

Abstract
γ-Secretase is a membrane-embedded aspartyl protease complex central in biology and medicine. How this enzyme recognizes transmembrane substrates and catalyzes hydrolysis in the lipid bilayer is unclear. Inhibitors that mimic the entire substrate transmembrane domain and engage the active site should provide important tools for structural biology, yielding insight into substrate gating and trapping the protease in the active state. Here we report transmembrane peptidomimetic inhibitors of the γ-secretase complex that contain an N-terminal helical peptide region that engages a substrate docking exosite and a C-terminal transition-state analog moiety targeted to the active site. Both regions are required for stoichiometric inhibition of γ-secretase. Moreover, enzyme inhibition kinetics and photoaffinity probe displacement experiments demonstrate that both the docking exosite and the active site are engaged by the bipartite inhibitors. The solution conformations of these potent transmembranemimetic inhibitors are similar to those of bound natural substrates, suggesting these probes are preorganized for high-affinity binding and should allow visualization of the active γ-secretase complex, poised for intramembrane proteolysis, by cryo-electron microscopy.
Graphical Abstract
γ-Secretase is a membrane-embedded aspartyl protease complex, with presenilin as the catalytic component,1 that hydrolyzes >90 known substrates,2–3 including the amyloid precursor protein (APP) of Alzheimer’s disease and the Notch family of developmental signaling receptors. How this enzyme recognizes substrate transmembrane domains and carries out intramembrane proteolysis has been mysterious. Advances in cryo-electron microscopy paved the way to the first detailed structure of the ~230 kDa complex,4 comprised of membrane proteins nicastrin, Aph-1 and Pen-2 along with presenilin. Most recently, structures of γ-secretase bound to Notch and APP substrates were reported,5–6 providing important insights into substrate recognition. Nevertheless, the active site was disabled through mutagenesis, and the substrates were artificially crosslinked to presenilin. To date, the enzyme has not been trapped in its active state, and the lateral gating pathway of substrate into the active site remains unclear.
To address this problem, we aimed to develop substrate TMD mimetics as chemical probes for structural analysis of γ-secretase. These probes would trap the active enzyme at the transition state, allowing acquisition of high-resolution snapshots of substrate recognition with the protease poised for catalysis of TMD cleavage. We and others previously reported peptidomimetic transition-state analogue inhibitors (TSAs) of γ-secretase7–9 and use of these as probes for active site binding pockets.10–13 We have also reported helical peptide inhibitors (HPIs) that interact with a substrate docking exosite distinct from but proximal to the active site.14–15 We recently demonstrated that substrate TMD is sufficient for high-affinity binding (Km < 100 nM)16 and therefore sought peptide-based inhibitors that would mimic the entire TMD and interact with both the docking site and the active site. Specifically, we worked to couple an HPI to a TSA through a variable linker (Fig. 1).
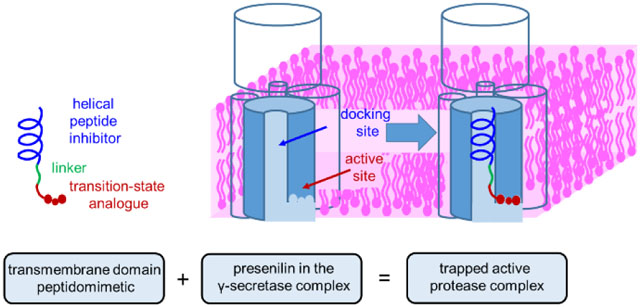
Figure 1. Design of inhibitors that mimic the transmembrane domain of γ-secretase substrates.
Helical peptide inhibitors (HPIs) directed to the substrate docking exosite were conjugated through a variable linker to transition-state analogue inhibitors (TSAs) directed to the active site. Presenilin (blue-grey) and other components of the γ-secretase complex (outlined) are shown schematically in the absence and presence of a hybrid HPI-TSA inhibitor.
We chose a pentapeptide TSA with a hydroxyethylurea moiety and spanning residues P2 through P3’ (TSA 1, see Table 1) that showed optimal activity in a cell-based assay for inhibiting γ-secretase-mediated production of the amyloid β-peptide (Aβ) from APP substrate.13 Residues P1’, P2’, and P3’ are especially important for substrate recognition and processing.17 In a purified enzyme assay, TSA 1 displayed an IC50 of 41 nM (Table 1). HPI 2, containing helix-inducing α-aminoisobutyric acid (Aib) residues spaced apart to arrange the Aib residues along one face of the helix and presenting APP TMD residues to the enzyme along the rest of the helix,14 showed comparable activity (IC50 of 58 nM). We aimed to connect these two compounds between HPI C-terminus and TSA N-terminus with intervening linkers of varying lengths. Coupling in this manner, with the TSA on the C-terminus of the TMD mimetic, is consistent with γ-secretase initially cleaving APP TMD on the C-terminal end three residues from the membrane-cytosol interface.18–19 To access these highly hydrophobic HPI-TSA conjugates, we generated hydroxyethylurea-containing tripeptide building blocks suitably protected for solid-phase peptide synthesis (Scheme S1). All synthesized peptides were purified to >95% by HPLC.
Table 1.
Inhibition of γ-secretase by helical peptide/transition-state analogue conjugates.
| Cmpd | Helical Peptide | Linker | Transition-State Analoguea | IC50b |
|---|---|---|---|---|
| APP transmembrane residues 707–717: | --------Optimized TSA----------- | |||
| ---Val-Gly-Gly-Val-Val-Ile-Ala-Thr-Val-Ile---- | ----P2 - P1 - P1′- P2′-P3′----- | |||
| 1 | Boc-Val-Phe-ψ-Phe-Leu-Val-NH2 | 41 ± 4 | ||
| 2 | Boc-Val-Gly-Aib-Val-Val-Ile-Aib-Phe-Val-Aib-OCH3 | 58 ± 6 | ||
| 3 | Boc-Val-Gly-Aib-Val-Val-Ile-Aib-Phe-Val-Aib- | - | -Val-Phe-ψ-Phe-Leu-Val-NH2 | 53 ± 1 |
| 4 | Boc-Val-Gly-Aib-Val-Val-Ile-Aib-Phe-Val-Aib- | -NH(CH2)2CO- | -Val-Phe-ψ-Phe-Leu-Val-NH2 | 12 ± 2 |
| 5 | Boc-Val-Gly-Aib-Val-Val-Ile-Aib-Phe-Val-Aib- | -NH(CH2)4CO- | -Val-Phe-ψ-Phe-Leu-Val-NH2 | 10 ± 1 |
| 6 | Boc-Val-Gly-Aib-Val-Val-Ile-Aib-Phe-Val-Aib- | -NH(CH2)8CO- | -Val-Phe-ψ-Phe-Leu-Val-NH2 | 0.80 ± 0.03 |
| 7 | - | BocNH(CH2)8CO- | -Val-Phe-ψ-Phe-Leu-Val-NH2 | 16 ± 1 |
| 8 | Boc-Val-Gly-Aib-Val-Val-Ile-Aib-Phe-Val-Aib- | -NH(CH2)8CO- | -Val-Phe - Phe-Leu-Val-NH2 | 18 ± 3 |
| 9 | Boc-Val-Gly-Aib-DVal-D Val-Ile-Aib-Phe-Val-Aib- | -NH(CH2)8CO- | -Val-Phe-ψ-Phe-Leu-Val-NH2 | 6 ± 2 |
ψ represents hydoxyethylurea replacement of peptide backbone;
concentration that inhibits 50% activity of 1 nM purified γ-secretase
HPI-TSA conjugate 3, containing no linker moiety, displayed an IC50 of 53 nM, with no improvement in potency compared to either TSA 1 or HPI 2 alone (Table 1). Inserting a 4-atom hydro-carbon linker gave 4, with an IC50 of 12 nM, while inserting a 6-atom spacer provided 5, with an IC50 of 10 nM, increasing potency 4-to-6-fold over TSA 1 or HPI 2. Finally, extending the linker to a 10-atom spacer gave 6, the most potent compound in the series, with an IC50 of 0.8 nM. This compound essentially titrates the enzyme, as the assay was done with 1 nM of purified γ-secretase.
Control peptide 7 containing TSA 1 and the 10-atom spacer alone was roughly twice as potent as 1, showing that the linker contributes to binding to the enzyme. Control peptide 8, with the transition-state mimicking isostere in 6 replaced with an amide bond, showed >20-fold reduced potency (IC50 = 18 nM). Moreover, MS analysis revealed that 8 was hydrolyzed by γ-secretase between the two Phe residues, validating the correct registry of binding and appropriate placement of the transition-state isostere in 6 (Fig. S1). When two internal L-Val residues of the potent 6 were exchanged with D-Val in compound 9, inhibitory potency decreased nearly eight-fold, presumably due to disruption of the helical conformation in this part of the TMD mimetic. A similar series of TMD mimetics were also synthesized with P2’ in the TSA moiety as Ala in place of Leu (10–16, Table S2), leading to identification of another stoichiometric inhibitor 16 (IC50 + 0.5 nM).
To confirm helicity of the HPI portion of 6 and disruption of this helix in 9, we performed 2D NMR experiments. Rotating frame nuclear Overhauser effect spectroscopy (ROESY) reveals spatial proximity between NH↔NH and CαH↔NH protons in peptides. NH↔NH cross-peak intensities between residues 5↔6, 6↔7, 7↔8 were found to be higher in 6 than in 9, suggesting that the HPI region of 6 is more helical than that of 9 (Fig. S3). In the CαH↔NH region of 6, medium-range NOEs indicated coupling between distal residues (e.g., 1↔4, 2↔5, 4↔7, 6↔9; Fig. S4). Such cross couplings are absent in 9, again suggesting that the HPI region of 6 is more helical than that of 9. Thus, the integrity of the helical peptide moiety is apparently important for maintaining stoichiometric inhibition of γ-secretase.
Enzyme inhibition kinetics experiments were carried out for the two most potent HPI-TSA inhibitors (6 and 16). Both compounds showed noncompetitive inhibition (Figs. S5, S6). Both HPI 2 and TSA 17 (identical to 1 but with C-terminal methyl ester) likewise showed noncompetitive inhibition on their own (Fig. S7), as has been reported before with other γ-secretase inhibitors.20 To determine whether the HPI and TSA moieties of 6 bind both docking and active sites on the enzyme, we performed cross-competition assays between two inhibitors.21 In these experiments, TSA 17 and HPI 2 did not compete with each other for binding, consistent with binding to distinct sites (active site for 17, docking site for 2)(Fig. 2a). However, HPI-TSA inhibitor (6) competed with both TSA 17 and HPI 2 for binding (seen as parallel lines in Fig. 2b,c), indicating that 6 occupies both docking site and active site on the enzyme.
Figure 2. Cross-competition kinetic experiment between two inhibitors.
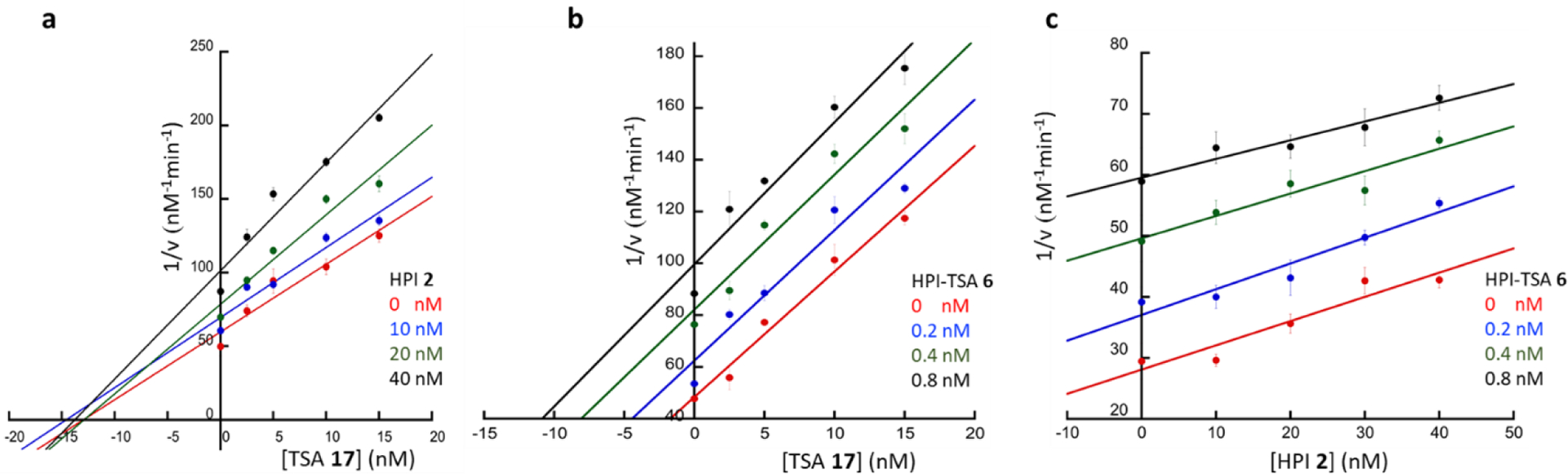
(a) TSA 17 and HPI 2 at [2] = 0 nM (●), 10 nM (●), 20 nM (●), and 40 nM (●). These two inhibitors do not compete, as lines converge near the x-axis. (b) TSA 17 and HPI-TSA 6 at [6] = 0 nM (●), 0.2 nM (●), 0.4 nM (●), and 0.8 nM (●). Parallel lines indicate cross-competition. (c) HPI 2 and HPI-TSA 6 at [6] = 0 nM (●), 0.2 nM (●), 0.4 nM (●), and 0.8 nM (●). Again, parallel lines indicate cross-competition, and HPI-TSA 6 competes with both TSA 17 and HPI 2.
To verify this, we performed competition experiments with biotinylated photoaffinity probes for γ-secretase:15, 22 active-site-directed photoprobe TSA-Bpa-Bt based on TSA 1 and docking-site-directed photoprobes HPI-Bpa-Bt based on HPI 2. We have previously shown that 10-residue HPIs do not inhibit labeling of presenilin by TSA-Bpa-Bt, whereas TSAs do not inhibit labeling by 10-residue HPI-Bpa-Bt.15 Solubilized lysates from γ-secretase-expressing human embryonic kidney 293 cells were incubated with each photoprobe in the presence and absence of the HPI-TSAs inhibitors and irradiated at 350 nM. Photolabelled proteins were pulled down with streptavidin beads, and the ~32 kDa band of the presenilin-1 (PS1) N-terminal fragment (NTF) subunit was detected by immunoblot. Lack of competition between TSA and HPI was confirmed (Fig. 3a, b). Decreased photolabelling of the PS1 NTF γ-secretase subunit by either probe was observed in the presence of HPI-TSA inhibitors 6 and 16 (Fig. 3c,d, lane 1 vs 2 and 3) and in a concentration-dependent manner (Fig. S8). These results indicate that stoichiometric inhibitors 6 and 16 interact with both active and docking sites on γ-secretase, as they compete with the binding of both photoprobes.
Figure 3: Competition of HPI-TSAs with photoaffinity probes for γ-secretase.
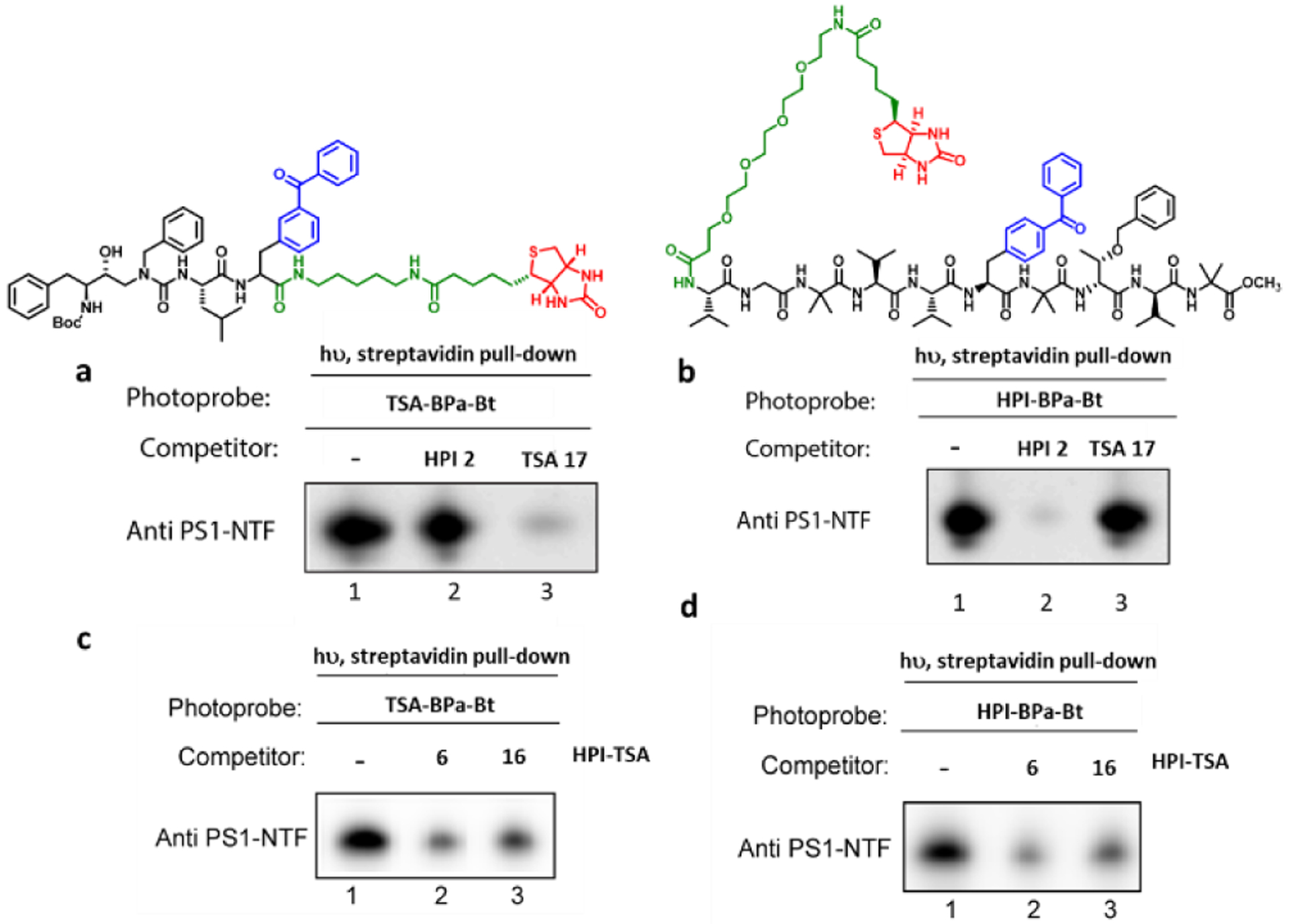
Photoprobes TSA-Bpa-Bt (left) and HPI-Bpa-Bt (right) covalently label presenilin-1 (PS1) N-terminal fragment (NTF) at the active site and docking site, respectively. (a) TSA (17) but not HPI (2) decreased the labelling of PS1 NTF by the TSA photoprobe. (b) HPI (2) but not TSA (17) decreased the labelling PS1 NTF by the HPI photoprobe. (c, d). HPI-TSAs 6 and 16 decreased labelling by both TSA photoprobe (c) and HPI photoprobe (d).
Finally, the conformation of these new inhibitors, determined using NMR constraints for 6, resembles the bound conformation of substrate in the new cryo-EM structures (Fig. 4), with a helical N-terminal region and an unfolded or extended C-terminal region. Thus, the new potent HPI-TSA inhibitors appear to be preorganized for ideal binding to γ-secretase and trapping the enzyme complex as it would be when poised for intramembrane proteolysis. Moreover, after initial substrate proteolysis, γ-secretase carries out successive carboxypeptidase trimming in intervals of three amino acids of the N-terminal cleavage product that contains most of the substrate TMD,19 and this trimming activity is deficient in γ-secretase complexes with Alzheimer-causing presenilin mutations.23–24 These new TMD mimetic inhibitors, with three P’ residues in the TSA moiety, should capture the enzyme as it would carry out this trimming reaction. Cryo-EM analysis of these new probes in complex with γ-secretase is in progress. Variations on these TMD inhibitors are under development to trap other stages of substrate interaction with γ-secretase (e.g., lateral gating, initial proteolysis). Together, these conjugates should be important structural probes for gaining insights into the mechanism of intramembrane proteolysis by γ-secretase and how mutations in the enzyme cause Alzheimer’s disease.
Figure 4: Solution conformation of designed HPI-TSA conjugate 6 resembles that of APP TMD substrate bound to γ-secretase.
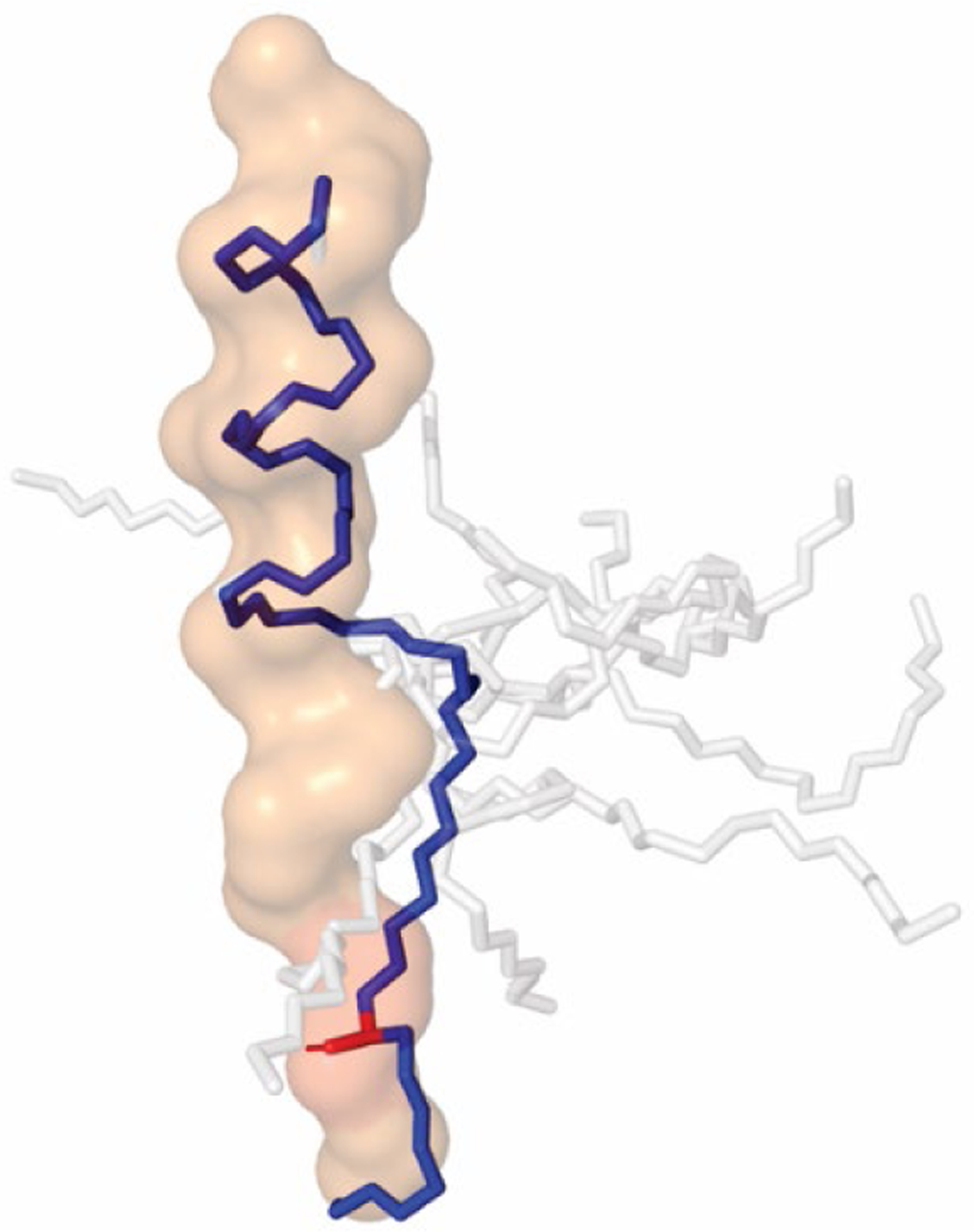
NMR constraints were used to determine low-energy conformations of 6. The top 10 conformers are shown as sticks, with the conformer closest to that of the bound substrate in blue. Structure rendered in Pymol using PDB file 6IYC for bound APP substrate, shown as surface outline. Transition-state mimicking hydroxyl group (red) of 6 overlaps with the scissile amide bond in the extended region of APP substrate when the helical moiety is aligned with the helical region of bound APP substrate.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Chamani Perera (KU Synthetic Chemical Biology Core) for technical help for solid-phase peptide synthesis, Dr. Benjamin Neuenswander (KU Purification Core) for peptide purity analysis, and Dr. Todd D. Williams (Director of the KU Mass Spectrometry Laboratory) for LC-MS/MS studies of the cleavage of control peptides by γ-secretase. Supported by NIH R01 grant GM 122894 to M.S.W. 2D NMR experiments were supported by NIH grant P30GM110761 (PI: Robert P. Hanzlik). This study made use of NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by NIH grant P41GM111135 (NIGMS).
Footnotes
Supporting Information
Detailed methods and results for (1) synthesis and characterization of hydroxyethylurea tripeptidomimetic building blocks and all HPI-TSA inhibitor peptidomimetics, (2) γ-secretase inhibition assays, (3) photoaffinity labelling experiments, (4) inhibition of γ-secretase by series 10–16 (P2’ = Ala); (5) LC-MS/MS study of proteolysis of peptides 8 and 12 by γ-secretase, (6) 1D- and 2D- NMR studies of 6 and 9, and (7) spectra of synthetic intermediates and final compounds are available in supporting information (95 pages).
REFERENCES
- 1.Wolfe MS, Structure and function of the γ-secretase complex. Biochemistry 2019, 58 (27), 2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hemming ML; Elias JE; Gygi SP; Selkoe DJ, Proteomic profiling of γ-secretase substrates and mapping of substrate requirements. PLoS Biol 2008, 6 (10), e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beel AJ; Sanders CR, Substrate specificity of γ-secretase and other intramembrane proteases. Cell Mol Life Sci 2008, 65 (9), 1311–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai XC; Yan C; Yang G; Lu P; Ma D; Sun L; Zhou R; Scheres SH; Shi Y, An atomic structure of human γ-secretase. Nature 2015, 525 (7568), 212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang G; Zhou R; Zhou Q; Guo X; Yan C; Ke M; Lei J; Shi Y, Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565 (7738), 192–197. [DOI] [PubMed] [Google Scholar]
- 6.Zhou R; Yang G; Guo X; Zhou Q; Lei J; Shi Y, Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363 (6428), eaaw0930. [DOI] [PubMed] [Google Scholar]
- 7.Wolfe MS; Citron M; Diehl TS; Xia W; Donkor IO; Selkoe DJ, A substrate-based difluoro ketone selectively inhibits Alzheimer’s γ-secretase activity. J Med Chem 1998, 41 (1), 6–9. [DOI] [PubMed] [Google Scholar]
- 8.Shearman MS; Beher D; Clarke EE; Lewis HD; Harrison T; Hunt P; Nadin A; Smith AL; Stevenson G; Castro JL, L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β-protein precursor γ-secretase activity. Biochemistry 2000, 39 (30), 8698–8704. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe MS; Xia W; Moore CL; Leatherwood DD; Ostaszewski B; Donkor IO; Selkoe DJ, Peptidomimetic probes and molecular modeling suggest Alzheimer’s γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 1999, 38, 4720–7. [DOI] [PubMed] [Google Scholar]
- 10.Moore CL; Leatherwood DD; Diehl TS; Selkoe DJ; Wolfe MS, Difluoro ketone peptidomimetics suggest a large S1 pocket for Alzheimer’s γ-secretase: implications for inhibitor design. J Med Chem 2000, 43 (18), 3434–3442. [DOI] [PubMed] [Google Scholar]
- 11.Li YM; Xu M; Lai MT; Huang Q; Castro JL; DiMuzio-Mower J; Harrison T; Lellis C; Nadin A; Neduvelil JG; Register RB; Sardana MK; Shearman MS; Smith AL; Shi XP; Yin KC; Shafer JA; Gardell SJ, Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000, 405 (6787), 689–94. [DOI] [PubMed] [Google Scholar]
- 12.Esler WP; Kimberly WT; Ostaszewski BL; Diehl TS; Moore CL; Tsai J-Y; Rahmati T; Xia W; Selkoe DJ; Wolfe MS, Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nature Cell Biology 2000, 2 (7), 428–34. [DOI] [PubMed] [Google Scholar]
- 13.Esler WP; Das C; Wolfe MS, Probing pockets S2-S4’ of the γ-secretase active site with (hydroxyethyl)urea peptidomimetics. Bioorg Med Chem Lett 2004, 14 (8), 1935–8. [DOI] [PubMed] [Google Scholar]
- 14.Das C; Berezovska O; Diehl TS; Genet C; Buldyrev I; Tsai JY; Hyman BT; Wolfe MS, Designed helical peptides inhibit an intramembrane protease. J Am Chem Soc 2003, 125 (39), 11794–5. [DOI] [PubMed] [Google Scholar]
- 15.Kornilova AY; Bihel F; Das C; Wolfe MS, The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc Natl Acad Sci USA 2005, 102 (9), 3230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolduc DM; Montagna DR; Gu Y; Selkoe DJ; Wolfe MS, Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc Natl Acad Sci USA 2016, 113 (5), E509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolduc DM; Montagna DR; Seghers MC; Wolfe MS; Selkoe DJ, The amyloid-β forming tripeptide cleavage mechanism of γ-secretase. eLife 2016, 5, pii: e17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weidemann A; Eggert S; Reinhard FB; Vogel M; Paliga K; Baier G; Masters CL; Beyreuther K; Evin G, A novel var ε-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 2002, 41 (8), 2825–2835. [DOI] [PubMed] [Google Scholar]
- 19.Takami M; Nagashima Y; Sano Y; Ishihara S; Morishima-Kawashima M; Funamoto S; Ihara Y, γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J Neurosci 2009, 29 (41), 13042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian G; Sobotka-Briner CD; Zysk J; Liu X; Birr C; Sylvester MA; Edwards PD; Scott CD; Greenberg BD, Linear non-competitive inhibition of solubilized human γ-secretase by pepstatin A methylester, L685458, sulfonamides, and benzodiazepines. J Biol Chem 2002, 277 (35), 31499–505. Epub 2002 Jun 18. [DOI] [PubMed] [Google Scholar]
- 21.Tian G; Ghanekar SV; Aharony D; Shenvi AB; Jacobs RT; Liu X; Greenberg BD, The mechanism of γ-secretase: multiple inhibitor binding sites for transition state analogs and small molecule inhibitors. J Biol Chem 2003, 278 (31), 28968–75. [DOI] [PubMed] [Google Scholar]
- 22.Kornilova AY; Das C; Wolfe MS, Differential effects of inhibitors on the γ-secretase complex. Mechanistic implications. J Biol Chem 2003, 278 (19), 16470–3. [DOI] [PubMed] [Google Scholar]
- 23.Quintero-Monzon O; Martin MM; Fernandez MA; Cappello CA; Krzysiak AJ; Osenkowski P; Wolfe MS, Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry 2011, 50 (42), 9023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez MA; Klutkowski JA; Freret T; Wolfe MS, Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem 2014, 289 (45), 31043–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.