

Abstract
Fostemsavir, a prodrug of human immunodeficiency virus attachment inhibitor temsavir (TMR), is in phase III development in combination with other antiretroviral agents for the treatment of human immunodeficiency virus type I (HIV‐1) infection in heavily treatment‐experienced adults with multidrug‐resistant HIV‐1 infection for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations. The proarrhythmic potential of fostemsavir was studied in a thorough QT study and exposure–response modeling was performed at therapeutic and supratherapeutic concentrations of TMR. Fostemsavir 1,200 mg b.i.d. did not result in a clinically meaningful change from placebo in baseline‐adjusted Fridericia‐corrected QTc (ddQTcF); however, at a supratherapeutic dose of 2,400 mg b.i.d., the upper bound of the two‐sided 90% confidence interval (CI) of ddQTcF was 13.2 msec, exceeding the clinically important 10 msec threshold. A linear model of ddQTcF as a function of TMR plasma concentrations described these observations. Based on simulations with this model, TMR concentrations up to 7,500 ng/mL are expected to have an upper 90% CI bound for QTcF ≤ 10 msec. This concentration is 4.2‐fold higher than the geometric mean TMR peak plasma concentration (Cmax) of 1,770 ng/mL in heavily treatment‐experienced HIV‐1 infected patients administered fostemsavir 600 mg b.i.d. in the phase III BRIGHTE study (NCT02362503).
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Temsavir (TMR), an attachment inhibitor of human immunodeficiency virus type 1, is in development as the active moiety of an oral prodrug, fostemsavir. Fostemsavir has shown QT interval prolongation risk at supratherapeutic dosing (or exposure), but not at therapeutic dosing (or exposure).
WHAT QUESTION DID THE STUDY ADDRESS?
☑ This study evaluated and modeled the effect of therapeutic and supratherapeutic TMR exposure on the QT interval to identify a concentration threshold for QT prolongations of clinical importance.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Therapeutic doses of fostemsavir are unlikely to result in a meaningful QT prolongation, even with concomitant use of medications that may inhibit CYP3A4, BCRP, and P‐gp, including pharmaco‐enhancing agents used to boost levels of other antiretrovirals that also elevate TMR exposure.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The data show that the QT effects of TMR are unlikely to be relevant at the therapeutic fostemsavir dose of 600 mg b.i.d. Corrected QT prolongation risk is confined to TMR exposures considerably greater than those likely to be attained at the recommended phase III fostemsavir dose with or without medications that may inhibit CYP3A4, BCRP, and P‐gp, including pharmaco‐enhancing agents.
Fostemsavir is a first‐in‐class oral human immunodeficiency virus type 1 (HIV‐1) attachment inhibitor prodrug that is hydrolyzed in vivo to its active moiety, temsavir (TMR).1 Fostemsavir is in phase III development in combination with other antiretroviral agents for the treatment of HIV‐1 infection in heavily treatment‐experienced adults with multidrug resistant HIV‐1 infection for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations. TMR binds directly to the HIV‐1 envelope glycoprotein gp120, close to the CD4 binding sites, locking it in a conformational state that prohibits the initial interaction between the virus and CD4 cell‐surface receptors to prevent viral attachment and subsequent entry into host CD4+ target cells.2 TMR has good membrane permeability but low solubility and a short half‐life, requiring its administration as an orally available extended‐release prodrug that undergoes alkaline phosphatase‐mediated conversion to TMR at the luminal surface of the small intestine.3, 4 Fostemsavir has a unique resistance profile with no in vitro cross‐resistance to other antiretroviral drug classes,5 and is active regardless of HIV‐1 tropism.2, 5, 6 The lack of cross‐resistance shown by TMR with other agents has made it an attractive prospect for treatment of multidrug‐resistant HIV‐1.7
Fostemsavir has been well‐tolerated in phase II and III studies at multiple doses up to 1,200 mg q.d. or 800 mg b.i.d. in combination with other antiretrovirals for 96 weeks.8 The proposed therapeutic dose in phase III is fostemsavir 600 mg b.i.d.9 Although nonclinical assessment of fostemsavir or TMR in dog telemetry studies indicated cardiovascular liability at maximum observed TMR concentrations ≥ 3,600 ng/mL (unpublished data), no clinically relevant cardiovascular abnormalities were observed in single‐ascending and multiple‐ascending dose studies in healthy volunteers who received a total daily fostemsavir dose of 20–2,400 mg.10 Corrected QT (QTc) prolongation risk at supratherapeutic doses is present for certain approved HIV medications (e.g., rilpivirine and lopinavir/ritonavir).11, 12 As per International Conference on Harmonization (ICH) E14 guideline,13 new pharmaceutical agents should be subjected to rigorous evaluation of QT interval prolongation to assess the potential to delay cardiac repolarization that leads to the development of ventricular arrhythmias (i.e., torsade de pointes), which may result in sudden death. In order to assess the pro‐arrhythmic potential of fostemsavir in humans, a thorough QT (TQT) study13 was performed in healthy volunteers and data from this study were used to derive a population exposure–response model of QT prolongation as a function of plasma TMR exposure. Simulations from this model were used to predict the effect of therapeutic exposures on QT prolongation with and without CYP3A4 inhibitors in heavily treatment‐experienced HIV‐1‐infected patients.
METHODS
Thorough QT/QTc study
This was a randomized, double‐blinded, two‐part, phase I study for evaluation of pro‐arrhythmic potential in non‐antiarrhythmic drugs.13 Eligible subjects were male and female adults aged 18–49 years with a body mass index of 18–32 kg/m2 inclusive, who were considered healthy according to medical history, physical examination, laboratory evaluation, and screening electrocardiogram (ECG) results. The study was conducted at a single clinical site in the United States from February 6, 2012, to May 27, 2012, in accordance with the ICH Code of Good Clinical Practice and with the ethical principles originating in the Declaration of Helsinki. All subjects provided informed consent. The protocol and informed consent forms were approved by the relevant institutional review board (Chesapeake Research Review, Columbia, MD).
Part 1 of the study was a sentinel cohort assessing the safety and pharmacokinetics (PKs) of the supratherapeutic dose of fostemsavir to be used in the main study (2,400 mg b.i.d.). Subjects were randomized 3:1 to receive oral fostemsavir or placebo for 7 days. Standard 12‐lead ECGs were conducted daily throughout part 1, and predose plasma concentrations of TMR were assessed on days 5–7, with 12‐hour serial sampling of TMR concentrations undertaken on day 7.
Part 2, the main study, was initiated after safety data from part 1 were reviewed and deemed acceptable by the sponsor and investigator. Part 2 had a double‐dummy, placebo‐controlled and active (moxifloxacin)‐controlled 4 × 4 Williams crossover design14 incorporating therapeutic and supratherapeutic doses of fostemsavir, with four treatments in four 7‐day treatment periods. Four treatments comprised 1,200 mg fostemsavir q.d. for 7 days (treatment A); 2,400 mg fostemsavir b.i.d. for 7 days (treatment B); a single dose of moxifloxacin 400 mg on the morning of day 7 (treatment C); or placebo b.i.d. for 7 days (treatment D). The 1,200 mg dose represents the clinically relevant dose assessed in phase II studies and was selected as providing the highest therapeutically relevant TMR peak plasma concentration (Cmax). Subjects were randomized 1:1:1:1 to treatment sequences ABCD, DABC, BCDA, or CDAB. Each treatment period within a sequence was followed by a 10‐day washout. All study drugs in parts 1 and 2 were administered under fed conditions: the morning dose followed a standard breakfast of ~ 400 kcal (36% from fat) and the evening dose followed a snack of ~ 350–400 kcal.
The primary end point was the difference from placebo in change from period‐specific baseline to day 7 in the Fridericia‐corrected15 QT interval (ddQTcF) at extraction times between 0.5 hour and 22.25 hours post‐morning dose in part 2. Secondary objectives included evaluating the effect of fostemsavir on other ECG end points—heart rate, PR interval, QRS interval, the uncorrected QT interval, and the Bazett‐corrected16 QT interval; evaluating the effect of single‐dose moxifloxacin 400 mg on the QT interval, and characterizing TMR PK.
Serial 12‐lead ECGs were acquired from 24‐hour Holter recordings on days 1 and 7 of each treatment period, and assessed centrally. Triplicate readings were obtained at 100, 70, and 40 minutes before the morning dose on both days of each treatment period, and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 11.5, 14, 15, 16, and 22.25 hours after the morning dose on day 7. The period‐specific baseline values were the mean of the three predose readings on day 1. A linear mixed model was applied to Fridericia‐corrected (QTcF) with fixed effects, including sequence, period, treatment group, time since dose, and time‐by‐treatment interaction, with baseline QTcF included as a covariate. Correlation between measurements from different periods or different times within the same period was enabled by use of a within‐subject variance covariance structure. The primary ddQTcF end point was derived from this model. Noninferiority assessments of each fostemsavir treatment vs. placebo were conducted using the intersection union test procedure with a model‐derived two‐sided 90% confidence interval (CI) for the estimated ddQTcF at each postdose time point and a 10 msec noninferiority bound. Noninferiority was concluded if the upper bound of the 90% CI was below 10 msec for all scheduled collection times. Assay sensitivity was assessed using the same procedures applied to the ddQTcF of moxifloxacin vs. placebo, with sensitivity confirmed if the multiplicity‐adjusted17 lower 90% CI boundary exceeded 5 msec at any of three post‐moxifloxacin dose time points (2, 3, or 4 hours) around the anticipated time to Cmax (Tmax).
In each treatment period in part 2, blood sampling for intensive PK assessment of TMR was undertaken at nominal collection points 10 minutes after scheduled predose and postdose serial ECG assessments on days 1 and 7 of each period. Plasma TMR in both parts of the study was analyzed using a validated liquid chromatography/mass spectrometry assay with a lower limit of quantitation of 5 ng/mL.18 Predose sampling times were set to zero for PK parameter assessments. Samples from subjects receiving placebo were not assayed. Cmax, Tmax, minimum plasma concentration (Cmin), and area under the concentration–time curve across the dosing interval (AUCtau) were calculated using noncompartmental methods in Kinetica 5.0 within the eToolbox (version 2.7; Thermo Electron, Philadelphia, PA).
For study part 1, administration of fostemsavir to six subjects in the sentinel cohort provided 80% probability of observing ≥ 1 occurrence of any adverse event (AE) with a 24% incidence rate in the population. For study part 2, complete data from 52 subjects would provide 90% power to conclude no clinically significant effect on ddQTcF, assuming a mean effect for fostemsavir 2,400 mg b.i.d. no greater than 4 msec. Power calculations were based on simulations after Hosmane and Locke,19 incorporating an assumed SD for ΔQTcF of 9.5 msec derived from conservative estimates from a published review.20
Exposure–response modeling and simulation
The relationship between ddQTcF and TMR plasma concentration was initially assessed graphically using data from part 2. Individual and mean PK and ddQTcF profiles and individual and mean hysteresis loops were plotted. A population exposure–response model was subsequently constructed to describe ddQTcF as a function of TMR plasma concentration with interindividual variability (IIV) and residual error models using NONMEM version 7.3 (ICON Development Solutions, Ellicott City, MD). A variety of linear and direct effect models with additive IIV and residual error models were explored. Model fit to the data was evaluated on goodness‐of‐fit criteria that included a minimum 3.84 point (P < 0.05) decrease in objective function value according to the log‐likelihood ratio test, good agreement between observed and predicted ddQTcF, uniformly scattered weighted residuals vs. predicted ddQTcF, increased precision of estimated parameters, and decreases in IIV and random residual variability. The statistical model included a term for interoccasional variability (IOV) to account for intrasubject differences between the two fostemsavir dosing periods. Different residual error models were also tested. Available covariates (age, race, and sex) were explored graphically for potential relationships.
The final concentration‐ddQTcF model was evaluated using a visual predictive check (VPC) and bootstrap analysis. VPC was performed with 1,000 simulations. Median and 5th and 95th percentile of simulated data were overlaid onto observed data to determine the appropriateness of the model to adequately describe the observed data. Bootstrap analysis was performed with 1,000 simulations and the mean and 90% CI compared with the model‐estimated parameters.
The final model simulated ddQTcF across the range of TMR concentrations observed in the TQT study using a step‐size of 200 ng/mL with 1,000 simulated trials and 100 virtual subjects per trial. The mean, 5th, and 95th percentiles for each trial were calculated at each TMR quartile with 32 partitions of the concentration range. The mean ddQTcF and 90% CI from all simulated trials were used to determine an estimated concentration threshold where the 90% CI upper bound of ddQTcF reached 10 msec.
To establish the risk of a QT effect of clinical importance with PK enhancers, model‐based simulations were used to determine the upper 90% CI boundary for ddQTcF at steady‐state TMR Cmax when fostemsavir 600 mg b.i.d. is given with or without cobicistat 150 mg q.d. to healthy adult subjects in a previous drug–drug interaction (DDI) study.21
RESULTS
Twenty‐three subjects were screened and eight were randomized in part 1 of the TQT study to assess the safety of supratherapeutic dose of fostemsavir. Randomized subjects received fostemsavir 2,400 mg b.i.d. (n = 6) or placebo (n = 2) for 7 days. Subjects were white (100%), predominantly Hispanic/Latino (87.5%), and female (62.5%); mean age was 35 years (range 19–48 years), and mean body mass index was 27. All subjects completed treatment. Safety data were unremarkable: all AEs (primarily dizziness and headache) were mild and there were no relevant cardiac signals. The main study (part 2) was, therefore, initiated, and 136 subjects were screened and 60 randomized to 1 of the 4 treatment sequences (n = 15 each). Subjects randomized as part of the main study were predominantly white (95%), Hispanic/Latino (92%), and male (63%); mean age was 34 years (range 18–49 years), and mean body mass index was 26.
Overall, 52 subjects completed all four treatment periods. Of the eight noncompleters, one was discontinued for a positive illicit drug screen; the remaining seven discontinued for AEs, including: one receiving fostemsavir 1,200 mg q.d. (diarrhea), five receiving fostemsavir 2,400 mg b.i.d. (two for vomiting, plus one each for elevated aminotransferases; rash; and headache/anxiety/dyspnea/pharyngeal edema), and one receiving placebo (pruritus).
Table 1 shows the statistical analysis of least‐squares mean changes in the QTcF15 from baseline for each treatment. Moxifloxacin assay sensitivity was confirmed, because the lower bound of the multiplicity‐adjusted 90% CI around the treatment difference vs. placebo for the change from period‐specific ddQTcF was above 5 msec at 2, 3, and 4 hours observed time points around the anticipated Tmax. There were no QTcF prolongations that reached clinical importance with fostemsavir 1,200 mg q.d.; the upper bound of the 90% CI around ddQTcF was below 10 msec at all time points: the maximum time‐matched ddQTcF was 4.3 msec at 6 hours postdose, with an upper bound of 6.3 msec for the two‐sided 90% CI. At 2,400 mg b.i.d., the upper bound of the 90% CI around ddQTcF exceeded 10 msec at 4, 5, and 6 hours after the am dose and at 3 and 4 hours after the pm dose. At this dose, the maximum time‐matched ddQTcF was 11.2 msec at 5 hours post‐am dose, with an upper bound of the two‐sided 90% CI of 13.3 msec. The ddQTcF–time data are shown graphically in Figure 1, and TMR concentration–time curves and exposure parameters in Figure S1 . Fostemsavir did not have a clinically meaningful effect on heart rate, QRS or PR intervals, or waveform morphology.
Table 1.
Statistical analysis of mean change from baseline in QTcF
| Time, hour |
Mean change from baseline, msec ddQTcF |
Mean difference vs. placebo (90% CI), msec ddQTcF |
|||||
|---|---|---|---|---|---|---|---|
|
FTR 1,200 mg q.d. (A) |
FTR 2,400 mg b.i.d. (B) |
MOX 400 mg (C) |
Placebo (D) |
FTR 1,200 mg q.d. (A–D) |
FTR 2,400 mg b.i.d. (B–D) |
MOX 400 mg (C–D) |
|
| 0.5 | –7.44 | –6.04 | –7.12 | –7.54 | 0.10 (–1.98, 2.18) | 1.50 (–0.63, 3.62) | 0.42 (–1.73, 2.58) |
| 1 | –7.25 | –6.15 | –4.82 | –7.76 | 0.51 (–1.57, 2.58) | 1.61 (–0.51, 3.73) | 2.95 (0.80, 5.10) |
| 2 | –6.78 | –6.90 | –1.93 | –10.62 | 3.84 (1.75, 5.93) | 3.72 (1.60, 5.84) | 8.69 (6.57, 10.82)a |
| 3 | –7.54 | –3.67 | 0.40 | –10.03 | 2.49 (0.40, 4.57) | 6.36 (4.23, 8.48) | 10.43 (8.32, 12.54)b |
| 4 | –3.37 | 1.64 | 3.30 | –7.03 | 3.66 (1.58, 5.75) | 8.67 (6.55, 10.79) | 10.33 (8.22, 12.44)c |
| 5 | –0.49 | 6.50 | 4.16 | –4.67 | 4.18 (2.11, 6.26) | 11.18 (9.05, 13.30) | 8.83 (6.72, 10.94) |
| 6 | –4.52 | 0.18 | –0.81 | –8.78 | 4.27 (2.19, 6.34) | 8.96 (6.84, 11.10) | 7.98 (5.87, 10.09) |
| 8 | –6.87 | –3.46 | –0.74 | –8.79 | 1.92 (–0.15, 3.40) | 5.33 (3.21, 7.46) | 8.05 (5.94, 10.17) |
| 10 | 0.76 | 1.51 | 5.78 | –3.18 | 3.94 (1.86, 6.01) | 4.69 (2.57, 6.81) | 8.96 (6.85, 11.07) |
| 11.5 | –5.31 | –3.88 | 1.83 | –6.92 | 1.61 (–0.48, 3.69) | 3.04 (0.92, 5.17) | 8.75 (6.64, 10.86) |
| 14 | –7.05 | –1.65 | 0.60 | –6.95 | –0.10 (–2.18, 1.972) | 5.30 (3.18, 7.43) | 7.55 (5.44, 9.66) |
| 15 | –3.84 | 4.45 | 1.49 | –4.15 | 0.30 (–1.78, 2.38) | 8.60 (6.47, 10.73) | 5.64 (3.52, 7.75) |
| 16 | –1.48 | 8.59 | 5.41 | –1.52 | 0.04 (–2.04, 2.12) | 10.10 (7.98, 12.23) | 6.92 (4.81, 9.03) |
| 22.25 | –3.18 | 0.16 | 2.73 | –3.46 | 0.29 (–1.79, 2.36) | 3.63 (1.51, 5.75) | 6.19 (4.08, 8.30) |
CI, confidence interval; ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; FTR, fostemsavir; MOX, moxifloxacin; QTcF, Fridericia‐corrected QT interval.
Multiplicity‐adjusted 90% CI for MOX: a(6.57; 10.82); b(7.70; 13.16); c(7.81; 12.84).
Figure 1.
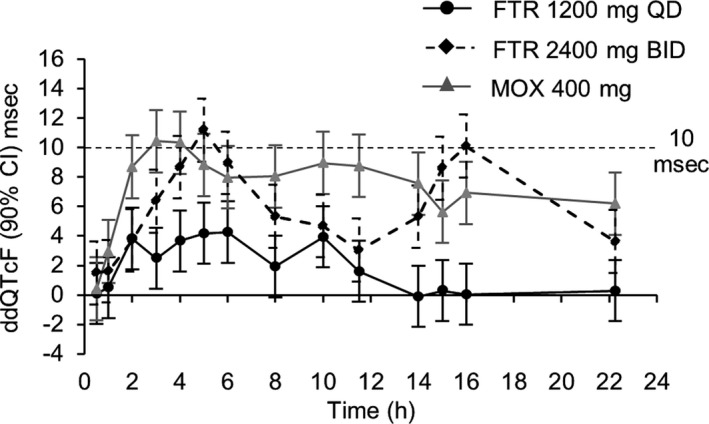
The ddQTcF over time for fostemsavir 1,200 mg q.d., fostemsavir 2,400 mg b.i.d., and moxifloxacin. CI, confidence interval; ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; FTR, fostemsavir; MOX, moxifloxacin.
Categorical analyses of maximum QTcF were undertaken for strata ≤ 450 msec, > 450 to 480 msec, > 480 to 500 msec, and > 500 msec; maximum change in QTcF was assessed in categories of ≤30 msec, > 30 to 60 msec, and > 60 msec. No subject had a maximum QTcF in either of the categories above 480 msec, and no one had a maximum change above 60 msec. Three subjects receiving fostemsavir 2,400 mg b.i.d., two receiving moxifloxacin, and one receiving placebo, had a maximum QTcF between 450 and 480 msec. Four subjects receiving fostemsavir 2,400 mg b.i.d. had a maximum change between 30 and 60 msec.
AEs were reported in 39 subjects (67%) receiving fostemsavir vs. 27 (47%) receiving placebo. Overall, AE rates were similar between fostemsavir 1,200 mg q.d. (53%) and fostemsavir 2,400 mg b.i.d. (56%). The most common AE was headache, which was more common on fostemsavir 2,400 mg b.i.d. (35%) than fostemsavir 1,200 mg q.d. (16%) or placebo (12%).
The mean time‐associated fostemsavir treatment effect on ddQTcF correlated with plasma TMR concentration at each extraction point (Figure S2 ), with a positive linear relationship (Figure 2). There was no evidence of a delayed effect at either dose in either mean or individual hysteresis plots (data not shown). The data were, therefore, consistent with a direct effect relationship. A variety of linear and direct‐effect maximum effect (Emax) models were evaluated with ddQTcF as a dependent variable expressed as a function of TMR plasma concentration. A summary of all tested models is shown in Table S1 .
Figure 2.
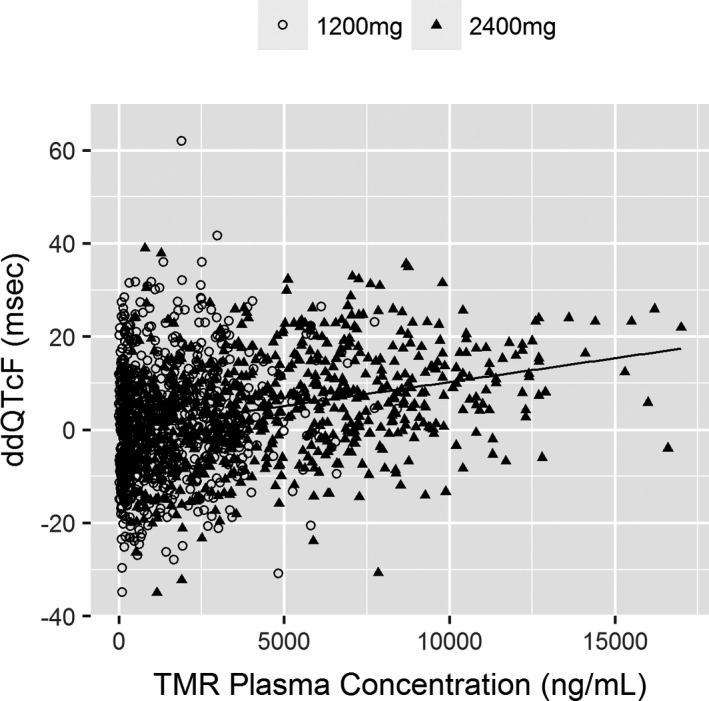
Scatterplot of ddQTcF vs. temsavir plasma concentrations for all measurements in subjects receiving fostemsavir 1,200 mg q.d. or 2,400 mg b.i.d. ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; TMR, temsavir.
A linear slope‐intercept model with both an IIV term on intercept and an intra‐individual IOV term for dosing period was selected as providing an adequate fit (Figure S3 ). Age, sex, and race showed no observable relationship as covariates with IIV (data not shown), although the caveats apply first that extremes of age were excluded from study eligibility with a narrow range of age representation from 19–48 years, second that sex is already accounted for in ddQTcF as baseline correction will nullify the effect of sex on QTcF, and, finally, that only three subjects were non‐white.
The final linear model was of the form:
where Cp is the plasma TMR concentration; DOSE1200 or DOSE2400 are 1 for a subject receiving fostemsavir 1,200 mg q.d. or 2,400 mg b.i.d., otherwise 0; θ1 is the intercept; θ2 is the slope; η1 is the IIV on the intercept; η2 and η3 are the IOV on intercept for 1,200 mg and 2,400 mg dosing, respectively; and ε is the additive residual variability. The parameter estimates for the final model are shown in Table 2, together with bootstrap parameter estimates from 1,000 simulations.
Table 2.
Final concentration‐ddQTcF model parameters
| Term | Parameter | Population mean estimate (% RSE) | Bootstrap parameter estimate for n = 1,000 (90% CI) |
|---|---|---|---|
| θ1 | Intercept (msec) | –0.783 (139) | –0.727 (–2.500, 1.072) |
| θ2 | Slope (msec per ng/mL TMR) | 0.00126 (7.9) | 0.00126 (0.00110, 0.00142) |
| η1 | Additive IIV_intercept (msec) | 6.05 (42.1) | 6.00 (3.50, 8.08) |
| η2, η3 | IOV on intercept for DOSE 1,200 mg and DOSE 2,400 mg (msec) | 6.92 (19.7) | 6.90 (5.75, 7.95) |
| ε | Additive residual variability (msec) | 6.74 (6.3) | 6.73 (6.39, 7.07) |
CI, confidence interval; ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; IIV, inter‐individual variability; IOV, inter‐occasion variability; RSE, relative standard error; TMR, temsavir.
The final model was evaluated by a VPC that plotted the median, 5th, and 95th percentiles of 1,000 Monte Carlo simulations compared with observed data. The plot (Figure 3) demonstrated acceptable predictivity for the central tendency and spread of observed ddQTcF. In total, ~ 10% of the observed data was outside the 90% CI of the simulated data.
Figure 3.
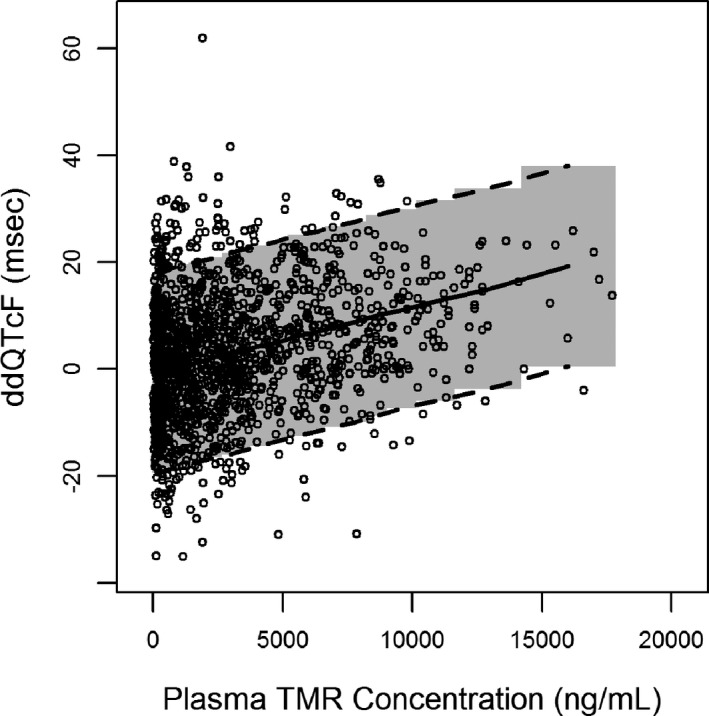
Visual predictive check of final model. Circles are observed data; lines are median, 5th, and 95th percentiles of simulated data; shaded area is the 90% CI of simulated data. CI, confidence interval; ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; TMR, temsavir.
Figure 4 shows the model‐simulated effect on ddQTcF across TMR concentration range observed in the TQT study (6.22 ng/mL to 17,900 ng/mL). The TMR plasma concentration at which the upper bound of the two‐sided 90% CI intersects 10 msec was ~ 7,500 ng/mL.
Figure 4.
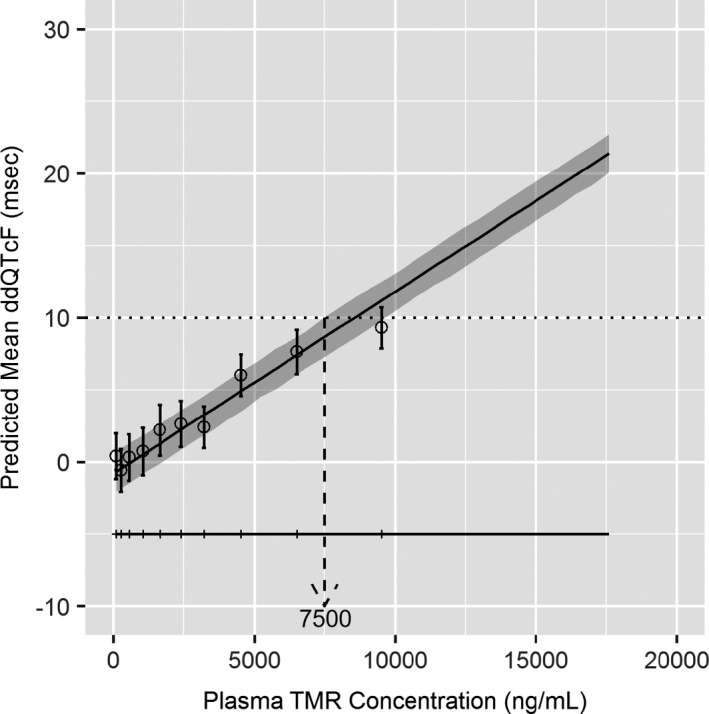
Model‐simulated effect on ddQTcF at increasing temsavir plasma concentrations. Solid line and shaded region represent model‐predicted mean ddQTcF and 90% CI as a function of plasma concentration. Horizontal line with tick marks shows range of concentrations divided into deciles. Circles and vertical bars denote observed means and 90% CIs for ddQTcF with each concentration decile. CI, confidence interval; ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; TMR, temsavir.
Table 3 shows model‐derived estimates of the upper 90% CI of ddQTcF at TMR Cmax when fostemsavir 600 mg b.i.d. is dosed with or without cobicistat 150 mg q.d. Observed TMR Cmax following co‐administration with cobicistat was comparable to the Cmax for fostemsavir 1,200 mg q.d. in this study, and well below the model‐derived TMR concentration associated with an upper 90% CI limit for ddQTcF of 10 msec.
Table 3.
Observed and predicted ddQTcF in healthy adults following administration of fostemsavir at 1,200 mg q.d., 2,400 mg b.i.d., and 600 mg b.i.d. ± cobicistat 150 mg q.d.
|
FTR 1,200 mg q.d. N = 57 |
FTR 2,400 mg b.i.d. N = 53 |
FTR 600 mg b.i.d. N = 16 |
FTR 600 mg b.i.d. + cobicistat 150 mg q.d. N = 15 |
|
|---|---|---|---|---|
| ddQTcF upper 90% CI boundary (msec) | 6.4 (at 6 hour) | 13.3 (at 5 hour) | 3.2a | 5.0a |
| Mean Cmax (ng/mL) | 3,584 | 8,900 | 2,096b | 3,515b |
CI, confidence interval; Cmax, peak plasma concentration; ddQTcF, difference vs. placebo for change from baseline in Fridericia‐corrected QT interval; FTR, fostemsavir.
Model‐predicted.
Data from ref. 21
DISCUSSION
This TQT study in healthy volunteers established that fostemsavir administered at 1,200 mg q.d. did not reach the clinically important QTc interval prolongation threshold, defined as the upper bound of the two‐sided 90% CI of ddQTcF exceeding 10 msec. By contrast, supratherapeutic dose of 2,400 mg b.i.d. for 7 days did exceed the 10 msec threshold, with a maximum upper bound of 13.3 msec at 5 hours postdose. These findings are consistent with data for the marketed antiretroviral rilpivirine and lopinavir/ritonavir, where QTc prolongations > 10 msec have been seen at supratherapeutic doses (one‐sided 95% CI upper bounds of 15.3 and 18.0 msec, at 75 mg and 800/200 mg doses, respectively) but not at therapeutic doses (one‐sided 95% CI upper bounds of 8.2 and 8.1 msec, at 25 mg and 400/100 mg, respectively). The threshold of 10 msec for the upper CI around a time‐matched mean treatment effect on the QTc interval is based on regulatory guidance as providing a reasonable assurance that the mean population‐level prolongation effect is not greater than ~ 5 msec, a degree of prolongation that does not appear to be associated with Torsades de pointes and elevated risk of cardiac arrest.13 Of note, a mean population‐level prolongation above 5 msec does not classify a drug as being pro‐arrhythmic, as a substantially increased risk of arrhythmia is not typically observed until QTc prolongation exceeds 20 msec.13
A linear relationship between TMR concentrations and ddQTcF was established through population exposure–response modeling, and the predicted TMR plasma concentration below which the upper bound of the two‐sided 90% CI remain below 10 msec was identified as 7,500 ng/mL. This concentration is 4.2‐fold greater than the mean TMR plasma Cmax 22 following fostemsavir 600 mg b.i.d. in heavily treatment‐experienced subjects in the phase III BRIGHTE study (1,770 ng/mL on average, NCT02362503). Furthermore, protocol‐defined study discontinuation due to QTc interval prolongation in BRIGHTE was infrequent. Less than 2% (7/371) of subjects treated for 96 weeks have been discontinued due to QT prolongation (> 450 msec) all of which were asymptomatic.23
Graphical assessment suggested a direct effect of TMR on the QT interval and showed an absence of a delayed response. Exposure–response assessment established a linear slope‐intercept model, containing terms for residual variability and both interindividual (within dose) and intra‐individual (between dose) variability. Simulations resulted in a virtual data set in agreement with the observed range and established the threshold TMR plasma concentration of 7,500 ng/mL.
Following therapeutic dosing of 600 mg fostemsavir b.i.d., the model‐derived upper boundaries of the 90% CI ddQTcF associated with observed mean TMR Cmax were ≤3.2 msec for administration without a pharmaco‐enhancer, and ≤ 5.0 msec for administration with cobicistat 150 mg q.d. These data, therefore, suggest that the clinically important threshold effects on the QT interval attributable to fostemsavir dosing are unlikely to occur at the therapeutic dose even with concomitant use of pharmacoenhancing protease inhibitors. Of note, ritonavir at its pharmaco‐enhancement dose of 100 mg (q.d. or b.i.d.) has been shown to have less impact on TMR plasma Cmax than cobicistat 150 mg q.d. (45–53% increase with ritonavir vs. 71% with cobicistat)18, 21 and, hence, these conclusions apply to the use of fostemsavir with both cobicistat and ritonavir‐boosted protease inhibitors and other medications that may inhibit CYP3A4, BRCP, and P‐gp. These findings can be interpolated to other clinical scenarios of DDIs in heavily treatment‐experienced populations given that coadministration with cobicistat represents a worst‐case increase in Cmax (1.71‐fold increase compared to without concomitant cobicistat).21, 22 Renal impairment has shown no effect on Cmax and, hence, no influence of renal impairment on QTc is anticipated.24 Further, the influence of hepatic impairment25 on Cmax is < 1.72‐fold, which will not be a concern as these exposures are expected to be lower than the established Cmax threshold based on exposure‐response analysis. Demographic factors, such as age and sex, do not have any clinically relevant impact on Cmax to be of concern for QT risk.9
In common with many model predictions derived from healthy volunteers, limitations accrue from the relatively homogeneous nature of the subjects. Potential interactions with age, race, or HIV disease could not be assessed, although there is no a priori expectation that these would significantly alter the relationship between ddQTcF and TMR concentration. Further limitations to the modeling process derive from the sparseness of observations above 10,000 ng/mL (~ 4% of all observations). Due to this sparseness, the use of observed concentrations for ddQTcF simulations would result in relatively few response values in the highest bin. The influence of binning at higher concentrations was mitigated by using intensive simulations for 1,000 trials of 100 subjects with a 200 ng/mL step size per trial across the observed concentration range.
In conclusion, a positive relationship was demonstrated between TMR concentrations and change in QTc interval with the clinically important threshold effect of 10 msec exceeded at a supratherapeutic fostemsavir dose of 2,400 mg b.i.d., but not at 1,200 mg q.d. Subsequent exposure–response modeling and simulations predict that for fostemsavir 600 mg b.i.d., with or without concomitant medications that may inhibit CYP3A4, BCRP, and P‐gp, including pharmaco‐enhancers, TMR concentrations are unlikely to result in a clinically important QT prolongation. The mean TMR plasma Cmax following phase III dosing in heavily treatment‐experienced subjects is 4.2‐fold below the concentration projected to be associated with an upper bound of 90% CI of ddQTcF prolongation that crosses 10 msec. The model‐defined concentration threshold is aligned with observed clinical data for fostemsavir and will be useful to apply to other clinical scenarios in the treatment‐experienced HIV‐1 infected population.
Funding
The thorough QT study described herein was funded by Bristol‐Myers Squibb prior to the acquisition of fostemsavir by ViiV Healthcare. The exposure–response analysis described was performed by GlaxoSmithKline and funded by ViiV Healthcare.
Conflict of Interest
All authors are employees of ViiV Healthcare (K.M., P.A., and C.L.) or GlaxoSmithKline (C.L. and M.M.).
Author Contributions
C.L., M.M., and K.M. wrote the manuscript. C.L., M.M., K.M., C.L., and P.A. designed the research. C.L., M.M., and K.M. performed the research. C.L. analyzed the data. C.L., M.M., and K.M. contributed to new reagents/analytical tools.
Supporting information
Figure S1. Concentration‐time plots and exposure parameters for fostemsavir (FTR) 1,200 mg q.d. and 2,400 mg b.i.d. in study part B.
Figure S2. Time‐from‐dose plots of mean temsavir concentration (left axis) and ddQTcF (right axis) for (A) fostemsavir 1,200 mg once daily, (B) fostemsavir 2,400 mg twice daily.
Figure S3. Goodness‐of‐fit plots for the final model.
Table S1. Models evaluated.
Acknowledgments
The authors thank all study participants and their families, and the study investigators. ViiV Healthcare acquired fostemsavir from Bristol‐Myers Squibb and is the sponsor. We thank all the Bristol‐Myers Squibb scientists for their contributions. Editorial assistance was provided by Nick Fitch, PhD, CMPP, of ArticulateScience (London, UK), funded by ViiV Healthcare.
References
- 1. Meanwell, N.A. et al Inhibitors of HIV‐1 attachment: the discovery and development of temsavir and its prodrug fostemsavir. J. Med. Chem. 61, 62–80 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Langley, D.R. et al Homology models of the HIV‐1 attachment inhibitor BMS‐626529 bound to gp120 suggest a unique mechanism of action. Proteins 83, 331–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown, J. et al Compartmental absorption modelling and site of absorption studies to determine feasibility of an extended‐release formulation of an HIV‐1 attachment inhibitor phosphate ester prodrug. J. Pharm. Sci. 102, 1742–1751 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Gorycki, P. et al Pharmacokinetics, metabolism and excretion of radiolabelled fostemsavir administered with or without ritonavir in healthy male subjects. 19th International Workshop on Clinical Pharmacology of Antiviral Therapy; Baltimore, MD, USA; May 22–24, Abstract 42 (2018). [DOI] [PubMed]
- 5. Nowicka‐Sans, B. et al In vitro antiviral characteristics of HIV‐1 attachment inhibitor BMS‐626529, the active component of the prodrug BMS‐663068. Antimicrob. Agents Chemother. 56, 3498–3507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li, Z. et al Activity of the HIV‐1 attachment inhibitor BMS‐626529, the active component of the prodrug BMS‐663068, against CD4‐independent viruses and HIV‐1 envelopes resistant to other entry inhibitors. Antimicrob. Agents Chemother. 57, 4172–4180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou, N. et al Genotypic correlates of susceptibility to HIV‐1 attachment inhibitor BMS‐626529, the active agent of the prodrug BMS‐663068. J. Antimicrob. Chemother. 69, 573–581 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Lalezari, J.P. et al Safety and efficacy of the HIV‐1 attachment inhibitor prodrug BMS‐663068 in treatment‐experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV 2, e427–e437 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Landry, I. et al Model‐based phase 3 dose selection for HIV‐1 attachment inhibitor prodrug BMS‐663068 in HIV‐1‐infected patients: population pharmacokinetics/pharmacodynamics of the active moiety, BMS‐626529. Antimicrob. Agents Chemother. 60, 2782–2789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nettles, R. et al Single and multiple dose pharmacokinetics and safety in non‐HIV‐infected healthy subjects dosed with BMS‐663068, an oral HIV attachment inhibitor. 12th International Workshop on Clinical Pharmacology of HIV Therapy; Miami, FL, USA; April 13–15, Abstract O_04 (2011).
- 11. Janssen Pharmaceutical Companies . Edurant®; (rilpivirine) [package insert]. U.S. Food and Drug Administration (Drugs@FDA.com). U.S. Food and Drug Administration Website. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202022s000lbl.pdf>. Accessed August 28, 2019.
- 12. Abbvie Inc. KALETRA® (lopinavir and ritonavir) [package insert]. U.S. Food and Drug Administration (Drugs@FDA.com). U.S. Food and Drug Administration Website. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021251s057,021906s052lbl.pdf>. Accessed August 28, 2019.
- 13. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs. <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf> (May 2005). Accessed March 11, 2019.
- 14. Williams, E.J. Experimental designs balanced for the estimation of residual effects of treatments. Aus. J. Sci. Res. 2, 149–168 (1949). [Google Scholar]
- 15. Fridericia, L.S. Die systolendauer im elektrokardiogramm bei normalen menschen und bei herzkranken. Acta. Med. Scand. 53, 469–486 (1920). [Google Scholar]
- 16. Bazett, H.C. An analysis of the time‐relations of the electrocardiograms. Heart 7, 353–370 (1920). [Google Scholar]
- 17. Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57, 289–300 (1995). [Google Scholar]
- 18. Zhu, L. et al Pharmacokinetic interactions between BMS‐626529, the active moiety of the HIV‐1 attachment inhibitor prodrug BMS‐663068, and ritonavir or ritonavir‐boosted atazanavir in healthy subjects. Antimicrob. Agents Chemother. 59, 3816–3822 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hosmane, B. & Locke, C. A simulation study of power in thorough QT/QTc studies and a normal approximation for planning purposes. Ther. Innov. Regul. Sci. 39, 447–455 (2005). [Google Scholar]
- 20. Natekar, M. et al Effect of number of replicate electrocardiograms recorded at each time point in a thorough QT study on sample size and study cost. J. Clin. Pharmacol. 51, 908–914 (2011). [DOI] [PubMed] [Google Scholar]
- 21. Vakkalagadda, B. et al HIV‐1 attachment inhibitor prodrug BMS‐663068: interactions with cobicistat and darunavir+cobicistat. ICAAC/ICC 2015; San Diego, CA, USA; September 17–21, Abstract 1676 (2015).
- 22. Moore, K. et al Fostemsavir drug‐drug interaction profile, an attachment inhibitor and oral prodrug of temsavir, for heavily treatment‐experienced HIV‐1 infected patients. ID Week 2019; Washington, DC, USA; October 2–6, Abstract 2500.
- 23. Lataillade, M. et al Week 96 safety and efficacy of the novel HIV‐1 attachment inhibitor prodrug fostemsavir in heavily treatment‐experienced participants infected with multi‐drug resistant HIV‐1 (BRIGHTE Study). International AIDS Conference on HIV Science (IAS) 2019; Mexico City, Mexico; July 21–24.
- 24. Moore, K. et al Impact of mild, moderate, and severe renal impairment and hemodialysis on temsavir pharmacokinetics following oral administration of fostemsavir, an attachment inhibitor for heavily treatment experienced HIV‐1 infected patients. HIV Drug Therapy 2018; Glasgow, UK; October 28–31. Poster 261.
- 25. Sevinsky, H. et al Pharmacokinetics of temsavir, the active moiety of the prodrug fostemsavir, in subjects with hepatic impairment. Open Forum Infect. Dis. 4 (suppl. 1), S430 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Concentration‐time plots and exposure parameters for fostemsavir (FTR) 1,200 mg q.d. and 2,400 mg b.i.d. in study part B.
Figure S2. Time‐from‐dose plots of mean temsavir concentration (left axis) and ddQTcF (right axis) for (A) fostemsavir 1,200 mg once daily, (B) fostemsavir 2,400 mg twice daily.
Figure S3. Goodness‐of‐fit plots for the final model.
Table S1. Models evaluated.