

Abstract
We report the development of photocatalytically patterned TiO2 arrays for selective on-plate enrichment and direct matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) analysis of phosphopeptides. A thin TiO2 nanofilm with controlled porosity is prepared on gold-covered glass slides by a layer-by-layer (LbL) deposition/calcination process. The highly porous and rough nanostructure offers high surface area for selective binding of phosphorylated species. The patterned arrays are generated by using an octadecyltrichlorosilane (OTS) coating in combination of UV irradiation with a photomask, followed by NaOH etching. The resulting hydrophilic TiO2 spots are thus surrounded by a hydrophobic OTS layer, which can facilitate the enrichment of low-abundance components by confining a large volume sample into a small area. The TiO2 arrays exhibit high specificity towards phosphopeptides in complex samples including phosphoprotein digests and human serum, and the detection can be made in the fmole range. Additional advantages of the arrays include excellent stability, reusability/reproducibility, and low cost. This method has been successfully applied to the analysis of phosphopeptides in non-fat milk. The patterned TiO2 arrays provide an attractive interface for performing on-plate reactions including selective capture of target species for MALDI-MS analysis, and can serve as a versatile lab-on-a-chip platform for high throughput analysis in phosphoproteome research.
Graphical Abstract
Considerable research interest has been invested in understanding and identification of post-translational modifications, in particular phosphorylation, due to its significant role in regulating diverse cellular biological processes and its relevance to several diseases including cancer.1 Recent advances in mass spectrometry (MS)-based techniques have greatly enhanced the phosphoproteomic study.2, 3 However, characterization of phosphorylated proteins and identification of phosphorylation sites still remain challenging. A main reason is that MS signal of phosphorylated species is frequently suppressed by the presence of abundant non-phosphopeptides. Therefore, isolation of phosphopeptides from various protein samples including proteolytic digests is a crucial step prior to MS analysis.4
To date, various strategies have been proposed for phosphopeptide enrichment and purification. Immunoaffinity purification provides satisfactory selectivity by using antibodies against phosphorylated amino acids,5 but the method is restricted by limited antibodies and their high cost. Immobilized metal ion affinity chromatography (IMAC), which utilizes the chelation interaction between metal ions (Fe3+, Ga3+, Ti4+, Zr4+ et al.) and phosphate groups, is another broadly used approach.6-10 The selectivity of this method, however, is usually limited due to nonspecific binding of acidic non-phosphorylated peptides.11 Other techniques such as strong cation/anion exchange chromatography (SCX/SAX),12-14,15 isoelectric focusing (IEF),16 hydrophilic interaction chromatography (HILIC)17 have been reported for preconcentration of phosphopeptides based on the difference in net charge, isoelectric point or polarity between phospho- and non-phosphopeptides. All these methods are less specific than affinity-based approaches.
In recent years, metal oxide affinity chromatography (MOAC) has been introduced as a promising approach in phosphoproteomic study because of the high selectivity towards phosphorylated species over other methods.18, 19 Currently, titanium oxide (TiO2) has become one of the most popular affinity materials to capture phosphopeptides in MOAC duo to its high specific affinity towards phosphopeptides, good stability and biocompatibility.11, 18 In addition to column, titania material has been prepared into pipette tips20 or coated on the magnetic nanoparticles (NPs)21 for enrichment before MALDI-MS analysis. These off-probe methods, however, are relatively time-consuming and suffer from the sample loss during multistep sample handling. In contrast, on-plate enrichment is fast and can minimize sample loss, and thus is more suitable for high throughput analysis. Several TiO2-based on-plate enrichment methods have been developed in recent years. A straightforward approach is to directly functionalize the commercial MALDI-plate with TiO2 NPs after calcination,22 but morphology and thickness of TiO2 spots are difficult to control due to the uneven dispersion and aggregation of NPs during solvent evaporation, which result in poor reproducibility in array preparation and MS analysis. The high cost of commercial MALDI-plate is another problem. To circumvent this problem, alternative substrates such as aluminum foil23 and glass slide24, 25 have been employed to replace the commercial plate. Different stencil methods, including screen-printing, rotogravure-printing and spraying deposition, were used to create arrays with TiO2-NPs on the substrate.23, 24 Though effective, these methods are not suitable for the fabrication of small-size TiO2-spots and high density arrays. Moreover, there lacks of effective control over pattern morphology and the thickness of TiO2 material. Recently, Torta et al.26 utilized the pulsed laser deposition (PLD) method to coat the standard MALDI plate with a TiO2 film, where the structure and thickness of film can be finely controlled within several hundred nanometers. However, the PLD technique is complex and requires expensive equipment, and the method has limited use in the fabrication of arrays.
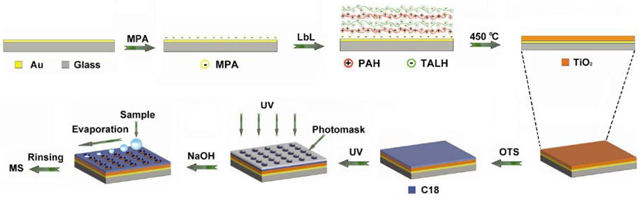
We report here the development of a simple but highly effective method to fabricate TiO2 array for on-plate selective enrichment of phosphopeptides prior to MALDI MS analysis. In this method, a TiO2 nanofilm is prepared on an Au-coated glass slide by a layer-by-layer (LbL) deposition/calcination process using titanium(IV) bis(ammonium lactato)dihydroxide (TALH) as the precursor (Figure 1). A relatively homogeneous film has been obtained on the substrate, and the film thickness and porosity can be finely controlled in nanometer scale, providing excellent reproducibility in film fabrication. By taking advantage of the photocatalytical property of the TiO2 material, an array is created on the octadecyltrichlorosilane (OTS)-modified TiO2 nanofilm by UV irradiation with a photomask and subsequent NaOH etching. The photocatalytically patterned TiO2 array (PPTA) is characterized by hydrophilic TiO2 spots surrounding by hydrophobic OTS-modified area, which simplifies on-target sample preparation and facilitates the enrichment of low-abundance samples. The selectivity, reproducibility and reusability of the PPTA-based on-plate approach in phosphopeptide analysis have been investigated, and the method is applied to analyzing phosphopeptides from complex samples such as phosphoprotein digests, human serum and non-fat milk samples. The results demonstrate high potential of the PPTA as a new and versatile lab-on-chip platform for high throughput phosphoproteomic analysis, in which the integration of on-plate reaction, analyte capture and MS detection could be envisioned.
Figure 1.

A schematic diagram of the fabrication of PPTA for on-plate enrichment and MS analysis of phosphopeptides
EXPERIMENTAL SECTION
Chemicals.
Titanium(IV) bis(ammonium lactato)dihydroxide solution (TALH, 50% in water), 3-mercaptopropionic acid (MPA, 99%), poly(allylamine hydrochloride) (PAH, average MW ~56,000), trifluoroacetic acid (TFA), superDHB, α-casein (≥70%, from bovine milk), β-casein (98%, from bovine milk), bovine serum albumin (BSA), trypsin (from bovine pancreas) and human serum (male AB plasma) were obtained from Sigma-Aldrich (St. Louis, MO). Octadecyltrichlorosilane (OTS) and phosphoric acid (H3PO4, 85%) were from Acros Organics (Fair Lawn, NJ). Fresh non-fat milk (protein content ~33 mg/mL) was obtained from local supermarket in Riverside. Acetonitrile (ACN), ethanol (EtOH) and toluene were from Fisher Scientific (Pittsburgh, PA).
Instruments.
Photocatalytic patterning of TiO2 was performed in CL-1000 254 nm UV Crosslinker with maximum intensity of 450 mW/cm2 (UVP Inc., Upland, CA). Scanning electron microscopy (SEM) was performed with Hitachi TM-1000 tabletop scanning electron microscope (Tokyo, Japan). Atomic force microscopy (AFM) images were collected by using Veeco Dimension 5000 AFM (Santa Barbara, CA). The scan rate was set as 1 Hz and the Rms surface roughness values were measured in defined 10 × 10 μm2 areas. The thickness profile of the nanofilm was measured by VEECO Dektak 8 surface profiler (Tucson, AZ).
Sample Preparation.
Digestion of α-/β-casein followed the reported work27. BSA digest preparation used the same procedure except for the denaturation at 100 °C for 5 min before digestion. Milk sample was prepared using a previously reported process.28 For in-solution digestion, 0.5 mL milk was dissolved in 0.5 mL of 50 mM NH4HCO3 solution, followed by incubation with 20 μL of 1 mg/mL trypsin at 37 °C for 18 h. Then the solution was diluted 10 times before PPTA enrichment. For on-target digestion, milk sample was firstly diluted by 20 times, and then 1 μL milk solution was directly dropped on TiO2 spots and dried in vacuum. Then, 4 μL of 1 mg/mL trypsin was added for digestion. the substrate was placed in a sealed petri dish and incubated at 37 °C for 2 h. The reaction was stopped by adding 2% TFA solution.
Preparation of TiO2 Nanofilm.
TiO2 nanofilms were prepared on gold-covered glass substrate by a LbL deposition/calcination process, which was modified based on our previous work29, 30. The gold substrates were immersed in a 10 mM 3-MPA ethanol solution overnight, followed by extensive rinsing with ethanol and drying under nitrogen gas. PAH (1mg/mL, pH 7.5) and TALH (5 wt %, pH 7.5) aqueous solution were sprayed on the substrates until the designated (PAH/TALH)n multilayers (n=4, 8, 12 and 16) were obtained. The substrates were then washed by D.I. water and dried in air, calcinated in a furnace by heating to 450 °C for 4 h (Figure 1).
Generation of PPTA.
TiO2 nanofilm was firstly immersed in a 10 mM OTS toluene solution to create a hydrophobic monolayer. Trace amounts of water was added to accelerate the hydrolysis of OTS so that the modification process could be completed within 10 min. Subsequently, the OTS-modified substrate was covered by a photomask and irradiated under 254 nm UV light for 1 h to photo-catalytically cleave the exposed OTS ligands. After rinsing with EtOH, 0.5 M NaOH was incubated on the patterned spots for 20 min to expose the TiO2 material. Finally, the substrate was rinsed with water and dried before use.
On-Plate Enrichment of Phosphopeptides.
Protein digests was loaded on the TiO2 spots and incubated for a certain time. The sample spots and the adjacent hydrophobic area were then rinsed intensively by 2% TFA solution and 2% TFA solution containing 60% ACN, respectively, to remove contaminants and nonspecifically adsorbed compounds. After that, 20 nL (for standard protein digests) or 100 nL (for milk digests), 10 mg/mL super-DHB in 50% ACN was deposited to TiO2 spots by using IBF Nanoliter (Nanoliter LLC, Henderson, NV). The use of super-DHB can avoid decomposition of phosphopeptide ions during mass analysis. To enhance phosphopeptide ion signals in MALDI-MS detection, 1% phosphoric acid was added as an additive in the matrix solution31. Super-DHB with 1% phosphoric acid could facilitate the release of phosphopeptides from TiO2 surface, thus alkaline eluting is not necessary prior to MS detection. A homemade sample stage was used to hold the substrate for MALDI-MS analysis.
MALDI-TOF MS and Data Analysis.
All MALDI-TOF mass spectra were collected using Voyager-DE STR MALDI-TOF mass spectrometer (Applied Biosystems, Framingham, MA). Experiments were performed in positive reflector mode at accelerating voltage of 20 kV. MS with an accumulation of 100 laser shots. Peptide mass mapping was carried out by using Findpept tools in ExPASy (Expert Protein Analysis system, http://www.uniprot.org).
RESULTS AND DISCUSSION
Characterization of the calcinated TiO2 nanofilm.
Layer-by-layer (LbL) assembly technique is one of the most promising methods for preparation of nanostructured film. The properties of LbL film such as thickness, composition and function can be precisely controlled by following a simple and low-cost procedure.32 In this work, TiO2 nanofilm was prepared on gold-covered glass slides using a LbL deposition/calcination process, which has been successfully utilized to prepare nanoscale silicate films to facilitate surface plasmon resonance (SPR) biosensing and MS detection in our previous work.29, 30, 33 Figure 2 shows SEM and AFM images of calcinated TiO2 films obtained with different (PAH/TALH)n (n=4, 8, 12 and 16) layers and the pore size distribution is given in supporting information. Highly porous films were formed on these substrates (a1-d1 in Figure 2) with the porous structure mainly consisting of two kinds of pores: micropores and nanopores. Both pores show relatively uniform size in distribution (a2-d2 in Figure 2). The highly porous structure is advantageous for increasing surface area of the TiO2 nanofilm, thus providing high capacity for analyte binding. The average size of micropores is about several μm and increases rapidly with the increase of film thickness (supporting information). For instance, in (PAH/TALH)4 film, the average size for micropores was only about 1.4 μm. The size increased approximately 10% with (PAH/TALH)8 film but rose rapidly with (PAH/TALH)12 and (PAH/TALH)16 films, resulting in 69% and 89% growth in size, respectively. In contrast, the diameter of nanopores was ca. 400 nm and remained relatively constant regardless of the film thickness. The two pore structures of the TiO2 nanofilm might be the results of two types of PAH aggregates (nanoscale and microscale) in LbL process. The nano-scale aggregates may relate to the compact coils of PAH generated in a relative basic deposition condition,34 while the generation of micro-scale ones may be attributed to the aggregation of polyelectrolytes during LbL deposition without water washing. The fast enlargement of micropores suggests the nonlinear growth of microscale aggregates with the increase in film thickness. In addition, the pore density for micropores increases with film thickness and becomes dominant for the (PAH/TALH)16 film, indicative of the increase of microscale PAH aggregates. The change of pore distribution reflects the exchange of species between nanoscale and microscale PAH aggregates, which apparently follows an Ostwald ripening process.35, 36 In addition to porous structure, particles and islands appeared with thick TiO2 films (n≥8) and scattered over the film surface, leading to enhancement of surface roughness. These nanostructures may stem from aggregation of electrolytes and become dominant for (PAH/TALH)12 and (PAH/TALH)16 films (c2 and d2 in Figure 2).
Figure 2.

SEM and AFM images of TiO2 nanofilms obtained from (a) (PAH/TALH)4, (b) (PAH/TALH)8, (c) (PAH/TALH)12 and (d) (PAH/TALH)16. SEM images are shown with magnification of 1,000 (a1-d1) and 10,000 (a2-d2) times, respectively. The dark dots are voids or pores, while the white ones are protruding particles or islands. AFM images (a3-d3) are obtained in a 10 μm × 10 μm area.
AFM was also applied to examine the surface of TiO2 films (a3-d3 in Figure 2). The (PAH/TALH)4 film consisted of multiple layers of compact NPs, leading to a relatively uniform and flat surface. The surface roughness (rms) was measured as 23.7 nm. With increasing of film thickness, a steep increase of roughness was observed where rms value for (PAH/TALH)8 film climbed to 116.8 nm. The increased rms appears to originate from the formation of large NPs and irregular islands. Large rms values were obtained for TiO2 film with more PAH/TALH layers, where the rms values for (PAH/TALH)12 and (PAH/TALH)16 films were 127.5 and 158.0 nm, respectively. We noticed that the size of NPs with these films has no significant change compared to that with (PAH/TALH)8 film, while the size for islands increased by about 2 to 3 times, suggesting that the increase of rms values was mainly caused by the growth of islands. Therefore, further increasing of film thickness can only promote the growth of islands and micropores, but will have little contribution to the increase of specific area. From these results, the (PAH/TALH)8 film shows a homogeneous porous surface with high rms and massive nanopores, providing increased specific surface area and binding capacity for phosphopeptides, and is therefore chosen for all of the following experiments. The growth of TiO2 nanofilm could be precisely controlled within nanometer scale, and the average thickness of (PAH/TALH)8 film was about 230 nm as measured by a profilometer.
Photocatalytic Patterning of TiO2 Array.
Surface patterning of the sample plate is effective to miniaturizing sample spots and increasing detection sensitivity in MALDI-MS analysis.37 Recently, IMAC microspots have been prepared on the plate for phosphopeptide analysis by UV-lithography of self-assemble monolayers (SAMs) on gold38 and in situ growth of polymer39. Photolithography has been used for producing patterns with precise shape of high resolution.40 However, this conventional method usually involves the use of vacuum system, polymeric photoresists and wet etching, which is laborious and of high cost. Here we utilized a simple and low cost photocatalytic approach to create TiO2 array for phosphopeptide analysis by taking advantage of photocatalysis of TiO2 material.41 As shown in Figure 3a, TiO2 surface showed highly hydrophobic property with coating of an OTS monolayer and gave a contact angle of ~162°. After exposed to UV irradiation, the contact angle decreased to ~69° (Figure 3b), demonstrating the change of surface property by photocatalytic cleavage of the OTS monolayer and feasibility of creating an array on the OTS modified-TiO2 nanofilm. Fig. 3c shows the photograph of 2 μL water droplets on a 5 × 5 PPTA with spots diameter of 1 mm prepared with UV irradiation and a photomask. The insert picture demonstrated the anchoring of water droplet within TiO2 spot by surrounding hydrophobic area, which is favorable for concentrating low-abundance sample with higher sample volume. This photocatalytic patterning approach avoids the use of polymer photoresists, and thereby is faster and cleaner than traditional methods.
Figure 3.

Photographs of water dropped on OTS-modified, 8-layer TiO2 nanofilm before (a) and after (b) UV irradiation, and on a 5 × 5 PPTA with spots diameter of 1 mm (c). A zoomed photograph of the droplet on PPTA spot is shown as insert in (c).
After the photo-patterning, NaOH solution was used to remove the residual Si-O bonds within the spots to completely expose the underlying TiO2 surface and reduce nonspecific interaction. Etching condition was optimized for subsequent enrichment experiments (supporting information), and incubation with 0.5 M NaOH for 20 min was found to provide good selectivity and minimum nonspecific binding, and thus chosen as the optimal condition for the rest of the study.
On-plate Enrichment of Phosphopeptides.
PPTA was firstly applied to on-plate capture of phosphopeptides from digests of phosphoproteins, including β-casein and α-casein. MS spectra are showed in Figure 4 (peptides see supporting information). Without enrichment, non-phosphopeptides showed high response in MS detection and were dominant in the spectrum with 1 pmol β-casein digest (Figure 4a). Ion signal for phosphopeptides was relatively low and swamped by the background of unmodified species. Only two mono-phosphopeptides, β1 and β2, were identified under this condition. After on-plate enrichment using PPTA, peptide β1 and β2 became dominant in spectrum and a very clean background was obtained (Figure 4b). Extra four phosphopeptides were detected after removal of non-phosphorylated species. Among these peptides, β3 was tetra-phosphopeptide from β-casein, and α2, α5 and α6 were assigned to phosphopeptides from low-abundance α-casein contaminants.11, 38 In the case of analysis of 4 pmol α-casein digests, only four phosphopeptides (α2, α4, α5 and α6) were identified in direct analysis (Figure 4c). These peptides were characterized with less phosphorylation sites (1 or 2). The signal of multiphosphopeptide with more phosphorylation sites was totally suppressed by unmodified ones due to their poor ionization efficiency42. After selective capture and enrichment, phosphopeptides became dominant in the spectrum and the four peptides mentioned above showed very high response in MS detection (Figure 4d). Additionally, seven more phosphopeptides, including two mono- (α1 and α3) and five multi-phosphopeptides (α7-11), were detected. These peptides either existed as relatively low abundance components in digests mixture or had poor ionization efficiency with multiphosphorylation, and thus difficult to detect without enrichment. PPTA-enrichment selectively captures these phosphopeptides and improves the detection sensitivity after removal of nonphosphorylated species, providing comparable and even better enrichment efficiency in terms of selectivity and reliability compared to the existing MOAC or IMAC approaches10, 24, 25, 38.
Figure 4.
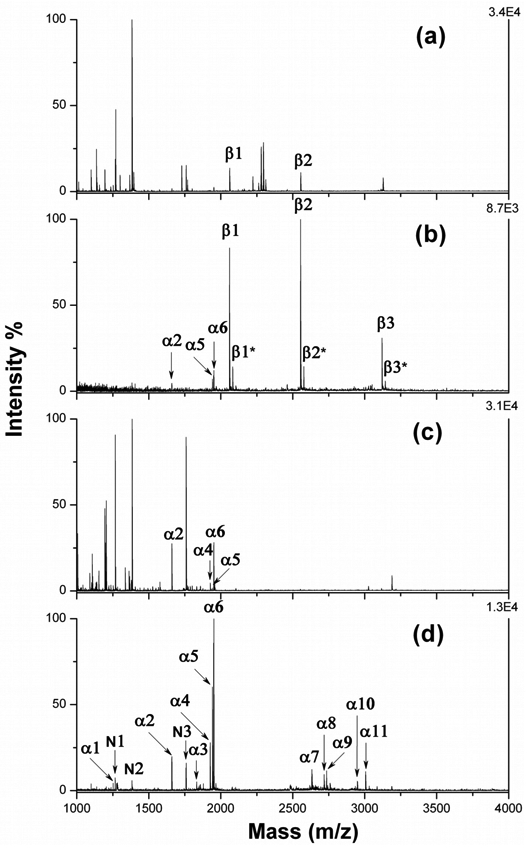
Mass spectra of 1 pmol β-casein digests before (a) and after (b) on-plate enrichment, and of 4 pmol α-casein digests before (c) and after (d) on-plate enrichment with PPTA. “α” and “β” represents identified phosphopeptides from α-casein and β-casein, respectively. “N” represents non-phosphopeptides, and “*” indicates Na+ adducted ions for phosphopeptides.
The capability of PPTA to capture phosphopeptides from a more complex mixture was tested with tryptic digests of BSA and β-casein at ratios of 1:1, 10:1 and 80:1 (mol/mol), respectively. The amount of β-casein digest was 1 pmol and remained constant in all samples. Compared to two phosphopeptides detected with pure β-casein digests (Figure 4a), only one monophosphopeptide (β1) was observed with addition of BSA digest at 1:1 ratio (supporting information). With further increase in BSA content, no phosphopeptide was detectable without enrichment. This result indicated the aggravation of signal suppression with increase of non-phophorylated species in the mixture. After PPTA enrichment, however, phosphopeptides and their related species were clearly detectable in all cases and they showed high response in MS spectra, which demonstrated good selectivity of the substrate towards phosphorylated peptides even in the presence of abundant nonphosphorylated species. In addition, no detectable nonphosphorylated species were found in these enriched samples, which suggest nonspecific binding from the hydrophobic OTS coating surrounding the spots was negligible under our condition. The ion signal for phosphopeptides tended to decrease with addition of high amount of BSA digest. For instance, ion intensities for three β-casein phosphopeptides (β1-3) decreased nearly 50% for the ratio of 10:1 (supporting information). For the 80:1 ratio, only tetra- phosphopeptide β3 was detectable with a relatively weak signal after enrichment. The results suggest that high concentration of non-phosphorylated peptides (i.e. acidic peptides) hinders the binding of phosphopeptides towards TiO2 material due to insufficient binding sites. This problem could be circumvented by using dilute samples or TiO2 spots of larger size.
The application of PPTA for analysis of complex biological samples was performed for selectively isolating endogenous phosphopeptides from human serum. Four phosphorylated fibrinogen fragments were successfully detected within 40 μL serum sample after on-plate enrichment (supporting information), which is consistent with the previous reports43, 44 and demonstrates the potential of PPTA for broader biomedical analysis.
Performance of PPTA for On-Plate Enrichment.
Binding capacity of 1 mm PPTA spots for phosphopeptides was evaluated by applying different amount of β-casein digests for on-plate enrichment. Figure 5 shows the ion signal for three PPTA-enriched β-casein phosphopeptides β1, β2 and β3 with different sample loadings. With 40 pmol of digest, only tetraphosphopeptide β3-related ions were observed in the MS spectrum, suggesting insufficient binding sites for the phosphopeptides. The protonated ion for β3 showed a very strong response, which was close to the saturation signal of the instrument. This phenomenon can be explained by the relatively high affinity of multiphosphopeptides towards TiO2 surface,45 which likely enable them to attach to the available binding sites more favorably than monophosphorylated species. Under this condition, multiphosphoryalted species can be selectively isolated from the peptide mixture with a high sample loading, which is useful for the study of multiple phosphorylation in proteins. The signal for β3 decreased with the reduction of sample amount. When sample amount was reduced by 10 times, β3 was still dominant in MS spectrum, but the ion intensity decreased by about 70%, corresponding to nearly 77% decrease of S/N ratio. Signals from two monophosphopeptides, β1 and β2, began to appear in the spectrum. With 1 pmol β-casein digest, β1 and β2 became dominant in the spectrum, suggesting the unsaturated binding at the TiO2 surface. Based on this result, the binding capacity for 1 mm spots was estimated to be 1 pmol for β-casein digest, which is comparable with the result reported with IMAC-spots.11, 39, 46 Compared to the results for 4 pmol digest, protonated ions of β1 and β2 increased by about 4.7 times in ion signal. In contrast, β3 signal continued to decrease, showing a nearly 3 times reduction in ion intensity and S/N ratio. Further decreasing the sample loading to 400 fmol led to β1 and β2 becoming dominant in MS spectrum, but ion signals for all three β-casein phosphopeptides diminished in similar proportion (ion intensity decreased by 3.8, 3.6 and 6.8 times for β1, β2 and β3 as compared to that with 1 pmol digest). When 40 fmol digest was applied, only β1 was detectable, which suggested the limit-of-detection for β-casein digest. This result demonstrated the existence of competition between phosphorylated species towards the limited binding sites within PPTA spots, which may enable the study of competitive binding property of phosphorylated species on TiO2 films in a quantitative fashion.
Figure 5.

3D bar graphic analysis for PPTA enrichment of β-casein phosphopeptides with different loadings. The diameter of TiO2 spots is 1 mm.
PPTA showed additional advantages with the unique configuration of hydrophilic TiO2 spots surrounded by a hydrophobic coating, which is favorable for concentrating sample into small areas. This patterning design allows loading of larger volumes that exceed those used for conventional dried droplet methods, and is more suitable for the concentration of low-abundance sample. Figure 6a shows that the loading volume could be expanded to 20 μL on a 1 mm TiO2 spot, much larger than a typical 1 μL loading, while the sample “stain” size was comparable (Figure 6b). The large volume enrichment (LVE) on phosphopeptides was further investigated by using β-casein digests with concentrations of 20 nM and 400 nM. Analysis using 20 nM sample with 1 μL loading failed to provide satisfactory result due to low sample amount on the surface. Detection can be improved by increasing either the analyte concentration or the loading volume, while the latter is desirable as it allows for direct use of diluted samples, which simplifies the process and avoids contamination in sample handling. Figure 6c and d show the MALDI-MS results for 400 nM sample with 1 μL loading and 20 nM with 20 μL loading for β-casein digests, respectively. No significant difference was observed in terms of ion intensity for three monophosphopeptides (β1, β2 and α8), indicating on-plate LVE with PPTA is an effective approach to preconcentrate low-concentration phosphopeptides within the spots. We noticed that the relative ion intensity of tetra-phosphopeptide β3 was slightly low with the diluted sample, which could be attributed to the nonspecific binding of β3 with the hydrophobic coating surrounding the spots. Incubation with rinsing solution (2% TFA in 60% ACN) after washing can alleviate this problem. Clearly on-plate LVE with PPTA facilitates the enrichment of low-concentration analyte in a simple manner, and thus presents a useful platform for coupling with micro-separation techniques, where the concentration process of dilute eluents usually required prior to MALDI-MS analysis can be omitted.
Figure 6.

Photographs of droplets with 1 μL and 20 μL sample solution on PPTA with 1 mm spots before (a) and after (b) evaporation, and mass spectra for 400 fmol β-casein digests with 1 μL (c) and 20 μL (d) loading on PPTA. The symbols are the same as in Figure 4.
The spot-to-spot reproducibility of PPTA for phosphopeptide enrichment was investigated with 400 fmol β-casein digests and 10 random spots. Phosphopeptides β1 and β2 were dominant in the spectra. The RSD of ion intensity for protonated β1 and β2 were ~5% and ~18% (n=10), respectively, demonstrating a good spot-to-spot reproducibility of PPTA. This good reproducibility may stem from the relatively homogenous structure of the TiO2 nanofilm and the effective control of the surface patterning.
The reusability of PPTA was also studied by using β-casein digests. The used TiO2 spots were regenerated and incubated with 0.5 M NaOH solution for 5 min, and then rinsed with water. This procedure was repeated 3 times. No peptide signal was found with MALDI-MS analysis with the regenerated spots, showing effective removal of peptide residues with NaOH washing. When the regenerated spot was applied for β-casein digest, similar enrichment efficiency was obtained with respect to the number and signal intensity for identified phosphopeptides, which demonstrated excellent reusability of the PPTA spots (supporting information).
On-Target Digestion with PPTA for Analysis of Complex Samples.
PPTA has attractive surface features and may serve as a useful platform for integrating multiple functions into a single chip for potentially high throughput analysis. We have carried out the on-target digestion of non-fat milk proteins within PPTA spots for phosphopeptide capture and MALDI-MS analysis. Table 1 summarizes the identified peptides from milk digests after PPTA enrichment with two digestion methods: in-solution and on-target digestion. In both cases, phosphorylated species were dominant in MS spectra, indicating good selectivity of TiO2 films towards phosphopeptides (supporting information). With in-solution digestion, seventeen phosphopeptides were detected after enrichment and assigned to eight groups of homogenous peptides from α-S1-casein, α-S2-casein and β-casein. On-target digestion yielded a relatively simple spectrum, allowing identification of thirteen phosphopeptides from the milk sample. Compared to in-solution results, all multiphosphopeptides were concentrated by PPTA after direct on-target digestion due to their high affinity for binding, though phosphorylation sites in peptide group 2, 4 and 7 were missed. This result may be attributed to the interference of excess protease used for on-target reaction. In our on-target digestion, a high amount of trypsin was used to accelerate the speed of reaction. The ratio of enzyme to protein was about 2.5, which was nearly 200 times higher than that for in-solution reaction. The existence of high abundant enzyme (~400 pmol/spot) and its autolytic peptides may hinder the binding of phosphopeptides with weak affinity. Therefore, high amount of enzyme is not recommended for on-target reaction/enrichment, and this problem could also be largely curbed by using TiO2 arrays with larger spots. Compared to traditional in-solution digestion, on-target reaction using an array simplifies sample preparation and avoids sample loss/contamination, and the reaction speed can be tuned by varying enzyme content to fit the throughput requirement. These attractive characteristics of PPTA make it possible for developing on-line hyphenate separation or optical techniques with mass spectrometry for rapid screening of phosphorylated species from complex biological samples.
Table 1.
Identified Phosphopeptides from Proteolytic Digests of Non-Fat Milka
| no.b | MH+ | amino acid sequence | protein | position | in-solution | on-plate |
|---|---|---|---|---|---|---|
| 1a | 1927.69 | DIG[pS]E[pS]TEDQAMEDIK | α-S1-casein | 58-73 | √ | |
| 1b | 1943.68 | DIG[pS]E[pS]TEDQA[oM]EDIK | α-S1-casein | 58-73 | √ | |
| 1c | 2935.16 | EKVNEL[pS]KDIG[pS]E[pS]TEDQAMEDIK | α-S1-casein | 50-73 | √ | |
| 2a | 1660.79 | VPQLEIVPN[pS]AEER | α-S1-casein | 121-134 | √ | |
| 2b | 1951.95 | YKVPQLEIVPN[pS]AEER | α-S1-casein | 119-134 | √ | |
| 3a | 2703.75 | *QMEAE[pS]I[pS][pS][pS]EEIVPN[pS]VEAQK | α-S1-casein | 74-94 | √ | √ |
| 3b | 2720.91 | QMEAE[pS]I[pS][pS][pS]EEIVPN[pS]VEAQK | α-S1-casein | 74-94 | √ | √ |
| 3c | 2736.91 | Q[oM]EAE[pS]I[pS][pS][pS]EEIVPN[pS]VEAQK | α-S1-casein | 74-94 | √ | √ |
| 4a | 1466.61 | TVDME[pS]TEVFTK | α-S2-casein | 153-164 | √ | |
| 4b | 1482.60 | TVD[oM]E[pS]TEVFTK | α-S2-casein | 153-164 | √ | |
| 5a | 2618.90 | NTMEHV[pS][pS][pS]EE[pS]IISQETYK | α-S2-casein | 17-36 | √ | √ |
| 5b | 2634.90 | NT[oM]EHV[pS][pS][pS]EESII[pS]QETYK | α-S2-casein | 17-36 | √ | √ |
| 6a | 3008.03 | NANEEEYSIG[pS][pS][pS]EE[pS]AEVATEEVK | α-S2-casein | 61-85 | √ | √ |
| 6b | 3088.00 | NANEEEY[pS]IG[pS][pS][pS]EE[pS]AEVATEEVK | α-S2-casein | 61-85 | √ | √ |
| 7a | 2061.83 | FQ[pS]EEQQQTEDELQDK | β-casein | 48-63 | √ | |
| 7b | 2556.09 | FQ[pS]EEQQQTEDELQDKIHPF | β-casein | 48-67 | √ | |
| 8a | 2966.16 | ELEELNVPGEIVE[pS]L[pS][pS][pS]EESITR | β-casein | 17-40 | √ | √ |
| 8b,8b*c | 3122.26 | RELEELNVPGEIVE[pS]L[pS][pS][pS]EESITR | β-casein | 16-40 | √ | √ |
| number of identified phosphopeptides | 17 | 10 |
“[pS]” represents phosphorylation on serine; “[oM]” represents oxidation on methionine; “*Q” presents pyroglutamylation on the N-terminal glutamine of peptide. 47
Phosphopeptides possess the same phosphorylation sites were classified into the same group and denoted with the same number (i.e. 1, 2, 3…). Letters (i.e. a, b, c…) were used to distinguish the different peptides in the same group.
8b* is [M+2H]2+ of peptide 8b.
CONCLUSIONS
We have demonstrated that PPTA is a very promising platform for analysis of phosphopeptides by mass spectrometry. High selectivity and large binding capacity towards phosphorylated species allow PPTA to specifically isolate and enrich phosphopeptides from complex biological samples including protein digests and human serum. The homogeneous and porous TiO2 nanofilm, prepared on gold-covered glass substrates by an LbL deposition/calcination process, has a number of advantages. The simple and low-cost preparation method offers good control over properties of the nanofilms such as thickness, porosity, composition and functions. Benefiting from the photocatalytical property of the film, well defined and precise patterns can be directly and conveniently generated on the OTS-modified films, producing analysis-ready arrays without the use of polymer photoresists and sophisticated instrument. We tested the PPTA chip with hydrophilic TiO2 spots surrounded by hydrophobic OTS-modified coating, which allowed for the enrichment of low-abundance components from a large volume solution. Excellent reproducibility and reusability has been demonstrated with PPTA. We believe PPTA can open new avenues for integration of multiple analytical functions such as on-target digestion, selective capture and MS detection into a single chip, promoting the development of hyphenated techniques for high throughput analysis in proteome research.
Supplementary Material
ACKNOWLEDGMNET
This research was supported by the National Science Foundation (CHE-0719224) and the National Institute of Health (1R21EB009551-01A2).
References
- (1).Hunter T Cell 2000, 100, 113–127. [DOI] [PubMed] [Google Scholar]
- (2).Reinders J; Sickmann A Proteomics 2005, 5, 4052–4061. [DOI] [PubMed] [Google Scholar]
- (3).Witze ES; Old WM; Resing KA; Ahn NG Nature Methods 2007, 4, 798–806. [DOI] [PubMed] [Google Scholar]
- (4).Thingholm TE; Jensen ON; Larsen MR Proteomics 2009, 9, 1451–1468. [DOI] [PubMed] [Google Scholar]
- (5).Kaufmann H; Bailey JE; Fussenegger M Proteomics 2001, 1, 194–199. [DOI] [PubMed] [Google Scholar]
- (6).Posewitz MC; Tempst P Analytical Chemistry 1999, 71, 2883–2892. [DOI] [PubMed] [Google Scholar]
- (7).Stensballe A; Andersen S; Jensen ON Proteomics 2001, 1, 207–222. [DOI] [PubMed] [Google Scholar]
- (8).Feng S; Ye ML; Zhou HJ; Jiang XG; Jiang XN; Zou HF; Gong BL Molecular & Cellular Proteomics 2007, 6, 1656–1665. [DOI] [PubMed] [Google Scholar]
- (9).Zhou HJ; Ye ML; Dong J; Han GH; Jiang XN; Wu RN; Zou HF J. Proteome Res 2008, 7, 3957–3967. [DOI] [PubMed] [Google Scholar]
- (10).Zhou HJ; Xu SY; Ye ML; Feng S; Pan C; Jiang XG; Li X; Han GH; Fu Y; Zou HJ Proteome Res. 2006, 5, 2431–2437. [DOI] [PubMed] [Google Scholar]
- (11).Dunn JD; Reid GE; Bruening ML Mass Spectrom. Rev 2010, 29, 29–54. [DOI] [PubMed] [Google Scholar]
- (12).Sui SH; Wang JL; Yang B; Song L; Zhang JY; Chen M; Liu JF; Lu Z; Cai Y; Chen S; Bi W; Zhu YP; He FC; Qian XH Proteomics 2008, 8, 2024–2034. [DOI] [PubMed] [Google Scholar]
- (13).Dai J; Wang LS; Wu YB; Sheng QH; Wu JR; Shieh CH; Zeng RJ Proteome Res. 2009, 8, 133–141. [DOI] [PubMed] [Google Scholar]
- (14).Han GH; Ye ML; Zhou HJ; Jiang XN; Feng S; Jiang XG; Tian RJ; Wan DF; Zou HF; Gu JR Proteomics 2008, 8, 1346–1361. [DOI] [PubMed] [Google Scholar]
- (15).Dong MM; Wu MH; Wang FJ; Qin HQ; Han GH; Gong J; Wu RA; Ye ML; Liu Z; Zou HF Analytical Chemistry 2010, 82, 2907–2915. [DOI] [PubMed] [Google Scholar]
- (16).Maccarrone G; Kolb N; Teplytska L; Birg I; Zollinger R; Holsboer F; Turck CW Electrophoresis 2006, 27, 4585–4595. [DOI] [PubMed] [Google Scholar]
- (17).McNulty DE; Annan RS Molecular & Cellular Proteomics 2008, 7, 971–980. [DOI] [PubMed] [Google Scholar]
- (18).Leitner A Trac-Trends in Analytical Chemistry 2010, 29, 177–185. [Google Scholar]
- (19).Kweon HK; Hakansson K Analytical Chemistry 2006, 78, 1743–1749. [DOI] [PubMed] [Google Scholar]
- (20).Hsieh HC; Sheu C; Shi FK; Li DT J. Chromatogr. A 2007, 1165, 128–135. [DOI] [PubMed] [Google Scholar]
- (21).Chen CT; Chen YC Analytical Chemistry 2005, 77, 5912–5919. [DOI] [PubMed] [Google Scholar]
- (22).Qiao L; Roussel C; Wan JJ; Yang PY; Girault HH; Liu BH J. Proteome Res 2007, 6, 4763–4769. [DOI] [PubMed] [Google Scholar]
- (23).Bi HY; Qiao L; Busnel JM; Devaud V; Liu BH; Girault HH Analytical Chemistry 2009, 81, 1177–1183. [DOI] [PubMed] [Google Scholar]
- (24).Niklew ML; Hochkirch U; Melikyan A; Moritz T; Kurzawski S; Schluter H; Ebner I; Linscheid MW Analytical Chemistry 2010, 82, 1047–1053. [DOI] [PubMed] [Google Scholar]
- (25).Eriksson A; Bergquist J; Edwards K; Hagfeldt A; Malmstrom D; Hernandez VA Analytical Chemistry 2010, 82, 4577–4583. [DOI] [PubMed] [Google Scholar]
- (26).Torta F; Fusi M; Casari CS; Bottani CE; Bachi AJ Proteome Res. 2009, 8, 1932–1942. [DOI] [PubMed] [Google Scholar]
- (27).Wang H; Duan JC; Zhang LH; Liang Z; Zhang WB; Zhang YK J. Sep. Sci 2008, 31, 480–487. [DOI] [PubMed] [Google Scholar]
- (28).Chang CK; Wu CC; Wang YS; Chang HC Analytical Chemistry 2008, 80, 3791–3797. [DOI] [PubMed] [Google Scholar]
- (29).Phillips KS; Han JH; Martinez M; Wang ZZ; Carter D; Cheng Q Analytical Chemistry 2006, 78, 596–603. [DOI] [PubMed] [Google Scholar]
- (30).Duan JC; Linman MJ; Cheng Q Analytical Chemistry 2010, 82, 5088–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kjellstrom S; Jensen ON Analytical Chemistry 2004, 76, 5109–5117. [DOI] [PubMed] [Google Scholar]
- (32).Wang Y; Angelatos AS; Caruso F Chem. Mat 2008, 20, 848–858. [Google Scholar]
- (33).Linman MJ; Culver SP; Cheng Q Langmuir 2009, 25, 3075–3082. [DOI] [PubMed] [Google Scholar]
- (34).Roiter Y; Minko S Journal of the American Chemical Society 2005, 127, 15688–15689. [DOI] [PubMed] [Google Scholar]
- (35).Krost A; Christen J; Oleynik N; Dadgar A; Deiter S; Blasing J; Krtschil A; Forster D; Bertram F; Diez A Appl. Phys. Lett 2004, 85, 1496–1498. [Google Scholar]
- (36).Thiel PA; Shen M; Liu DJ; Evans JW Journal of Physical Chemistry C 2009, 113, 5047–5067. [Google Scholar]
- (37).Tu T; Gross ML Trac-Trends in Analytical Chemistry 2009, 28, 833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hoang T; Roth U; Kowalewski K; Belisle C; Steinert K; Karas M Analytical Chemistry 2010, 82, 219–228. [DOI] [PubMed] [Google Scholar]
- (39).Wang WH; Bruening ML Analyst 2009, 134, 512–518. [DOI] [PubMed] [Google Scholar]
- (40).Matsuda A; Sasaki T; Tadanaga K; Tatsumisago M; Minami T Chem. Mat 2002, 14, 2693–2700. [Google Scholar]
- (41).Lai YK; Lin CJ; Wang H; Huang HY; Zhuang HF; Sun L Electrochemistry Communications 2008, 10, 387–391. [Google Scholar]
- (42).Kim S; Choi R; Park ZY Molecules and Cells 2007, 23, 340–348. [PubMed] [Google Scholar]
- (43).Hu LH; Zhou HJ; Li YH; Sun ST; Guo LH; Ye ML; Tian XF; Gu JR; Yang SL; Zou HF Analytical Chemistry 2009, 81, 94–104. [DOI] [PubMed] [Google Scholar]
- (44).Lin HY; Chen WY; Chen YC Anal. Bioanal. Chem 2009, 394, 2129–2136. [DOI] [PubMed] [Google Scholar]
- (45).Li QR; Ning ZB; Tang JS; Nie S; Zeng RJ Proteome Res. 2009, 8, 5375–5381. [DOI] [PubMed] [Google Scholar]
- (46).Dunn JD; Igrisan EA; Palumbo AM; Reid GE; Bruening ML Analytical Chemistry 2008, 80, 5727–5735. [DOI] [PubMed] [Google Scholar]
- (47).Zhao LA; Wu RA; Han GH; Zhou HJ; Ren LB; Tian RJ; Zou HF Journal of the American Society for Mass Spectrometry 2008, 19, 1176–1186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.