

Abstract
RNA repeat expansions cause more than 30 neurological and neuromuscular diseases with no known cures. Since repeat expansions operate via diverse pathomechanisms, one potential therapeutic strategy is to rid them from disease-affected cells, using bifunctional small molecules that cleave the aberrant RNA. Such an approach has been previously implemented for the RNA repeat that causes myotonic dystrophy type 1 [DM1, r(CUG)exp] with Cugamycin, which is a small molecule that selectively binds r(CUG)exp conjugated to a bleomycin A5 cleaving module. Herein, we demonstrate that, by replacing bleomycin A5 with deglycobleomycin, an analogue in which the carbohydrate domain of bleomycin A5 is removed, the selectivity of the resulting small-molecule conjugate (DeglycoCugamycin) was enhanced, while maintaining potent and allele-selective cleavage of r(CUG)exp and rescue of DM1-associated defects. In particular, DeglycoCugamycin did not induce the DNA damage that is observed with high concentrations (25 μM) of Cugamycin, while selectively cleaving the disease-causing allele and improving DM1 defects at 1 μM.
Graphical Abstract
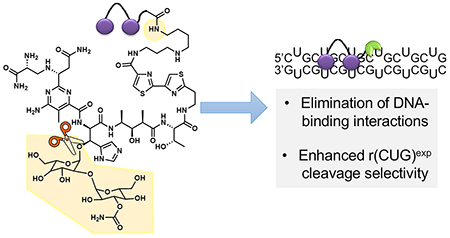
Developing small-molecule chemical probes that modulate RNA function is increasingly important, because of the numerous mechanisms by which RNA can cause disease. One way to target RNAs is the recognition of unstructured regions by antisense (ASOs) and other oligonucleotide-base modalities that bind via base pairing interactions. However, many RNAs have structured regions that directly influence biological function. Small molecules can target these biologically important structures, by matching the RNA’s three-dimensional binding pocket, in terms of size, shape, and complementarity in the display of functional groups and/or surfaces.1,2
One class of disease-causing RNAs that form stable structures is repeat expansions that cause more than 30 microsatellite disorders, including Huntington’s disease3 [HD, r(CAG)exp, where the repeating nucleotides are given in parentheses and “exp” denotes expansion] and myotonic dystrophy types 14 [DM1, r(CUG)exp] and 25 [DM2, r(cCUG)exp]. DM1 is the most common form of adult onset muscular dystrophy, which presently has no cure. This neuromuscular disorder is caused by an expanded CTG repeat, ranging in size from 75 to thousands, harbored in the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene.4 When transcribed into RNA, r(CUG)exp forms a hairpin structure with repeating 1 × 1 U/U internal loops. These loops provide high-affinity binding sites for RNA-binding proteins such as muscleblind-like 1 (MBNL1), which are sequestered in nuclear foci (Figure 1A).6 Thus, r(CUG)exp operates by a gain-of-function mechanism. MBNL1 regulates the alternative splicing of a subset of pre-mRNAs, and its sequestration by r(CUG)exp in nuclear foci6 results in pre-mRNA splicing defects that contribute to the phenotypes found in DM1 (Figure 1B).7,8
Figure 1.
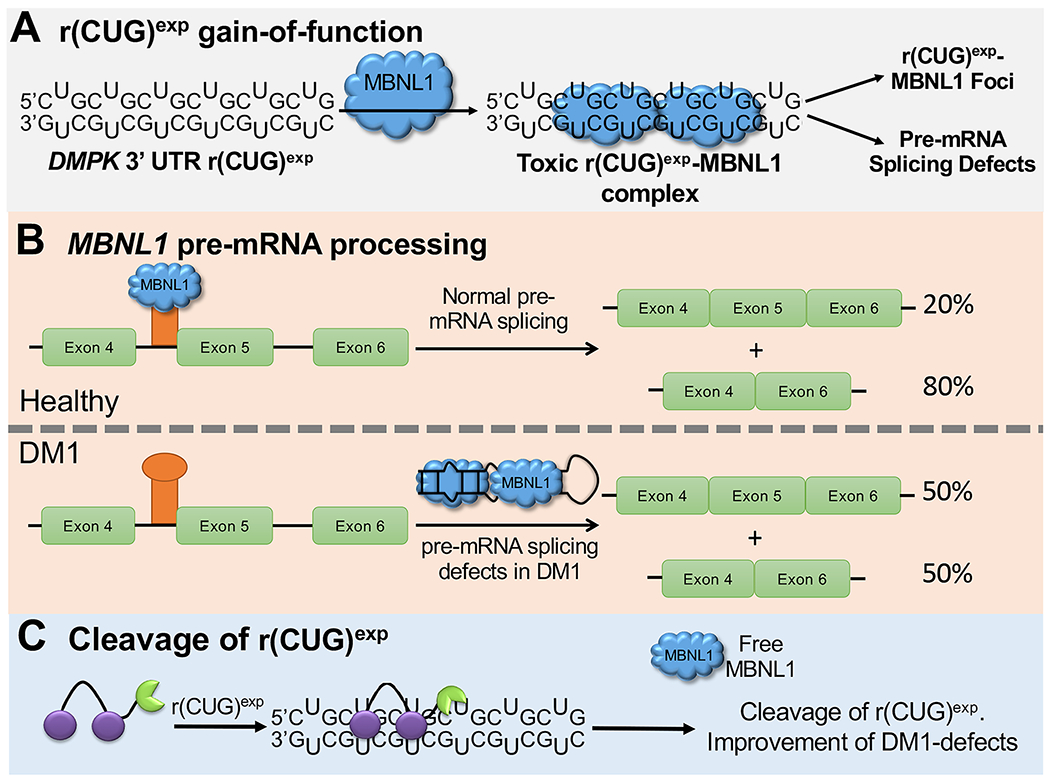
Small-molecule cleavage of r(CUG)exp. (A) DM1 is caused by r(CUG)exp, located in the 3′ UTR of the DMPK gene, which forms a structure with repeating 1 × 1 U/U internal loops. The loops bind and sequester MBNL1, resulting in nuclear foci and pre-mRNA splicing defects. (B) MBNL1 protein regulates the splicing of its own pre-mRNA. When MBNL1 is sequestered by r(CUG)exp, its exon 5 is included too frequently. (C) Scheme of small-molecule cleavage of r(CUG)exp, resulting in improvement of DM1-associated defects.
One approach to alleviate DM-associated defects is to utilize small molecules that recognize the structure of r(CUG)exp, thereby liberating bound MBNL1 or preventing its binding.9–11 Alternatively, the expression of r(CUG)exp has been reduced or eliminated by using RNA targeted-Cas9 editing of r(CUG)exp,12 ASOs,13,14 DNA-binding small molecules that inhibit transcription,15,16 and small molecules that bind and directly cleave r(CUG)exp.9,17,18 The latter approach (Figure 1C) has been accomplished with Cugamycin, which is a small molecule that selectively binds the r(CUG)exp structure conjugated to the natural product bleomycin A5 (BLM) (see Figure 2, as well as Figure S1 in the Supporting Information). Indeed, Cugamycin broadly improved disease associated defects with no off-target effects in a mouse model of DM1.17
Figure 2.
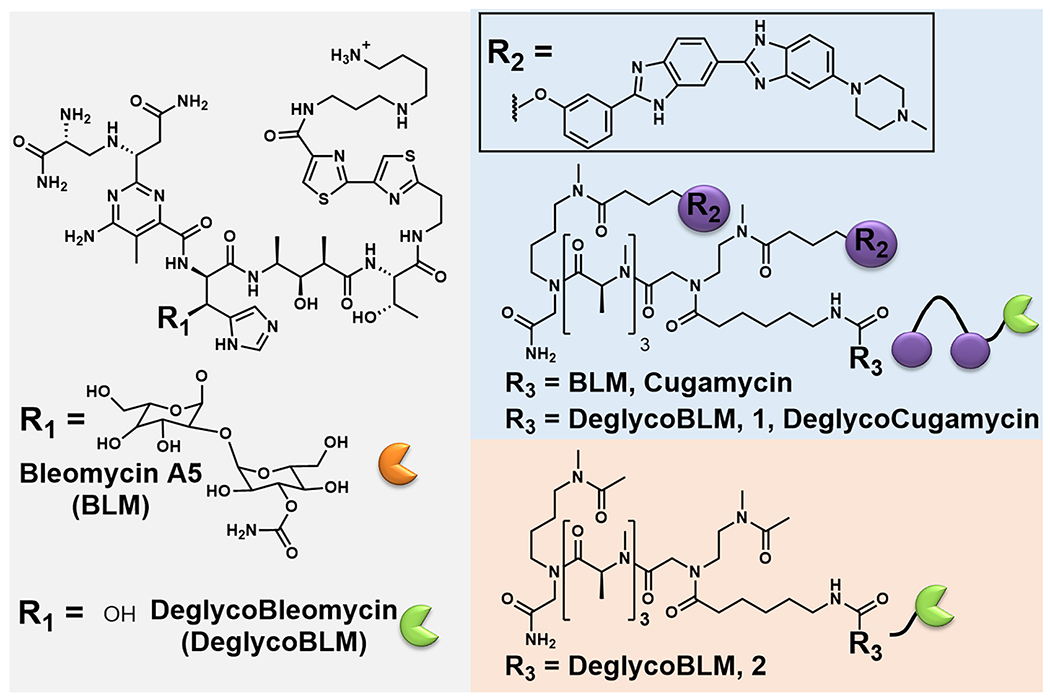
Chemical structures of bleomycin A5 (BLM), deglycobleomycin (DeglycoBLM), Cugamycin, 1, and 2.
BLM is an anticancer natural product that cleaves DNA and RNA through H atom abstraction and the production of a radical species by the metal binding core.19,20 Extensive structure–activity relationship (SAR) evaluations of BLM derivatives19,21 and structural data for DNA-bound BLM22,23 have revealed structural components that are essential for metal coordination, oxygen activation, DNA binding, and subsequent cleavage. This information has been used to guide attachment of RNA-binding small molecules at the C-terminal amine of BLM, eliminating a charge critical to DNA binding and producing BLM-conjugated compounds that specifically cleave a target RNA.17,24 SAR studies of BLM can guide the selection of analogues to further enhance RNA selectivity by eliminating DNA-binding interactions.19 One such analogue is deglycobleomycin (DeglycoBLM; see Figure 2, as well as Figure S1), in which the disaccharide moiety of BLM is removed. The carbohydrate domain can contribute to DNA binding affinity by participating in hydrogen bonding interactions with the DNA backbone, and DeglycoBLM cleaves DNA between 2- and 5-fold less efficiently than BLM.19,23 This disaccharide also contributes to the cellular permeability of BLM.25 Collectively, the attachment of DeglycoBLM to small molecules targeting r(CUG)exp could further reduce its affinity for DNA to enhance RNA selectivity in cells, provided the compound retains cellular permeability. The examination of such features is the subject of this report.
DeglycoBLM was synthesized via HF-pyridine cleavage of the carbohydrate of BLM26 and conjugated to a dimeric compound that recognizes r(CUG)exp (2H–K4NMeS, 3; see Figure S1)9 to afford compound 1 (DeglycoCugamycin; see Figure 2, as well as Figure S1). A control compound that does not contain the RNA-binding modules in 1 and, thus, has no affinity for the RNA target, was also synthesized (compound 2; see Figure 2, as well as Figure S1). To assess the molecular recognition of 1, its affinity for r(CUG)12, r(GC)8, and DNA was measured in the absence of Fe(II). Compound 1 only bound avidly to r(CUG)12 (Kd = 610 ± 150 nM) (see Figure 3A, as well as Figure S2 in the Supporting Information), which is comparable to the affinity of Cugamycin (Kd = 365 ± 75 nM).9 Thus, removal of the carbohydrate domain does not affect the ability to bind r(CUG)exp in vitro.
Figure 3.
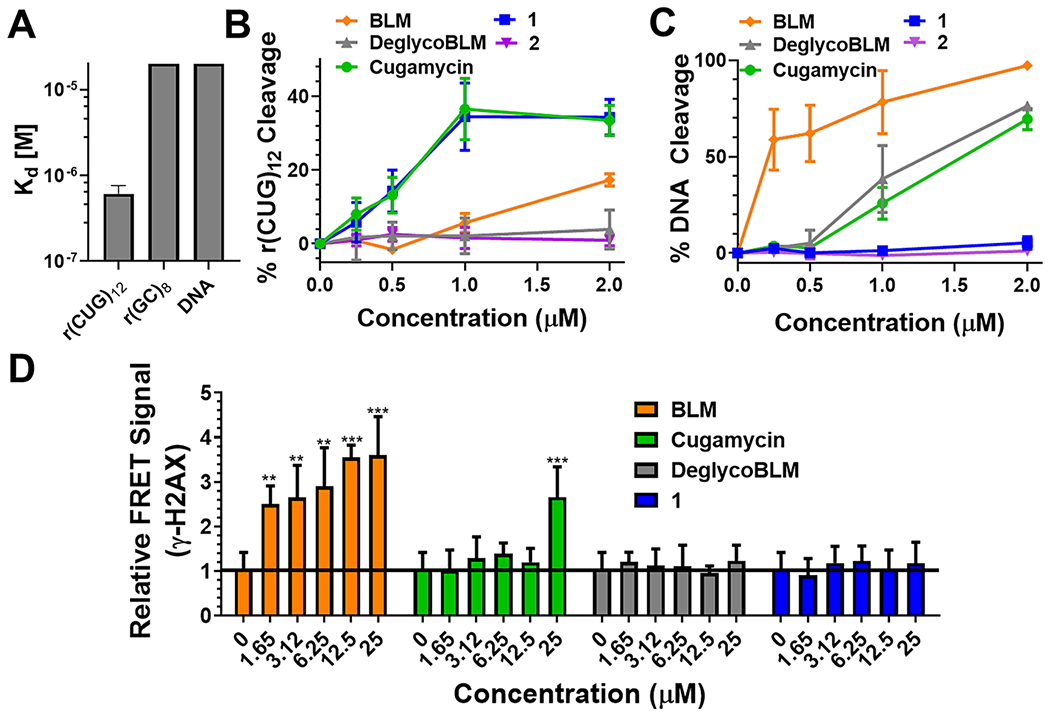
Cleaving capacity and selectivity of small-molecule cleavers. (A) Binding affinity of 1 for r(CUG)12 (Kd = 610 ± 150 nM), r(GC)8 (Kd > 20 μM), and DNA (Kd > 20 μM); n = 3. (B) Quantification of cleavage of r(CUG)10 by 1, 2, Cugamycin, BLM, and DeglycoBLM; n = 3. (C) Quantification of cleavage of DNA by 1, 2, Cugamycin, BLM, and DeglycoBLM; n = 3. (D) Effects of 1, Cugamycin, BLM, and DeglycoBLM on γ-H2AX, a marker of DNA damage, in C2C12 cells. n = 8, (**) P < 0.01, (***) P < 0.001, as determined by comparison to untreated cells by a one-way analysis of variance (ANOVA). Error bars indicate the standard deviation (SD) for all panels.
Next, the ability of 1 to cleave r(CUG)10 and DNA was assessed in vitro. Cugamycin and 1 cleaved r(CUG)10 to a similar extent at the same concentrations (~35% cleavage at 1 μM), while BLM only cleaved r(CUG)10 by 15% at 2 μM (see Figure 3B, as well as Figure S3 in the Supporting Information). In contrast, DeglycoBLM and 2 (lacks RNA-binding modules) were unable to cleave r(CUG)10 at the concentrations tested (up to 2 μM; see Figure 3B, as well as Figure S3), as expected, since DeglycoBLM alone is 5-fold less efficient at cleaving nucleic acids than BLM.19 Thus, functional RNA cleavage by 1 is not affected through removal of the disaccharide. The selectivity of the observed cleavage was assessed by measuring DNA cleavage (see Figure 3C, as well as Figure S4 in the Supporting Information). While BLM efficiently cleaved DNA in vitro with >50% cleavage observed at all concentrations (from 250 nM to 2 μM), DeglycoBLM cleaved DNA ~5-fold less efficiently, with >50% cleavage only observed at 2 μM (Figure 3C), consistent with previous studies.19 We previously showed that Cugamycin does not cleave DNA when r(CUG)12 is present17 and, thus, is selective for cleaving the RNA target. However, when incubated in the absence of r(CUG)12, Cugamycin cleaved DNA at concentrations >500 nM (Figure 3C). In contrast, 1 and 2 did not significantly cleave DNA at any of the concentrations tested (from 250 nM to 2 μM; see Figure 3C). Thus, by eliminating two key DNA-binding interactions through removal of the disaccharide and attachment of the r(CUG)-binding compound at the C-terminal amine, DNA cleavage is further ablated and selectivity for r(CUG)exp is enhanced.
To study potential off-target binding to DNA in cells, we measured the amount of phosphorylated histone H2A variant H2AX (γ-H2AX), which forms foci in response to DNA double strand breaks, induced by compound treatment in the rapidly growing mouse myoblast cell line C2C12 and in DM1 patient-derived myotubes. In C2C12 cells, we used a fluorescence resonance energy transfer (FRET) assay to quantify γ-H2AX foci after treating with the compound of interest for 24 h. BLM caused a significant increase in γ-H2AX at all concentrations tested (1.65–25 μM), as expected from previous studies27 (Figure 3D). In contrast, Cugamycin only induced DNA damage at 25 μM, which is a concentration that is ~10-fold higher than its bioactive concentration in DM1 myotubes,17 while no increase in γ-H2AX foci was observed for 1 or DeglycoBLM upon treatment with up to 25 μM compound (Figure 3D). Importantly, and consistent with these studies in C2C12 cells, neither 1 nor DeglycoBLM induced DNA damage in DM1 patient-derived myotubes, as determined from immunostaining and imaging by fluorescence microscopy (see Figure S5 in the Supporting Information). [Note: the signal:noise observed in the FRET assay described above for C2C12 cells was not sufficient for quantification in DM1 myotubes.] Thus, 1 further diminished off-target DNA cleavage in cells, compared to Cugamycin, in agreement with in vitro DNA cleavage analysis.
To probe if the difference in DNA damage in cells is due to changes in cellular uptake, as the disaccharide has previously been implicated in cell permeability,25 the concentration of Cugamycin, 1, and the dimer from which they are derived (3;9 see Figure S1) taken up into DM1 myotubes was determined by measuring the fluorescence of the RNA-binding modules after washing and lysing treated cells. Cugamycin and 1 had similar cell permeabilities, and both compounds were only ~3-fold less permeable than 3 (see Figure S6 in the Supporting Information). To confirm these results, permeability and localization were studied by using live-cell fluorescence microscopy. Both Cugamycin and 1 localized in the nucleus where r(CUG)exp is sequestered in foci to a similar extent (see Figure S6). Thus, although the carbohydrate domain has been shown to affect the permeability of DeglycoBLM itself and may account for its lack of DNA damage in cells (Figure 3D), the disaccharide did not affect permeability of conjugate compounds, as determined by comparing Cugamcyin and 1.
Since 1 ablated DNA damage observed for Cugamycin without reducing cell permeability, its ability to cleave r(CUG)exp and improve DM1-associated defects in cells was measured. In DM1 patient-derived myotubes,28 1 cleaved ~30% of r(CUG)exp-containing DMPK at low micromolar concentrations (Figure 4A), which is comparable to the cleaving activity of Cugamycin.17 Importantly, 2, which lacks RNA-binding modules, did not affect DMPK levels (see Figure S7A in the Supporting Information). To demonstrate that the reduction in DMPK levels was due to direct cleavage of the RNA, a competition experiment was performed in which cells were co-treated with 1 and 3; 3 binds r(CUG)exp but does not affect DMPK mRNA levels (Figure 4B). Indeed, upon cotreatment, cleavage by 1 was inhibited by 3, and DMPK levels were restored to levels similar to untreated samples or samples treated with 3 alone (Figure 4B). Notably, 1 was selective for cleaving r(CUG)exp, as DMPK levels were not affected in wild-type cells expressing r(CUG)20 (see Figure 4C, as well as Figure S8A in the Supporting Information), nor were mRNAs containing short nonpathogenic r(CUG) repeats (Figure 4C). We have previously shown that this selectivity is due to structural differences in transcripts containing short r(CUG) repeats versus r(CUG)exp, as the small molecule recognizes the structure formed by the repeat expansion.17 Indeed, compounds that recognize the structure of r(CUG)exp can be selective for the toxic disease-driving repeat expansion; that is, structure-targeting ligands can be allele-selective.17 In contrast, an ASO complementary to the r(CUG) repeats is not able to discriminate between short and long repeats and, thus, has off-target effects.17
Figure 4.
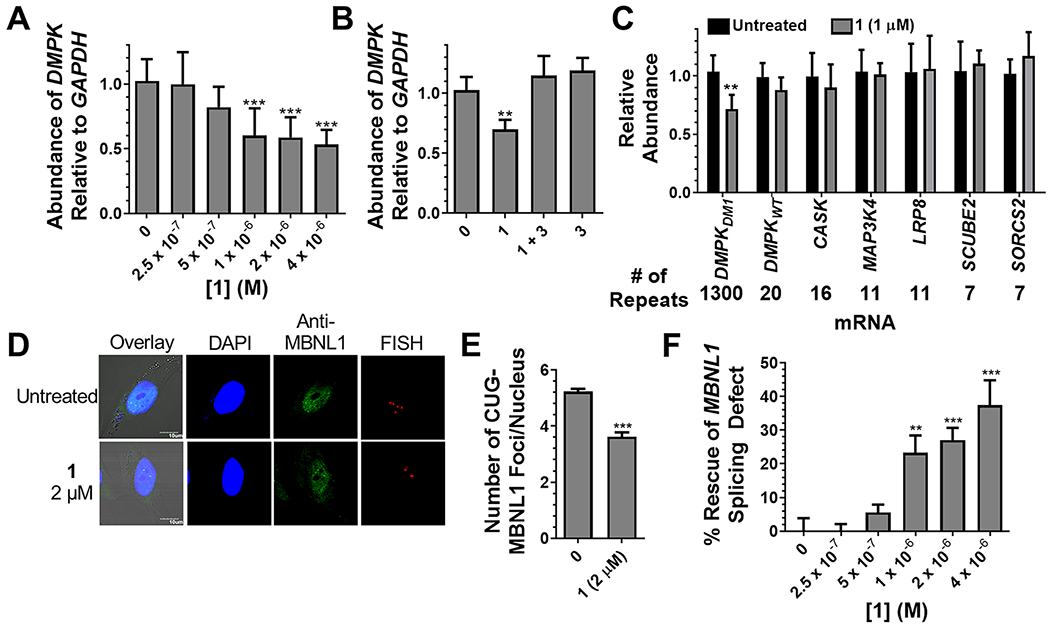
Activity of 1 in DM1 myotubes. (A) Cleavage of r(CUG)exp-containing DMPK by 1 as determined by RT-qPCR. n = 6, (***) P < 0.001, as compared to untreated cells; determined by a one-way ANOVA. (B) Competitive cleavage experiment between 1 (1 μM) and 3 (5 μM) in which 3 prevents the cleavage of DMPK. n = 3, (**) P < 0.01, as compared to untreated cells; determined by a one-way ANOVA. (C) Effect of 1 on r(CUG)n-containing transcripts. n = 3, (**) P < 0.01, as compared to untreated cells; determined by a Student’s t-test. (D) Representative images of r(CUG)exp–MBNL1 foci imaged by RNA fluorescence in situ hybridization (FISH) and anti-MBNL1 immunostaining. (E) Quantification of nuclear foci. n = 3, 40 nuclei quantified/replicate, (***) P < 0.001, as compared to untreated cells; determined by a Student’s t-test. (F) Improvement of the MBNL1 exon 5 splicing defect in DM1 myotubes upon treatment with 1. n = 6, (**) P < 0.01, (***) P < 0.001, as compared to untreated cells; determined by a one-way ANOVA. Error bars indicate the SD for all panels.
After confirming that 1 cleaved r(CUG)exp with similar selectivity and potency as Cugamycin, the ability of 1 to rescue DM1 defects, including the formation of r(CUG)exp–MBNL1 nuclear foci6 and MBNL1-regulated splicing defects,7 was assessed. At 2 μM, 1 reduced the number of r(CUG)exp–MBNL1 nuclear foci by ~40% (see Figures 4D and 4E), similar to Cugamycin,17 while 2, which lacks RNA-binding modules, had no effect (see Figure S7 in the Supporting Information). In DM1 myotubes, MBNL1 exon 5 splicing is dysregulated (Figure 1B), as MBNL1 regulates the alternative splicing of its own pre-mRNA29 Cleavage of r(CUG)exp by 1 resulted in an ~30% improvement in this splicing defect (see Figure 4F, as well as Figure S9 in the Supporting Information), which is an improvement that is similar to that observed for Cugamycin.17 Compound 2 had no effect on MBNL1 exon 5 splicing, as expected (see Figure S7). Importantly, 1 did not affect MBNL1 exon 5 splicing in wild-type myotubes (see Figure S8 in the Supporting Information) nor the NOVA-dependent splicing of MAP4K4 exon 22a30 (see Figure S9). Thus, rescue of the MBNL1 exon 5 splicing defect can be traced specifically to cleavage of the r(CUG)exp. Collectively, these studies show that the removal of the carbohydrate domain of BLM allows for enhanced selectivity by further ablating DNA damage without affecting cellular permeability or activity.
Small molecules that selectively cleave a target RNA are attractive chemical probes, because they can more potently improve disease-associated defects than simple binding compounds.17,31 Furthermore, RNA cleavage, either through direct cleavage as demonstrated herein or through recruitment of a cellular nuclease,31 can be used to profile molecular recognition of RNA-binding small molecules. The use of BLM analogues to specifically cleave r(CUG)exp offers an attractive method to enhance RNA cleavage selectivity by further diminishing off-target DNA cleavage. Although the carbohydrate domain of BLM is necessary for its efficient cleavage of DNA19 and cellular permeability,25 the disaccharide is not essential for permeability or cleavage of r(CUG)exp when attached to r(CUG)exp-binding small molecules. Thus, by using BLM analogues, RNA cleavage and the ability to improve DM1-associated defects is retained while further enhancing selectivity by reducing DNA damage that occurs with high concentrations of BLM-conjugated small molecules.
The most common way to target RNA for destruction is by using oligonucleotide-based target recognition elements. These approaches recognize unstructured regions in RNA. The ability to design ligands that target structured regions in an RNA and cleave them selectively provides an alternative approach to probe the biology of RNA in general and RNA structure in particular. Although bleomycin–small-molecule conjugates have higher molecular weights than orally bioactive drugs, they still have lower molecular weight than oligonucleotides and significantly lower molecular weight than CRISPR approaches that are packed into viruses.12 In addition, medicinal chemistry approaches may be more broadly applicable to these compound sets as the RNA-binding modules and linkers that tether them can be therapeutically optimized. It is likely that, as more information is accumulated on the RNA folds that bind small molecules and on the small molecules that bind RNA folds, the deglycobleomycin cleavage module described herein could be broadly deployed. Furthermore, the ability to effect cleavage of an RNA target can allow for more diverse modes of action. Small molecules can now target an RNA for destruction in cells via three mechanisms: (i) direct cleavage by using bleomycin conjugates;9,17 (ii) nuclease recruitment by using ribonuclease targeting chimeras (RIBOTACs);31,32 and (iii) shunting introns with toxic expanded repeats to decay pathways.3 Some targets may be more amendable to one strategy than the others. The ability to minimize off-target effects by using the deglycobleomycin cleavage module described here could have broad implications in this emerging area.
METHODS
A detailed description of methods can be found in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the U.S. Department of Defense Peer Reviewed Medical Research Program [No. W81XWH-18–0718 (to M.D.D.)], the National Institutes of Health F31 NS110269 (to A.J.A.), and R01 CA042056 (to D.L.B.)], the National Science Foundation [No. NSF/DGE-1346837 (to C.M.G.)], and a Shelton Endowment Scholarship (C.M.G.). We thank D. Furling at the Centre de Recherche en Myologie for the generous gift of the cell lines used in this study. We thank P. Dawson for experimental advice with the synthesis of deglycobleomycin.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00036.
Table S1; Figures S1–S9; experimental methods; synthetic methods and characterization (PDF)
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
Contributor Information
Alicia J. Angelbello, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Mary E. DeFeo, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Christopher M. Glinkerman, Department of Chemistry, The Scripps Research Institute, La Jolla, California 92037, United States
Dale L. Boger, Department of Chemistry, The Scripps Research Institute, La Jolla, California 92037, United States
Matthew D. Disney, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
REFERENCES
- (1).Ursu A, Vezina-Dawod S, and Disney MD (2019) Methods to identify and optimize small molecules interacting with RNA (SMIRNAs). Drug Discov. Today 24, 2002–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Disney MD (2019) Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 141, 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).MacDonald ME, Ambrose CM, Duyao MP, Myers RH, and Lin C et al. (The Huntington’s Disease Collaborative Research Group). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- (4).Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, and Housman DE (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. [DOI] [PubMed] [Google Scholar]
- (5).Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, and Ranum LPW (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867. [DOI] [PubMed] [Google Scholar]
- (6).Taneja KL, McCurrach M, Schalling M, Housman D, and Singer RH (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol 128, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jiang H, Mankodi A, Swanson MS, Moxley RT, and Thornton CA (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet 13, 3079–3088. [DOI] [PubMed] [Google Scholar]
- (8).Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson M, Moxley RT, and Thornton CA (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol 74, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, and Disney MD (2017) Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 13, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, and Disney MD (2012) Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol 7, 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nakamori M, Taylor K, Mochizuki H, Sobczak K, and Takahashi MP (2016) Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol 3, 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, Chaim IA, Thomas JD, Zhang N, Nguyen V, Aigner S, Markmiller S, Xia G, Corbett KD, Swanson MS, and Yeo GW (2017) Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell 170, 899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, and Thornton CA (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jauvin D, Chretien J, Pandey SK, Martineau L, Revillod L, Bassez G, Lachon A, MacLeod AR, Gourdon G, Wheeler TM, Thornton CA, Bennett CF, and Puymirat J (2017) Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther.—Nucleic Acids 7, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Siboni RB, Nakamori M, Wagner SD, Struck AJ, Coonrod LA, Harriott SA, Cass DM, Tanner MK, and Berglund JA (2015) Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep. 13, 2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lee J, Bai Y, Chembazhi UV, Peng S, Yum K, Luu LM, Hagler LD, Serrano JF, Chan HYE, Kalsotra A, and Zimmerman SC (2019) Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci U. S. A 116, 8709–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Angelbello AJ, Rzuczek SG, Mckee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET, and Disney MD (2019) Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U. S. A 116, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Guan L, and Disney MD (2013) Small-molecule-mediated cleavage of RNA in living cells. Angew. Chem., Int. Ed. Engl 52, 1462–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Boger DL, and Cai H (1999) Bleomycin: synthetic and mechanistic studies. Angew. Chem., Int. Ed. Engl 38, 448–476. [DOI] [PubMed] [Google Scholar]
- (20).Abraham AT, Lin J-J, Newton DL, Rybak S, and Hecht SM (2003) RNA cleavage and inhibition of protein synthesis by bleomycin. Chem. Biol 10, 45–52. [DOI] [PubMed] [Google Scholar]
- (21).Madathil MM, Bhattacharya C, Yu Z, Paul R, Rishel MJ, and Hecht SM (2014) Modified bleomycin disaccharides exhibiting improved tumor cell targeting. Biochemistry 53, 6800–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wu W, Vanderwall DE, Turner CJ, Kozarich JW, and Stubbe J (1996) Solution structure of Co-bleomycin A2 green complexed with d(CCAGGCCTGG). J. Am. Chem. Soc 118, 1281–1294. [Google Scholar]
- (23).Goodwin KD, Lewis MA, Long EC, and Georgiadis MM (2008) Crystal structure of DNA-bound Co(III)-bleomycin B2: Insights on intercalation and minor groove binding. Proc. Natl. Acad. Sci. U. S. A 105, 5052–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li Y, and Disney MD (2018) Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem. Biol 13, 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schroeder BR, Ghare MI, Bhattacharya C, Paul R, Yu Z, Zaleski PA, Bozeman TC, Rishel MJ, and Hecht SM (2014) The disaccharide moiety of bleomycin facilitates uptake by cancer cells. J. Am. Chem. Soc 136, 13641–13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wanner J, Tang D, McComas CC, Crowley BM, Jiang W, Moss J, and Boger DL (2003) A new and improved method for deglycosidation of glycopeptide antibiotics exemplified with vancomycin, ristocetin, and ramoplanin. Bioorg. Med. Chem. Lett 13, 1169–1173. [DOI] [PubMed] [Google Scholar]
- (27).Burma S, Chen BP, Murphy M, Kurimasa A, and Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem 276, 42462–42467. [DOI] [PubMed] [Google Scholar]
- (28).Arandel L, Polay Espinoza M, Matloka M, Bazinet A, De Dea Diniz D, Naouar N, Rau F, Jollet A, Edom-Vovard F, Mamchaoui K, Tarnopolsky M, Puymirat J, Battail C, Boland A, Deleuze J-F, Mouly V, Klein AF, and Furling D (2017) Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech 10, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gates DP, Coonrod LA, and Berglund JA (2011) Autoregulated splicing of muscleblind-like 1 (MBNL1) pre-mRNA. J. Biol. Chem 286, 34224–34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, and Darnell RB (2005) Nova regulates brain-specific splicing to shape the synapse. Nat. Genet 37, 844–852. [DOI] [PubMed] [Google Scholar]
- (31).Costales MG, Matsumoto Y, Velagapudi SP, and Disney MD (2018) Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 140, 6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Costales MG, Suresh B, Vishnu K, and Disney MD (2019) Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol 26, 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Benhamou RI, Angelbello AJ, Wang ET, and Disney MD (2020) A Toxic RNA catalyzes the cellular synthesis of its own inhibitor, shunting it to endogenous decay pathways. Cell Chem. Biol 27, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.