

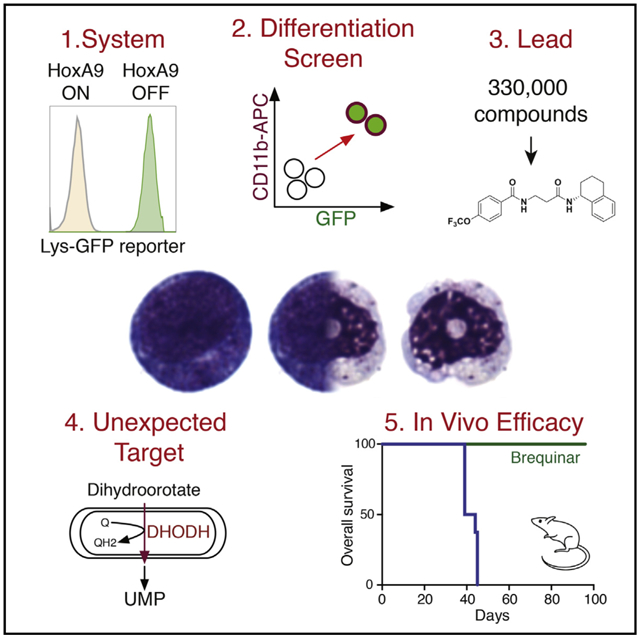
SUMMARY
While acute myeloid leukemia (AML) comprises many disparate genetic subtypes, one shared hallmark is the arrest of leukemic myeloblasts at an immature and self-renewing stage of development. Therapies that overcome differentiation arrest represent a powerful treatment strategy. We leveraged the observation that the majority of AML, despite their genetically heterogeneity, share in the expression of HoxA9, a gene normally downregulated during myeloid differentiation. Using a conditional HoxA9 model system, we performed a high-throughput phenotypic screen and defined compounds that overcame differentiation blockade. Target identification led to the unanticipated discovery that inhibition of the enzyme dihydroorotate dehydrogenase (DHODH) enables myeloid differentiation in human and mouse AML models. In vivo, DHODH inhibitors reduced leukemic cell burden, decreased levels of leukemia-initiating cells, and improved survival. These data demonstrate the role of DHODH as a metabolic regulator of differentiation and point to its inhibition as a strategy for overcoming differentiation blockade in AML.
Graphical Abstract
In Brief
Inhibition of a metabolic enzyme involved in pyrimidine biosynthesis induces differentiation of leukemic cells, identifying a potential therapeutic approach for treating a range of acute myeloid leukemias, independent of their oncogenic driver.
INTRODUCTION
Acute myeloid leukemia (AML) is a clinically devastating disease. Even with improvements in diagnosis and supportive care, the 5-year survival rate of an adult with AML is only 30%, with an even more dismal prognosis in patients over the age of 65. While these disappointing outcomes highlight the need for improved therapies, the chemotherapy backbone—a combination of cytarabine and an anthracycline—has remained unchanged for more than 40 years (Yates et al., 1973).
One hallmark of AML is that the leukemic blast is arrested at an early stage of differentiation. Prior to the development of karyotyping and genetic analysis, morphologic hallmarks of immaturity were used to classify a patient’s disease histologically. The recognition that leukemic blasts were frozen at an immature stage of development suggested that new therapies might be directed at promoting differentiation.
In the small subset (10%) of patients with acute promyelocytic leukemia (APL), recurrent chromosomal translocations result in fusion oncoproteins involving the retinoic acid receptor. Exploiting this dependency by treating patients with all-trans retinoic acid (ATRA) and arsenic trioxide releases the cells from differentiation arrest, allowing the leukemic blasts to resume their normal maturation to terminally differentiated neutrophils. The dramatic success and clinical impact of this differentiation therapy inverted the survival curve for patients with APL; where APL was once among the worst prognostic subsets of AML, it now has the best outlook for cure, with overall survival rates in excess of 85% (Lo-Coco et al., 2013). An unmet challenge is to identify similar differentiation therapy strategies for the remaining 90% of AML patients.
Efforts to identify new therapeutic targets to overcome myeloid differentiation blockade have been largely unsuccessful. Small-molecule inhibitors of mutant isocitrate dehydrogenase (IDH)2 (IDH2) (Wang et al., 2013) or IDH1 (Okoye-Okafor et al., 2015) may be capable of inducing cellular differentiation among that subset (15%) of patients with IDH1/2 mutations. However, the remainder of AML cases involve complex and heterogeneous combinations of chromosomal alterations and gene mutations (Cancer Genome Atlas Research Network, 2013), highlighting the difficulty in developing mutation-specific therapies.
Reasoning that diverse mutagenic events that affect differentiation funnel through common molecular pathways, we sought to define and target pathways of differentiation shared across a range of genetic subtypes of AML. We were intrigued by the observation that homeobox transcription factor HoxA9 expression is upregulated in 70% of patients with AML (Golub et al., 1999), likely reflecting that the leukemic blasts are halted at a common stage of differentiation arrest. HoxA9 is critical to normal myelopoiesis, and its expression must be downregulated to permit normal differentiation (Sauvageau et al., 1994). Furthermore, HoxA9 is essential to the maintenance of leukemias driven by mixed-lineage leukemia (MLL) translocations such as MLL/AF9 (reviewed in Collins and Hess, 2016), HoxA9 is upregulated during the transition in chronic myeloid leukemia patients to blast-phase disease (Tedeschi and Zalazar, 2006), and HoxA9 expression itself is an independent risk factor in children with leukemia (Adamaki et al., 2015). Therefore, we reasoned that the persistent expression of HoxA9 might represent a commonly dysregulated node suitable for therapeutic targeting across a range of disparate AML subtypes.
We developed a cellular model of HoxA9-enforced myeloid differentiation arrest to use in an unbiased phenotypic screen. As persistent expression of HoxA9 results in myeloid differentiation arrest (Kroon et al., 1998), we used an estrogen receptor-HoxA9 (ER-HoxA9) fusion protein to conditionally immortalize cultures of primary murine bone marrow. ER-HoxA9 cells were generated from the bone marrow of a mouse with GFP knocked into the lysozyme locus. Lysozyme is a myeloid granule protein expressed only in differentiated cells (Faust et al., 2000), permitting phenotypic screening of small molecules for those capable of triggering differentiation (indicated by GFP expression) in the presence of active HoxA9.
We identified dihydroorotate dehydrogenase (DHODH) as the target of our most active compounds. DHODH is the enzyme responsible for the fourth step of de novo pyrimidine biosynthesis (Löffler et al., 1997), and its modulation has not previously been shown to induce AML differentiation. We demonstrate that DHODH inhibitors exert potent differentiation activity in vitro and in vivo in both murine and human models of AML. The anti-leukemic activity of DHODH inhibitors points toward a novel link between uridine biosynthesis and cell-fate decisions and may offer a much-needed new therapeutic option for treatment of patients with AML.
RESULTS
An ER-HoxA9 Fusion Establishes Conditional Myeloid Differentiation Arrest
High-throughput myeloid differentiation assays are challenging; specific measures of differentiation are cumbersome and rely on morphologic or enzymatic assays or on changes in gene expression. Furthermore, myeloid differentiation is typically assessed in AML cell lines where the mechanism of differentiation arrest is not clearly defined.
We used an estrogen receptor (ER)-HoxA9 (ER-HoxA9) fusion protein to conditionally immortalize cultures of primary murine bone marrow. The persistent expression of HoxA9 is sufficient to enforce myeloid differentiation arrest; fusion of the hormone-binding domain of the human ER to the N terminus of HoxA9 results in a protein that is constitutively translated but inactive in the absence of beta-estradiol (E2). Upon binding beta-estradiol, the ER-HoxA9 translocates to the nucleus, where it retains its wild-type activity. We used the G400V variant of the human ER, which is insensitive to physiologic concentrations of estrogen or to the trace estrogens that are found in fetal bovine serum (FBS; Figure S1A) (Tora et al., 1989).
Primary murine bone marrow cells transduced with ER-HoxA9 proliferate as stem-cell-factor-dependent myeloblast cell lines, with the cell-surface receptor profile consistent with granulocyte-macrophage progenitors (GMPs) (Figure S1B). In the presence of E2 and active ER-HoxA9 protein, these cells proliferate indefinitely as immature myeloblasts, while upon withdrawal of E2, the cells undergo synchronous and terminal neutrophil differentiation over 5 days, demonstrating the expected changes in cell-surface CD11b and Gr-1 expression (Figure 1A). This normal differentiation was confirmed by assaying changes in cell cycle (Figure 1B) and morphology (Figure 1C) and by functional assays of neutrophil function, including phagocytosis (Figure 1D) and superoxide production (Figure S1C).
Figure 1. ER-HoxA9 Cells Undergo Conditional Myeloid Differentiation.
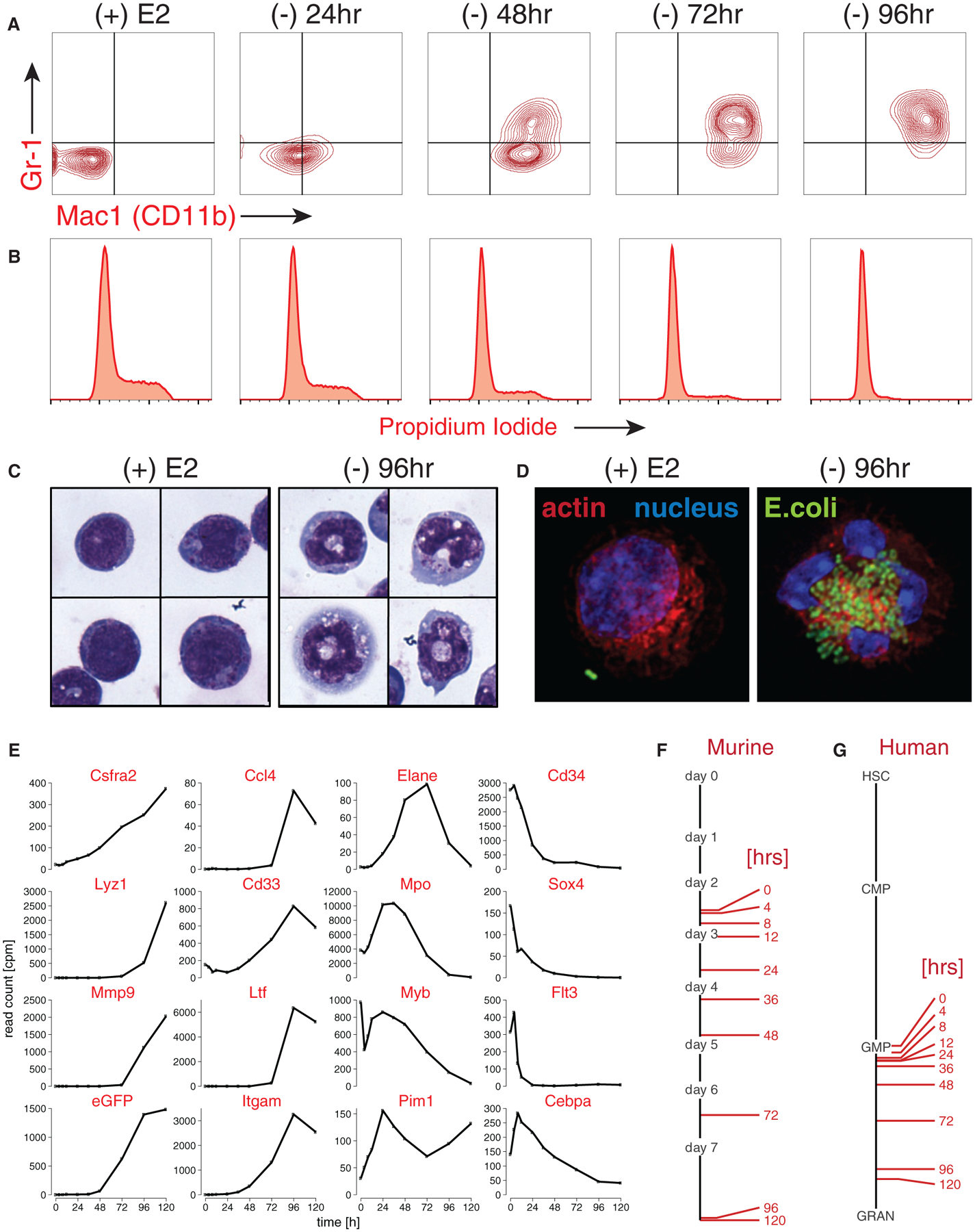
(A) Primary murine bone marrow cells transduced with MSCVneo-ER-HoxA9 grow as lineage-negative cells in the presence of beta-estradiol, (+) E2. Removal of E2 and inactivation of ER-HoxA9 result in the synchronous upregulation of the myeloid differentiation markers CD11b and Gr-1, as demonstrated by flow cytometry.
(B) Terminal differentiation of the ER-HoxA9 cells is accompanied by exit from the cell cycle.
(C) Morphologic changes that accompany myeloid differentiation are confirmed by Wright-Giemsa staining of cells in the presence and absence of E2.
(D) Terminally differentiated cells, but not undifferentiated cells, are capable of phagocytosis of fluorescently labeled E. coli.
(E-G) In (E), Lys-GFP-ER-HoxA9 cells demonstrate expected changes in myeloid gene expression over a 5-day differentiation time course. Their stepwise gene expression parallels the patterns of unmanipulated murine bone marrow myeloid cells (F) as well as purified populations of primary human bone marrow cells (G). HSC, hematopoietic stem cell; CMP, common myeloid progenitor; GMP, granulocyte-monocyte progenitor; GRAN, granulocytes.
See also Figure S1.
A Phenotypic Screen of AML Differentiation Using the Lysozyme-GFP-ER-HoxA9 Model
To facilitate a small-molecule differentiation screen, an ERHoxA9 GMP cell line was derived from the bone marrow of the lysozyme-GFP knockin mouse, in which the expression of GFP is limited to mature myeloid cells (Faust et al., 2000).
Time-course gene-expression analysis by RNA sequencing (RNA-seq) was performed on undifferentiated cells as well as at nine time points following the removal of E2. The cells demonstrated the expected expression changes of key transcription factors (Myb and Sox4), primary and secondary granule genes (Elane, Mpo, and Ltf), and HoxA9 target genes (Cd34 and Flt3) (Figure 1E). Gene expression was compared to that of unmanipulated cultures of primary murine myeloblasts allowed to differentiate in vitro over 7 days (Figures 1F, S1D, and S1E) (Yuan et al., 2007), as well as to freshly sorted subsets of human myeloid cells (Figure 1G) (Novershtern et al., 2011). The changes in gene expression during the differentiation of ER-HoxA9 cells are remarkably similar to those of unmanipulated murine and human primary myeloblasts.
In the Lys-GFP-ER-HoxA9 cell line, GFP expression accompanied the normal process of myeloid differentiation (Figures 1E and 2A) and paralleled the expression of the myeloid markers CD11b and Gr-1 (Figure 2B). Imaging flow cytometry demonstrated morphologic changes associated with differentiation accompanied by increased CD11b staining, decreased CD117 (CKIT) staining, and increased GFP expression (Figure S2A). The Lys-GFP-ER-HoxA9 cells had a doubling time of ~12 hr and underwent four to five doublings prior to terminal differentiation when cultured out of E2 (Figure S2B).
Figure 2. High-Throughput Screening and Medicinal Chemistry Identifies ML390 as an Inducer of Myeloid Differentiation.
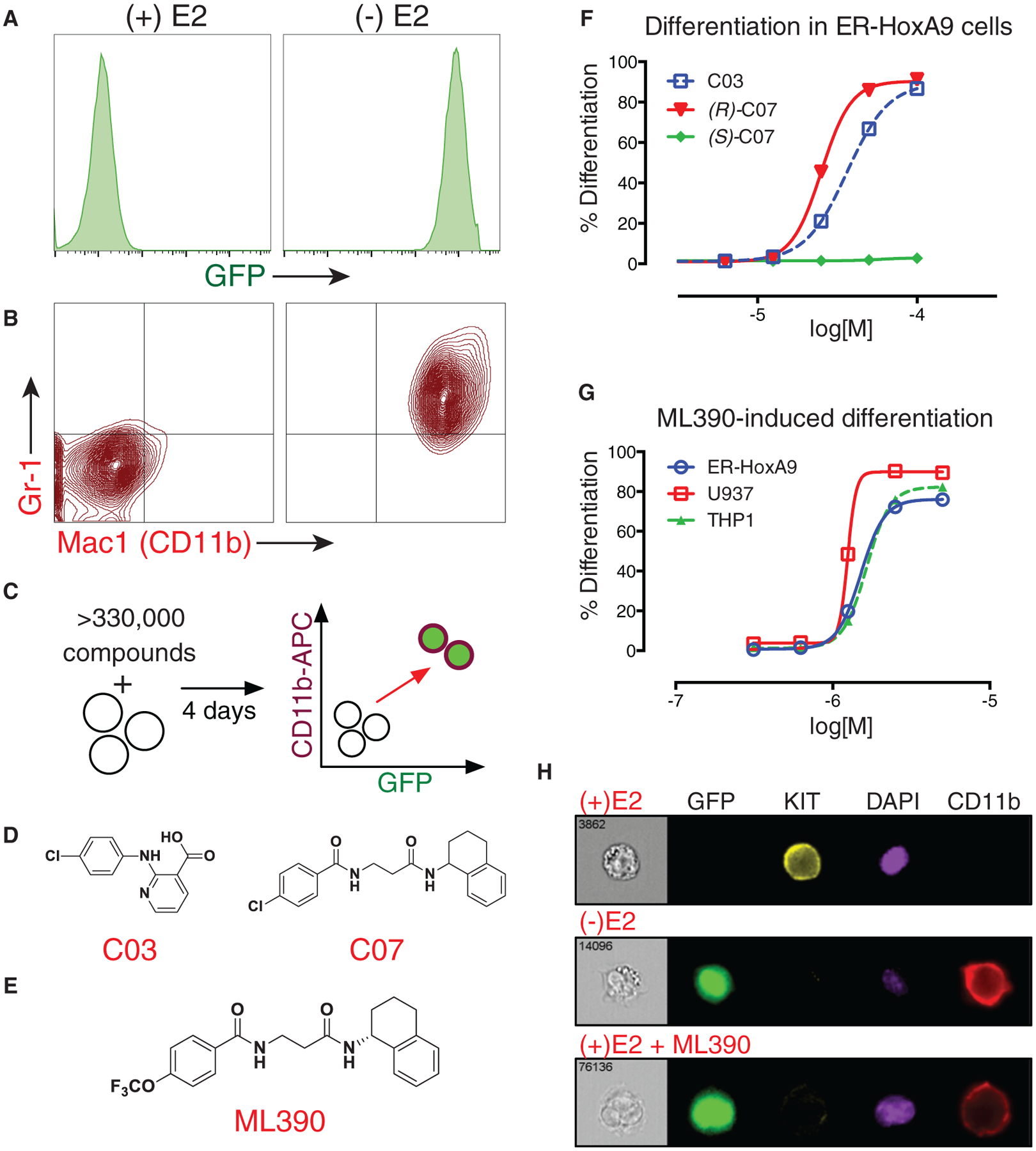
(A and B) In (A), upon differentiation (following the removal of estradiol), Lys-GFP-ER-HoxA9 GMPs upregulate GFP fluorescence and (B) the cell-surface markers CD11b and Gr-1.
(C) A high-throughput flow cytometry phenotypic screen was established to identify compounds that could trigger differentiation of Lys-GFP-ER-HoxA9 cells as monitored by the upregulation of GFP and CD11b.
(D) Twelve biologically active compounds were identified, including compound 3 (C03) and compound 7 (C07).
(E) The most potent small molecule was a derivative of (R)-C07, designated ML390.
(F) C03 and (R)-C07 triggered myeloid differentiation, while (S)-C07 lacked activity.
(G) ML390 is capable of causing myeloid differentiation in murine (ER-HoxA9) and human (U937 and THP1) AML models.
(H) Imaging flow cytometry demonstrates upregulation of GFP and CD11b expression as well as the downregulation of KIT expression in Lys-GFP-ER-HoxA9 cells during differentiation in the absence of estradiol (-E2) or as the result of treatment with ML390.
See also Figure S2.
A High-Throughput Screen Identifies 12 Small-Molecule Mediators of Myeloid Differentiation
Using the Lys-GFP-ER-HoxA9 GMPs, we performed a small-molecule phenotypic screen to identify compounds that could trigger myeloid differentiation in the presence of active HoxA9. After 4 days of compound treatment, cells were assessed by high-throughput flow cytometry (Haynes et al., 2009). Viability was evaluated by forward and side-scatter, and differentiation by GFP expression and cell-surface CD11b (allophycocyanin [APC] fluorescence; Figure 2C). We assessed the differentiation potential of the 330,000 small molecules within the NIH Molecular Library Program’s Molecular Library Small-Molecule Repository (MLSMR) (Schreiber et al., 2015). Active compounds were validated in concentration-response experiments to eliminate toxic compounds, autofluorescent compounds, and estrogen antagonists. Twelve compounds demonstrated reproducible myeloid differentiation in multiple clones of ER-HoxA9 and wild-type HoxA9 murine GMP cell lines.
ML390 Is a Potent Derivative of C07
Two compounds with distinct chemical scaffolds (designated C03 and C07; Figure 2D) were chosen for compound optimization based on their cross-species activity in both murine and human AML models (U937 and THP1). Compound C03 is struc turally similar to non-steroidal anti-inflammatory compounds (NSAIDs) and to known inhibitors of aldo-keto reductase 3 (AKR3). However, treatment of the Lys-GFP-ER-HoxA9 cells with confirmed NSAIDs or AKR3 inhibitors did not result in myeloid differentiation, suggesting that this was not the mechanism of action.
Testing derivatives of C07 demonstrated that the biological activity was specific to that of the (R)-enantiomer (Figure 2F). The (R)-C07 halide group was varied, resulting in the lead compound, designated ML390, having increased potency (Figure 2E) (Sykes et al., 2015). ML390 was active with an ED50 (effective concentration triggering 50% of its maximal differentiation activity) of ~2 μM in murine and human AML cell lines (Figure 2G). The differentiation triggered by ML390 was similar to the normal differentiation accompanying ER-HoxA9 inactivation (Figure 2H).
Analysis of Resistant Cell Lines Identifies DHODH as the Target of ML390
Given that the library was largely un-annotated, the protein targets of the small molecules were unknown. We succeeded in target identification by generating compound-resistant cell lines, an approach previously successful in our laboratory (Chattopadhyay et al., 2015). To generate resistance, the murine Lys-GFP-ER-HoxA9 and human U937 leukemia cells were cultured in slowly escalating (5 μM to 50 μM) concentrations of DMSO, C03, or (R)-C07.
While the compound-treated cells initially grew slowly and were more differentiated, resistant cells emerged after 6 months that were undifferentiated and proliferated at the same rate as control cells cultured in DMSO (Figure 3A). Resistance developed along a similar time frame in both the (R)-C07 and C03-treated cultures and in both murine and human cell lines. Despite their seemingly unrelated chemical structures, cross-resistance to (R)-C07 and to C03 was observed, suggesting a similar resistance mechanism. Cells retained their resistance for more than 6 weeks after discontinuing treatment, suggesting a stable genetic alteration as the mechanism of resistance.
Figure 3. Resistant Cell Lines Identify DHODH as the Target of ML390.
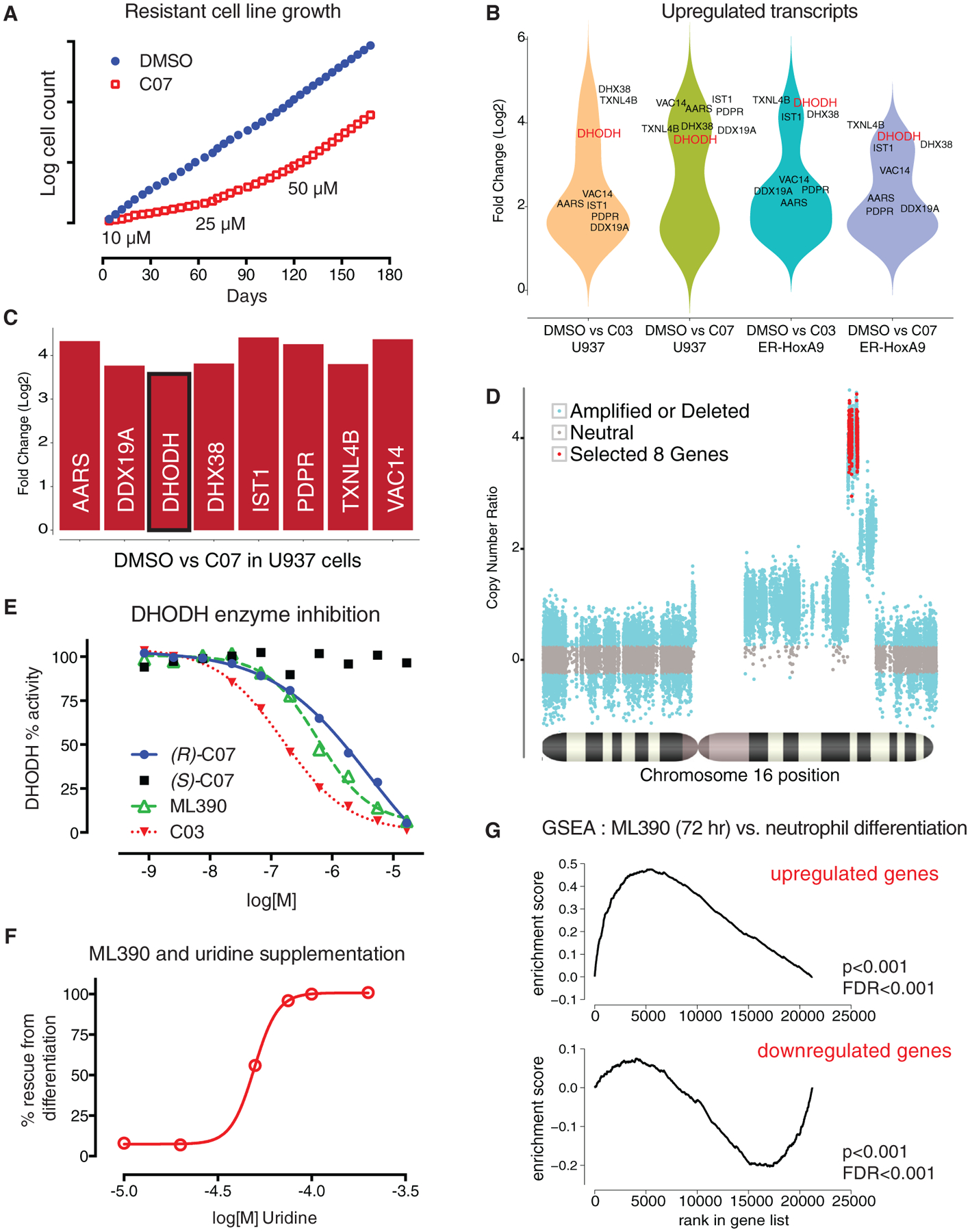
(A) ER-HoxA9 and U937 cell lines resistant to C03 and (R)-C07 were generated by continuous culture in slowly increasing concentration of compound.
(B) A comparison of gene expression between DMSO control and compound-resistant cells demonstrated that only eight overexpressed genes (more than 2-fold upregulated, p < 0.01) were shared across the four resistant cell lines.
(C) The eight genes were gene syntenic gene neighbors on chromosome 16 (human) and chromosome 8 (mouse).
(D) Analysis of the whole-exome sequencing data revealed an increased coverage over a narrow region of chromosome 16, consistent with chromosomal amplification as the mechanism underlying increased gene expression.
(E and F) In (E), DHODH was confirmed as the target of C03, C07, and ML390 using an in vitro enzyme inhibition assay, as well as (F) by demonstrating that the differentiating effects of C03 and ML390 could be abrogated by supplementation of uridine in the cell-culture media.
(G) Treatment of the Lys-GFP-ER-HoxA9 cells with ML390 demonstrated gene-expression changes consistent with myeloid differentiation by gene set enrichment analysis.
See also Figure S3.
We noted strikingly similar gene expression changes in cells resistant to C03 and (R)-C07 across species (Figure S3A). RNA-seq analysis revealed that only eight shared genes were upregulated (>2-fold) in both C03-resistant and (R)-C07-resistant populations, and in both Lys-GFP-ER-HoxA9 and U937 cells (Figures 3B and 3C). Interestingly, these eight transcripts were gene neighbors within a 100-kb region of the long-arm of chromosome 16 (human) or chromosome 8 (mouse). Analysis of whole-exome sequencing (WES) data confirmed chromosomal amplification as the mechanism of resistance (Figure 3D).
One of the amplified genes encoded DHODH, a critical enzyme in the intracellular de novo synthesis of pyrimidines. DHODH is highly conserved between human and mouse, consistent with the ability of C03 and (R)-C07 to trigger differentiation in both murine and human AML models. We confirmed that C03 and (R)-C07 were inhibitors of DHODH using recombinant human DHODH protein in an in vitro enzyme inhibition assay. The enzyme inhibitory activity (half maximal inhibitory concentration; IC50) paralleled the biological differentiation effect (ED50) (Figure 3E). Likewise, known inhibitors of DHODH, including leflunomide, its active metabolite teriflunomide, and brequinar sodium (BRQ), were active in enzyme-inhibition and cellular-differentiation assays (Figures S3B and S3C).
While cells depend on DHODH for intracellular uridine synthesis, they can also salvage extracellular uridine through nucleo-side transporters. Increasing concentrations of uridine abrogated the differentiation effect of C03 and (R)-C07 in the Lys-GFP-ERHoxA9 cells (Figure 3F). This ‘‘uridine rescue’’ demonstrated that myeloid differentiation was completely due to interference with uridine monophosphate (UMP) synthesis (Figure 5A) and does not involve additional mechanisms unrelated to DHODH inhibition.
Figure 5. Inhibition of DHODH Leads to an Accumulation of Upstream Metabolites and to a Depletion of Downstream Metabolites.
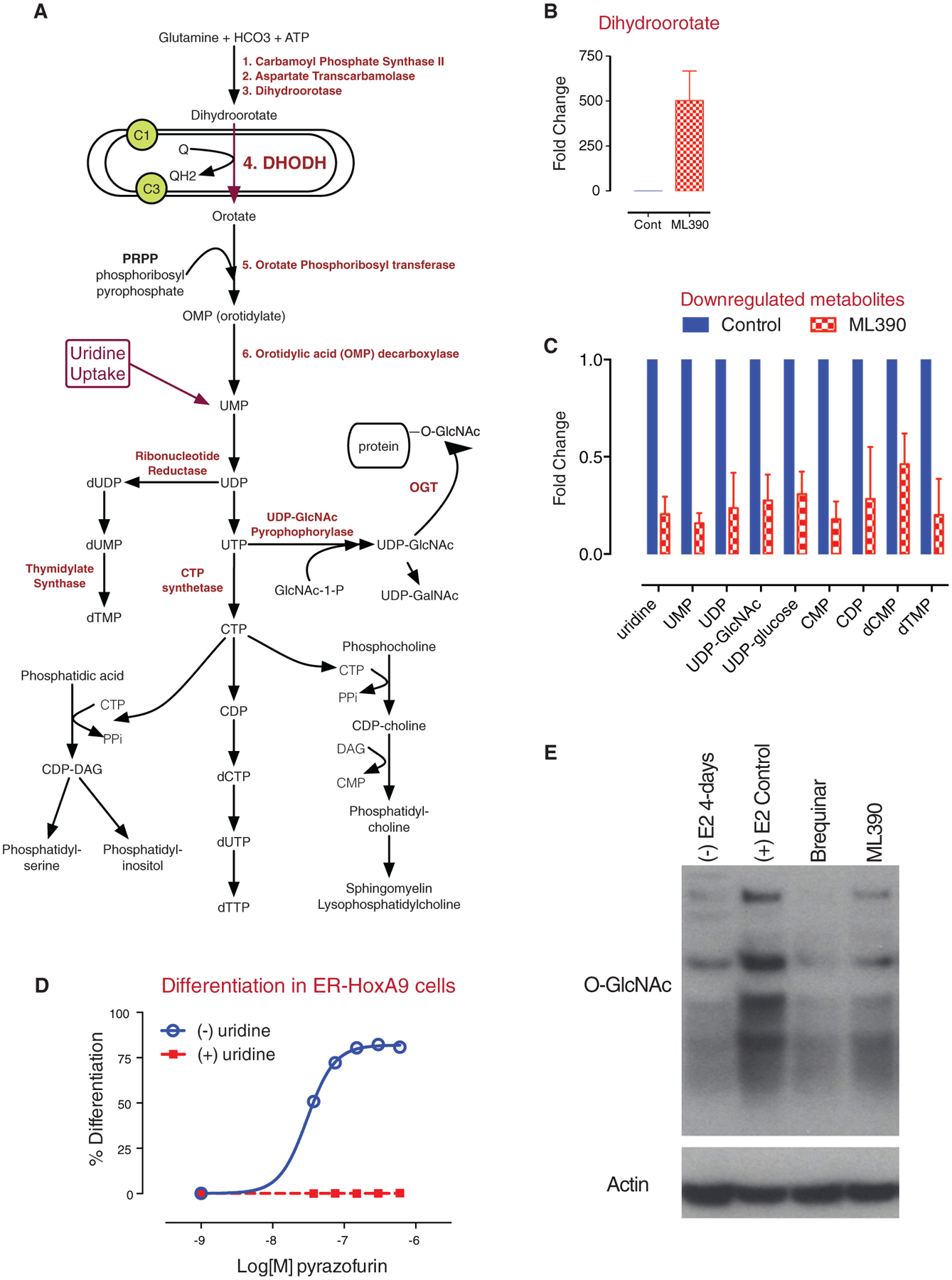
(A) A model of the enzymatic steps involved in de novo pyrimidine synthesis as well as lipid biogenesis. DHODH is located in the mitochondrial inner membrane and passes electrons to ubiquinone.
(B and C) In (B), treatment of ER-HoxA9 cells with ML390 results in an accumulation of dihydroorotate and (C) to a depletion of downstream metabolites including UMP, uridine, UDP, UDP-GlcNAc, and UDP-glucose.
(D) Treatment of ER-HoxA9 cells with pyrazofurin, an inhibitor of OMP decarboxylase, results in differentiation that can be rescued by uridine supplementation.
(E) Treatment of cells with ML390 or BRQ, followed by immunoblotting, demonstrates a global decrease in the degree of protein N-acetyl glycosylation (GlcNAc). Data in (B) and (C) are represented as the mean ± SD.
DHODH Inhibition Triggers Gene Expression Consistent with Myeloid Differentiation
The gene-expression changes in Lys-GFP-ER-HoxA9 cells treated with ML390 for 12, 36, and 72 hr resembled primary neutrophil differentiation (Figure 3G). However, these patterns were less pronounced compared to those following inactivation of ER-HoxA9 (Figures 1F, S1D, and S3F). Gene-expression changes following treatment with ML390 that were not observed during normal differentiation (Figure S3E) were likely related to decreased pyrimidine availability and a global suppression of RNA and DNA synthesis.
BRQ, a Potent and Selective Inhibitor of DHODH, Is Suitable for In Vivo Studies
The low solubility and bioavailability of ML390 limited its potential as an in vivo tool compound. BRQ is an inhibitor of DHODH, originally developed by DuPont Pharmaceuticals (DUP 785; NSC 368390). BRQ inhibits DHODH activity in vitro with an IC50 of ~20 nM (Figure S3B) and triggers differentiation in the ER-HoxA9, U937, and THP1 cells with an ED50 of ~1 μM (Figure S3D). The potency of BRQ depends on extracellular concentrations of uridine; cells cultured in 50% FBS (to better approximate the extracellular plasma concentrations of uridine in vivo) showed an ~2-fold increase in their ED50 (Figure S3G).
BRQ has a half-life of ~12 hr in vivo (Figure S4A) and is highly protein bound (98%–99%), consistent with published literature (Cramer, 1995). To assess the possibility that BRQ inhibited enzymes other than DHODH, we profiled BRQ against a panel of >400 kinases (DiscoverX KinomeScan). BRQ showed a near-complete absence of kinase inhibitory activity at 100 nM and 1 μM concentrations (Figure S4B).
The maximum tolerated dose (MTD) of BRQ was evaluated in C57Bl/6 mice. When administered daily, BRQ was tolerated at doses up to 5 mg/kg. Mice receiving higher doses exhibited weight loss, anemia, and thrombocytopenia (Figure S5A). This toxicity was reversible, and mice recovered fully after discontinuation of treatment. Measurements of plasma BRQ concentration after a single intraperitoneal (i.p.) dose of 15 mg/kg or 25 mg/kg suggested that an intermittent dosing schedule could maintain concentrations above the in vitro cellular ED50 of ~1 μM (Figures S3D and S4A). This intermittent schedule was well tolerated, and mice given 25 mg/kg or 50 mg/kg every 3 days (Q3D) for 24 doses (72 days) showed normal weight gain and only a mild anemia without leukopenia or thrombocytopenia (Figures S5C–S5G).
DHODH Inhibition Demonstrates Anti-leukemia Activity and Differentiation In Vivo in Xenograft Models of AML
THP1 cells were implanted subcutaneously into the flank of NOD.SCID mice (NOD.CB17-Prkdcscid) and allowed 10 days to engraft (tumor size of ~40 mm2). Treatment with BRQ slowed tumor growth at both 15 mg/kg Q3D and 5 mg/kg daily dosages (Figure 4A). THP1 tumors were explanted for differentiation analysis; THP1 cells from mice treated with BRQ exhibited marked differentiation, as evidenced by increased CD11b expression (depicted in Figure 4B and by mean fluorescence intensities in Figure 4C).
Figure 4. BRQ Causes Differentiation and Shows Anti-tumor Activity in In Vivo Xenotransplant Models of AML.
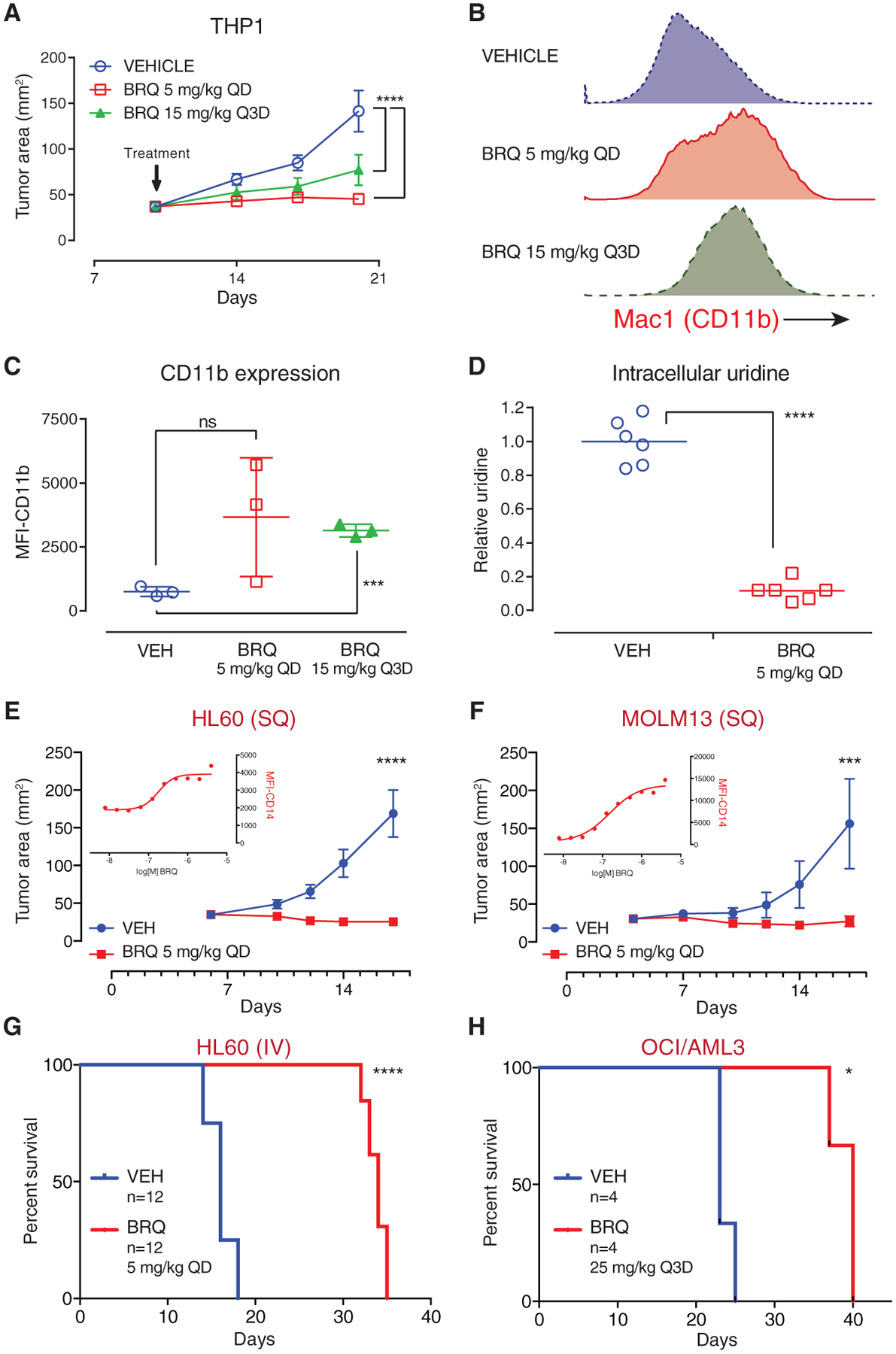
(A) THP1 cells were implanted subcutaneously in the flank of SCID mice; treatment with brequinar (BRQ) decreased tumor growth. QD, once every day.
(B) Explanted THP1 tumors analyzed by flow cytometry demonstrated that BRQ causes the upregulation of the myeloid differentiation marker CD11b.
(C) The geometric mean fluorescence intensity of CD11b-APC expression was compared for the explanted tumors from three mice per group.
(D) Metabolites from explanted tumors were extracted into methanol, and levels of intracellular uridine were measured by mass spectroscopy.
(E) HL60 cells were implanted subcutaneously in the flank of SCID mice, and the mice were treated with vehicle or BRQ. BRQ causes differentiation of HL60 cells in culture, as demonstrated by the upregulation of CD14 (inset graph).
(F) MOLM13 cells were implanted subcutaneously in the flank of SCID mice, and the mice were treated with vehicle or BRQ. BRQ causes differentiation of MOLM13 cells in culture, as demonstrated by the upregulation of CD14 (inset graph).
(G) BRQ prolongs survival in an intravenous HL60 leukemia model.
(H) BRQ prolongs survival in an intravenous OCI/AML3 leukemia model.
Data in (A), (C), (E), and (F) are represented as the mean ± SD. *p % 0.05; ***p % 0.001; ****p %0.0001; ns, not significant.
See also Figure S4.
In addition, BRQ arrested tumor growth in HL60 (Figure 4E) and MOLM13 (Figure 4F) subcutaneous xenograft models of AML in NOD.SCID mice. The inset graphs demonstrate the differentiation (upregulation of CD14) of HL60 and MOLM13 cells following treatment with BRQ in culture. Treatment with BRQ prolonged survival in disseminated (intravenous) HL60 (Figure 4G) and OCI/AML3 (Figure 4H) models of AML.
DHODH Inhibition Depletes Uridine and UDP Metabolites In Vitro and In Vivo
DHODH catalyzes the conversion of dihydroorotate (DHO) to orotate in the endogenous synthesis of UMP (Figure 5A). In vitro, treatment of Lys-GFP-ER-HoxA9 cells with ML390 for 48 hr inhibited DHODH activity, leading to the dramatic (>500-fold) accumulation of the upstream metabolite DHO (Figure 5B) and the depletion of uridine and other downstream metabolites (Figure 5C).
While DHODH catalyzes the fourth step of uridine biosynthesis, the enzyme OMP decarboxylase (OMPD) catalyzes the sixth step (Figure 5A). Pyrazofurin is a potent small-molecule inhibitor of OMPD (Dix et al., 1979), and treatment of the Lys-GFPER-HoxA9 cells with pyrazofurin phenocopied the differentiation effect of DHODH inhibition (Figure 5D).
Uridine supplementation abrogated the differentiation effect of ML390 and pyrazofurin (Figures 3G and 5D). Uridine supplementation also reversed the depletion of downstream metabolites but did not reverse the accumulation of DHO. Together, these findings demonstrate that inhibition of UMP synthesis at two points along the pathway, but not the accumulation of DHO, leads to myeloid differentiation. Thus, DHO is a marker of enzyme inhibition, not an oncometabolite like 2-hydroxyglutarate (2HG) in patients with IDH mutant leukemias (Ward et al., 2010).
To confirm DHODH inhibition in vivo, we performed cellular metabolite analysis of subcutaneous THP1 cells and HoxA9-Meis1 bone marrow leukemia cells. THP1 cells isolated from BRQ-treated mice showed a significant reduction in cellular uridine levels compared to vehicle-treated controls (Figure 4D). HoxA9-Meis1 leukemia cells isolated from the bone marrow of BRQ-treated mice had similar decreases in cellular uridine and in uridine diphosphate (UDP) and UDP-glycoconjugates (e.g., UDP-GlcNAc [N-acetyl-D-glucosamine], UDP-glucose).
DHODH and OMPD inhibitors lead to depletion of uridine and to myeloid differentiation. However, downstream inhibitors of DNA and RNA biosynthesis (e.g., methotrexate and hydroxyurea) or DNA-damaging agents (e.g., cytarabine and daunorubicin) caused cytotoxicity without differentiation. We hypothesized that part of the differentiation effect may be due to the depletion of UDP-GlcNAc, leading to decreased O-linked N-acetylglycosylation (GlcNAc) post-translational modification of proteins. DHODH inhibition with ML390 or BRQ led to a global reduction of protein O-GlcNAc modification (Figure 5E).
DHODH Inhibitors Cause Differentiation and a Loss of Leukemia-Initiating Cell Activity ina HoxA9 Model of AML
We addressed the efficacy of BRQ in a retroviral transduction model of HoxA9 + Meis1 AML. Bone marrow leukemia cells were transduced with lentivirus expressing the Venus fluorescent protein, expanded in culture, and used in subsequent experiments. Leukemic cells were transplanted intravenously into recipient mice, and BRQ or vehicle treatment was initiated after 14 days (Figure 6A). Mice were treated with BRQ on an every 2-day schedule (Figure S6) or a twice-weekly schedule (days 1 + 4). The twice-weekly schedule was better tolerated in terms of weight loss and hematologic parameters (Figures S5A and S5B).
Figure 6. BRQ Causes Differentiation, Shows Anti-leukemia Activity, and Leads to Depletion of Leukemia Initiating Cell Activity in an In Vivo Syngeneic Model of HoxA9 AML.
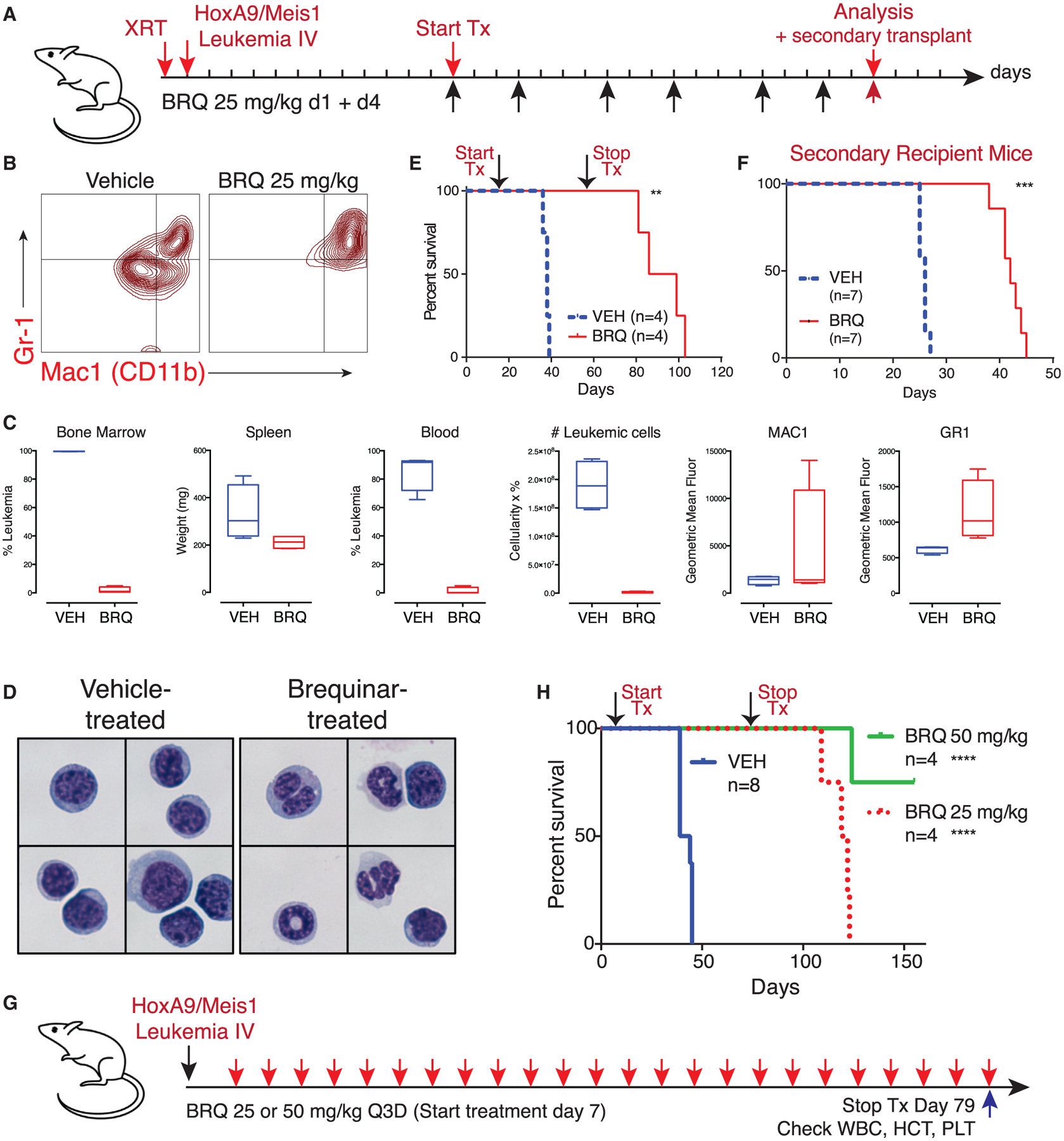
(A) Experimental outline of a syngeneic model of HoxA9 and Meis1-driven acute myeloid leukemia.
(B) Flow-cytometric analysis of bone marrow leukemic cells from mice treated with BRQ demonstrates an increase in the expression of differentiation markers CD11b and Gr-1.
(C) Mice treated with 25 mg/kg BRQ given on days 1 and 4 of a 7-day schedule, for a total of six doses, show a decrease in leukemia burden and increase in differentiation markers. Data are box and whisker plots where the mean, the minimum, and the maximum are indicated.
(D) Leukemia cells from mice that were treated with vehicle or BRQ were purified via FACS. Cytospin preparations stained with Wright-Giemsa showed signs of granulocytic maturation, including nuclear condensation and cytoplasmic clearing, in leukemic cells isolated from BRQ-treated mice.
(E) Treatment with 12 doses of BRQ prolongs overall survival.
(F) The same number of purified live leukemia cells from mice treated with vehicle or BRQ was introduced into recipient mice as a functional assay for leukemia-initiating cell activity. These secondary recipient mice were not treated. BRQ treatment leads to a decrease in the frequency of leukemia-initiating cells.
(G) HoxA9+Meis1 leukemia was introduced into mice without pre-conditioning, and the mice were treated with BRQ given every 3 days.(H) The extended treatment of mice with BRQ leads to prolonged survival. **p % 0.01; ***p % 0.001; ****p % 0.0001; ns, not significant.
See also Figure S5.
Treatment with BRQ led to differentiation in vivo, as evidenced by the upregulation of differentiation markers CD11b (Mac-1) and Gr-1 (Ly6C/G) (Figure 6B). Mice treated with BRQ showed a dramatic decrease in the leukemic involvement of their bone marrow, spleen, and peripheral blood (Figures 6C and S6B). FACS (fluorescence-activated cell sorting)-sorted leukemia cells demonstrated evidence of morphologic granulocytic differentiation in the BRQ-treated mice (Figure 6D). Treatment with BRQ for 6 weeks led to a prolongation of survival, though the mice eventually relapsed approximately 4 weeks following discontinuation of treatment (Figures 6E and S6C).
Next, we addressed the efficacy and tolerability of extended treatment with BRQ (Figure 6G). Leukemic cells were introduced without radiation pre-conditioning, and the mice were treated with BRQ every 3 days for 24 doses (72 days) of therapy. The mice exhibited normal activity, weight gain, and only a mild anemia (Figures S5C–S5G). This Q3D regimen of BRQ resulted in dramatically prolonged survival compared with vehicle-treated mice (Figure 6H). Of note, the survival of the remaining mice continued well beyond discontinuation of BRQ therapy, a durability of response not previously seen with other interventions in this aggressive leukemia model.
DHODH Inhibitors Cause Differentiation and Depletionof Leukemia-Initiating Cell Activity In Vivo
Leukemic cells isolated from the BRQ-treated mice were more differentiated by CD11b and Gr-1 cell-surface expression (Figure 6B) and morphology (Figure 6D). To assess their potential as leukemia-initiating cells, live Venus-positive leukemic cells were freshly isolated from vehicle or BRQ-treated mice by FACS. The same number of leukemia cells, isolated from vehicle-or BRQ-treated mice, was re-introduced into new recipient mice. Cells from mice that received BRQ 25 mg/kg every other day, for four doses (Figure S6D), and from mice that received BRQ on days 1 + 4 of a 7-day cycle, for a total of six doses (Figure 6F), were used. In both cases, mice that received the same number of live leukemic cells from BRQ-treated primary donors took longer to develop symptoms of leukemia, consistent with a decrease in leukemia-initiating cell potential.
DHODH Inhibitors Cause Differentiation and a Lossof Leukemia-Initiating Cell Activity in an MLL/AF9 Model of AML
Next, we tested BRQ in the context of an aggressive syngeneic model of MLL/AF9 leukemia (Figure 7A). Here, we compared the effects of BRQ to cytarabine and doxorubicin induction chemotherapy (iCT), as previously described (Zuber et al., 2009). Treatment of the mice with four doses of BRQ led to reduced leukemia burden (Figure 7B) and differentiation, as evidenced by CD11b (Figure 7C), GR1 (Figure 7D), CKIT expression (Figure 7E), and morphology (Figure 7H). Continued treatment of these animals prolonged survival beyond the benefit gained from iCT (Figure 7I).
Figure 7. BRQ Causes Differentiation, Shows Anti-leukemia Activity, and Leads to Depletion of Leukemia-Initiating Cell Activity in an In Vivo Syngeneic Model of MLL/AF9 AML.
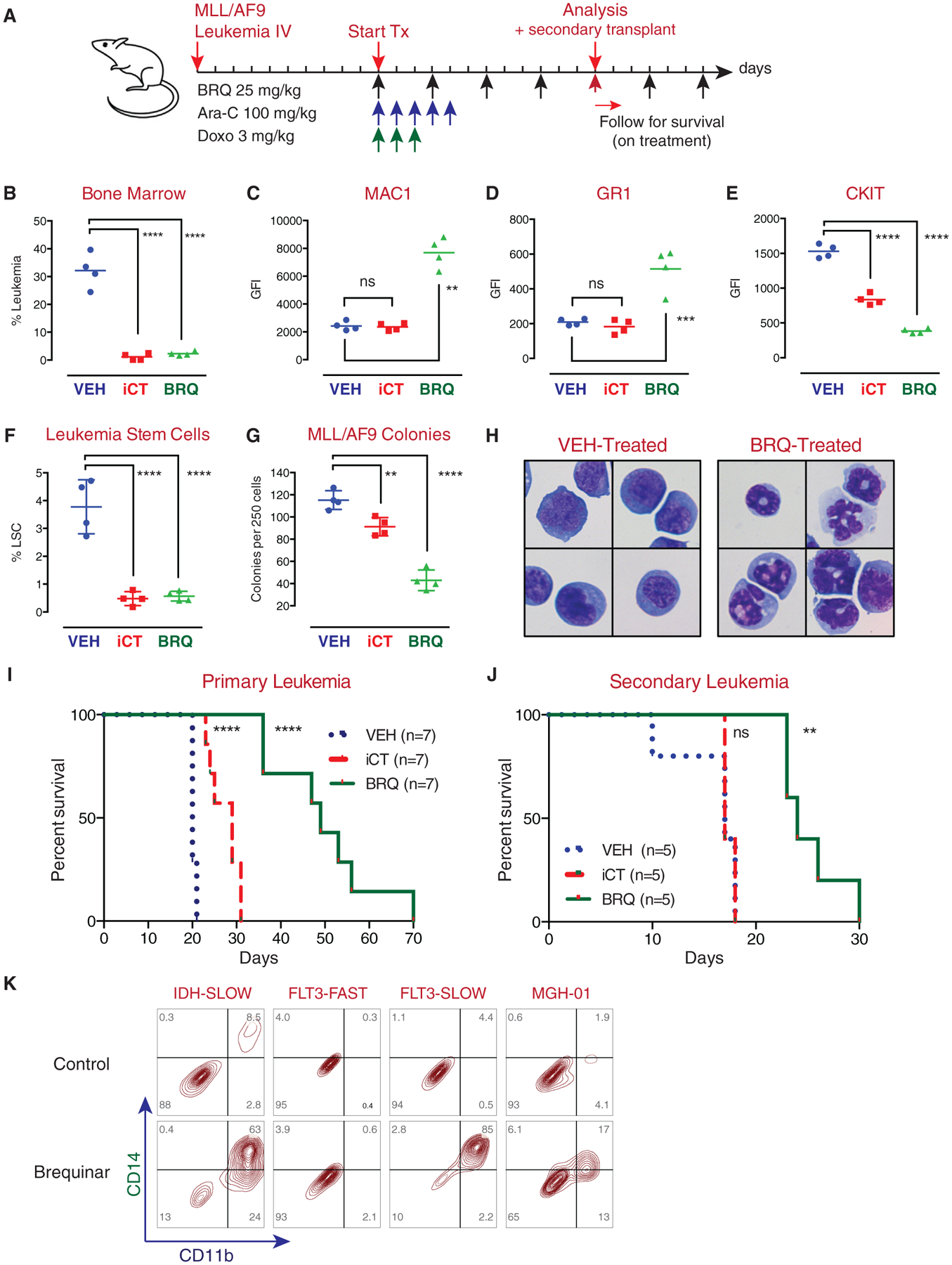
(A) Experimental outline of MLL/AF9 leukemia; mice were treated with standard-of-care chemotherapy (Ara-C and doxorubicin) or BRQ.
(B-E) Treatment with BRQ decreased leukemia burden in the bone marrow (B), increased expression of MAC1 (C), increased expression of Gr-1 (D), and decreased expression of CKIT (E) cell-surface markers.
(F) Treatment with BRQ or chemotherapy led to a decrease in the number of phenotypic leukemic stem cells.
(G) Leukemic cells isolated from mice treated with BRQ or chemotherapy were placed into methylcellulose colony formation assays. Treatment of the mice with BRQ or chemotherapy led to a decrease in colony formation activity.
(H) Leukemia cells isolated from mice treated with BRQ show morphologic evidence of differentiation.
(I) Treatment with BRQ leads to prolonged survival in the primary recipient mice beyond the benefit seen with chemotherapy.
(J) Treatment with BRQ leads to a decrease in the frequency of leukemia-initiating cells, as evidenced in secondary transplant assays. In this assay, treatment with chemotherapy does not lead to a decrease in the frequency of leukemia-initiating cells.
(K) Treatment of patient-derived xenograft (PDX) leukemia samples with BRQ in culture shows an increase in myeloid differentiation markers in three out of four models.
Data in (F) and (G) are represented as the mean ± SD. **p % 0.01; ****p % 0.0001; ns, not significant.
See also Figure S7.
These differentiation changes were accompanied by a loss of phenotypic leukemia stem cells (Figure 7F), as previously defined (Krivtsov et al., 2006), as well as a decrease in colony formation (Figure 7G). These changes resulted in prolonged survival in secondary recipient animals (Figure 7J), confirming a loss of leukemia-initiating cell activity in the MLL/AF9 model. Importantly, treatment of the mice with cytarabine and doxorubicin led to a decrease in leukemia burden but no loss of leukemia-initiating cell activity, pointing toward an important functional difference between iCT and BRQ.
We performed gene expression analysis (RNA-seq) on FACS-purified leukemia cells isolated from vehicle and BRQ-treated mice; treatment with BRQ led to in vivo gene expression changes consistent with normal neutrophil differentiation (Figure S7A).
DHODH Inhibition Is Active in a Patient-Derived Xenotransplant Model of FLT3-ITD AML
Four patient-derived xenotransplant (PDX) leukemia samples were cultured and were treated with BRQ ex vivo; all models exhibited growth inhibition, while three of the four models also exhibited differentiation as assessed by CD11b and CD14 expression (Figure 7K).
Only the FLT3-ITD SLOW PDX sample engrafted in recipient NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/Sz) mice. In this model, treatment of two cohorts of mice with four doses of BRQ (Figures S7B and S7C) or two doses of BRQ 25 mg/kg (Figures S7D and S7E) led to a reduction of leukemia burden in the peripheral blood and bone marrow. BRQ triggered myeloid differentiation in vivo, albeit to a lesser degree than the ex vivo effect (Figure S7F).
DISCUSSION
Here, we describe a model of conditional myeloid differentiation arrest using an estrogen-dependent form of HoxA9. The expression of ER-HoxA9 in primary murine bone marrow mononuclear cells generates factor-dependent GMP cell lines that undergo terminal neutrophil differentiation upon the inactivation of ERHoxA9. The cells provide an inexhaustible source of progenitors for the study of normal myelopoiesis or to identify small molecules that overcome differentiation arrest. Using these cells, we performed an unbiased phenotypic screen for compounds that could trigger myeloid differentiation.
In order to determine the mechanism of action, we generated cell lines with acquired resistance to the differentiation effects of our small molecules. We found that our most highly active compounds were inhibitors of the enzyme DHODH. In fact, 11 of the 12 hits from a library of 330,000 compounds were DHODH inhibitors, reinforcing the importance of the pyrimidine biosynthetic pathway as a regulator of myeloid differentiation. Our study highlights an advantage of phenotypic screening in the identification of new and therapeutically relevant targets that also offer unexpected insights into new biology.
DHODH in Human Disease
DHODH catalyzes the fourth step in pyrimidine synthesis, the conversion of DHO to orotate. The enzyme is located in the inner mitochondrial membrane and transfers electrons between DHO and complex III of the electron-transport chain via the reduction of its ubiquinone (coenzyme Q10) cofactor. Its role in electron transport is not associated with the myeloid differentiation effect; inhibition of the downstream enzyme OMPD (which is not involved in electron transport) phenocopies the differentiation effects of DHODH inhibition.
DHODH is a ubiquitous enzyme, and a complete lack of activity is not compatible with life. The Miller syndrome is a rare autosomal recessive disorder in which patients have inherited hypomorphic mutations in both alleles of DHODH, resulting in multi-organ dysfunction (Ng et al., 2010). DHODH is not known to be mutated or overexpressed in patients with cancer.
Two inhibitors of human DHODH are approved for clinical use. Leflunomide, a pro-drug, is used in the treatment of patients with rheumatoid arthritis. Its active form, teriflunomide, is approved for multiple sclerosis. Leflunomide and teriflunomide are weak inhibitors of DHODH (IC50, ~5 μM) and are likely to have additional anti-kinase effects (Doscas et al., 2014) or effects as aryl hydrocarbon antagonists (O’Donnell et al., 2010).
Leflunomide affects erythroid differentiation of K562 cells in vitro, via the depletion of uridine triphosphate (UTP) and cytidine triphosphate (CTP) ribonucleotides (Huang et al., 2002). Leflunomide was active in a zebrafish model of melanoma, where the proposed mechanism of action was inhibition of transcriptional elongation (White et al., 2011). The combination of leflunomide and the BRAF inhibitor vemurafenib were studied in a phase I/II clinical trial of patients with BRAFV600 mutant meta-static melanoma (https://ClinicalTrials.gov/ identifier NCT01611675). In our hands, leukemic mice treated with leflunomide (25 mg/kg daily) treatment showed a very mild increase in the expression of CD11b but no reduction in leukemic burden. Furthermore, it was poorly tolerated, with mice showing weight loss and lethargy.
Brequinar is a potent and specific inhibitor of DHODH. Given encouraging pre-clinical activity, BRQ was previously evaluated in the phase-1 and -2 trials of patients with advanced solid tumor malignancies (Arteaga et al., 1989; Burris et al., 1998; Noe et al., 1990; Schwartsmann et al., 1990). BRQ was not effective at the doses and schedules evaluated in these trials. Of note, BRQ was not studied in the context of patients with leukemia or other hematologic malignancies.
The lack of efficacy of BRQ in these clinical trials should be interpreted with caution. In our systems, sustained exposure to BRQ was required for its myeloid differentiation effect; brequinar pulses shorter than 48 hr had almost no effect. In the human trials, BRQ was most often administered as a single infusion given once every 2 or 3 weeks. One trial evaluated daily dosing for 5 days but then allowed 3 weeks off of treatment, and this trial did not include patients with hematologic malignancies (Noe et al., 1990). We hypothesize that these schedules would be unlikely to lead to the prolonged suppression of uridine production that would be required to eliminate cancer cells or to induce AML differentiation.
The Potential of DHODH Inhibitors as Differentiation Therapy in the Treatment of AML
In this study, we describe how brequinar and other DHODH inhibitors trigger myeloid differentiation in vitro and in vivo and lead to the depletion of functional leukemia-initiating cells. Wild-type mice bearing syngeneic leukemias (HoxA9+Meis1 or MLL/AF9) and immunocompromised mice implanted with human xenografts (THP1, HL60, MOLM13, and OCI/AML3) tolerated long periods of treatment with BRQ, suggesting a differential sensitivity to DHODH inhibition between normal and malignant cells. This observation points to the potential of a metabolic therapeutic window in the treatment of patients with AML.
What could explain this therapeutic window, given that DHODH is ubiquitously expressed in normal and malignant cells? DHODH inhibition leads to the depletion of pyrimidine precursors and inhibition of nucleic acid synthesis. Unlike many chemotherapies that lead to cumulative DNA damage, DHODH inhibition results in periods of nucleotide depletion, driving a dependency on salvage pathways. We hypothesize that the efficacy of brequinar results from a difference in the ability of malignant cells to tolerate intermittent periods of nucleotide ‘‘‘starvation.’’ This hypothesis is consistent with the importance of dose schedule in our mouse models, where BRQ administered every 3 days demonstrates an anti-leukemia effect without the weight loss and thrombocytopenia observed with daily dosing.
DHODH and the Mechanism of Myeloid Differentiation
The mechanism through which a reduction in de novo pyrimidine biosynthesis modulates myeloid differentiation is not clear. The differentiation effect of DHODH inhibitors is likely to involve a combination of inhibition of nucleic acid synthesis, cell-cycle arrest, and changes in the post-translational glycosylation of important protein targets. Case reports suggest that low-dose cytarabine in the treatment of patients with AML induces differentiation in rare circumstances (Wisch et al., 1983). In our model systems, the observation that the differentiation effect can be phenocopied by pyrazofurin—an inhibitor of OMPD—but not by cytarabine, hydroxyurea, or methotrexate implicates upstream depletion of uridine/UMP/UDP as being of specific importance in the differentiation effect.
One intriguing potential mechanism of differentiation is the alteration of O-linked N-acetylglycosylation, a common protein post-translational modification (Bond and Hanover, 2015). The enzyme O-GlcNAc transferase (OGT) is a ubiquitous enzyme that transfers GlcNAc from UDP-GlcNAc to serine and threonine residues, and this modification can compete with other modifications, including phosphorylation in the regulation of protein function. Particularly interesting proteins that undergo GlcNAc post-translational modification include Akt, the TET family of proteins, and c-Myc (reviewed in Hanover et al., 2012; Jóźwiak et al., 2014). We have demonstrated that inhibition of DHODH leads to a global decrease in protein N-acetylglycosylation; future experiments will elucidate whether specific protein modifications are critical to differentiation.
The rationale for differentiation therapy in the treatment of leukemia is supported by the overwhelming benefits of ATRA and arsenic trioxide in patients with AML. Here, we highlight the potential of DHODH as a therapeutic target for AML differentiation. Our work stresses the importance of phenotypic screens in identifying previously unrecognized molecular pathways relevant in malignant cell biology. The in vivo efficacy of brequinar raises the possibility that a better understanding of its mechanism of action will allow for a more rational dosing schedule in future clinical trials using novel, optimized DHODH inhibitors in the treatment of patients with AML.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the corresponding author, David B. Sykes (dbsykes@partners.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human AML Cell Lines
The human AML cell lines were purchased from the American Type Culture Collection (ATCC; http://www.atcc.org) or the Leibniz-Institute DSMZ (http://www.dsmz.de). The compound-resistant U937 cell lines were authenticated using the ATCC Cell Authentication Testing Service.
Mice and Animal Housing
The lysozyme 2-GFP knock-in mouse was generated by Dr. Thomas Graf (Faust et al., 2000) and purchased from the Mutant Mouse Resource and Research Centers (MMRRC; strain name B6.129(Cg)-Lyz2tm1.1Graf). Other mice were purchased from Jackson Laboratories. Mice were maintained under pathogen-free conditions; experiments were approved by the Massachusetts General Hospital (MGH) Institutional Animal Care and Use Committee (IACUC).
Subcutaneous Xenograft Tumor Models
Female NOD/SCID (NOD.CB17-Prkdcscid) recipient mice 6–8 weeks of age were used for the THP1, HL60, and MOLM13 models. Cells (5 × 106) were implanted subcutaneously in a Matrigel matrix under the right flank of each mouse and allowed to grow to the pre-specified size of 40 mm2. Mice were treated with vehicle or BRQ via i.p. injection in a final volume of 250 μl (10 ml/kg). BRQ was given at 5 mg/kg daily or 15 mg/kg every 3 days. The experiment was terminated when tumors in the control group reached the pre-specified size of 150 mm2.
Disseminated Intravenous Xenograft Leukemia Models
Female NOD/SCID (NOD.CB17-Prkdcscid) recipient mice 6–8 weeks of age were used for the HL60 model. Female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) recipient mice 6–8 weeks of age were used for the OCI/AML3 model. Cells (5 × 106) were introduced intravenously by tail vein injection.
Syngeneic Leukemia Experiments
A retroviral transduction model of HoxA9-Meis1 leukemia was generated by the infection of bone marrow mononuclear cells by an MSCV-HoxA9-IRES-Meis1 construct (originally designed by Dr. Guy Sauvageau). Leukemic bone marrow cells from a mouse were harvested and expanded ex vivo in media supplemented with SCF and IL-3 (as described earlier). Cells were infected with lentivirus expressing the Venus fluorescent protein and sorted twice for purity to allow for in vivo tracking.
The MLL/AF9 leukemia model was established by crossing MLL/AF9 knockin mice (JAX strain 009079) with mice expressing GFP under the control of the ubiquitin promoter as well as mice expressing luciferase under the control of the beta-actin promoter. The primary leukemias derived from these mice were also expanded in culture and used in subsequent experiments.
Age-matched recipient male mice (age 10–12 weeks) were injected intravenously (tail vein or retroorbital) with 5 × 106 leukemia cells in a volume of 250 μl of PBS. Mice were treated with vehicle (70% PBS, 30% polyethylene glycol (PEG)-400, pH 7.2) or BRQ delivered via 250 μl i.p. injection using a 25G needle.
The mice in Figure 6A and Figure S6A received 450 cGy of radiation 24 hr prior to injection of leukemia cells. The mice in Figure 6G and Figure 7A did not receive pre-conditioning radiation.
PDX Models
Mice engrafted with patient AML samples were purchased from Jackson Laboratories. These three models were designated FLT3-ITD-FAST, FLT3-ITD-SLOW, and IDH1-SLOW based on their hallmark mutations and rate of engraftment. Human cells were isolated from the mice (STEMCELL Technologies EasySep Mouse/Human Chimera Isolation Kit) and treated with BRQ in culture for 5 days prior to flow-cytometric analysis of differentiation.
Male NSG mice were irradiated (300 cGy) 24 hr prior to the intravenous transplantation of bone marrow cells from the FLT3-ITD-SLOW mouse. Engraftment, as evidenced by the appearance of human CD45-positive cells in the peripheral blood, was detectable by 8 weeks.
METHOD DETAILS
Generation of ER-HoxA9 GMP Cell Lines
Primary murine bone marrow was harvested from a male mouse by crushing the femur and tibia bones. The cells were filtered through a 40-mm filter and layered over a Ficoll-Paque PLUS gradient to collect live mononuclear cells. Cells were cultured for 48 hr in RPMI supplemented with 10% FBS, penicillin/streptomycin, stem cell factor (SCF; 10 ng/ml), interleukin(IL)-3 (10 ng/ml), and IL-6 (10 ng/ml). Tissue-culture-treated 12-well plates were pre-coated with human plasma fibronectin (1 ml per well, overnight at 37°C, at a final concentration of 10 μg/ml in PBS). Fibronectin was aspirated prior to the addition of cells. At ~48 hr, 2.5 × 105 cells in a volume of 500 ml were transferred to each well. Polybrene was added, and the cells were transduced via spinoculation (1,000 × g, 90 min, 22°) with 1 ml of ecotropic retrovirus (MSCVneo-EE-ER-HoxA9; the EE peptide denoting a GLU-GLU epitope tag). The transduction volume was, therefore, 1.5 ml, with a polybrene concentration of 8 mg/ml. Following the transduction, 3 ml of fresh media was added to each well to dilute the polybrene to a less toxic concentration.
ER-HoxA9 cells were maintained in RPMI supplemented with 10% FBS, penicillin/streptomycin, and SCF. The source of SCF was conditioned media generated from a Chinese hamster ovary (CHO) cell line that stably secretes SCF. Conditioned medium was added at a final concentration of 1%–2% (depending on the batch, the final concentration of SCF was approximately 100 ng/ml as measured by ELISA). Beta-estradiol (abbreviated E2; Sigma, E2758) was added to a final concentration of 0.5 μM from a 10-mM stock dissolved in 100% ethanol. The media was stable for at least 4 weeks when maintained at 4°C.
GMP cell lines emerged after 3–4 weeks of culture. This was in contrast to uninfected cells maintained in the same conditions that differentiated and ceased proliferating over this same period.
Cell Culture
The U937 and THP1 human AML cell lines, as well as all other human AML cell lines, were purchased from the American Type Culture Collection (ATCC; http://www.atcc.org). The cells were maintained in RPMI supplemented with 10% FBS and penicillin/streptomycin.
The HoxA9+Meis1 or MLL/AF9 primary leukemia cells were expanded in culture with the same SCF media used earlier to which recombinant IL-3 (10 ng/ml) was added.
Flow Cytometry and FACS
Antibodies (mouse: CD11b, Gr-1, CD48, CD150, c-Kit, Sca-1; human: CD14) were all purchased from BioLegend. Cells were suspended in FACS buffer (PBS + 2% FBS + 1 mM EDTA) and stained for 45 min at 4°C in the dark. 7-AAD or propidium iodide was included as a viability dye to help identify dead cells. Flow cytometry data was collected on either a BD FACSCalibur or BD LSR2 flow cytometer and analyzed using FlowJo software. FACS (cell sorting) was performed on a BD Aria 2, with the support of the HSCI-CRM Flow Cytometry Facility.
Analysis of Cell Cycle
For cell-cycle analysis, cells were resuspended in 300 μl of PBS. While gently vortexing the cells, 700 μl of ice-cold 100% ethanol was added in a drop-wise fashion to a final concentration of 70%. The cells were fixed at −20°C for a minimum of 24 hr. Following ethanol fixation, the cells were washed and resuspended in PBS. A 103 staining buffer (RNase A, Triton X-100, propidium iodide) was added, and the cells were incubated at 37°C prior to analysis of DNA content on a BD LSR2 flow cytometer. Cell-cycle analysis was performed using FlowJo and ModFit LT software.
Cytospins and Wright-Giemsa Staining
Cells were prepared in PBS at a concentration of approximately 2 million/ml. Cytospin (Thermo Scientific Shandon) preparations were made (1,000 rpm, 60 s), and the cells were allowed to air dry. Cells were stained in 100% Wright-Giemsa (Siemens) for 2 min, and in 20% Wright-Giemsa diluted in buffer for 12 min. Stained cells were rinsed in deionized water, and coverslips were affixed with Permount prior to microscopy.
Phagocytosis Assay
ER-HoxA9 cells were differentiated out of beta-estradiol for a period of 4 days. Cells were resuspended in fresh media along with fluorescein-labeled heat-killed Escherichia coli BioParticles (Molecular Probes). Cells and bioparticles were agitated gently at 37°C for 60 min, and cytospin preparations of the cells were made. Cells were labeled with anti-actin to stain the cytoplasm and with DAPI to stain nucleic acid prior to fluorescence microscopy.
Isolation of RNA and DNA
For RNA-seq, total RNA was isolated using QIAGEN RNeasy-Plus Mini columns, with additional on-column DNase treatment to eliminate traces of genomic DNA. For WES, genomic DNA was prepared using the QIAGEN DNeasy Blood & Tissue columns. Nucleic acid concentration was quantified using a NanoDrop (Thermo Scientific).
Gene-Expression Analysis: Compound-Resistant Cell Lines
Next-generation sequencing of RNA-seq samples was performed (Illumina HiSeq), resulting in approximately 40 million pairs of 75-bp reads per sample. These reads were mapped to hg19 and mm9 genomes using Tophat2, followed by the analysis of differential expression using the Cufflinks and Cuffdiff packages. For WES analysis, reads were mapped to the hg19 genome using BWA (Burrows-Wheeler Transform Aligner), followed by the analysis of copy number variation using VarScan2.
Gene-Expression Analysis: Differentiation Time Course
RNA-seq of the Lys-GFP-ER-HoxA9 differentiation time course upon beta-estradiol withdrawal, and of the ML390 treatment, was performed by the Genomics Platform at the Broad Institute. RNA-seq and WES of the C03- and C07-resistant cell lines was performed by the Partners HealthCare Personalized Medicine Translational Genomics Core. In both cases, Poly(A)-selected libraries were generated using the Illumina TruSeq protocol and sequenced on an Illumina HiSeq.
Alignment
Reads were aligned to the mouse GRCm38 primary assembly extended with the sequence of EGFP (http://www.ebi.ac.uk/ena/data/view/AHK23750) using STAR (Dobin et al., 2013) and the GENCODE annotation M5 (http://www.gencodegenes.org). Differential expression analysis was performed with DESeq2 version 1.8.1 (Love et al., 2014) in R version 3.2.1.
Microarray expression data for a primary mouse neutrophil differentiation time course (Yuan et al., 2007) were normalized with GC Robust Multi-array Average (GCRMA; Wu et al., 2004) and corrected for batch effects using COMBAT (Johnson et al., 2007). Differential expression analysis was performed with limma, version 3.24.14 (Ritchie et al., 2015). Genes were only included in the analysis if they exceeded a mean expression level of 2.244 across all samples (threshold determined using the genefilter package, version 1.50.0) (Bourgon et al., 2010). Normalized microarray expression data for human primary hematopoietic cell populations were obtained from the Differentiation Map (DMap) portal (http://www.broadinstitute.org/dmap/home) (Novershtern et al., 2011).
Time course mapping
Log2-FPKM (fragments per kilobase of exon per million fragments mapped) values were calculated for the E2 RNA-seq samples to estimate relative expression levels for each gene using DESeq2, version 1.8.1.
DMap human hematopoiesis differentiation data
Mouse ensemble gene IDs for the E2 RNA-seq samples were mapped to human gene symbols using biomaRt, version 2.24.0 (Durinck et al., 2005). The log2-FPKM values were combined with the normalized DMap data, keeping only the genes that occurred in both datasets, and were subsequently quantile normalized to yield comparable distributions of expression values. Linear discriminant analysis (LDA) was then performed only on the DMap samples to obtain a one-dimensional projection (first linear discriminant, LD1; capturing 55.1% of the total data variance) of the samples along the differentiation axis. E2 RNA-seq samples were then projected onto this axis by predicting their values for LD1.
Murine neutrophil differentiation data
For the neutrophil time course, E2 RNA-seq samples were normalized by combining the microarray and log2-FPKM data and performing quantile normalization. Again, only genes measured in both datasets were included. Because the neutrophil time course is associated with a quantitative measure of time (days after induction of differentiation), we used linear regression (LR) rather than LDA to map the E2 time course onto the neutrophil differentiation time course. First, principal-component analysis (PCA) was performed only on the neutrophil data to reduce data dimensionality. The first two principal components (capturing 52.1% total variance), as well as their interaction, were then used to build an LR model, thus mapping the expression data to a linear axis of time (measured in days). The E2 time course data were mapped onto this axis by predicting the differentiation time in days from their normalized expression values.
Gene set enrichment analysis
We compared the changes in normal neutrophil development to the changes observed in our HoxA9-ER model of differentiation using gene set enrichment analysis (GSEA) (Subramanian et al., 2005). Differentially expressed genes between the first and last time points (day 0 and day 7) for the neutrophil microarray data were identified with limma, version 3.24.14, at a false discovery rate (FDR) of 10%. We used GSEA, version 2.2.0 (preranked mode with default parameters), to test whether the resulting sets of up- and downregulated genes were enriched at the top or bottom of FPKM values obtained for the E2 and ML390 RNA-seq time course data.
Similarity matrices
Similarity matrices between the neutrophil time course and the E2 and ML390 time course data represent pairwise Pearson correlation values between log-fold changes of indicated time points compared to the respective zero time point (day 0 for neutrophil and 0 hr for E2/ML390). Only genes measured in both datasets were used for the comparison. All figures were generated using ggplot2, version 1.0.1.
Superoxide Production Analysis
Superoxide production was measured in a luminescence assay using the LumiMax Superoxide Anion Detection Kit (Agilent Technologies). ER-HoxA9 cells cultured in the presence or absence of beta-estradiol for a period of 4 days were harvested, counted, and resuspended in assay buffer according to the manufacturer’s instructions.
High-Throughput Flow Cytometry Screen
The primary screen was performed at the University of New Mexico Center for Molecular Discovery. Details of the primary and counterscreens have been described (Sykes et al., 2015). In brief, Lys-GFP-ER-HoxA9 cells (2,500) were seeded in a volume of 50 μl in each well of a tissue-culture-treated 384-well plate. Compounds were added to the well by robotic pinning to a final concentration of 4 μM. Given that there is no known small-molecule inhibitor of HoxA9, we took advantage of the ER-HoxA9 fusion protein and used the estrogen antagonist fulvestrant as a positive control at a concentration of 10 μM. Following a 4-day incubation, 10 μl of staining solution (anti-mouse-CD11b-APC antibody at a concentration of 1 to 100; Clone M1/70, BioLegend) and inert marker beads (Spherotech) were added to each well. Cells were analyzed by high-throughput flow cytometry (HyperCyt system, IntelliCyt). Live cells were distinguished from dead cells on the basis of forward and side scatter properties. Differentiation was assayed by measuring GFP (FL-1) and APC (FL-4) fluorescence.
In Vitro Differentiation Assay
Lys-GFP-ER-HoxA9, U937, and THP1 cells were used in the in vitro differentiation assay. Cells (2,500 to 5,000) were plated in 100 ml of media in 96-well round-bottom plates. Compounds were added from DMSO stocks using the D300 digital dispenser (Hewlett Packard/Tecan), and the cells were incubated with compound for 5 days. Cells were washed in the 96-well plate, resuspended in FACS buffer, and stained with CD11b-APC and propidium iodide. Samples were analyzed in a 96-well format using a BD LSR2 flow cytometer with high-throughput sampler or using an iQue Screener (IntelliCyt).
Imaging Flow Cytometry
Cells were stained with CKIT-PE and CD11b-APC and fixed with 1% paraformaldehyde. Fixed cells were stained with DAPI, and data were collected with the help of Scott Mordecai in the Department of Pathology Flow and Image Cytometry Core (Amnis ImageStream, EMD Millipore).
Monitoring Proliferation via CFSE Assay
Cells were stained with CFSE (CellTrace Far Red, ThermoFisher) per the manufacturer’s instructions, though we used 25% of the recommended CFSE concentration, which improved the viability of our cells. Cell fluorescence was analyzed every 24 hr by flow cytometry (BD LSR2).
Generation of Resistant Cell Lines
Lys-GFP-ER-HoxA9 cells and U937 cells were cultured in DMSO or C03 or C07 at a starting concentration of 10 μM. The cell number was normalized, and cells were passaged to fresh media and compound every 4 days. Over the course of 6 months, cells became resistant to the differentiating effects of increasing concentration of compound, up to 50 μM.
DHODH Enzymatic Assay
The enzymatic assay couples DHODH activity with bleaching of the dye 2,6-dichlorophenolindophenol (DCIP) (Knecht and Löffler, 1998; Miller et al., 1968). The assay was conducted in aqueous buffer containing 50 mM Tris, 0.1% Triton X-100, 150 mM KCl, 0.4 mg/ml DHODH, 1 mM DHO, 0.1 mM decylubiquinone, 0.06 mM DCIP, and 0.17% DMSO (pH 8.0) at room temperature, in a 384-well plate format. Compounds were added via pin transfer or via D300 digital dispenser, and the reaction was initiated by addition of substrates. Enzyme activity was monitored kinetically by the reduction in DCIP absorbance at 600 nm over the course of 1 hr. Purified recombinant human DHODH (full-length, C-terminal MYC/DDK-tag) enzyme was purchased from Origene (catalog no. TP309034). Other chemicals, including leflunomide and teriflunomide, were purchased from Sigma-Aldrich. Absorbance measurements were obtained using a Molecular Devices Spectramax M5 plate-reading spectrophotometer.
Uridine Rescue In Vitro
Uridine (Sigma-Aldrich) was dissolved in DMSO or in water at a concentration of 100 mM and stored in frozen aliquots. Uridine was supplemented into the tissue culture media at the described concentrations up to 200 μM.
Chemical Synthesis
The synthesis of ML390 has been previously described (Sykes et al., 2015). Compound 7, (R)-C07, and (S)-C07 were prepared similarly by Dr. Timothy Lewis at the Broad Institute. BSQ was prepared by Dr. Steven Ferrara at the Broad Institute as previously described according to the published procedure (Hesson, 1987). (See details in Data File S1.)
All reactions were carried out under a nitrogen (N2) atmosphere. All reagents and solvents were purchased from commercial vendors and used as received. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker (1H, 300 MHz; 13C, 75 MHz) spectrometer. Flash chromatography was performed using 40–60 μm silica gel (60-Å mesh) on the Teledyne Isco CombiFlash Rf 4x System. Tandem liquid chromatography/mass spectrometry (LC/MS) was performed on a Waters 2795 Separations Module and 3100 Mass Detector with a Waters Symmetry C18 Column (3.5 μm, 4.6 × 100 mm), with a gradient of 0%–100% CH3CN in water over 2.5 min with constant 0.1% formic acid. All compounds were >95% pure as determined by high-performance liquid chromatography (HPLC) and 1H NMR analysis.
C03
C03 was made using a literature procedure (Wolf et al., 2006). To 4 ml of n-butanol was added 1.0 g of 2-chloronicotinic acid(6.4 mmol), 1.6 g of 4-chloro aniline (13 mmol), 1.3 g of K2CO3 (9.5 mmol), and 40 mg of copper powder (640 mmol), and the mixture was heated at 130°C overnight. After cooling, 20 ml of water was added, the mixture was filtered, and the filtrate was made acidic with HCl. The tan precipitate was filtered off and recrystallized from EtOH, yielding 380 mg of tan solid (24%), which was identical to purchased material (ChemBridge catalog no. 9209980) by LC/MS analysis.
(R)-C07
To 2.00 g of (R)-1-amino-1,2,3,4-tetrahydronaphthalene (13.6 mmol) in 20 ml of DMF was added 2.58 g of N-tBOC-β-alanine(13.6 mmol), 14 ml of Et3N (140 mmol), 1.84 g of HOBt (13.6 mmol), and 3.90 g of EDAC , HCl (10.2 mmol), and the mixture was stirred overnight at room temperature. Water and CH2Cl2 were added and separated, the water was rinsed several times with CH2Cl2, and the combined CH2Cl2 layers were rinsed several times with brine before drying (MgSO4), filtering, and concentration. Chromatography with 25%–50% EtOAc in hexane yielded 3.58 g of product as a white solid (83%).
To 212 mg of this product (0.67 mmol) dissolved in 10 ml CH2Cl2 was added 2 ml of trifluoroacetic acid (TFA, 27 mmol), and the solution was stirred 1 hr before concentration. The crude product was dissolved in 10 ml CH2Cl2, and to it was added 0.25 ml of Et3N(1.8 mmol) and 136 mg (0.78 mmol) of 4-chlorobenzoyl chloride, and the mixture was stirred overnight. More CH2Cl2 was added and was rinsed with NaHCO3 (aq), and the CH2Cl2 was dried and concentrated. Recrystallization from CH2Cl2 and hexane yielded 178 mg of white solid product (75%).
These reactions were run analogously starting with (S)-1-amino-1,2,3,4-tetrahydronaphthalene to produce compound (S)-CO7.
LCMS analyses of both (R)-CO7 and (S)-CO7 were identical to that of commercially available racemic CO7 material (Enamine catalog no. Z26396336).
ML390
The initial tBOC-amine product above (3.58 g, 10.1 mmol) was dissolved in 150 ml of CH2Cl2 and cooled in an ice bath before addition of 40 ml of TFA and removing the ice bath. After stirring 2 hr, the reaction was concentrated to an oil; the addition of ether produced white solids that were filtered and rinsed with ether, 3.70 g (99%). 1H NMR (300 MHz, DMSO-d6) d 8.48 (d, J = 8.5, 1H), 7.24–7.02 (m, 4H), 4.99 (dd, J = 5.9, 13.7, 1H), 3.05 (s, 2H), 2.73 (d, J = 5.9, 2H), 1.95–1.79 (m, 2H), 1.79–1.60 (m, 2H). To 304 mg of this TFA salt(0.916 mmol) stirred in 10 ml of CH2Cl2 was added 280 mg of Et3N (2.79 mmol), dissolving the solids, then 226 mg (1.01 mmol) of 4-trifluoromethoxy benzoyl chloride, and the reaction was stirred overnight. Aqueous NaHCO3 and CH2Cl2 were added and separated; and the CH2Cl2 was dried, concentrated, and chromatographed with 20%–70% EtOAc in hexanes before recrystallization from CH2Cl2 and hexane, which yielded 292 mg of product (78%).
BSQ
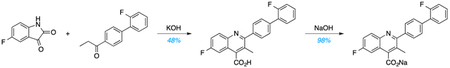
Utilizing a Pfitzinger reaction, 5-fluoroisatin was condensed with 4-(2-fluorophenyl)propiophenone in the presence of potassium hydroxide (KOH) to produce the free-acid form of BSQ, which was recrystallized from dimethylformamide (DMF). Subsequent salt formation using sodium hydroxide produced BSQ (47% yield) over two steps. LC and quantitative 1H-NMR spectroscopy studies confirmed >95% purity. For in vitro experiments, BSQ was dissolved in DMSO at a concentration of 10 mM. For in vivo experiments, BSQ was dissolved in aqueous solution (PBS/30% PEG-400, pH 7.2) and delivered by i.p. injection in a final volume of 250 μl (10 ml/kg).
Kinase-Inhibition Assays
The potential of ML390 as a kinase inhibitor was assayed at a concentration of 10 μM against a panel of more than 300 kinases using the commercially available Merck Millipore KinaseProfiler service. The potential of BSQ as a kinase inhibitor was assayed at concentrations of 100 nM and 1 μM against a panel of more than 450 kinases through the commercially available DiscoverX KinomeScan service.
Analysis of Intracellular Uridine
Frozen THP1 tumor tissues were pulverized using the Bessman Tissue Pulverizer (Spectrum Laboratories). The pulverized tissues were weighed and kept on dry ice. Metabolites were extracted into 80% methanol and homogenized 2 × 15 s at 5,800 rpm (Precellys24, Precellys Ceramic Kit 1.4mm, Paqlab). N,N-di-methylphenylalanine (50 ng/ml) was included as an internal standard at 4 μl/mg of tumor. The supernatant was cleared by centrifugation. Chromatographic separation was performed on an Acquity UPLC I-class system (Waters Corporation). Metabolite separation was performed at 40°C using an Acquity UPLC BEH Amide column (1.7 μm, 2.1 × 150 mm, Waters). The mobile phase was composed of (A) 90% acetonitrile with 10% H20 and 10 mM ammonium acetate (pH 9, adjusted with NH3) and (B) H20 with 10 mM ammonium acetate (pH 9, adjusted with NH3). The gradient elution was as follows: 0–8 min linear elution from 95% to 50% A, 8–9 min from 95% to 50% A and hold at 95% A for 5 min for column equilibration (total run time, 14 min, flow rate constant at 0.4 ml/min). Uridine detection were carried out using a Waters Xevo TQ-S micro Triple Quadrupole tandem mass spectrometer with electrospray ionization (ESI) source (Waters) operating both in the positive- and negative-ion modes. The spectra were acquired with a source temperature of 150°C, a desolvation temperature of 600°C, a cone gas flow of 20 l/min, and a desolvation gas flow of 1,000 l/hr. Multiple reaction monitoring (MRM) transitions of 243.04 > 199.96 and 243.04 > 109.89 were tracked in negative-ion mode for uridine. The raw data were acquired and processed with MassLynx 4.1 software.
Analysis of Intracellular Metabolites
Lys-GFP-ER-HoxA9 cells were treated in culture with 10 μM ML390 for 48 hr. The cells were washed three times in normal saline, and metabolites were extracted into 80% ice-cold methanol. Metabolites were analyzed with the help of Dr. Kerry Pierce and Dr. Clary Clish at the Broad Institute. Negative ionization mode analyses of polar metabolites were acquired using an LC-MS system comprising an Acquity UPLC System (Waters) and a 5500 QTrap triple quadrupole mass spectrometer (AB SCIEX). Samples for negative ion mode analyses of polar metabolites were achieved using the HILIC (hydrophilic interaction chromatography) method under basic conditions as described previously (Mayers et al., 2014), and MS data were acquired over m/z 70–750. MS data were processed using MultiQuant (version 2.1, AB SCIEX).
Immunoblotting
Lys-GFP-ER-HoxA9 cells were treated with ML390, BSQ, or DMSO for 72 hr. Cell lysates were harvested in RIPA buffer supplemented with protease inhibitors and with PUGNAc at a concentration of 10 μM. Total protein was quantified (BioRad) and 50 mg of protein was loaded per lane. Immunoblotting was performed with anti-O-GlcNAc or anti-actin antibodies (Cell Signaling Technology).
In Vivo Leukemia Analysis
Mice exhibiting signs of distress were anesthetized (isoflurane) for retro-orbital peripheral blood sampling and then euthanized (CO2 asphyxiation). Complete blood counts were performed. The spleen was removed, weighed, and macerated through a 40-μM filter using the rubber plunger from a 10-cc syringe. The long bones of the arms and legs were removed and crushed into PBS containing 2% FBS and 1 mM EDTA (FACS buffer). The splenocytes and bone marrow mononuclear cells were isolated over a Ficoll-Paque PLUS density gradient, counted (using acridine orange and the Cellometer instrument to avoid counting contaminating red blood cells), and antibody stained prior to analysis by flow cytometry (BD LSR2).
Transplant for Leukemia-Initiating Cell Analysis
Leukemic cells from control and BRQ-treated mice were isolated (as described earlier) and purified by FACS on the basis of their Venus fluorescent protein expression. An equivalent number (50,000) of live cells were injected into recipient mice (either conditioned with 450 Gy 24 hr prior to injection for the HoxA9 + Meis experiment or without conditioning for the MLL/AF9 experiment).
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad PRISM 7 software was used to perform statistical analyses. The Student’s t test was used for pairwise comparisons of significance. The log-rank (Mantel-Cox) test was used for the survival curves analyses. The analyses of gene expression datasets is described earlier.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the gene expression of MLL/AF9 leukemia cells sorted from mice treated with or without BSQ, time course of myeloid differentiation in the lysozyme-GFP ER-HoxA9 cells following estradiol withdrawal, and gene expression analysis of parental cell lines and cell lines with acquired resistance to compound 3 and compound 7 are GEO: GSE84873, GSE84874, GSE84875, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD11b (MAC1) (M1/70) | BioLegend | Cat#: 101222; RRID: AB_493705 |
| CD117 (CKIT) (2B8) | BioLegend | Cat#: 105807; RRID: AB_313216 |
| GR1 (Ly6C/G) (RB6–8C5) | BioLegend | Cat#: 108431; RRID: AB_10896783 |
| CD48 (HM48–1) | BioLegend | Cat#: 103431; RRID: AB_2561462 |
| CD150 (TC15–12F12.2) | BioLegend | Cat#: 115914; RRID: AB_439797 |
| Sca-1 (D7) | BioLegend | Cat#: 108127; RRID: AB_10898327 |
| CD14 (HCD14) | BioLegend | Cat#: 325603; RRID: AB_830676 |
| TER119 (TER119) | BioLegend | Cat#: 116211; RRID: AB_313712 |
| O-GlcNAc (mouse monoclonal) | Cell Signaling Technology | Cat#: 9875S; RRID: AB_10950973 |
| Actin (rabbit monoclonal) | Cell Signaling Technology | Cat#: 4970S; RRID: AB_10694069 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Beta-estradiol | Sigma-Aldrich | E2758 |
| Polybrene | Millipore | TR-1003-G |
| Interleukin (IL)-3 | Peprotech | 200–03 |
| IL-6 | Peprotech | 200–06 |
| Fulvestrant | Sigma-Aldrich | I4409 |
| Leflunomide | Sigma-Aldrich | L5025 |
| Teriflunomide | Sigma-Aldrich | SML0936 |
| PuGNAc | Sigma-Aldrich | A7229 |
| Fibronectin (Human plasma) | Millipore | FC010 |
| 7-AAD | BD Pharmingen | 559925 |
| Propidium iodide | Life Technologies | P3566 |
| DAPI | Molecular Probes | D21490 |
| RNase A (PureLink) | Invitrogen | 12091021 |
| Uridine | Sigma-Aldrich | U3750 |
| DHODH (recombinant human protein) | Origene | TP309034 |
| Critical Commercial Assays | ||
| E. coli bioparticles (Molecular Probes) | Molecular Probes | E2861 |
| QIAGEN RNeasy Plus Mini Kit | QIAGEN | 74134 |
| CellTrace Far Red Cell Proliferation Kit | Thermo Fisher | C34564 |
| Wright-Giemsa | Siemens | 239020 |
| DNeasy Blood & Tissue Kit | QIAGEN | 69504 |
| LumiMax Superoxide Anion Detection Kit | Agilent Technologies | 204525 |
| Deposited Data | ||
| GEO: Gene expression of MLL/AF9 leukemia cells sorted from mice treated with or without brequinar | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84873 | GEO: GSE84873 |
| GEO: Time course of myeloid differentiation in the ysozyme-GFP ER-HoxA9 cells following estradiol withdrawal | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84874 | GEO: GSE84874 |
| GEO: Gene expression analysis of parental cell lines and cell lines with acquired resistance to compound 3 and compound 7 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84875 | GEO: GSE84875 |
| Experimental Models: Cell Lines | ||
| ER-HoxA9 | This paper | N/A |
| Lys-GFP-ER-HoxA9 | This paper | N/A |
| U937 | ATCC | CRL-1593.2 |
| THP1 | ATCC | TIB-202 |
| HL60 | ATCC | CCL-240 |
| MOLM13 | DSMZ | ACC 554 |
| Experimental Models: Organisms/Strains | ||
| Lysozyme2-GFP knockin mouse | MMRRC | B6.129(Cg)-Lyz2tm1.1Graf |
| C57BI/6J | Jackson Laboratories | 000664 |
| NOD/SCID (NOD.CB17-Prkdcscid) | Jackson | 001303 |
| Software and Algorithms | ||
| FlowJo, version 10 | FlowJo | N/A |
| ModFit LT, version 4 | Verity | N/A |
Highlights.
A phenotypic differentiation screen identifies DHODH as a target in AML
DHODH is a key link between pyrimidine synthesis and myeloid differentiation
DHODH represents a metabolic vulnerability across a range of AML subtypes
DHODH inhibitors show therapeutic potential in preclinicalAML models.
ACKNOWLEDGMENTS
The authors would like to thank Alexa Carver at Massachusetts General Hospital (MGH); Michael Churchill at Columbia University; and Anthony Arvanites, Lance Davidow, and Lee Rubin at the Harvard Stem Cell Institute for their help in optimizing the primary small-molecule screen. We thank Thomas Graf for the lysozyme GFP knockin mouse; Timothy Ley for providing the murine myeloid differentiation microarray data; David Tan, who created the mouse graphic; and Mark Kamps, who provided insight and support throughout the project. We thank Kathleen Higgins and Scott Vafai of the Broad Institute for their help with the Seahorse oxygen consumption experiment. We thank Amir Schajnovitz for sharing his model of MLL/AF9 leukemia for the in vivo comparison of brequinar with standard chemotherapy. We thank Sven Christian for his work profiling the human AML cell lines and their response to DHODH inhibition. We were supported by the HSCI-CRM Flow Cytometry Facility, by Scott Mordecai in the Department of Pathology Flow and Image Cytometry Core (NIH 1S10OD012027-01A1), by the Partners HealthCare Personalized Medicine Translational Genomics Core, and by the Genomics Platform at the Broad Institute. The small-molecule screen was supported by NIH grant 1R03DA032471-01 (to D.B.S.) and the NIH Molecular Library Program (U54 HG005032-1 awarded to S.L.S. and U54 MH084690-1 awarded to L.A.S.). D.B.S. was supported by grants from the American Society of Hematology, Alex’s Lemonade Stand Foundation, the Leukemia and Lymphoma Society, Harvard Catalyst, and an American Cancer Society Institutional Research Grant. D.T.S. was supported by the Harvard Stem Cell Institute and the Ludwig Center at Harvard, as well as the Amelia Peabody Charitable Fund, and the Gerald and Darlene Jordan Chair of Medicine at Harvard University. H.M., A.L.E., D.S., and A.J. are employees of Bayer Pharma AG. The work was supported in part by a collaboration with Bayer Pharma AG.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.08.057.
REFERENCES
- Adamaki M, Lambrou GI, Athanasiadou A, Vlahopoulos S, Papavassiliou AG, and Moschovi M (2015). HOXA9 and MEIS1 gene overexpression in the diagnosis of childhood acute leukemias: significant correlation with relapse and overall survival. Leuk. Res 39, 874–882. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Brown TD, Kuhn JG, Shen HS, O’Rourke TJ, Beougher K, Brentzel HJ, Von Hoff DD, and Weiss GR (1989). Phase I clinical and pharmacokinetic trial of Brequinar sodium (DuP 785; NSC 368390). Cancer Res 49, 4648–4653. [PubMed] [Google Scholar]
- Bond MR, and Hanover JA (2015). A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol 208, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgon R, Gentleman R, and Huber W (2010). Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. USA 107, 9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris HA 3rd, Raymond E, Awada A, Kuhn JG, O’Rourke TJ, Brentzel J, Lynch W, King SY, Brown TD, and Von Hoff DD (1998). Pharmacokinetic and phase I studies of brequinar (DUP 785; NSC 368390) in combination with cisplatin in patients with advanced malignancies. Invest. New Drugs 16, 19–27. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Stewart AL, Mukherjee S, Huang C, Hartwell KA, Miller PG, Subramanian R, Carmody LC, Yusuf RZ, Sykes DB, et al. (2015). Niche-based screening in multiple myeloma identifies a kinesin-5 inhibitor with improved selectivity over hematopoietic progenitors. Cell Rep 10, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CT, and Hess JL (2016). Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene 35, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer DV (1995). Brequinar sodium. Pediatr. Nephrol 9 (Suppl ), S52–S55. [DOI] [PubMed] [Google Scholar]
- Dix DE, Lehman CP, Jakubowski A, Moyer JD, and Handschumacher RE (1979). Pyrazofurin metabolism, enzyme inhibition, and resistance in L5178Y cells. Cancer Res 39, 4485–4490. [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doscas ME, Williamson AJ, Usha L, Bogachkov Y, Rao GS, Xiao F, Wang Y, Ruby C, Kaufman H, Zhou J, et al. (2014). Inhibition of p70 S6 kinase (S6K1) activity by A77 1726 and its effect on cell proliferation and cell cycle progress. Neoplasia 16, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Faust N, Varas F, Kelly LM, Heck S, and Graf T (2000). Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96, 719–726. [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. (1999). Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Krause MW, and Love DC (2012). Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol 13, 312–321. [DOI] [PubMed] [Google Scholar]
- Haynes MK, Strouse JJ, Waller A, Leitao A, Curpan RF, Bologa C, Oprea TI, Prossnitz ER, Edwards BS, Sklar LA, and Thompson TA (2009). Detection of intracellular granularity induction in prostate cancer cell lines by small molecules using the HyperCyt high-throughput flow cytometry system. J. Biomol. Screen. 14, 596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesson DP (1987). 2-phenyl-4-quinolinecarboxylic acids and pharmaceutical compositions thereof. U.S. patent US4680299 A http://www.google.com/patents/US4680299. [Google Scholar]
- Huang M, Wang Y, Collins M, Mitchell BS, and Graves LM (2002). A77 1726 induces differentiation of human myeloid leukemia K562 cells by depletion of intracellular CTP pools. Mol. Pharmacol 62, 463–472. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jóźwiak P, Forma E, Bryś M, and Krześlak A (2014). O-GlcNAcylation and metabolic reprograming in cancer. Front. Endocrinol. (Lausanne) 5, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht W, and Löffler M (1998). Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives. Biochem. Pharmacol 56, 1259–1264. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, and Sauvageau G (1998). Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 17, 3714–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, et al. ; Gruppo Italiano Malattie Ematologiche dell’Adulto; German-Austrian Acute Myeloid Leukemia Study Group; Study Alliance Leukemia (2013). Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med 369, 111–121. [DOI] [PubMed] [Google Scholar]
- Löffler M, Jöckel J, Schuster G, and Becker C (1997). Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol. Cell. Biochem 174, 125–129. [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, et al. (2014). Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med 20, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RW, Kerr CT, and Curry JR (1968). Mammalian dehydroorotateubiquinone reductase complex. Can. J. Biochem 46, 1099–1106. [DOI] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet 42, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe DA, Rowinsky EK, Shen HS, Clarke BV, Grochow LB, McGuire WB, Hantel A, Adams DB, Abeloff MD, Ettinger DS, et al. (1990). Phase I and pharmacokinetic study of brequinar sodium (NSC 368390). Cancer Res 50, 4595–4599. [PubMed] [Google Scholar]
- Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, et al. (2011). Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell EF, Saili KS, Koch DC, Kopparapu PR, Farrer D, Bisson WH, Mathew LK, Sengupta S, Kerkvliet NI, Tanguay RL, and Kolluri SK (2010). The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS ONE 5, e13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, Rominger C, Quinn C, Smallwood A, Wiggall KJ, et al. (2015). New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol 11, 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, Largman C, Lawrence HJ, and Humphries RK (1994). Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl. Acad. Sci. USA 91, 12223–12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Kotz JD, Li M, Aubé J, Austin CP, Reed JC, Rosen H, White EL, Sklar LA, Lindsley CW, et al. ; NIH Molecular Libraries Project Team (2015). Advancing biological understanding and therapeutics discovery with small-molecule probes. Cell 161, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartsmann G, Dodion P, Vermorken JB, ten Bokkel Huinink WW, Joggi J, Winograd B, Gall H, Simonetti G, van der Vijgh WJ, van Hennik MB, et al. (1990). Phase I study of Brequinar sodium (NSC 368390) in patients with solid malignancies. Cancer Chemother. Pharmacol 25, 345–351. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes DB, Haynes MK, Waller A, Garcia M, Ursu O, Gouveia KE, Sklar L, Lewis TA, Dandapani S, Munoz B, et al. (2015). Discovering Small Molecules that Overcome Differentiation Arrest in Acute Myeloid Leukemia (Bethesda, MD: National Center for Biotechnology Information; ). [PubMed] [Google Scholar]
- Tedeschi FA, and Zalazar FE (2006). HOXA9 gene expression in the chronic myeloid leukemia progression. Leuk. Res 30, 1453–1456. [DOI] [PubMed] [Google Scholar]
- Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, and Chambon P (1989). The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J 8, 1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. [DOI] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, et al. (2011). DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisch JS, Griffin JD, and Kufe DW (1983). Response of preleukemic syndromes to continuous infusion of low-dose cytarabine. N. Engl. J. Med 309, 1599–1602. [DOI] [PubMed] [Google Scholar]
- Wolf C, Liu S, Mei X, August AT, and Casimir MD (2006). Regioselective copper-catalyzed amination of bromobenzoic acids using aliphatic and aromatic amines. J. Org. Chem 71, 3270–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA, and Gentleman R (2004). A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc 99, 909–917. [Google Scholar]
- Yates JW, Wallace HJ Jr., Ellison RR, and Holland JF (1973). Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother. Rep 57, 485–488. [PubMed] [Google Scholar]
- Yuan W, Payton JE, Holt MS, Link DC, Watson MA, DiPersio JF, and Ley TJ (2007). Commonly dysregulated genes in murine APL cells. Blood 109, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Radtke I, Pardee TS, Zhao Z, Rappaport AR, Luo W, McCurrach ME, Yang M-M, Dolan ME, Kogan SC, et al. (2009). Mouse models of human AML accurately predict chemotherapy response. Genes Dev 23, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the gene expression of MLL/AF9 leukemia cells sorted from mice treated with or without BSQ, time course of myeloid differentiation in the lysozyme-GFP ER-HoxA9 cells following estradiol withdrawal, and gene expression analysis of parental cell lines and cell lines with acquired resistance to compound 3 and compound 7 are GEO: GSE84873, GSE84874, GSE84875, respectively.