

Abstract
Deutetrabenazine (Austedo, Teva Pharmaceuticals) is a deuterated form of tetrabenazine. It is the first deuterated drug to receive US regulatory approval and is approved for treatment of chorea in Huntington’s disease and tardive dyskinesia. Two oral single dose studies comparing deutetrabenazine (25 mg) with tetrabenazine (25 mg) in healthy volunteers evaluated the impact of deuteration on pharmacokinetics of the active metabolites, alpha‐dihydrotetrabenazine (α‐HTBZ) and beta‐dihydrotetrabenazine (β‐HTBZ), metabolite profile, safety, and tolerability. In the two‐way, cross‐over study, the mean elimination half‐life of deuterated total (α + β)‐HTBZ was doubled compared with nondeuterated total (α + β)‐HTBZ, with a twofold increase in overall mean exposure (area under the concentration‐time curve from zero to infinity (AUC0–inf)) and a marginal increase in mean peak plasma concentration (Cmax). In the mass balance and metabolite profiling study, there were no novel plasma or urinary metabolites of [14C]‐deutetrabenazine relative to [14C]‐tetrabenazine. Specific deuteration in deutetrabenazine resulted in a superior pharmacokinetic profile and an increased ratio of active‐to‐inactive metabolites, attributes considered to provide significant benefits to patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Deutetrabenazine is the first deuterated drug to receive regulatory approval.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The studies described herein are the first clinical studies evaluating the effects of deuteration on tetrabenazine metabolism and on pharmacokinetics of the active metabolites, alpha‐dihydrotetrabenazine (α‐HTBZ) and beta‐dihydrotetrabenazine (β‐HTBZ).
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The two studies confirm that the metabolic pathway of deutetrabenazine is identical to that of tetrabenazine and no new metabolites are formed. The specific deuteration in deutetrabenazine slows conversion of the active metabolites to inactive products, resulting in a nearly doubled mean half‐life and a more than twofold increase in overall exposure (area under the concentration‐time curve from zero to infinity (AUC0–inf)) with only a marginally increased peak plasma concentration (Cmax) relative to the nondeuterated form, tetrabenazine. This permits achievement of the desired overall exposure and a lower Cmax with half the dose. The longer half‐lives of the deuterated active metabolites further increases concentrations at trough, resulting in reduced fluctuation and the ability to dose less frequently; attributes thought to potentially provide clinical benefit.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The studies confirm that specific deuteration provides a tool to modify the metabolism of a known drug in a targeted manner and that the observed changes in in vitro metabolism can be translated into significant changes of the pharmacokinetics in humans, which are expected to be clinically meaningful.
Tetrabenazine, a vesicular monoamine transporter 2 (VMAT2) inhibitor was approved in 2008 as a treatment to reduce chorea that can occur in Huntington’s disease.1 Originally synthesized in the 1950s, tetrabenazine (cis‐rac –1,3,4,6,7,11b‐hexahydro‐9,10‐dimethoxy‐3‐(2‐methylpropyl)2H‐benzo[a]quinolizin‐2‐one), following oral administration, is rapidly and extensively converted during first‐pass hepatic metabolism by carbonyl reductase to its active metabolites alpha‐dihydrotetrabenazine (α‐HTBZ) and beta‐dihydrotetrabenazine (β‐HTBZ).2, 3 Plasma concentrations of tetrabenazine are, in fact, near the limit of detection, such that the α‐HTBZ and β‐HTBZ metabolites are primarily responsible for conferring pharmacological activity. These metabolites act as reversible high‐affinity inhibitors of monoamine uptake into vesicles of presynaptic neurons by binding selectively to the VMAT2. Together, they are considered to represent the active moieties for tetrabenazine (Xenazine (Lundbeck) Prescribing Information). As a result of reversible binding of α‐HTBZ and β‐HTBZ to synaptic vesicles, monoamine degradation in the neuronal cytoplasm is augmented, with correspondingly depleted monoamine content in the storage synapses. In turn, reduced release of monoamines during synaptic transmission, particularly dopamine,4 is widely accepted as the mechanism of action for the therapeutic efficacy of orally administered tetrabenazine.
The active metabolites have short elimination half‐lives; 4–8 hours for α‐HTBZ and 2–4 hours for β‐HTBZ, due to O‐demethylation at their two methoxy groups, catalyzed primarily by cytochrome P450 2D6 (CYP2D6).5 The products of O‐demethylation have substantially less affinity for VMAT2.6 Accordingly, tetrabenazine often has to be administered three times a day to maintain efficacy over a 24‐hour period. Such frequent dosing may lead to poor compliance7 and the attendant fluctuations in circulating α‐HTBZ and β‐HTBZ have the potential for adverse events (AEs) associated with high peak concentrations as well as loss of efficacy due to low trough concentrations. Consistent with suboptimal peak‐to‐trough fluctuations, 44% of patients receiving tetrabenazine in a pivotal randomized controlled study required dose reduction for intolerability.1 In addition, metabolism by CYP2D6 leads to potential drug interactions and daily dose limits when tetrabenazine is taken with strong CYP2D6 inhibitors.8
Deuteration is a chemical reaction process whereby any covalently bonded hydrogen atom is replaced by a deuterium atom.9 Replacement of hydrogen by deuterium theoretically has the potential to affect the pharmacokinetics (PKs) of drugs whose metabolism is mediated by oxidative processes of cytochrome P450 enzymatic activity and involves the cleavage of a hydrogen‐carbon bond.10, 11 Thus, with minimal changes on lipophilicity or other properties that could change target protein binding, interactions with enzymes or transporters, or binding to plasm proteins in a nonspecific manner that could influence distribution,12 deuteration has long been studied as a way to stabilize and improve drug metabolism. Deutetrabenazine (Austedo; Teva Pharmaceuticals) is the first approved deuterated product and is approved as a treatment for chorea in Huntington’s disease and tardive dyskinesia.13, 14
Deutetrabenazine was specifically developed to provide patients and prescribers with an effective treatment for chorea in Huntington’s disease with an improved pharmacokinetic and tolerability profile compared with tetrabenazine.15 It was rationally designed using deuteration at a selected position with the goal of slowing the CYP‐dependent metabolism of α‐HTBZ and β‐HTBZ to their less potent O‐desmethyl metabolites.10, 11 To that end, the two methoxy groups in tetrabenazine, the targets of CYP2D6 oxidation, were replaced with deuteromethyl moieties. This placement of deuterium notably did not influence the reduction of deutetrabenazine to the HTBZ metabolites. Accordingly, the deuterated HTBZ metabolites are formed in a similar manner as they are with tetrabenazine (Teva Pharmaceuticals, data on file). In contrast, the presence of C‐D bonds in deutetrabenazine was shown to attenuate the metabolism of α‐HTBZ and β‐HTBZ in human hepatic microsomes to their O‐demethylated products. The potential clinical advantages of slower substrate metabolism through deuterium substitution include a reduced dose to achieve desired systemic exposures, longer half‐life, reduced peak concentrations, and reduced impact of drug interactions along with unchanged binding properties.16, 17
In this paper, we report two of the first clinical studies performed with deutetrabenazine in healthy volunteers, both of which test the hypothesis that deuterium substitution will increase the plasma half‐life and systemic exposure to the clinically relevant HTBZ metabolites. The first study (AUS‐SD‐809‐CTP‐06) was a randomized, double‐blind, single‐dose crossover study to compare the pharmacokinetics, safety, and tolerability of deutetrabenazine with tetrabenazine. The second study (SD‐809‐C‐12) was an open‐label, two‐period study designed to evaluate the mass balance recovery and metabolite profile of [14C]‐deutetrabenazine in comparison to that of [14C]‐tetrabenazine.
METHODS
Study approvals
The studies were conducted at Q‐Pharm Pty Ltd (Brisbane, Queensland, Australia; Study AUS‐SD‐809‐CTP‐06) and Quotient Clinical (Nottingham, UK; Study SD‐809‐C‐12) in full accordance with the International Conference on Harmonization Good Clinical Practice Consolidated Guideline (E6) consistent with the principles that have their origin in the Declaration of Helsinki. Before study initiation, the protocols were submitted to the local Ethics Committees. Administration of radioactive substances was carried out following approval of the protocol by the Medicines and Healthcare Products Regulatory Agency (MHRA) and the Administration of Radioactive Substances Advisory Committee (ARSAC).
Study drug
Deutetrabenazine and tetrabenazine (powder in gelatin capsules) for Study AUS‐SD‐809‐CTP‐06 were manufactured according to the Good Manufacturing Practice regulations. For Study SD‐809‐C‐12, deutetrabenazine (batch number CCS‐1209d6/STG‐05/00111) and tetrabenazine (batch number CCS‐1209d0/STG‐05/00111) were supplied by Inogent Laboratories Private Limited (Hyderabad, India) to Quotient Bioresearch (Rushden, UK) for carbon‐14‐labeling. Capsules containing [14C]‐deutetrabenazine and capsules containing [14C]‐tetrabenazine were manufactured by Quotient Clinical (Nottingham, UK). The radioactive dose of the labeled drugs was not more than 2.92 MBq (79 μCi).
Study design
Study AUS‐SD‐809‐CTP‐06 was a randomized, double‐blind, single‐dose, two‐period crossover study designed to evaluate the pharmacokinetic, safety and tolerability profiles of deutetrabenazine (25 mg) and tetrabenazine (25 mg) administered in the fasted state to 21 healthy male and female volunteers. The study consisted of a screening visit within 21 days prior to first dosing followed by two treatment periods consisting of a 5‐day inpatient stay and an end of study assessment at the last day of the second period. Dosing was separated by a minimum 7‐day washout period. The order of the two treatments was assigned via a randomization scheme in a 1:1 ratio such that half of the subjects received deutetrabenazine treatment first and the other half received tetrabenazine treatment first. Thus, subjects received either deutetrabenazine or tetrabenazine with 250 mL of water on day 1 of each period. The study pharmacist was unblinded to prepare study treatment for dosing. Prior to study drug administration, the container with the subject’s assigned capsule was properly labeled in a blinded manner to ensure that the investigator, clinic staff, and subjects remained blinded during the trial. Blood samples for pharmacokinetic measurements were collected at predose, 20 and 40 minutes, then at 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60, and 72 hours postdose. Twelve‐lead echocardiogram (ECG) recordings were collected predose and 1, 2, and 24 hours postdose. Blood and urine samples were collected for clinical laboratory testing (hematology, serum chemistries, and urinalysis) at admission and after at least 10 hours fasting at ~ 24 and 72 hours postdose.
Study SD‐809‐C‐12 was a two‐period, two‐cohort, open‐label study designed to compare the mass balance and metabolite profile following single oral doses of either [14C]‐deutetrabenazine or [14C]‐tetrabenazine administered in the fasted state. Healthy male adult volunteers, who were extensive or intermediate CYP2D6 metabolizers and who met inclusion and exclusion criteria, were enrolled into one of two cohorts, each comprising six subjects, age 35–65 years (body mass index (BMI) ≥ 18.5 and ≤ 30.0 kg/m2). The two cohorts ran consecutively in periods 1 and 2 to avoid test article cross contamination. A screening visit took place within 28 days prior to dosing and a follow‐up call was done 3 to 5 days following the final collection period. Participants were dosed with a single hard gelatin capsule containing 25 mg [14C]‐deutetrabenazine in cohort 1 and a single hard gelatin capsule containing 25 mg [14C]‐tetrabenazine in cohort 2. Immediately following oral administration, subjects were given 240 mL of water. Blood samples for pharmacokinetic analysis in plasma and total radioactivity measures were collected at predose, 20 and 40 minutes, then at 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 96, 120, 144, 168, 192, and 216 hours postdose. Additional blood samples were collected at predose, 2, 2.5, 6, 12, 24, and 48 hours postdose for metabolite profiling and identification. Urine and fecal samples for total radioactivity, and urine samples for metabolite profiling and identification were collected for the following periods: predose, 0–6, 6–12, and 12–24 hours postdose; then daily through 168 hours postdose.
Quantification of drug and metabolite concentrations in plasma
Deutetrabenazine and tetrabenazine are used as racemic mixtures and the active and nonactive metabolites were analyzed and treated in pharmacokinetic analyses as racemates in both studies. The plasma concentrations of deutetrabenazine, tetrabenazine, and their respective metabolites were determined by liquid‐chromatography tandem mass spectrometry (LC‐MS/MS) analysis. This was performed on a Sciex API 4000 instrument, equipped with a Turbo Ion Spray ion source operated in the positive ion mode and interfaced to an Accela HPLC system fitted with a Gemini‐NX C18 column (3 µm; 150 mm × 4.6 mm i.d.) and eluted with a gradient mobile phase consisting of acetonitrile/100 mM ammonium acetate (pH 8.0). Quantitation of analytes was based on selected reaction monitoring procedures in which the precursor ions in all cases were the 12C‐containing protonated molecular ion species (MH+); in the case of the unlabeled molecules, these were at m/z 318 (tetrabenazine), m/z 320 (HTBZ), m/z 306 (O‐desmethyl‐HTBZ), m/z 334 (monohydroxy tetrabenazine), and m/z 350 (2‐methylpropanoic acid‐β‐HTBZ). Product ions monitored (from both unlabeled and deuterated compounds) corresponded to those formed by the following neutral losses from their respective MH+ precursors: tetrabenazine – C6H10O (98 Da); α/β‐HTBZ and O‐desmethyl α/β‐HTBZ – C2H6O (46 Da); monohydroxy tetrabenazine – C2H4O2 (60 Da); and 2‐methylpropanoic acid‐β‐HTBZ – C3H8O3 (92 Da). For quantification of the unlabeled analytes, the corresponding deuterium‐labeled compounds served as internal standards, whereas the reverse was true for measurement of the deuterated analytes. The lower limit of quantification for both unlabeled and deuterated analytes was 0.100 ng/mL for the parent drug, 0.200 ng/mL for monohydroxy tetrabenazine and 2‐methylpropanoic acid‐β‐HTBZ, and 0.500 ng/mL for the remaining metabolites.
Determination of total radioactivity
In Study SD‐809‐C‐12, total radioactivity was determined by quantitative radiochemical analysis by liquid scintillation counting. The characterization of metabolites relative to total radioactivity was derived by pooling four plasma samples over 12 hours per subject, a time course based on the previous limit of detection of metabolites by liquid scintillation counting. An additional analysis was conducted in which the percent of total radioactivity for metabolites in human plasma from retained plasma samples was quantified using validated LC‐MS/MS bioanalytical methods. The assay sensitivity supported analysis of all samples collected over the entire pharmacokinetic time course (23 samples over 216 hours). Following administration of [14C]‐deutetrabenazine, the limit of detection for total radioactivity in plasma was 0.007ng equivalents/mL, and in whole blood was 0.019 ng equivalents/g. Following administration of [14C]‐tetrabenazine, the limit of detection for total radioactivity in plasma was 0.008 ng equivalents/mL, and in whole blood was 0.017 ng equivalents/g.
Metabolite profiling and chemical structure identification
Metabolite profiling and chemical structure identification were performed from plasma and urine samples in Study SD‐809‐C‐12 by Quotient Metabolism using high performance liquid chromatography with online radio‐detection and LC‐MS/MS. The proposed structures of major metabolites were confirmed by comparison with authentic standards prepared by synthesis.
Pharmacokinetic evaluation
The pharmacokinetic parameters determined from plasma concentrations following administration of deutetrabenazine and tetrabenazine in Study AUS‐SD‐809‐CTP‐06 included the following: area under the plasma concentration‐time curve from time 0 to the time of the last quantifiable concentration (AUC0–t), area under the plasma concentration–time curve from time 0 to infinity (AUC0–inf), maximum observed plasma drug concentration (Cmax), time to Cmax (Tmax), terminal elimination half‐life (t 1/2), terminal elimination rate constant, percentage of AUC0–inf extrapolated (%AUCextrap), apparent total body clearance, and apparent volume of distribution.
The primary pharmacokinetic analysis in Study AUS‐SD‐809‐CTP‐06 included only subjects who had been identified as extensive CYP2D6 metabolizers by genotyping. A secondary analysis was performed on pooled data from all subjects who completed both periods and who were defined as being evaluable for the purpose of pharmacokinetic assessment.
For Study SD‐809‐C‐12, Cmax, Tmax, AUC0–t, AUC0–inf, %AUCextrap, terminal elimination rate constant, t 1/2, MRT0–t (mean residence time based on AUC0–t), and MRT0–inf were determined for total radioactivity, parent drug, and metabolite concentrations in plasma. The amounts of radioactivity excreted in urine and feces were determined as total and percent of administered dose, per time period and cumulatively. The 14C‐labelled urinary metabolites were identified and quantified from urine samples pooled over 0–72 hours postdose. The 14C‐labelled plasma metabolites were identified and quantified from samples pooled in a time‐proportional manner18 from the samples taken 2, 2.5, 6, and 12 hours postdose. All plasma PK parameters were calculated using Phoenix WinNonlin version 6.3 (Certara, Princeton, NJ) with noncompartmental analysis.
Statistical analyses
In Study AUS‐SD‐809‐CTP‐06, pharmacokinetic parameters for deutetrabenazine and tetrabenazine were compared using an analysis of variance model for a two‐period crossover trial with sequence, treatment, period, and subject included as factors; all values, except Tmax, were log‐transformed before analysis. Point estimates and confidence intervals on the log scale were back‐transformed to give estimates for ratio comparisons (deutetrabenazine/tetrabenazine (parent drugs and metabolites)). For Tmax, a nonparametric signed rank test was used and estimates of the median differences together with the corresponding 90% confidence intervals were calculated. Descriptive post hoc analyses were conducted on the ratio of inactive to active metabolites for subjects treated with deutetrabenazine and tetrabenazine. No formal statistical analysis was performed for Study SD‐809‐C‐12, the results were summarized using descriptive statistics. For safety and tolerability analyses, AEs were summarized by the MEDRA system organ class and preferred term. All other safety data (laboratory data, vital signs, ECG, physical examination, and concomitant medication) were summarized by treatment.
RESULTS
Subject demographics and disposition
All 21 subjects (10 men and 11 women) enrolled into Study AUS‐SD‐809‐CTP‐06 received at least one investigational product and were included in the safety and tolerability evaluation. The study occurred from July 2011 to September 2011. The mean age of the subjects was 24 years (range 18–39 years), mean weight was 70 kg (range 47–89 kg), and mean BMI was 23 kg/m2 (range 18.5–26.6 kg/m2). All subjects were white except one who was of other race. One subject, a 22‐year‐old woman who withdrew due to mild AEs (headache, nausea, feeling hot, agitation, and photophobia) following the deutetrabenazine dose during period 1 was excluded from the primary pharmacokinetic analysis. Seven subjects identified as poor (4; containing either no active alleles or only one partially active allele) or intermediate (3; containing one wild type allele and one partially active allele) CYP2D6 metabolizers were excluded from the primary pharmacokinetic analysis; the remaining 13 subjects who were identified as extensive (2; wild‐type alleles with no duplication) CYP2D6 metabolizers were included in the primary analysis.
In Study SD‐809‐C‐12, four extensive and two intermediate CYP2D6 metabolizers with a body weight of 81 kg (range 67–102 kg), BMI of 26 kg/m2 (range 23.3–29.9 kg/m2), and age 52 years (range 45–62 years) were enrolled into cohort 1 ([14C]‐deutetrabenazine), and five extensive and one intermediate CYP2D6 metabolizers with a body weight of 86 kg (range 75–95 kg), BMI of 27.4 kg/m2 (range 21.7–30.0 kg/m2), and age 46 years (range 37–53 years) were enrolled into cohort 2 ([14C]‐tetrabenazine). The study occurred from November 2012 to December 2012. All 12 subjects received their allocated investigational product and were included in the analyses.
Plasma Pharmacokinetics: Study AUS‐SD‐809‐CTP‐06
Plasma concentrations of both deutetrabenazine and tetrabenazine were below the lower limit of quantification (< 0.100 ng/mL) in the majority of samples and where quantifiable, were transient relative to the concentrations observed for their active metabolites α‐HTBZ and β‐HTBZ (Figure 1). There was no observable difference in exposure (AUC0–t and Cmax) or Tmax for the parent compounds (Table 1). Elimination half‐lives for the majority of subjects were not calculable.
Figure 1.
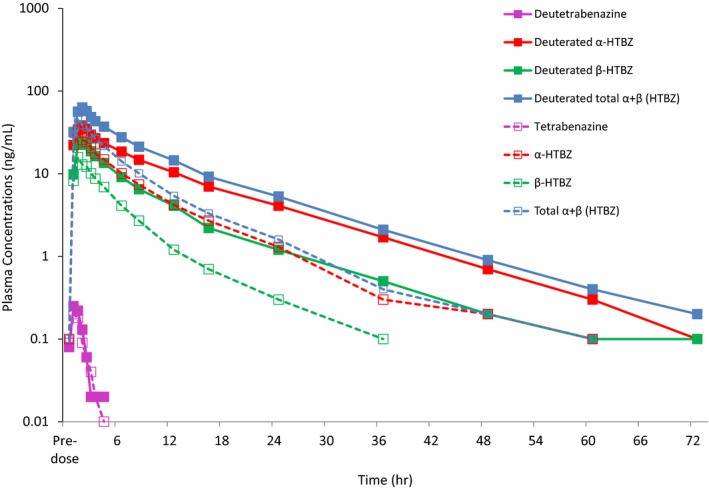
Mean plasma concentrations of deutetrabenazine, tetrabenazine, and their metabolites. HTBZ, dihydrotetrabenazine.
Table 1.
Pharmacokinetic parameters for deutetrabenazine and tetrabenazine and their active metabolites (Study AUS‐SD‐809‐CTP‐06)
|
Deutetrabenazine (n = 13) Mean (% CV) Geometric mean |
Tetrabenazine (n = 13) Mean (% CV) Geometric mean |
LS geometric mean ratio (95% CI) [% Deutetrabenazine/Tetrabenazine]a |
|
|---|---|---|---|
| Parent compound | |||
| Cmax, ng/mL | 0.30 (59.8) 0.27 | 0.29 (89.7) 0.29 | – |
| AUC0–t, hour*ng/mL | 0.26 (79.0) 0.20 | 0.18 (105.2) 0.15 | – |
| AUC0–inf, hour*ng/mL | Not calculable | Not calculable | – |
| t 1/2, hour | Not calculable | Not calculable | – |
| Tmax, hourb | 0.67 (0.33–1.50) | 0.67 (0.33–1.52) | – |
| Total (α + β)‐dihydrotetrabenazine (HTBZ) | |||
| Cmax, ng/mL | 67.9 (36.3) 63.6 | 60.1 (41.9) 55.0 | 113.4 (96.8, 132.9) |
| AUC0–t, hour*ng/mL | 405 (46.1) 368.1 | 171 (50.2) 154.1 | 234.9 (217.4, 253.8) |
| AUC0–inf, hour*ng/mL | 414 (45.1) 378.5 | 177 (50.3) 159.2 | 234.1 (216.3, 253.3) |
| t 1/2, hour | 7.61 (37.8) 7.1 | 4.05 (57.4) 3.58 | – |
| Tmax, hourb | 1.50 (0.67–2.00) | 1.00 (0.67–2.00) | – |
| α‐dihydrotetrabenazine (α‐HTBZ) | |||
| Cmax, ng/mL | 44.4 (29.9) 42.4 | 42.0 (39.6) 39.0 | 106.7 (91.8, 123.9) |
| AUC0–t, hour*ng/mL | 306 (41.4) 283.2 | 130 (46.5) 119.0 | 234.1 (217.3, 252.1) |
| AUC0–inf, hour*ng/mL | 316 (40.1) 294.0 | 137 (46.7)124.6 | 232.2 (214.9, 251.0) |
| t 1/2, hour | 7.95 (34.6) 7.5 | 4.51 (57.4) 4.0 | – |
| Tmax, hourb | 1.50 (0.67–2.00) | 1.00 (0.67–2.00) | – |
| β‐dihydrotetrabenazine (β‐HTBZ) | |||
| Cmax, ng/mL | 24.8 (49.3) 21.8 | 18.2 (55.1) 15.6 | 138.6 (115.3, 166.7) |
| AUC0–t, hour*ng/mL | 96 (61.6) 81.2 | 40 (63.7) 33.0 | 242.3 (213.0, 275.7) |
| AUC0–inf, hour*ng/mL | 100 (61.3) 84.7 | 42.0 (61.7) 35.5 | 235.5 (208.3, 266.4) |
| t 1/2, hour | 3.51 (61.5)3.2 | 2.36 (35.9) 2.2 | – |
| Tmax, hourb | 1.50 (1.00–2.07) | 1.00 (0.67–2.00) | – |
%CV, percentage of coefficient of variation; AUC0–inf, area under the concentration‐time curve from zero to infinity; AUC0–t, area under the concentration‐time profiles; CI, confidence interval; Cmax, peak plasma concentration; LS, least squares; t1/2, terminal half‐life; Tmax, time of maximum plasma concentration.
Geometric mean ratios of deutetrabenazine/tetrabenazine by analysis of variance performed on log‐transformed parameters using SAS Proc GLM with model: <parameter> = Treatment Period Sequence Subject (Sequence), then back‐transformed, included only extensive metabolizers.
Median (range).
The results of the statistical comparison (primary analysis) of the PK parameters AUC0–t, AUC0–inf, Cmax, and t 1/2 listed in Table 1 demonstrated significant differences in exposure to and elimination of the deuterated metabolites, α‐HTBZ and β‐HTBZ individually, and as a sum, following administration of deutetrabenazine compared with the nondeuterated metabolites following tetrabenazine. The mean half‐life of deuterated total (α + β)‐HTBZ was nearly doubled when compared with nondeuterated total (α + β)‐HTBZ, resulting in a more than twofold increase in overall exposure (AUC0–inf). The Cmax of deuterated total (α + β)‐HTBZ was marginally increased (~ 13%). There was a slight decrease in intersubject variability in the Cmax, AUC0–t, and AUC0–inf for the deuterated metabolites α‐HTBZ and β‐HTBZ and total (α + β)‐HTBZ. The effect of deuteration on the pharmacokinetics of individual metabolites α‐HTBZ and β‐HTBZ was comparable regarding increased AUC (increases of 132.2% to 142.3% for both metabolites) but slightly different comparing Cmax and t 1/2. Deuterated α‐HTBZ Cmax after deutetrabenazine administration was similar (within 6%) to that of nondeuterated α‐HTBZ after tetrabenazine, with a 76% increase in t 1/2. In contrast, the Cmax of deuterated β‐HTBZ slightly increased (~ 36%) with a smaller increase in t 1/2 (49%) compared with the increase with deuterated α‐HTBZ. Comparison of Tmax values using a signed rank test showed that deuteration statistically significantly increased the median Tmax for β‐HTBZ and total (α + β)‐HTBZ from 1.00 hours to 1.50 hours. Although a similar increase in median Tmax was observed for α‐HTBZ, the difference was not statistically significant.
Inclusion of poor and intermediate CYP2D6 metabolizers in the secondary analysis did not change the overall findings as described above for the primary analysis despite the higher exposure and slower elimination of parent and active metabolite in these subjects (see results from the secondary analysis in Table S1 and Figure S1 ). Descriptive analyses were used to compare, for each subject, the amount of active metabolites relative to inactive O‐desmethyl metabolites. As shown in Figure 2, after treatment with deutetrabenazine, circulating levels of the active metabolites were consistently higher relative to the inactive metabolites, whereas after treatment with tetrabenazine, in most cases, the active metabolites represented a minor fraction of the circulating metabolites (AUC0–t values for active and inactive metabolites are shown in Table S2 ).
Figure 2.
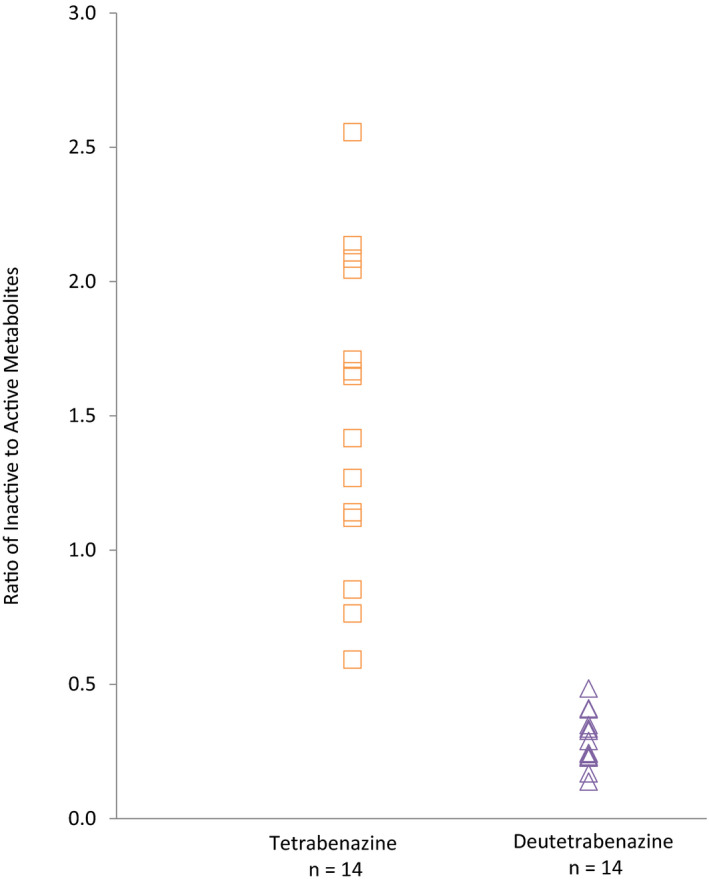
Ratio of exposure (area under the concentration‐time profiles) for inactive and active metabolites for tetrabenazine and deutetrabenazine in healthy subjects.
Plasma Pharmacokinetics: Study SD‐809‐C‐12
For Study SD‐809‐C‐12, the pharmacokinetic evaluations for total (α + β)‐HTBZ and their O‐desmethyl HTBZ metabolites in plasma, as assessed by nonradioactive bioanalytical methods, indicate that systemic exposure to the deuterated metabolites α‐HTBZ and β‐HTBZ after [14C]‐deutetrabenazine administration was higher than systemic exposure to nondeuterated α‐HTBZ and β‐HTBZ after [14C]‐tetrabenazine administration (Table S3 ). The slower clearance of deuterated α‐HTBZ and β‐HTBZ, an expected outcome of deuterium‐substituted O‐methyl groups, led to longer t 1/2 values of deuterated α‐HTBZ and β‐HTBZ and extended presence of these primary metabolites in the systemic circulation (longer MRT). Rate of formation and exposure to the downstream O‐desmethyl HTBZ metabolites (9 and 10‐O‐desmethyl‐α‐and β‐HTBZ) were lower after [14C]‐deutetrabenazine relative to [14C]‐tetrabenazine administration.
Evaluation of total radioactivity and mass balance recovery: Study SD‐809‐C‐12
Following oral administration of 25 mg [14C]‐deutetrabenazine and [14C]‐tetrabenazine, plasma concentrations of total radioactivity rose rapidly after a short lag time, reaching a maximum at 2 hours postdose with tetrabenazine and 3 hours postdose with deutetrabenazine (Table 2). Cmax was lower and AUC0–t was greater after administration of [14C]‐deutetrabenazine than after [14C]‐tetrabenazine. MRT of total plasma radioactivity was longer for [14C]‐deutetrabenazine than for [14C]‐tetrabenazine. The higher concentrations of total plasma sample radioactivity (observed starting 6 hours postdosing) after [14C]‐deutetrabenazine paralleled the increase in deuterated α‐and β‐HTBZ exposure, relative to the nondeuterated metabolites of tetrabenazine. The whole blood to plasma total radioactivity ratios indicated limited binding of radioactivity to the cellular component of whole blood, with no differences observed between the investigational products. The geometric mean estimate for whole blood to plasma total radioactivity ratio for Cmax was 0.75 for [14C]‐deutetrabenazine and 0.63 for [14C]‐tetrabenazine.
Table 2.
Pharmacokinetic parameters for plasma total radioactivity and total (α + β)‐ HTBZ metabolite following administration of radiolabeled deutetrabenazine and tetrabenazine (Study SD‐809‐C‐12)
| PK parametera |
Total radioactivity after [14C]‐deutetrabenazine (n = 6) |
Total radioactivity after [14C]‐tetrabenazine (n = 6) |
Total (α + β)‐HTBZb metabolites after [14C]‐deutetrabenazine (n = 6)c |
Total (α + β)‐HTBZb metabolites after [14C]‐tetrabenazine (n = 6)c |
|---|---|---|---|---|
| Tmax, hourd | 3.0 (0.67–6.00) | 2.0 (1.00–4.20) | 1.50 (0.67–4.00) nc | 1.50 (0.67–3.00) nc |
| Cmax | 128; 126e (16.5) | 164; 154e (33.2) | 46.6; 44.6f (35) | 27; 24.4f (44.3) |
| AUC0–t | 3290; 3190e (25) | 2230; 2220e (9.7) | 735; 652f (49.7) | 142; 136f (33.6) |
| AUC0–inf | nc | nc | 672; 598 (59) [n = 5] | 159; 152 (36) [n = 4] |
| MRT0–t, hour | 23.9; 22.9 (27) | 18.0; 16.8 (39) | 16.5; 15.7 (36) | 7.1; 6.6 (45) |
AUC0–inf, area under the concentration‐time curve from zero to infinity; AUC0–t, area under the concentration‐time profiles; Cmax, peak plasma concentration; HTBZ, dihydrotetrabenazine; MRT, mean residence time; nc, not calculated; PK, pharmacokinetic; Tmax, time of maximum plasma concentration.
Unless otherwise indicated, the PK parameters are mean; geometric mean (geometric percentage of coefficient of variation).
(α + β)‐Dihydrotetrabenazine.
Nonradioactive bioanalytical assay results.
Median (range).
Cmax (ng equiv/mL) and AUC0–t (ng equiv*h/mL).
Cmax (ng/mL) and AUC0–t (ng*h/mL).
The recovery of total radioactivity (urine + feces) was similar after 25 mg doses of [14C]‐deutetrabenazine (92.2%) and [14C]‐tetrabenazine (91.4%). For [14C]‐deutetrabenazine‐treated subjects, 74.8–86.5% of the dose was excreted in the urine up to 240 hours after administration with the majority of urinary radioactivity being recovered within 48 hours of dosing. Overall, 7.7–11.3% of the total dose was recovered in the feces with the majority of fecal radioactivity being recovered within 144 hours of dosing (Figure 3). For [14C]‐tetrabenazine‐treated subjects, 76.4–82.9% of the dose was excreted in the urine up to 216 hours after administration with the majority of urinary radioactivity being recovered within 48 hours of dosing. Overall, 7.8–13.5% of the total dose was recovered in the feces with the majority of fecal radioactivity being recovered within 96 hours of dosing.
Figure 3.
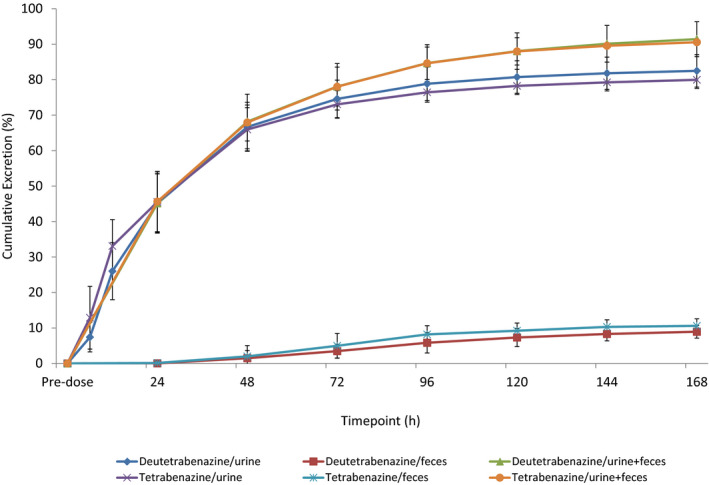
Mean (SD) excretion and recovery of total radioactivity.
Analysis of metabolites: Study SD‐809‐C‐12
In a radiochromatographic analysis for Study SD‐809‐C‐12, the characterization of metabolites relative to total radioactivity was derived by pooling four plasma samples over 12 hours per subject, a time course based on the previous limit of detection of metabolites by liquid scintillation counting. For the metabolite characterization of the urine samples, 0.5% of total urine sample weight for each subject was collected during multiple sample periods to create a 0 to 72 hour pool. Radioactive chromatograms for the pooled plasma and urine samples are shown in Figures S2 and S3 . Following [14C]‐deutetrabenazine administration there were no novel metabolites in plasma or urine relative to metabolites identified after [14C]‐tetrabenazine dosing. Deutetrabenazine and tetrabenazine were both present at similarly low levels near the limit of detection throughout the sampling period. Plasma sample radioactivity ascribed to metabolites > 1% of the total radioactivity accounted for between 77.3% and 86.0% of the deutetrabenazine and between 62.8% and 91.2% of the tetrabenazine radiochromatograms.
Evaluation of the plasma chromatograms for all 12 subjects found that six metabolites (depicted as M1–M6 in Table 3) were present at levels of at least 10% of plasma sample radioactivity in at least one individual in either cohort or in pooled samples during the course of the metabolite profiling analysis. Both deutetrabenazine and tetrabenazine underwent rapid reduction to α‐HTBZ and β‐HTBZ, which were subsequently O‐demethylated at the 9 and 10‐positions. The resulting circulating O‐desmethyl HTBZ metabolites were conjugated with sulphate. A mono‐hydroxy metabolite of deutetrabenazine and tetrabenazine was formed (M4), accounting for 12.9% and 15.6%, respectively, of total plasma sample radioactivity in the pooled samples. Additional minor metabolites in plasma, which represented between ~ 1% and 10% of sample radioactivity and were quantified in aggregate for each subject included the following: sulphates of O‐desmethyl HTBZ, a glucuronide of HTBZ, mono‐hydroxy HTBZ, 9‐O‐desmethyl‐β‐HTBZ, and mono‐hydroxy O‐desmethyl tetrabenazine. These same minor components observed following administration of [14C]‐deutetrabenazine were detected following administration of [14C]‐tetrabenazine. Subsequently the mono‐hydroxy metabolite (M4) of deutetrabenazine and tetrabenazine (oxidized at the subterminal position of the isobutyl group) as well as the 2‐methylpropanoic acid metabolite of β‐HTBZ (M1) were requantified in individual (non‐pooled) samples with a validated LC‐MS/MS bioanalytical method. The geometric mean AUC0–inf for M4, deuterated M4, M1, and deuterated M1 were 5.9%, 5.1%, 1.5%, and 9.0% of total plasma radioactivity, respectively, thus redefining these as minor metabolites (< 10% of total drug‐related exposure within cohort). In the case of M4 and deuterated M4 as well as M1, the time‐proportional pooling of the 4 samples within 12 hours over‐represented the contribution of the metabolites with respect to the total drug‐related radioactivity.
Table 3.
Summary of plasma and urinary metabolites (Study SD‐809‐C‐12)
| Metabolite | Percentage of total sample radioactivity (Mean ± SD) | |
|---|---|---|
| [14C]‐Deutetrabenazine (n = 6) | [14C]‐Tetrabenazine (n = 6) | |
| Plasma metabolites | ||
| α‐HTBZ (M6) | 13.0 ± 4.6 | 4.0 ± 1.4 |
| β‐HTBZ (M5) | 8.3 ± 4.2 | 1.8 ± 1.5 |
| Mono‐hydroxy metabolite of tetrabenazine (M4)a | 12.9 ± 3.2 | 15.6 ± 4.9 |
| 2‐methylpropanoic acid metabolite of β‐HTBZ (M1)b | 9.2 ± 3.6 | 4.1 ± 2.0 |
| Sulfate of O‐desmethyl α‐HTBZ (M3) | 4.0 ± 1.5 | 16.4 ± 5.6 |
| Sulfate of O‐desmethyl β‐HTBZ (M2) | 2.5 ± 1.1 | 6.4 ± 2.9 |
| Subtotal for metabolites M1–M6 | 49.8 ± 8.1 | 48.2 ± 5.1 |
| Additional minor metabolites (total)c | 31.9 ± 7.1 | 30.0 ± 8.7 |
| Total for plasma metabolites | 81.7 ± 3.0 | 78.2 ± 12.4 |
| Urine metabolites (cohort pool) | ||
| 2‐methylpropanoic acid metabolite of β‐HTBZ (M1) | 13.9 | 2.7 |
| Mono‐hydroxy β‐HTBZ (2 isomers) | 9.3 | 1.2 |
| Sulfate of O‐desmethyl β‐HTBZ | 10.5 | 14.2 |
| Co‐elution: Glucuronide of α‐HTBZ plus sulphate of O‐desmethyl HTBZd plus mono‐hydroxy α‐HTBZ | 15.3 | 3.8 |
| Sulphate of O‐desmethyl α‐HTBZ | 16.7 | 27.7 |
| Subtotal for major metabolites | 65.7 | 49.6 |
| Additional minor metabolites (total)e | 27.2 | 30.6 |
| Total for urine metabolites | 92.9 | 80.2 |
HTBZ, dihydrotetrabenazine.
In subsequent analyses, M4 was determined to be a minor metabolite constituting 5.1 and 5.9% of total drug‐related material in plasma for [14C]‐deutetrabenazine and [14C]‐tetrabenazine, respectively.
Also referred to as carboxylic acid.
Additional minor plasma metabolites included a sulfate of O‐desmethyl HTBZ, a glucuronide of HTBZ, monohydroxy HTBZ, 9‐O‐desmethyl β‐HTBZ and mono‐hydroxy O‐desmethyl tetrabenazine.
Due to the very low intensity of the protonated molecular ion, no product ion spectra were obtained for this compound.
Additional minor urinary metabolites included mono‐hydroxy tetrabenazine, β‐HTBZ, α‐HTBZ, mono‐hydroxy β‐HTBZ, sulphate of O‐desmethyl HTBZ, glucuronide of α‐HTBZ and mono‐hydroxy α‐HTBZ.
Plasma exposure to deuterated HTBZ metabolites was greater than exposure to the corresponding nondeuterated HTBZ metabolites, as predicted from in vitro results that demonstrated the operation of a kinetic deuterium isotope effect in the O‐demethylation reactions. The increased elimination half‐life of deuterated HTBZ was accompanied by a concomitant decrease in exposure to O‐demethylated metabolites, both free and conjugated (information on the unconjugated metabolites is provided in Table S3 ). Other biotransformation products of the deuterated HTBZ substrates were increased in plasma or urine, namely metabolites arising from their oxidation (mono‐hydroxy and carboxylic acid; e.g., M1) and O‐glucuronidation (Table 3).
The profile of major metabolites in urine confirmed the attenuation of the O‐demethylation of deuterated (α + β)‐HTBZ after oral dosing of [14C]‐deutetrabenazine. Urinary levels of the deuterated metabolites of HTBZ (carboxylic acid and monohydroxy derivatives) and conjugates of HTBZ were increased in parallel with a reduction in conjugates of deuterated‐O‐desmethyl HTBZ metabolites relative to their nondeuterated counterparts (Table 3).
Safety and tolerability evaluation
In both studies, single doses of deutetrabenazine and tetrabenazine or their 14C‐labeled analogues were well‐tolerated and had a comparable safety profile. In Study AUS‐SD‐809‐CTP‐06, the number and percentage of subjects with at least one treatment‐emergent adverse event (TEAE) was 10 (46%) for the deutetrabenazine group and 9 (45%) for the tetrabenazine group. The most common TEAEs were somnolence, nausea, and headache. AEs occurring in two more subjects following a single dose of either deutetrabenazine or tetrabenazine were somnolence (four (19%) and two (10%)), nausea (two (9.5%) and three (15%)), and headache (two (9.5%) and three (15%)), respectively. In the tetrabenazine group, there were three reported events of dizziness and two reports of bruising at the site of venipuncture. In Study AUS‐SD‐809‐C‐12, three subjects reported a total of six TEAEs. Most TEAEs were mild in severity, and there were no serious or severe TEAEs in either study. AEs led to study discontinuation for one subject in the Study AUS‐SD‐809‐CTP‐06 who withdrew consent for study participation ~ 4 hours following dosing with deutetrabenazine in period 1. There were no clinically significant findings in clinical laboratory assessments, vital signs parameters, ECG measurements, or physical examinations following either treatment.
DISCUSSION
The studies presented herein provide the first full in vivo human characterization of deutetrabenazine and confirm that deuterium substitution enhances the pharmacokinetic profile of the pharmacologically active species. As expected, the deuteration process did not affect the metabolism of the parent drugs to the active metabolites, but slowed down the metabolism of the active metabolites to the less potent O‐desmethyl metabolites. Both deutetrabenazine and tetrabenazine were shown to be highly permeable and readily absorbed as 80% of the radioactive dose was recovered. This is consistent with the classification of tetrabenazine as BDDCS Class II compound19 and preclinical data, which showed deutetrabenazine to be highly permeable through a rat perfusion model (Teva Pharmaceuticals, data on file).
As illustrated in Figure 4, the observed metabolic pathway of deutetrabenazine was found to be identical to that of tetrabenazine20 and no new metabolites were identified compared with tetrabenazine. The carboxylic acid metabolite (M1) had not been reported previously as a human metabolite of tetrabenazine but was found with both tetrabenazine and deutetrabenazine in this study. The targeted placement of the C‐D bond in deutetrabenazine resulted in a shift to increased active metabolites relative to inactive metabolites, and slowed elimination of the active metabolites without a significant increase of Cmax compared with the same dose of tetrabenazine; attributes that are expected to reflect in patient benefits in clinical use.
Figure 4.
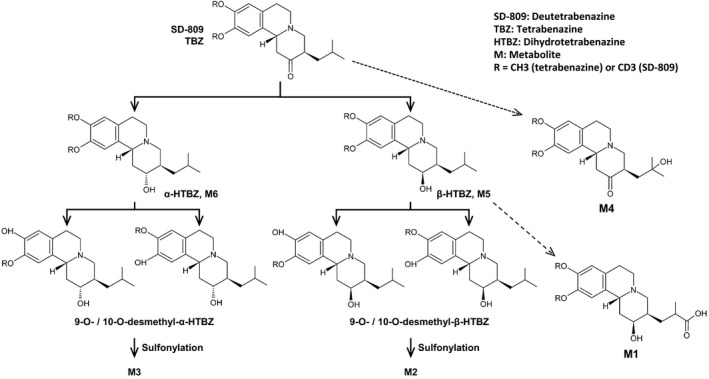
Metabolic pathways of tetrabenazine and deutetrabenazine. RR enantiomer depicted.
The comparable exposures to the administered parent compounds (deutetrabenazine and tetrabenazine) are consistent with an enzymatic reduction to their respective active α‐HTBZ and β‐HTBZ metabolites that is not influenced by changes in the bond cleavage rate in the remote methoxy groups. As predicted, deuterium substitution in the O‐methyl groups resulted in prolonged half‐lives of deuterated α‐HTBZ and β‐HTBZ, with a concomitant reduced exposure to the free and conjugated circulating O‐demethylated metabolites of HTBZ in circulation. The attenuated metabolism of α‐HTBZ and β‐HTBZ that was observed in vitro in incubations with human liver microsomes (kH/kD 1.51 and 2.44, respectively) was associated in vivo with an increase of the mean half‐lives of 76% and 49%, respectively (Teva Pharmaceuticals, data on file). The increase of urine metabolites that are not formed by O‐demethylation of HTBZ and the decrease of O‐methylation products in urine after deutetrabenazine compared with tetrabenazine show that the reduced rate of demethylation also decreased the extent of metabolism via this reaction.
The specificity of the reaction is confirmed not only by the similar parent metabolism but also by the similar exposure to the mono‐hydroxy tetrabenazine metabolite (M4) whose formation is unrelated to O‐demethylation. Similarly, for the M1 metabolite, no downstream metabolites were noted and a large presence in urine confirmed that it is likely directly excreted. Instead, the slower reaction rate of the metabolic degradation of β‐HTBZ to O‐desmethyl‐β‐HTBZ shifts the metabolism to the alternative pathway. The increased formation of M1 reflects the commonly observed phenomenon of metabolic switching. In addition, and consistent with the predicted limited effect of deuterium substitution on protein binding, in vitro binding assays in rats confirmed that deuteration did not change the binding of tetrabenazine to VMAT2, or its distribution, or its routes of excretion (Teva Pharmaceuticals, data on file).
The crossover design in Study AUS‐SD‐809‐CTP‐06 enabled a within‐subject comparison and calculation of the actual magnitude of the effect of deuteration on systemic exposure. The primary analysis of Study AUS‐SD‐809‐CTP‐06, which included only the extensive CYP2D6 metabolizers indicated that the administration of 25 mg of deutetrabenazine achieved a 2.3‐fold greater systemic exposure to the deuterated active metabolites based on AUC, whereas the increase in Cmax was only 1.1‐fold when compared with that following tetrabenazine 25 mg administration. The secondary analysis, which included all evaluable subjects regardless of their CYP2D6 phenotype, showed similar results; deutetrabenazine doubled the AUC but Cmax remained similar to that following tetrabenazine administration. Evaluation of individual subject AUC values of inactive to active metabolites suggests that deutetrabenazine treatment resulted in a consistent increase in the active metabolites relative to inactive metabolites, whereas tetrabenazine treatment resulted in variable ratios of active to inactive metabolites. The results of the nonradioactive bioanalytical assay in Study SD‐809‐C‐12 also confirmed the prolongation of t 1/2 for deuterated (α + β)‐HTBZ compared with nondeuterated (α + β)‐HTBZ and a corresponding increase in overall exposure (AUC0–t).
The bioanalytical‐derived plasma pharmacokinetics in Study SD‐809‐C‐12 are supportive in that they showed that the point estimates of the treatment effect are essentially the same for extensive CYP2D6 metabolizers and all evaluable subjects, demonstrating that the inclusion of subjects with reduced CYP2D6 function did not affect the outcome. In deutetrabenazine, the stronger C‐D bond, which attenuates the rate‐limiting O‐demethylation reaction catalyzed by CYP2D6, was found to reduce variability in drug metabolism and increase the terminal elimination half‐life. This kinetic deuterium isotope effect exhibited by deutetrabenazine means that the active metabolites α‐HTBZ and β‐HTBZ are eliminated more slowly in the tested poor (containing either no active alleles or only one partially active allele), intermediate (containing one wild type allele and one partially active allele), and extensive (containing two wild type alleles) CYP2D6 metabolizers.
Together, these studies characterize the pharmacokinetics and metabolism of deutetrabenazine in direct and indirect comparison to tetrabenazine. The results confirm that the observed metabolic pathway of deutetrabenazine is qualitatively identical to that of tetrabenazine and no new metabolites were identified compared with tetrabenazine. The attenuated metabolism attributable to the specific deuteration in deutetrabenazine doubles the exposure to the active metabolites and slows elimination compared with an equal dose of tetrabenazine, allowing for the desired exposure to be achieved with half the dose load and, thus, a lower Cmax. The longer half‐life further results in reduced fluctuation. In addition, the reduced rate of CYP2D6 metabolism with deutetrabenazine reduces interindividual variability in metabolism, and has the potential to reduce the impact of genetic or acquired CYP2D6 deficiency.
Funding
The studies described in this report were sponsored by Auspex Pharmaceuticals, Inc. (La Jolla CA), which was acquired by Teva Pharmaceutical Industries Ltd. (Petah Tikva Israel).
Conflicts of Interest
F.S., E.H., L.R.G., E.H., and P.S.L. are employees of Teva Pharmaceuticals who acquired deutetrabenazine from Auspex Pharmaceuticals, Inc., La Jolla, CA. M.B., D.S., D.C., and J.M.S. are former employees of Teva Pharmaceuticals and D.S. and M.B. were employees at Auspex Pharmaceuticals at the time the studies were conducted. T.A.B. was a paid consultant to Auspex Pharmaceuticals and Teva Pharmaceuticals.
Author Contributions
F.S., M.B., T.A.B., P.S.L., and J.M.S. wrote the manuscript. M.B., T.A.B., and D.S. designed the research. M.B., D.S., and E.H. performed the research. F.S., M.B., T.A.B., E.H., D.C., P.S.L., J.M.S., and L.R.G. analyzed the data.
Supporting information
Supplementary Tables S1–S3 and Figures S1–S3.
Acknowledgments
The authors appreciate the participation of the volunteer subjects and the work of the investigators of these studies: Joanne Marjason (Q‐Pharm Pty, Ltd Australia), Iain Shaw, Philip Bond, and Shran Sidhu (Quotient Clinical United Kingdom) who conducted these studies under the sponsorship of Auspex Pharmaceuticals, subsidiary of Teva Pharmaceutical Industries Ltd.
References
- 1. Huntington Study Group . Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology 66, 366–372 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Paleacu, D. Tetrabenazine in the treatment of Huntington's disease. Neuropsychiatr. Dis. Treat. 3, 545–551 (2007). [PMC free article] [PubMed] [Google Scholar]
- 3. Yao, Z. et al Preparation and evaluation of tetrabenazine enantiomers and all eight stereoisomers of dihydrotetrabenazine as VMAT2 inhibitors. Eur. J. Med. Chem. 46, 1841–1848 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pettibone, D.J. , Totaro, J.A. & Pflueger, A.B. Tetrabenazine‐induced depletion of brain monoamines: characterization and interaction with selected antidepressants. Eur. J. Pharmacol. 102, 425–430 (1984). [DOI] [PubMed] [Google Scholar]
- 5. Yero, T. & Rey, J.A. Tetrabenazine (Xenazine), an FDA‐approved treatment option for Huntington's disease‐related chorea. P T 33, 690–694 (2008). [PMC free article] [PubMed] [Google Scholar]
- 6. Goswami, R. , Ponde, D.E. , Kung, M.P. , Hou, C. , Kilbourn, M.R. & Kung, H.F. Fluoroalkyl derivatives of dihydrotetrabenazine as positron emission tomography imaging agents targeting vesicular monoamine transporters. Nucl. Med. Biol. 33, 685–694 (2006). [DOI] [PubMed] [Google Scholar]
- 7. Claxton, A.J. A systematic review of the associations between dose regimens and medication compliance. Clin. Ther. 23, 1296–1310 (2001). [DOI] [PubMed] [Google Scholar]
- 8. Kaur, N. , Kumar, P. , Jamwal, S. , Deshmukh, R. & Gauttam, V. Tetrabenazine: spotlight on drug review. Ann. Neurosci. 23, 176–185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaur, S. & Gupta, M. Deuteration as a tool for optimization of metabolic stability and toxicity of drugs. Glob. J. Pharmaceut. Sci. 1, GJPPS.MS.ID.555566 (2017). [Google Scholar]
- 10. Baillie, T.A. The use of stable isotopes in pharmacological research. Pharmacol. Rev. 33, 81–132 (1981). [PubMed] [Google Scholar]
- 11. Kushner, D.J. , Baker, A. & Dunstall, T.G. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can. J. Physiol. Pharmacol. 77, 79–88 (1999). [PubMed] [Google Scholar]
- 12. Tayar, N.E. , van de Waterbeemd, H. , Gryllaki, M. , Testa, B. & Trager, W.F. The lipophilicity of deuterium atoms. A comparison of shake‐flash and HPLC methods. Inter. J. Pharmaceut. 19, 271–281 (1984). [Google Scholar]
- 13. Huntington Study Group. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA 316, 40–50 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Morrow, T. Two new drugs for tardive dyskinesia hit the market. Manag. Care 27, 35–36 (2018). [PubMed] [Google Scholar]
- 15. Armstrong, M.J. & Miyasaki, J.M. American Academy of Neurology: evidence‐based guideline: pharmacologic treatment of chorea in Huntington disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology 79, 597–603 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Timmins, G.S. Deuterated drugs: where are we now? Expert. Opin. Ther. Pat. 24, 1067–1075 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gant, T.G. Using deuterium in drug discovery: leaving the label in the drug. J. Med. Chem. 57, 3595–3611 (2014). [DOI] [PubMed] [Google Scholar]
- 18. Hamilton, R.A. , Garnett, W.R. & Kline, B.J. Determination of mean valproic acid serum level by assay of a single pooled sample. Clin. Pharmacol. Ther. 29, 408–413 (1981). [DOI] [PubMed] [Google Scholar]
- 19. Benet, L.Z. , Broccatelli, F. & Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 13, 519–547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwartz, D.E. , Bruderer, H. , Rieder, J. & Brossi, A. Metabolic studies of tetrabenazine, a psychotropic drug in animals and man. Biochem. Pharmacol. 15, 645–655 (1966). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables S1–S3 and Figures S1–S3.