

Abstract
Infants born extremely preterm are at a high risk of developing bronchopulmonary dysplasia (BPD) which is characterized by large, simplified alveoli, increased inflammation, disrupted and dysregulated vasculogenesis, decreased cell proliferation, and increased cell death in the lungs. Due to lack of specific drug treatments to combat this condition, BPD and its long-term complications have taken a significant toll of healthcare resources. AVR-25, a novel immune modulator experimental compound, was able to partially recover the pulmonary phenotype in the hyperoxia-induced experimental mouse model of BPD. We anticipate that AVR-25 will have therapeutic potential for managing human BPD.
Keywords: bronchopulmonary dysplasia, interleukin-10, anti-inflammatory
Introduction
Bronchopulmonary dysplasia (BPD) is a complex disease with significant adverse outcomes for infants born at < 30 weeks gestational age. There remains a big gap in understanding the pathophysiology of this neonatal lung disease because the injury pathways are complex. 1 There are few effective targeted therapies to prevent this devastating neonatal disease 2 and management takes a significant toll on healthcare resources, costing an average of $116,000 per infant discharged. 3 4 Additionally, BPD is associated with significant pulmonary and neurodevelopmental sequelae that continue to have health ramifications into adulthood. Surfactant, vitamin D, corticosteroids, and non-invasive respiratory support have all been incorporated into standard of care, but the incidence of BPD has remained the same (43%) 5 or has even increased over the years. 6
The pathologic hallmarks of BPD are enlarged simplified alveoli, pulmonary inflammation, 7 increased cell death, 8 and dysregulated angiogenic factors 9 that culminate in impaired alveolarization and disrupted vascularization of the lungs, usually secondary to an immature lung. A variety of inflammatory molecules, for example, tumor necrosis factor-α (TNF-α), interleukin 1-β (IL-1β), IL-6, inducible nitric oxide synthase (iNOS), are implicated and/or associated with BPD.
Chitin and chitosans are high molecular weight oligosaccharides with diverse biological activities having hypocholesterolemic, antimicrobial, immunostimulating, antitumorigenic, calcium and iron absorption acceleration, anti-inflammatory, and antioxidant properties. 10 We have previously shown that 11 a low molecular weight chitin analog AVR-25 (N-((2R,3R,4R,5S,6R)-5-(2S,3R,4R,5S,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)-2-(cyclohexyloxy)-4-hydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-3-yl)acetamide) modulates macrophages in a manner similar to its parent compound chitohexaose. 12 This modulation (via alternate activation) to a noninflammatory phenotype occurs by interacting with the host immune receptor, toll-like receptor 4 (TLR4). In doing so, AVR-25 increases the production of IL-10 and inhibits polymicrobial infection-induced inflammatory mediators such as TNF-α, IL-1β, IL-6, and iNOS in a cecal ligation and puncture (CLP)-induced sepsis model of mice. 11 It is known that alternatively activated macrophages are known to control inflammatory response, enhance phagocytic activity, and repairtissue damage. 13 Because BPD is a complex multifactorial inflammatory disease of the lung, we took advantage of this therapeutic property of AVR-25 and tested it in an experimental mouse model of BPD and found AVR-25 to be significantly efficacious in mitigating, at least in part, the BPD pulmonary phenotype.
Most preclinical research on BPD is performed in term neonatal rodents exposed to excessive hyperoxia because term pups are born in the saccular stage of lung development, morphologically comparable to moderate to very preterm, and are instead of, human neonates. Even in the absence of hyperoxia, oxidative stress might play an important role in impairing lung development after preterm birth. 14 Also, RA contains a supraphysiological oxygen pressure in comparison to the uterine environment, and preterm lungs have weaker antioxidant defense mechanisms. 15 Based on these assumptions, we chose the mouse as our preclinical model to test the efficacy of AVR-25 as a future target for preventing human BPD.
Methods
The neonatal mice BPD experiments were done following the methods described in Leary et al. 16 C57Bl/6 strain (Jackson Labs; Bar Harbor, Maine, United States) was used in the present study. The mouse model of experimental BPD used was exposed to 100% O 2 from postnatal day 0 (PN0) to PN4, followed by recovery in room air (RA) for 10 days. 8 16 AVR-25, in the dose of 10 mg/kg (30 µL), was injected intraperitoneally (IP) on PN2 and PN4. Mice were sacrificed on PN14. All the pups in a litter ( n = 6–7) were used for the subsequent analysis for histology, morphometry, immunostaining, Western blotting, and enzyme-linked immunosorbent assay (ELISA). All animal experiments were approved by the institutional animal care committee of Drexel University.
Bronchoalveolar lavage (BAL) fluid total cell and protein count, morphometry, cell counting, Western blotting, histology, and immunostaining were done as per Leary et al. 16 Individual macrophage and neutrophil cell populations were counted on cytosmears of BAL fluid after differential staining with HEMA (Fisher Scientific: Waltham, MA, United States); the absolute number of neutrophil and macrophage population was counted on each individual slide and further multiplied by the total cell count. Transferase dUTP nick end labeling (TUNEL) staining (Roche) was done following the manufacturer's instructions. Multiplex ELISA for inflammatory markers in the lungs and serum was done using the highly sensitive MSD multispot assay system (Rockville, Maryland, United States); fluorescent immunostaining was done for Ki67 (Abcam, 1:10) and von Willebrand factor (vWF) (Dako, 1:100). Western blotting was done for interferon gamma (IFN-γ), IL-1β, IL-6, and TNF-α (1:1000; Cell Signaling Technology, Massachusetts, United States). Densitometric quantification of the Western blots was done using ImageJ.
Statistical Analysis
All statistical analyses were performed using Graph Pad Prism version 8.0 (GraphPad Software; San Diego, California, United States). The sample size was based on our previous publications. 8 16 17 Using chord length as the outcome of alveolarization, we noted that this is abnormal in > 99% of wild-type BPD mice lungs. Using our targeted molecular therapy, we expected a 70% improvement. Hence, with an α = 0.005, β = 0.2, and a power of 80%, we need n = 6 in each group. The data are expressed as the mean ± standard error of mean with n = 5 to 7 mice in each group for morphometry, n = 4 to 5 mice for Western blotting, n = 4 to 5 mice for total cell count, and total protein estimation in BAL, n = 8 to 10 fields on hematoxylin and eosin sections for morphometry, n = 4 to 5 mice for immunostaining and TUNEL assay, and n = 4 to 5 mice for serum ELISA. Groups were compared with the two-tailed unpaired t -test and one- or two-way analysis of variance, as appropriate. A p- value < 0.05 was considered statistically significant.
Results and Discussion
We found AVR-25 to be significantly efficacious in mitigating the BPD pulmonary phenotype at 10 mg/kg, via IP dosing on PN2 and PN4, when the immature lungs are in the saccular stage of development. On PN14, the pups were sacrificed to collect the BAL fluid, blood, and lung tissues to assess for changes in various cytokine and chemokine levels, cell death, cell proliferation, and vascularization.
Histologically, BPD is characterized by large simplified alveoli (measured as chord length; Fig. 1A , 1B ), increased septal thickness ( Fig. 1A , 1C ), decreased radial alveolar count ( Fig. 1A , 1D ), and mechanistically, by decreased cell proliferation and increased cell death. After treatment with AVR-25, all these parameters were restored toward RA control values ( Fig. 1A–D ). In addition, after treatment with AVR-25, there was increased cell proliferation (evident from Ki67 staining, Fig. 2A ), improved vasculogenesis associated with sprouting angiogenesis of terminal blood capillaries and tiny blood vessels (evident from vWF staining, Fig. 2B ), and decreased cell death (evident from TUNEL staining, Fig. 2C ).
Fig. 1.
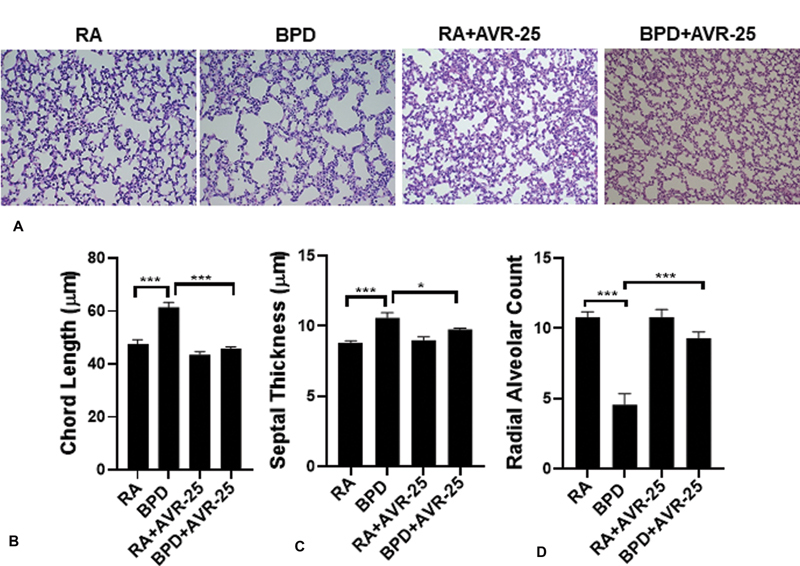
Histology, morphology: ( A ) hematoxylin and eosin (H&E) staining showing histology of mouse lungs in room air (RA), bronchopulmonary dysplasia (BPD) (100% O 2 from postnatal or PN day 0-PN4; recovery in RA for next 10 days till PN14), (RA + AVR-25) and (BPD + AVR-25) treated groups. In the BPD group, the alveoli are larger and simplified as compared with RA controls. After treatment with AVR-25 (10 mg/kg), intraperitoneally (IP) (30 µL), at PN2 and PN4, the alveolar architecture recovers, in part, similar to those of RA controls. There was no change in the histology in the (RA + AVR-25) treated group. Bottom panel shows improvement in ( B ) chord length, ( C ) septal thickness and ( D ) radial alveolar counts or radial alveolar count (RAC) after treatment. (X100). * p < 0.05, *** p < 0.0001, one-way analysis of variance (ANOVA), n = 3–6.
Fig. 2.
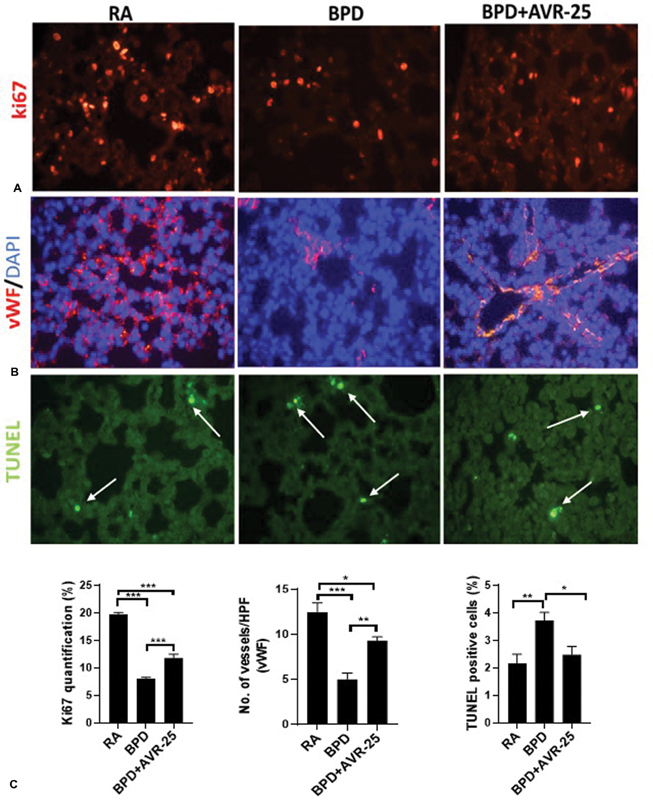
Cellular responses (proliferation and death), and vascularization of neonatal lungs after AVR-25 treatment: immunostaining showing ( A ) restoration of cell proliferation by Ki67 staining, ( B ) improved vascularization by von Willebrand factor (vWF) staining, and ( C ) decrease in cell death by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining with corresponding quantification in the bottom panel. Arrows (white) point to the fragmented/apoptotic nuclei within a cell. X400. * p < 0.05, ** p < 0.001, *** p < 0.0001, one-way analysis of variance (ANOVA), n = 3–6. BPD, bronchopulmonary dysplasia; HPF, high power field; RA, room air.
In BPD, several populations of circulating inflammatory cells infiltrate into the lungs. 18 At the same time, due to injury to the alveolar epithelium and damage of the endothelial wall, proteins leak into the BAL fluid and increase the total protein content. However, after treatment with AVR-25, the total immune cell numbers as well as the total protein content were significantly decreased and were close to RA control values ( Fig. 3A , 3B ). As pulmonary inflammation in BPD is characterized by the presence of inflammatory cells, for example neutrophils and monocytes, 1 we also counted the individual neutrophil and macrophage population in the BAL fluid cytosmears and found that both neutrophils and macrophages were significantly increased in the BPD group as compared with the RA and RA + AVR-25 group. After treatment with AVR-25, there was no change in the neutrophil population between the RA, RA + AVR-25, and BPD + AVR-25 groups; but the macrophage population was significantly decreased in the BPD + AVR-25 as compared with BPD group but higher than the RA group ( Fig. 3C , 3D ).
Fig. 3.
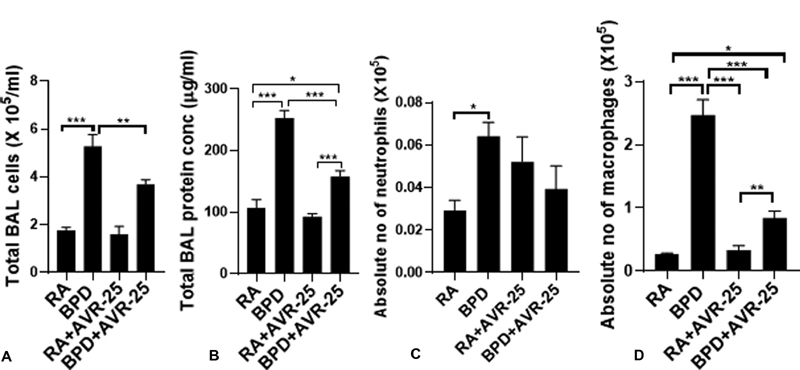
Biochemical changes in the neonatal lungs after AVR-25 treatment: ( A ) total immune and inflammatory cells in bronchoalveolar lavage (BAL) fluid and ( B ) total protein content in BAL fluid. After treatment with AVR-25 (10 mg/kg, intraperitoneally (IP) (30 µL), at PN2 and PN4, there is a significant decrease in inflammatory cells as well as total protein in the treated group as compared with bronchopulmonary dysplasia (BPD) alone group. ( C ) Individual neutrophil and ( D ) macrophage population in the BAL fluid as evident from cytosmears by differential HEMA staining. Neutrophils are modestly increased while macrophages are robustly increased in the BPD group as compared with room air (RA) controls. After treatment with AVR-25, there is no change in the neutrophil population in both the RA and in the BPD group. However, although the macrophage population did decrease after treatment in the BPD group, it was still higher when compared with RA only or RA + AVR-25 groups. * p < 0.05, ** p < 0.001, *** p < 0.0001, one-way analysis of variance (ANOVA), n = 5–7.
Using Western blot in the lung tissues ( Fig. 4A ) and multiplex ELISA in the serum ( Fig. 4B , 4C ), we confirmed that after treatment with AVR-25 there was decreased expression of proinflammatory cytokines IFN-γ, IL-1β, IL-6, TNF-α, and an increase in anti-inflammatory cytokine IL-10, in both the lungs and in the serum ( Fig. 4B , 4C ) of the treated groups as compared with the control groups. These cytokines were selected based on our previous observations. 19 We also found a decrease in macrophage inflammatory protein-1 (MIP-1), monocyte chemoattractant protein-1 (MCP-1), and induced protein-10 (IP-10) in mouse lungs and serum after treatment. It is to be noted that lung inflammation is triggered by the activation of the innate immunity system that in turn activates the cell surface receptors. 20 These receptors play a key role in the activation of the inflammasome, and it is for this reason that this pathway may be an active target for drug development to combat inflammation. 21 MCP-1 recruits inflammatory cells to the area of injury, while TNF-α enhances the expression of other proinflammatory cytokines. IL-10, on the other hand, suppresses inflammatory response by inhibiting the nuclear factor kappa B pathway. 1
Fig. 4.
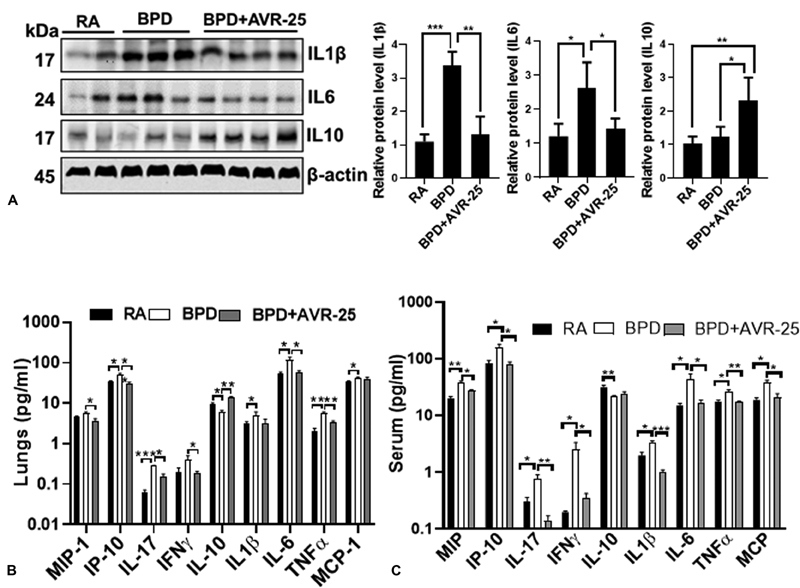
Suppression of inflammation after AVR-25 treatment: ( A ) representative Western blotting showing the expression of interleukin-1β (IL-1β), IL-6, and IL-10 in the lung tissues. β actin is the loading control. Both IL-1β and IL-6 are increased in bronchopulmonary dysplasia (BPD) that is markedly decreased after AVR-25 treatment; in contrast, the anti-inflammatory IL-10 is upregulated following treatment with AVR-25. n = 3–5. Similar results were also obtained in a multiplex enzyme-linked immunosorbent assay (ELISA) assay on ( B ) lung lysates and ( C ) serum; * p < 0.05, ** p < 0.001, *** p < 0.0001, one-way analysis of variance (ANOVA), n = 3–6. For cytokine ELISA assays, two-way ANOVA (Tukey's multiple comparison test) was done. IP-10, induced protein-10; MCP-1, monocyte chemoattractant protein-1; MIP-1, macrophage inflammatory protein-1; RA, room air; TNF-α, tumor necrosis factor-α.
The hyperoxia-induced mouse model of experimental BPD does have some similarities to human BPD. Specifically, the term mouse lung is in the saccular stage of lung development and postnatal day (PN0) at birth is ∼26 to 28 weeks of human equivalent gestational age. 22 Furthermore, while the term mouse is surfactant-sufficient, it can be considered akin to a preterm infant who has received a full complement of antenatal steroids to mature the lung. Lack of surfactant (or presence of respiratory distress syndrome) is not a prerequisite to develop BPD. BPD is a disease of genetic–environmental interactions; among the latter contributors, hyperoxia per se (depending upon dose and duration of exposure) is sufficient to initiate and result in persistent inflammation on the path to BPD. 1 23 In addition, ante (chorioamnionitis) and postnatal sepsis, in conjunction with invasive ventilation ± hyperoxia exposure has also been associated with BPD. 24 25
Although the mechanism of action of AVR-25 has not been studied in detail, from the preliminary data generated from this current study, we believe that AVR-25 modulates the TLR4 pathway by decreasing the proinflammatory cytokines and chemokines while simultaneously increasing the expression of the anti-inflammatory IL-10. AVR-25 selectively binds to TLR4 but not to TLR2 protein (data not shown) and in doing so, stimulates macrophages to produce the compensatory cytokine IL-10. 11 The depletion of IL-10 is observed along with a significant increase in inflammatory cytokines IL-6, IL-8, and IL-1β in preterm neonates between 3 and 21 days 26 after birth. It is also known that IL-10 negatively regulates the production of IL-1β, 27 28 29 thus potentially protecting from lung injury, which is evident from our results. More importantly, AVR-25 did not have any toxic effect on other organ systems in the neonatal pups (data not shown). Hence, we believe that stimulation of IL-10 by AVR-25 contributes towards an improved lung phenotype, which is otherwise compromised in BPD.
Conclusion
We report that compound AVR-25 partially prevents the development of the hyperoxia-induced experimental BPD pulmonary phenotype in neonatal mice pups. This property of AVR-25 can be harnessed as a potential therapeutic agent for the prevention of a neonatal disease for which there is no available treatment, to date. The detailed mechanism of action of AVR-25 and structure activity optimization is currently in progress.
Funding Statement
Funding This work was partly supported by a research contract grant received from AyuVis Research Inc.
Conflict of Interest None declared.
Current affiliation: Cooper Health University and Hospital, Suite 206, 401 Haddon Avenue, Education and Research Building, Camden, NJ 08103, USA.
These authors contributed equally to this article.
Authors' Contributions
P.D., S.A., B.A., and V.B. conceptualized and designed the study, drafted the initial manuscript, and submitted as approved; P.D., S.A., D.S., B.A., and V.P. performed the initial experiments. All the authors approved the final manuscript.
References
- 1.Balany J, Bhandari V. Understanding the impact of infection, inflammation, and their persistence in the pathogenesis of bronchopulmonary dysplasia. Front Med (Lausanne) 2015;2:90. doi: 10.3389/fmed.2015.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhandari V. Drug therapy trials for the prevention of bronchopulmonary dysplasia: current and future targets. Front Pediatr. 2014;2:76. doi: 10.3389/fped.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell R B, Green N S, Steiner C A et al. Cost of hospitalization for preterm and low birth weight infants in the United States. Pediatrics. 2007;120(01):e1–e9. doi: 10.1542/peds.2006-2386. [DOI] [PubMed] [Google Scholar]
- 4.Jobe A H. Mechanisms of lung injury and bronchopulmonary dysplasia. Am J Perinatol. 2016;33(11):1076–1078. doi: 10.1055/s-0036-1586107. [DOI] [PubMed] [Google Scholar]
- 5.Smith V C, Zupancic J A, McCormick M C et al. Trends in severe bronchopulmonary dysplasia rates between 1994 and 2002. J Pediatr. 2005;146(04):469–473. doi: 10.1016/j.jpeds.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 6.Trembath A, Laughon M M. Predictors of bronchopulmonary dysplasia. Clin Perinatol. 2012;39(03):585–601. doi: 10.1016/j.clp.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gentner S, Laube M, Uhlig U et al. Inflammatory mediators in tracheal aspirates of preterm infants participating in a randomized trial of permissive hypercapnia. Front Pediatr. 2017;5:246. doi: 10.3389/fped.2017.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sureshbabu A, Syed M, Das P et al. Inhibition of regulatory-associated protein of mechanistic target of rapamycin prevents hyperoxia-induced lung injury by enhancing autophagy and reducing apoptosis in neonatal mice. Am J Respir Cell Mol Biol. 2016;55(05):722–735. doi: 10.1165/rcmb.2015-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syed M A, Choo-Wing R, Homer R J, Bhandari V. Role of nitric oxide isoforms in vascular and alveolar development and lung injury in vascular endothelial growth factor overexpressing neonatal mice lungs. PLoS One. 2016;11(01):e0147588. doi: 10.1371/journal.pone.0147588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia W S, Liu P, Zhang J L, Chen J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011;25(02):170–179. [Google Scholar]
- 11.Das P, Panda S K, Agarwal B et al. Novel chitohexaose analog protects young and aged mice from CLP induced polymicrobial sepsis. Sci Rep. 2019;9(01):2904. doi: 10.1038/s41598-019-38731-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panda S K, Kumar S, Tupperwar N C et al. Chitohexaose activates macrophages by alternate pathway through TLR4 and blocks endotoxemia. PLoS Pathog. 2012;8(05):e1002717. doi: 10.1371/journal.ppat.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair M G, Cochrane D W, Allen J E. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol Lett. 2003;85(02):173–180. doi: 10.1016/s0165-2478(02)00225-0. [DOI] [PubMed] [Google Scholar]
- 14.Salaets T, Aertgeerts M, Gie A et al. Preterm birth impairs postnatal lung development in the neonatal rabbit model. Respir Res. 2020;21(01):59. doi: 10.1186/s12931-020-1321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank L, Sosenko I R. Failure of premature rabbits to increase antioxidant enzymes during hyperoxic exposure: increased susceptibility to pulmonary oxygen toxicity compared with term rabbits. Pediatr Res. 1991;29(03):292–296. doi: 10.1203/00006450-199103000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Leary S, Das P, Ponnalagu D, Singh H, Bhandari V. Genetic strain and sex differences in a hyperoxia-induced mouse model of varying severity of bronchopulmonary dysplasia. Am J Pathol. 2019;189(05):999–1014. doi: 10.1016/j.ajpath.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choo-Wing R, Syed M A, Harijith A et al. Hyperoxia and interferon-γ-induced injury in developing lungs occur via cyclooxygenase-2 and the endoplasmic reticulum stress-dependent pathway. Am J Respir Cell Mol Biol. 2013;48(06):749–757. doi: 10.1165/rcmb.2012-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Um-Bergström P, Hallberg J, Pourbazargan M et al. Pulmonary outcomes in adults with a history of bronchopulmonary dysplasia differ from patients with asthma. Respir Res. 2019;20(01):102. doi: 10.1186/s12931-019-1075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson A, Bhandari V. Pulmonary biomarkers of bronchopulmonary dysplasia. Biomark Insights. 2008;3:361–373. doi: 10.4137/bmi.s834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolls J K. Commentary: understanding the impact of infection, inflammation and their persistence in the pathogenesis of bronchopulmonary dysplasia. Front Med (Lausanne) 2017;4:24. doi: 10.3389/fmed.2017.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim H Y, Chang Y J, Subramanian Set al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity J Allergy Clin Immunol 201212901216–27.e1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol. 2014;307(12):L936–L947. doi: 10.1152/ajplung.00159.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harijith A. Switzerland: Springer International Publishing; 2016. Hyperoxia in the pathogenesis of bronchopulmonary dysplasia. In Bronchopulmonary Dysplasia; pp. 3–26. [Google Scholar]
- 24.Aghai Z H, Camacho J, Saslow J G et al. Impact of histological chorioamnionitis on tracheal aspirate cytokines in premature infants. Am J Perinatol. 2012;29(07):567–572. doi: 10.1055/s-0032-1311980. [DOI] [PubMed] [Google Scholar]
- 25.Nayeri U A, Buhimschi C S, Zhao G, Buhimschi I A, Bhandari V. Components of the antepartum, intrapartum, and postpartum exposome impact on distinct short-term adverse neonatal outcomes of premature infants: a prospective cohort study. PLoS One. 2018;13(12):e0207298. doi: 10.1371/journal.pone.0207298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ambalavanan N, Carlo W A, D'Angio C T et al. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics. 2009;123(04):1132–1141. doi: 10.1542/peds.2008-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vento G, Capoluongo E, Matassa P G et al. Serum levels of seven cytokines in premature ventilated newborns: correlations with old and new forms of bronchopulmonary dysplasia. Intensive Care Med. 2006;32(05):723–730. doi: 10.1007/s00134-006-0138-1. [DOI] [PubMed] [Google Scholar]
- 28.Garingo A, Tesoriero L, Cayabyab R et al. Constitutive IL-10 expression by lung inflammatory cells and risk for bronchopulmonary dysplasia. Pediatr Res. 2007;61(02):197–202. doi: 10.1203/pdr.0b013e31802d8a1c. [DOI] [PubMed] [Google Scholar]
- 29.Sanin D E, Prendergast C T, Mountford A P. IL-10 production in macrophages is regulated by a TLR-driven CREB-mediated mechanism that is linked to genes involved in cell metabolism. J Immunol. 2015;195(03):1218–1232. doi: 10.4049/jimmunol.1500146. [DOI] [PMC free article] [PubMed] [Google Scholar]