019. Methods to Study Monocyte and Macrophage Trafficking in Atherosclerosis Progression and Resolution. Methods Mol Biol 1951, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JW, Randolph GJ, Zinselmeyer BH 2017. A Polecat’s View of Patrolling Monocytes. Circ Res 120, 1699–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T, Omelchenko I, Foster S, Zhang Y, Shi X, Nuttall AL 2014. JAK2/STAT3 inhibition attenuates noise-induced hearing loss. PLoS One 9, e108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AC, Ryan AF 2015. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front Aging Neurosci 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Cai Q, Vethanayagam RR, Wang J, Yang W, Hu BH 2016. Immune defense is the primary function associated with the differentially expressed genes in the cochlea following acoustic trauma. Hear Res 333, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Vethanayagam RR, Dong Y, Cai Q, Hu BH 2015. Activation of the antigen presentation function of mononuclear phagocyte populations associated with the basilar membrane of the cochlea after acoustic overstimulation. Neuroscience 303, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Kristiansen A, Liberman MC 1999. Heat stress and protection from permanent acoustic injury in mice. J Neurosci 19, 10116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB 2010. Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–80. [DOI] [PubMed] [Google Scholar]