

Abstract
Autophagy is associated with the cytoprotection of physiological processes against inflammation and oxidative stress. Salvia miltiorrhiza possesses cardiovascular protective actions and has powerful anti-oxidative and anti-inflammatory effects; however, whether and how Salvia miltiorrhiza-derived microRNAs (miRNAs) protect vascular smooth muscle cells (VSMCs) by inducing autophagy across species are unknown. We first screened and identified Sal-miR-58 from Salvia miltiorrhiza as a natural autophagy inducer. Synthetic Sal-miR-58 suppresses chronic angiotensin II (Ang II) infusion-induced abdominal aortic aneurysm (AAA) formation in mice, as well as induces autophagy in VSMCs and attenuates the inflammatory response elicited by Ang II in vivo and in vitro. Mechanistically, Sal-miR-58 downregulates Krüppel-like factor 3 (KLF3) expression through direct binding to the 3′ UTR of KLF3, which in turn relieves KLF3 repression of E3 ubiquitin ligase neural precursor cell-expressed developmentally downregulated 4-like (NEDD4L) expression, whereas NEDD4L upregulation increases the ubiquitination and degradation of the platelet isoform of phosphofructokinase (PFKP), subsequently leading to a decrease in the activation of Akt/mammalian target of rapamycin (mTOR) signaling and facilitating VSMC autophagy induced by Sal-miR-58 in the context of chronic Ang II stimulation and aneurysm formation. Our results provide the first evidence that plant-derived Sal-miR-58 induces autophagy and attenuates inflammation in VSMCs through cross-species modulation of the KLF3/NEDD4L/PFKP regulatory pathway.
Keywords: autophagy, AAA, KLF3, NEDD4L, PFKP, Sal-miR-58, vascular smooth muscle cell
Graphical Abstract
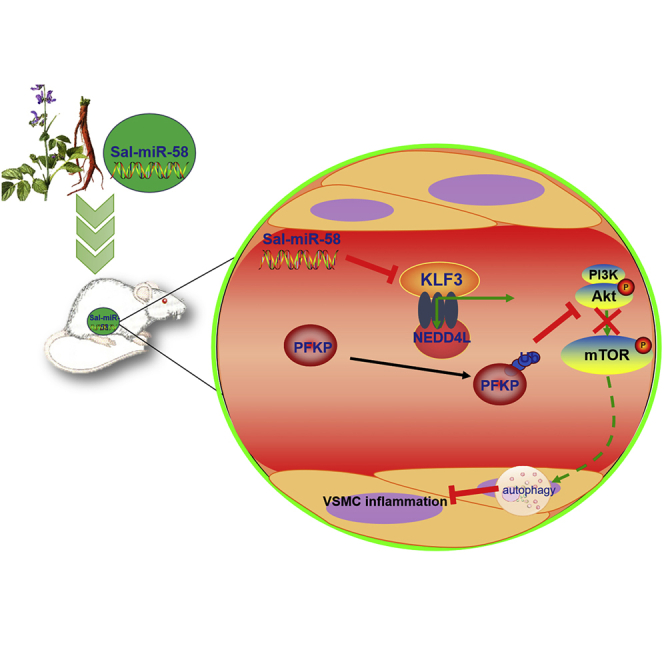
This study identified Salvia miltiorrhiza-derived Sal-miR-58 as a novel vasoprotective agent and demonstrated that Sal-miR-58 induces autophagy and attenuates inflammation in VSMCs through cross-species modulation of the KLF3/NEDD4L/PFKP regulatory pathway.
Introduction
Autophagy and inflammation are two fundamental biological processes that are present in a number of physiological and pathological conditions.1 Autophagy selectively degrades damaged proteins and removes dysfunctional organelles under basal conditions, maintaining normal cellular homeostasis and energy balance.2 Under pathological conditions, autophagy can be activated or inhibited in most tissues by multiple stimuli and various stresses, including metabolic stress, reactive species, cytokines, and drugs.3 In the vasculature, defective autophagy has been implicated in a variety of vascular diseases, including hypertension, vascular aging, atherosclerosis, and interventional restenosis.2 Activation of inflammation has been shown to promote the recruitment of immune cells and secretion of cytokines and chemokines, which in turn help recruit other immune cells for activation of adaptive immune responses, thus playing defense against microorganisms and other pathogens.1 However, chronic inflammation is associated with multiple vascular diseases such as atherosclerosis, restenosis, and abdominal aortic aneurysm (AAA).4,5 Emerging evidence has shown that autophagy has important effects on the inflammatory reaction. In atherosclerosis, excessively stimulated autophagy can lead to endothelial cell death that can result in plaque destabilization, maintaining the inflammatory status of the plaque.6 A recent study demonstrated that autophagy deficiency in vascular smooth muscle cells (VSMCs) promotes atherosclerosis at least in part through enhancing the inflammatory response.2 These data suggest that autophagy plays a critical role in vascular diseases through regulating vascular cell functions, and the induction of autophagy can be beneficial in decreasing the inflammatory response in arteries. Therefore, manipulating autophagy through pharmacological approaches represents an attractive strategy to treat or prevent arterial diseases.
An increasing number of studies have shown that most nutritional and pharmacological approaches that are known to extend lifespan induce autophagy, and thus autophagy seems to be a causal effector of pro-longevity drugs, such as phytochemical resveratrol,7 rapamycin, metformin, and nicotinamide adenine dinucleotide (NAD+) precursors.8 These longevity drugs lose their efficacy when autophagy is suppressed.9 These results indicate that autophagy is closely associated with general cytoprotection properties and overall health. Recently, Carmona-Gutierrez et al.10 identified flavonoid 4,4′-dimethoxychalcone (DMC) as a natural compound with anti-aging properties in Asian traditional medicine and demonstrated that DMC promotes autophagy-dependent longevity across species.
Salvia miltiorrhiza (Danshen) is a well-known medicinal herb that possesses broad cardiovascular protective actions and has been widely used in Asian countries. The chemical components of Salvia miltiorrhiza identified to date include two main types, that is, lipophilic compounds that have a diterpene quinone structure, and hydrophilic compounds that mainly have a phenolic acid structure.11 Accumulating evidence suggests that this multi-component cardiovascular herbal medicine has pleiotropic effects including anti-inflammation, anti-oxidation, anti-thrombosis, lipid lowering, and improvement of endothelial function.12,13 Previous studies have shown that there are many microRNAs (miRNAs) unique to Salvia miltiorrhiza, and these miRNAs regulate the biosynthesis of major medicinal components and development and responses to abiotic stress.14 However, it is still unclear whether miRNAs derived from Salvia miltiorrhiza, as the effective components, participate in the regulation of cardiovascular diseases, especially autophagy and inflammation. Remarkably, Zhang et al.15 found plant-derived miRNAs in human serum and tissues, and they reported that exogenous plant miR168a can regulate the expression of low-density lipoprotein receptor adaptor protein 1 (LDLRAP1) in humans and mice, revealing an evidence of cross-kingdom regulation by miRNAs. Plant miRNAs are 2′-O-methyl modified on their terminal nucleotide,16 and thus they were more stable than mammalian miRNAs.15 Therefore, it is of great significance to identify Salvia miltiorrhiza-derived miRNAs with cardiovascular protection properties and to investigate the mechanism underlying the induction of autophagy by Salvia miltiorrhiza-derived miRNAs.
Autophagy is tightly controlled by a family of autophagy regulators, autophagy-related proteins (Atgs), and their homologs and regulated by different signaling pathways such as mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and mammalian target of rapamycin (mTOR),17,18 which coordinate autophagy through regulating autophagosome formation and autophagosome-lysosome fusion. In this study, we screened and identified miRNAs from Salvia miltiorrhiza and investigated whether and, if so, how the identified miRNA regulates autophagy in angiotensin II (Ang II)-stimulated VSMCs and mouse AAA models. In this study, we report the identification of Sal-miR-58 as a natural autophagy inducer and show that Sal-miR-58 induces autophagy and attenuates inflammation in VSMCs through cross-species modulation of the Krüppel-like factor 3 (KLF3)/neural precursor cell-expressed developmentally down-regulated 4-like (NEDD4L)/platelet isoform of phosphofructokinase (PFKP) regulatory pathway.
Results
Sal-miR-58 Specifically Present in Salvia miltiorrhiza Might Enter the Mouse Body after Exogenous Administration to Mice
We first used a high-throughput sequencing method to identify the miRNAs highly expressed in Salvia miltiorrhiza. As a result, 187 miRNAs, including ppt-miR-414, hbr-miR-156, gra-miR-172b-5p, and Sal-miR-58, were identified from sequence data. Among them, 41 miRNAs were specifically present in Salvia miltiorrhiza, and Sal-miR-58 was the most highly enriched miRNA compared with other miRNAs (Figures 1A and 1B). Moreover, sequence analysis revealed no significant homology between the Sal-miR-58 and mammalian miRNAs, and the expression of Sal-miR-58 was undetectable in mouse VSMCs (Figure 1B). In order to determine whether the miRNAs derived from Salvia miltiorrhiza could be absorbed in mice, Salvia miltiorrhiza was intragastrically administered to mice, and the expression of Sal-miR-58 in different tissues of mice was measured 6 h later. The results showed that Sal-miR-58 was detected in the blood vessels, liver, spleen, stomach, and small intestine of mice (Figure 1C). Because Sal-miR-58 is the most abundant miRNA in Salvia miltiorrhiza (Figure 1A), and it is widely distributed after oral administration in many tissues and organs of mice (Figure 1C), we therefore focused on the roles of Sal-miR-58 in all subsequent experiments.
Figure 1.

Sal-miR-58 May Enter the Mouse Body and Induces VSMC Autophagy and Inhibits the Inflammatory Response in a Mouse AAA Model
(A) The strategy and result analysis of high-throughput second-generation deep sequencing of Salvia miltiorrhiza-derived miRNAs. (B) qRT-PCR analysis of four miRNAs in Salvia miltiorrhiza and mouse VSMCs. ∗∗∗p < 0.001 versus VSMCs (n = 3). (C) qRT-PCR detected the expression of Salvia miltiorrhiza-derived miRNAs in different mouse tissues after Salvia miltiorrhiza was orally administered to ApoE−/− mice for 6 h. ∗∗∗p < 0.001 versus ppt-miR-414 (n = 3 in each group). (D) Sal-miR-58 was determined by qRT-PCR in the serum of mouse AAA models after exogenous administration of Sal-miR-58 for 28 days. ∗∗∗p < 0.001 versus ApoE−/− (n = 5 in each group). (E) miRNAs isolated from mouse serum were treated with/without sodium periodate, and Sal-miR-58 was detected by qRT-PCR. (F) Aortic ultrasonography was used to detect the diameter of abdominal aortas. The data represent the mean ± SEM. ∗∗∗p < 0.001 versus ApoE−/−; ###p < 0.001 versus ApoE−/−+Ang II (n = 5 in each group). (G) Representative photographs of ApoE−/− mouse abdominal aortic dilation after exogenous administration of Sal-miR-58 for 28 days. The data represent the mean ± SEM. ∗∗∗p < 0.001 versus ApoE−/−; ###p < 0.001 versus ApoE−/−+Ang II (n = 5 in each group). (H) Western blot analysis of the inflammatory factors and autophagy markers in mouse AAA tissues. (I) Levels of IL-1β, IL-6, and TNF-α in the serum of mouse AAA models were determined by the ELISA. ∗∗∗p < 0.001 versus ApoE−/−; ###p < 0.001 versus ApoE−/−+Ang II (n = 5 in each group). (J) Immunofluorescence staining of α-SMA (SMA; green), Beclin1 (red) and the nucleus (DAPI; blue) in the injured aortas of control and ApoE−/− mice exposed to Ang II for 28 days. Scale bars, 100 μm. (K) Immunofluorescence staining of α-SMA (SMA; green), LC3B (red) and the nucleus (DAPI; blue) in the injured aortas of control and ApoE−/− mice exposed to Ang II for 28 days. Scale bars, 100 μm.
Synthetic Sal-miR-58 Induces VSMC Autophagy and Inhibits the Inflammatory Response in AAA
One of the important mechanisms underlying the beneficial effects of Salvia miltiorrhiza is its anti-inflammation.19 To determine whether Sal-miR-58 exerts a vasoprotective effect by anti-inflammation, apolipoprotein E (ApoE)−/− mice were infused with Ang II (1,000 ng/kg/min) for 4 weeks to induce AAA formation. First, we demonstrated that high levels of Sal-miR-58 could be detected in the serum of ApoE−/− mice regardless of treatment with Ang II (Figure 1D) and showed that Sal-miR-58 was a genuine plant miRNA, as evidenced by the fact that Sal-miR-58 isolated from mouse serum was resistant to sodium periodate (oxidizing agent) (Figure 1E), which is characteristic of plant miRNAs.16 Then, we found that compared with saline-infused control mice, infusion of Ang II for 4 weeks resulted in a significant increase in the diameter of the abdominal aortas above the renal artery (diameter of abdominal aortas, 2.383 ± 4.721 mm versus 1.042 ± 3.103 mm, ∗∗∗p < 0.001); however, the abdominal aorta diameter of AAA in ApoE−/− mice treated with synthetic Sal-miR-58 was significantly lower than that of the ApoE−/− mice infused with Ang II alone (diameter of abdominal aortas, 1.832 ± 5.176 mm versus 2.383 ± 4.721 mm, ###p < 0.001), as seen by ultrasound as well as by the naked eye (Figure 1F). The mean expansion rate of AAA in ApoE−/− mice treated with synthetic Sal-miR-58 was significantly lower than that of the ApoE−/− mice infused with Ang II alone (expansion rate, 25.620% ± 1.789% versus 69.924% ± 2.143%, ###p < 0.001) (Figure 1G). These findings suggest that Salvia miltiorrhiza-derived Sal-miR-58 may suppress chronic Ang II infusion-induced AAA formation in mice. Because AAA pathogenesis is known to be a multifactorial inflammatory process,20 we determined whether Sal-miR-58 inhibits Ang II infusion-induced AAA formation through its anti-inflammation action. Thus, we examined the effect of Sal-miR-58 on nuclear factor κB (NF-κB) p65 and p50, which play an essential role in the inflammatory response through regulation of genes encoding inflammatory cytokines (interleukin 1β [IL-1β], tumor necrosis factor α [TNF-α], IL-6).21,22 The results showed that the expression levels of NF-κB p65 and p50 in the aortas of Ang II-infused ApoE−/− mice were significantly higher than those in control mice, while Sal-miR-58 treatment significantly reduced the expression of NF-κB p65 and p50 (Figure 1H). Consistently, compared with Ang II-infused ApoE−/− mice, the serum levels of inflammatory factors IL-1β, IL-6, and TNF-α also declined significantly after Sal-miR-58 administration to Ang II-infused ApoE−/− mice, as detected by ELISA (Figure 1I).
Because autophagy is closely related to inflammatory response, and loss of autophagy is a crucial factor in triggering inflammatory response,23 we investigated the effect of Sal-miR-58 on autophagy. We found that the expression of autophagy-related genes Atg5 and Beclin1 was clearly reduced in the aortas of Ang II-infused ApoE−/− mice, whereas treating Ang II-infused ApoE−/− mice with Sal-miR-58 markedly upregulated the expression levels of these two genes. Moreover, the ratio of LC3BII/LC3BI was significantly increased following Sal-miR-58 treatment (Figure 1H). Furthermore, we used a VSMC marker (smooth muscle α-actin [α-SMA]) to label VSMCs in the injured aortas to observe the localization of Beclin1 and LC3B in VSMCs. Immunofluorescence staining showed that Beclin1 (red) and LC3B (red) co-localized with α-SMA in the aortic VSMCs injured by Ang II, and the fluorescence intensity of Beclin1 and LC3B was significantly increased after Sal-miR-58 treatment compared with that injured by Ang II alone (Figures 1J and 1K). These results suggest that Sal-miR-58 induces autophagy in ApoE−/− mouse aortas injured by chronic Ang II infusion.
Sal-miR-58 Inhibits Ang II-Induced Inflammation in VSMCs
We next investigated whether Sal-miR-58 also has an anti-inflammatory effect on the cultured VSMCs. First, we used Ang II to treat mouse VSMCs and assessed Ang II-induced inflammation via the production of pro-inflammatory factors. Western blot analysis showed that Ang II treatment obviously enhanced the expression of NF-κB p65 and p50 in a time- and concentration-dependent manner (Figures 2A and 2B). Correspondingly, in Ang II-treated VSMCs, NF-κB-regulated inflammatory genes IL-1β, IL-6, and TNF-α were also upregulated at the transcriptional level in a concentration- and time-dependent manner (Figures 2C and 2D). Moreover, parallel with alterations in their mRNAs, Ang II time- and concentration-dependently increased the levels of IL-1β, IL-6, and TNF-α proteins in the medium (Figures 2E and 2F). We then transfected VSMCs with Sal-miR-58 and confirmed that Sal-miR-58 was successfully transduced into cells and was resistant to oxidation by sodium periodate (Figures 2G and 2H). Transfecting VSMCs with Sal-miR-58 before Ang II stimulation largely abrogated the upregulation of the above-mentioned inflammatory genes induced by Ang II, as shown by western blotting, qRT-PCR, and ELISA assays (Figures 2I–2K). Also, immunofluorescence staining revealed that the fluorescence intensity of IL-1β, IL-6, and TNF-α was strengthened in Ang II-treated VSMCs, whereas Ang II-induced upregulation of the inflammatory factors was attenuated by Sal-miR-58 transfection (Figures S1A–S1C). These data suggest that Sal-miR-58 suppresses Ang II-induced inflammatory response in VSMCs.
Figure 2.

Sal-miR-58 Inhibits Ang II-Induced Inflammation in VSMCs
(A) Western blot analysis for NF-κB p50 expression. (B)Western blot analysis for NF-κB p65 expression.(C) Relative expression of IL-6, TNF-α, and IL-1β mRNA was examined by qRT-PCR and presented after normalizing to 18S rRNA (mean ± SEM; n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus 0 h Ang II. (D) Relative expression of IL-6, TNF-α, and IL-1β mRNA was examined by qRT-PCR and presented after normalizing to 18S rRNA (mean ± SEM; n = 3). ∗∗p < 0.01, ∗∗∗p < 0.001 versus 0 M Ang II.(E ) ELISA analysis of IL-1β, IL-6, or TNF-α in the culture medium of VSMCs. ∗∗p < 0.01, ∗∗∗p < 0.001 versus 0 h Ang II (n = 3). (F) ELISA analysis of IL-1β, IL-6, or TNF-α in the culture medium of VSMCs. ∗∗p < 0.01, ∗∗∗p < 0.001 versus 0 M Ang II (n = 3). (G) Sal-miR-58 was determined by qRT-PCR in VSMCs transfected with Sal-miR-58. ∗∗∗p < 0.001 versus miR-Ctl (n = 3). (H) qRT-PCR detected Sal-miR-58 in VSMCs transfected with Sal-miR-58 after treatment with sodium periodate. ∗∗∗p < 0.001 versus miR-Ctl+Unoxidized (n = 3). (I) VSMCs were treated with Ang II alone or combined with Sal-miR-58 for 24 h, and the expression of NF-κB p65 and p50 was examined by western blotting. (J) Relative expression of IL-6, TNF-α, and IL-1β mRNA was determined by qRT-PCR in VSMCs treated as in (I) and presented after normalizing to 18S rRNA. ∗∗p < 0.01, ∗∗∗p < 0.001 versus miR-Ctl; ###p < 0.001 versus miR-Ctl+Ang II (n = 3). (K) Levels of IL-1β, IL-6, and TNF-α were determined by ELISA in the culture medium of VSMCs treated as in (I). ∗∗p < 0.01, ∗∗∗p < 0.001 versus miR-Ctl; ##p < 0.01, ###p < 0.001 versus miR-Ctl+Ang II (n = 5).
Sal-miR-58-Induced Autophagy Attenuates VSMC Inflammation Elicited by Ang II
Because it has been demonstrated that autophagy has important effects on the induction of the inflammatory reaction,6,24 we investigated the relationship between Sal-miR-58-induced autophagy and inflammation in Ang II-treated VSMCs. Western blot analysis showed that treating VSMCs with Ang II clearly reduced the expression of Beclin1 and Atg5, which was reversed by Sal-miR-58 transfection, as evidenced by increased expression of Beclin1 and Atg5 as well as by enhanced conversion of LC3BI to LC3BII (Figure 3A). To further validate these results, we infected VSMCs with red fluorescent protein (RFP)-green fluorescent protein (GFP)-LC3B double-labeled adeno-associated virus (AAV) to label autophagosomes and their maturation to autolysosomes and examined GFP-LC3B (green puncta) and mRFP-LC3B (red puncta) formation by a fluorescence microscope. The results showed that autophagy was markedly increased in Sal-miR-58-transfected VSMCs regardless of treatment with Ang II, as shown by increased bright fluorescent spots (Figure 3B), indicating that LC3B protein is translocated to the lysosomal membrane and showing that Sal-miR-58 induces VSMC autophagy.
Figure 3.

Sal-miR-58-Induced Autophagy Attenuates VSMC Inflammation Elicited by Ang II
(A) VSMCs were treated with Ang II alone or combined with Sal-miR-58 for 24 h, and the expression of Belin1, Atg5, and LC3B was examined by western blotting. (B) VSMCs treated as in (A) were infected with RFP-GFP-LC3B double-labeled adeno-associated virus, and autophagosomes and autolysosomes were observed by a fluorescence microscope. Yellow fluorescent spots indicate autophagosome, and red fluorescent spots indicate autolysosome. Scale bar, 100 μm. (C) VSMCs were treated with rapamycin (25 nM) and/or Ang II for 24 h, and the protein was harvested from VSMCs and analyzed by western blotting using antibodies against Beclin1, Atg5, and LC3B. (D) VSMCs were treated as in (C) and infected with RFP-GFP-LC3B double-labeled adeno-associated virus, and autophagosomes and autolysosomes were observed as in (B). Scale bar, 25 μm. (E) VSMCs were treated as in (C), and the expression of NF-κB p65 and p50 was examined by western blotting. (F) VSMCs were treated as in (C), and the expression of IL-1β, IL-6, and TNF-α was examined by ELISA in the culture medium of VSMCs. ∗∗p < 0.01, ∗∗∗p < 0.001 versus miR-Ctl; #p < 0.05, ##p < 0.01, ###p < 0.001 versus miR-Ctl+Ang II, n = 5. (G) VSMCs were treated with Sal-miR-58 and Ang II or combined with chloroquine (50 μM) or bafilomycin A1 (25 nM), and the expression of Beclin1, Atg5, or LC3B was examined by western blotting. (H) VSMCs were treated with Sal-miR-58 or bafilomycin A1 alone or together and infected with RFP-GFP-LC3B double-labeled adeno-associated virus to detect autophagosomes and autolysosomes by a method described as in (B). Scale bar, 100 μm. (I) VSMCs were treated as in (G), and the expression of NF-κB p50 and p65 was examined by western blotting. (J) VSMCs were treated as in (G), and the expression of IL-1β, IL-6, or TNF-α was examined by ELISA in the culture medium of VSMCs. ∗p < 0.05 versus miR-Ctl; ##p < 0.01, ###p < 0.001 versus Sal-miR-58+Ang II (n = 5).
In the further experiments, we used rapamycin, an inducer of autophagy, to treat VSMCs and confirmed that rapamycin (25 nM) treatment for 2 h markedly upregulated Beclin1 and Atg5 expression and promoted the conversion of LC3BI to LC3B II (Figure 3C). Also, in RFP-GFP-LC3B double-labeled AAV-infected VSMCs, rapamycin treatment clearly increased the formation of autolysosomes (Figure 3D). Remarkably, rapamycin largely abrogated the upregulation of NF-κB and its downstream genes IL-1β, IL-6, and TNF-α by Ang II, as shown by western blotting and ELISA analysis (Figures 3E and 3F). These results indicate that the autophagy triggered by rapamycin decreases the inflammatory response induced by Ang II in VSMCs. In addition, we showed that treating VSMCs with autophagic inhibitors bafilomycin A1 (25 nM) or chloroquine (50 μM) could abolished the inducing effect of Sal-miR-58 on VSMC autophagy, as shown by decreased Beclin1 and Atg5 expression as well as by the reduced ratio of LC3BII/LC3BI and LC3B-positive vesicles (Figures 3G and 3H). Correspondingly, bafilomycin A1 or chloroquine treatment largely abrogated the inhibitory effects of Sal-miR-58 on the expression of NF-κB and its downstream genes IL-1β, IL-6, and TNF-α (Figures 3I and 3J). Taken together, these results indicate that Sal-miR-58-induced VSMC autophagy is accompanied by decreased inflammation in VSMCs.
Sal-miR-58 Inhibits Ang II-Induced Inflammation by Downregulating KLF3 in VSMCs
Because plant-derived miRNAs, through dietary intake, could regulate mammalian gene expression by binding to the 3′ UTR of mammalian target genes,15,25 we used the bioinformatics databases miRanda, TargetScan, and RNAhybrid to predict Sal-miR-58 targets. The results showed that the transcriptional factor KLF3 contains a potential Sal-miR-58-binding site in its 3′ UTR. Next, we tested whether Sal-miR-58 regulates the expression of KLF3 and found that although the mRNA expression of KLF3 and other members of the KLF family, such as KLF4 and KLF5, had no significant change in Sal-miR-58-transfected VSMCs (Figure 4A), the protein level of KLF3 was significantly downregulated, as shown by western blotting. The specificity of Sal-miR-58-targeting KLF3 was confirmed by the fact that transfecting VSMCs with Sal-miR-58 did not affect the protein level of KLF4 and KLF5, both of which play important roles in the regulation of VSMC function (Figure 4B). Also, immunofluorescence staining showed that the fluorescence intensity of KLF3 in Sal-miR-58-transfected VSMCs was significantly attenuated, and Sal-miR-58 transfection abrogated the translocation of KLF3 to the nucleus (Figure 4C). These results suggest that KLF3 may be a direct target of Sal-miR-58. Furthermore, a dual luciferase reporter assay was performed and showed that Sal-miR-58 significantly attenuated the luciferase activity mediated by wild-type KLF3 3′ UTR but had no significant effect on the luciferase activity mediated by its mutants (Figure 4D). The above results indicate that Sal-miR-58 directly targets the 3′ UTR of KLF3.
Figure 4.

Sal-miR-58 Inhibits Ang II-Induced Inflammation by Downregulating KLF3 in VSMCs
(A) mRNAs for KLF3, KLF4, and KLF5 were determined by qRT-PCR in VSMCs transfected with Sal-miR-58. nsp > 0.05 versus miR-Ctl (n = 3). ns, not significant. (B) Western blot analysis of KLF3, KLF4, and KLF5 in VSMCs transfected with Sal-miR-58 or miR-Ctl. (C) Immunofluorescence staining of KLF3 (red) and the nucleus (DAPI; blue) in VSMCs treated with Sal-miR-58 or Sal-miR-58+Ang II. Scale bar, 100 μm. (D) Luciferase reporter assays were performed in HEK293 cells co-transfected with Sal-miR-58 and a wild-type or mutant KLF3 3′ UTR-luciferase reporter. ∗p < 0.05 versus miR-Ctl; ##p < 0.01 versus Sal-miR-58+wild-type. The Sal-miR-58-binding site is shown in green; the mutated sites are shown in red. (E) Relative expression of KLF3 mRNA was examined by qRT-PCR in VSMCs treated with different doses of Ang II and presented after normalizing to 18S rRNA (mean ± SEM; n = 3). ∗p < 0.05 versus 0 M Ang II. (F) VSMCs were treated with different doses of Ang II for 24 h, and expression of KLF3 was examined by western blotting. (G) Expression of KLF3 in Ang II-treated VSMCs was examined by immunofluorescence staining. Red and blue stainings indicate KLF3 and the nuclei, respectively. Scale bar, 100 μm. (H) VSMCs were treated with different doses of Ang II for 24 h, and expression of KLF3 in the nucleus and the cytoplasm was examined by western blotting. (I) VSMCs were transfected with Sal-miR-58 or pcDNA3.1-KLF3 alone or together for 24 h, and expression of NF-κB p50 and p65 was determined by western blotting. (J) VSMCs were transfected as in (I), and levels of IL-1β, IL-6, and TNF-α in the culture medium were determined by ELISA. ∗∗p < 0.01, ∗∗∗p < 0.001 versus pcDNA3.1; #p < 0.05 versus pcDNA3.1-KLF3.
We next investigated the relationship between KLF3 expression and Ang II-induced inflammation in VSMCs and found that Ang II treatment concentration-dependently upregulated the expression of KLF3 both at the mRNA (Figure 4E) and protein (Figures 4F and 4G) levels, with translocation of KLF3 from the cytoplasm to the nucleus, as revealed by increased KLF3 nuclear localization (Figures 4G and 4H). Importantly, overexpression of KLF3 in VSMCs dramatically upregulated the expression of NF-κB p65, p50, IL-1β, IL-6, and TNF-α, which was largely abolished by Sal-miR-58 transfection (Figures 4I and 4J). These findings suggest that Sal-miR-58 inhibits the inflammatory response by downregulating KLF3 protein expression.
Sal-miR-58 Induces VSMC Autophagy by Relieving KLF3 Repression of NEDD4L Expression
Using a transcription factor prediction database, we predicted the target genes regulated by KLF3 and found that there exist putative KLF3 binding sites in the proximal promoter region of E3 ubiquitin (Ubi) ligase NEDD4L, which is closely related to autophagy and inflammation.26 Chromatin immunoprecipitation (ChIP) with KLF3 antibodies followed by qPCR showed that KLF3 was recruited to the –380 to +34 bp region of the NEDD4L promoter in KLF3-overexpressing VSMCs (Figure 5A). The dual luciferase reporter assay revealed that KLF3 significantly inhibited the transcription of NEDD4L gene, as evidenced by decreased luciferase activity in KLF3-overexpressing cells (Figure 5B). To further clarify whether NEDD4L plays a role in Sal-miR-58-regulated VSMC autophagy and inflammatory responses, we examined the effect of Sal-miR-58 on NEDD4L expression in VSMCs. The results showed that Ang II reduced, whereas Sal-miR-58 upregulated, the mRNA (Figure 5C; Figure S2A) and protein (Figure 5D; Figure S2B) level of NEDD4L. Moreover, the upregulation of NEDD4L by Sal-miR-58 was partly abrogated by Ang II treatment, as shown by qRT-PCR (Figure 5C), western blotting (Figure 5D), and immunofluorescence staining (Figure 5E). Knockdown of KLF3 expression by si-KLF3 resulted in changes similar to those seen in Sal-miR-58-transfected VSMCs (Figures S2C, S2D, and S2F–S2H), whereas enforced expression of KLF3 in VSMCs had opposite effects on NEDD4L expression (Figures S2C and S2E). These results clearly suggest that Sal-miR-58 exerts an inhibitory effect on the expression of KLF3, which in turn relieves KLF3 repression of the expression of NEDD4L and thus elevates the NEDD4L expression level.
Figure 5.

Sal-miR-58 Induces VSMC Autophagy by Relieving KLF3 Repression of NEDD4L Expression
(A) Schematic representation of the −1880 to +34 bp region of the NEDD4L promoter and primers for amplifying the different regions of the NEDD4L promoter. ChIP analysis of KLF3 binding to the NEDD4L promoter was performed in VSMCs transfected with pcDNA3.1-KLF3. Data are the mean ± SEM (n = 3). ∗∗∗p < 0.001 versus pcDNA3.1. (B) A luciferase reporter controlled by the NEDD4L promoter was transfected into 293A cells along with pcDNA3.1 or pcDNA3.1-KLF3. Luciferase activity was measured using the dual luciferase reporter assay system. Data represent the relative NEDD4L promoter activity normalized to pRL-TK activity. ∗∗∗p < 0.001 versus pGL3-basic. (C) VSMCs were treated with Ang II or Sal-miR-58 (100 nM) alone or together, and the total RNA was harvested and analyzed by qRT-PCR. ∗p < 0.05 versus 0 M Ang II or miR-Ctl; #p < 0.05 versus 0 M AngII+Sal-miR-58. (D) VSMCs were treated as in (C), and the protein was harvested and analyzed by western blotting using anti-KLF3 or anti-NEDD4L antibodies. (E) Expression of KLF3 and NEDD4L in VSMCs treated as in (C) was examined by immunofluorescence staining. Green, red, and blue staining indicates NEDD4L, KLF3, and the nuclei, respectively. Scale bar, 100 μm. (F and G) VSMCs were transfected with Sal-miR-58 or pcDNA3.1-KLF3 alone or together for 24 h, and expression of KLF3 and NEDD4L was determined by (F) qRT-PCR and (G) western blot analysis. ∗p < 0.05 versus pcDNA3.1; #p < 0.05 versus pcDNA3.1+Sal-miR-58. (H) VSMCs were transfected with pcDNA3.1-NEDD4L and treated with/without Ang II, the total RNA was harvested, and Atg5, Beclin1, and LC3B expression levels were analyzed by qRT-PCR. ∗p < 0.05, ∗∗p < 0.01 versus pcDNA3.1. (I) VSMCs were treated as in (H), and the protein was harvested and analyzed by western blotting using anti-Beclin1, anti-Atg5, and anti-LC3B. (J) VSMCs were treated as in (H) and infected with RFP-GFP-LC3B double-labeled adeno-associated virus, and autophagosomes and autolysosomes were examined as described above. Scale bar, 100 μm. (K) VSMCs were transfected with Sal-miR-58 or si-NEDD4L alone or together for 24 h, and expression of Beclin1, Atg5, and LC3B was determined by western blotting. (L) VSMCs were treated as in (K) and infected with RFP-GFP-LC3B double-labeled adeno-associated virus, and autophagosomes and autolysosomes were examined as described above. Scale bar, 100 μm.
To provide additional evidence that Sal-miR-58 upregulated the expression of NEDD4L by downregulating KLF3, we observed the effect of KLF3 overexpression alone or combined with Sal-miR-58 transfection on NEDD4L expression. The results showed that overexpression of KLF3 partially abolished the upregulation of NEDD4L induced by Sal-miR-58 both at the mRNA and protein levels (Figures 5F and 5G). NEDD4L plays a key role in the early stage of autophagy induction through ubiquitinating autophagy-regulated proteins.27 To further investigate whether NEDD4L participates in the induction of autophagy in Ang II-treated VSMCs, we forcedly expressed NEDD4L in VSMCs and found that NEDD4L overexpression significantly increased the expression of Beclin1, Atg5, and LC3B at the levels of transcription and translation, accompanied by enhanced conversion of LC3BI to LC3BII regardless of treatment with Ang II (Figures 5H and 5I). Similarly, immunofluorescence staining of VSMCs infected with RFP-GFP-LC3B double-labeled AAV showed that enforced expression of NEDD4L in VSMCs facilitated the formation of autophagosomes and autolysosomes, as revealed by increased bright fluorescent spots (Figure 5J). In contrast, knockdown of NEDD4L partially abolished VSMC autophagy induced by Sal-miR-58, as evidenced by western blotting (Figure 5K) and immunofluorescence staining (Figure 5L). These results indicate that NEDD4L plays an important role in Sal-miR-58-induced VSMC autophagy by a mechanism whereby Sal-miR-58 relieves KLF3 repression of the expression of NEDD4L.
NEDD4L Ubiquitinates PFKP
To determine which substrates regulated by NEDD4L are involved in Sal-miR-58-induced autophagy, NEDD4L was knocked down or overexpressed in VSMCs, and label-free quantitative analysis of ubiquitylation in these two cells was performed according to the strategy shown in Figure 6A. As a result, 3,391 ubiquitination sites located on 1,456 proteins were identified in the protein extract of VSMCs, of which 2,689 sites of 983 proteins contained quantitative information. Compared with NEDD4L-knocked down cells, the levels of ubiquitination at 142 sites were upregulated, and 78 sites were downregulated in NEDD4L-overexpressed cells (Figure 6B). Based on the above data, we performed a systematic bioinformatics analysis of proteins containing quantitative information sites, including protein annotation, functional classification, functional enrichment, and enrichment-based clustering analysis (Figures 6C–6E), and selected five candidate proteins, including PFKP, eEF1A1, MYH9, VDAC3, and integrin β1, to further investigate the effects of NEDD4L on their expression. Western blot analysis showed that the knockdown of NEDD4L significantly upregulated the protein level of PFKP, whereas the overexpression of NEDD4L had the opposite effects compared with their corresponding control, but there was no significant effect of NEDD4L knockdown or overexpression on the expression of eEF1A1, MYH9, VDAC3, and integrin β1 (Figures S3A and S3B). Also, NEDD4L knockdown or overexpression did not significantly affect PFKP mRNA level (Figures S3C and S3D).
Figure 6.

NEDD4L Interacts with and Ubiquitylates PFKP
(A–E) VSMCs were transfected with pcDNA3.1-NEDD4L or si-NEDD4L, and label-free quantitative ubiquitylation analysis was performed (A). The numbers of differentially expressed genes (B and C) and the results of functional enrichment analysis (D and E) are shown. (F) Western blot analysis detected PFKP expression in VSMCs transfected with HA-PFKP or treated with MG132 for 24 h. (G) VSMCs were transfected with si-NEDD4L for 24 h, and cell lysates were immunoprecipitated with anti-NEDD4L or anti-PFKP antibodies and the resulting precipitates were analyzed by western blotting using anti-PFKP or anti-NEDD4L antibodies. (H) NEDD4L and PFKP in the VSMCs treated with Sal-miR-58 or Ang II alone or together were examined by immunofluorescence staining. Green, red, and blue staining indicates NEDD4L, PFKP, and the nuclei, respectively. Yellow staining indicates co-localization of NEDD4L with PFKP. Scale bar, 100 μm. (I) HEK293 cells were transfected with HA-PFKP and Flag-NEDD4L for 24 h. The cell lysates were immunoprecipitated with anti-Flag antibody, and the resulting precipitates were analyzed by western blotting using anti-PFKP or anti-NEDD4L antibody. (J) VSMCs were transfected with pcDNA3.1-NEDD4L and/or treated with MG132 for 24 h. The cell lysates were immunoprecipitated with anti-NEDD4L or anti-PFKP antibody and the resulting precipitates were analyzed by western blotting using anti-PFKP or anti-ubiquitin (Ubi) antibody. (K) VSMCs were transfected with pcDNA3.1-NEDD4L or HA-PFKP and treated with MG132 for 24 h. The cell lysates were immunoprecipitated with anti-PFKP antibody, and the resulting precipitates were analyzed by western blotting using anti-Ubi or anti-PFKP antibody. (L) VSMCs were transfected with si-NEDD4L or HA-PFKP and treated with MG132 for 24 h. The cell lysates were immunoprecipitated with anti-PFKP antibody and the resulting precipitates were analyzed as indicated in (K). (M) VSMCs were transfected with Flag-NEDD4L, HA-PFKP, and HA-Ubi and treated with/without MG132 for 24 h. The cell lysates were immunoprecipitated with anti-Ubi antibody and the resulting precipitates were analyzed by western blotting using anti-PFKP antibody.
Furthermore, we examined PFKP stability by a cycloheximide (CHX) chase experiment. Treatment with CHX reduced the protein level of PFKP in a time-dependent manner, and the half-life of PFKP in VSMCs was about 6 h. However, pretreatment of VSMCs with the proteasome inhibitor MG132 led to prolonged PFKP protein stability (Figure 6F), implying that PFKP undergoes ubiquitin-mediated degradation in VSMCs. To further confirm that there exists a physical interaction between NEDD4L and PFKP, co-immunoprecipitation (coIP) experiments were carried out, and we found that the NEDD4L was co-immunoprecipitated by anti-PFKP antibody. Likewise, a similar result was obtained by reciprocal immunoprecipitation with anti-NEDD4L antibody, indicating that there is an interaction between NEDD4L and PFKP in VSMCs (Figure 6G). The specificity of this interaction was confirmed by decreased association of NEDD4L with PFKP in NEDD4L-knocked down cells (Figure 6G). Furthermore, confocal immunofluorescence staining of NEDD4L and PFKP showed that PFKP and NEDD4L were co-localized in the cytoplasm of VSMCs, and the upregulation of NEDD4L by Sal-miR-58 clearly reduced PFKP expression (Figure 6H). To further examine whether NEDD4L downregulates the protein level of PFKP by promoting ubiquitination of PFKP, we co-transfected HEK293 cells with hemagglutinin (HA)-PFKP and Flag-NEDD4L expression plasmids and showed that PFKP was detected in the sediment precipitated by anti-Flag antibody (Figure 6I), again suggesting that there is an interaction between NEDD4L and PFKP. We further determined the effects of NEDD4L knockdown or overexpression on PFKP ubiquitination. As expected, overexpression of NEDD4L obviously increased the level of ubiquitinated PFKP and thus attenuated the protein level of PFKP (Figures 6J and 6K). Reversely, NEDD4L knockdown by small interfering RNA (siRNA) decreased the level of ubiquitinated PFKP and increased PFKP stability (Figure 6L). In addition, we co-transfected HEK293 cells with Flag-NEDD4L and HA-PFKP expression plasmids and confirmed that co-expression of NEDD4L and PFKP markedly promoted the ubiquitination of PFKP. The proteasome inhibitor MG132 suppressed ubiquitin-mediated degradation of PFKP and enhanced the accumulation of ubiquitinated PFKP (Figure 6M). Collectively, these findings suggest that NEDD4L interacts with and ubiquitylates PFKP, leading to NEDD4L-mediated ubiquitination and degradation of PFKP.
PFKP Mediates Sal-miR-58/KLF3/NEDD4L-Induced Autophagy and Anti-inflammatory Effects in VSMCs
The above results demonstrated that NEDD4L mediates the ubiquitination and degradation of PFKP and is involved in Sal-miR-58-induced autophagy. We next investigated whether PFKP plays a role in Sal-miR-58-mediated autophagy and anti-inflammatory effects. First, we showed that although Sal-miR-58 transfection did not significantly affect PFKP mRNA levels regardless of treatment with Ang II (Figure 7A), the level of PFKP protein was dramatically downregulated in Sal-miR-58-transfected VSMCs (Figure 7B). Moreover, knockdown of PFKP (Figures S4A and S4B) significantly induced VSMC autophagy, as shown by increased Beclin1 and Atg5 expression and conversion of LC3BI to LC3BII (Figure 7C) as well as by increased formation of autolysosomes (Figure 7D). Correspondingly, the upregulation of IL-1β, IL-6, and TNF-α induced by Ang II was greatly abrogated by PFKP knockdown (Figure 7E). Therefore, it can be concluded that PFKP is at least partially responsible for Sal-miR-58-mediated autophagy and anti-inflammatory effects.
Figure 7.

PFKP Mediates Sal-miR-58/KLF3/NEDD4L-Induced Autophagy and Anti-inflammatory Effects
(A) VSMCs were transfected with Sal-miR-58 and treated with/without Ang II for 24 h. Expression of PFKP mRNA was determined by qRT-PCR and presented after normalizing to 18S rRNA (mean ± SEM; n = 3). (B) VSMCs were treated as in (A), and expression of PFKP was examined by western blotting. (C) VSMCs were transfected with si-PFKP and treated with/without Ang II, and expression of Beclin1, Atg5, and LC3B was examined by western blotting. (D) VSMCs were treated as in (C) and infected with RFP-GFP-LC3B double-labeled adeno-associated virus, and autophagosomes and autolysosomes were observed by a fluorescence microscope. Scale bars, 50 μm. (E) VSMCs were treated as in (C), and levels of IL-1β, IL-6, and TNF-α in the culture medium were examined by ELISA. ∗∗∗p < 0.001 versus si-Ctl; ###p < 0.001 versus si-Ctl+Ang II. (F and I) Expression levels of Beclin1, Atg5, LC3B, phosphorylated (p-)Akt, Akt, p-mTOR, and mTOR were examined by western blotting after PFKP-knocked down VSMCs were treated with Ang II and SC-79 (F) or MHY1485 (I) for 24 h. (G) RFP-GFP-LC3B double-labeled adeno-associated virus was used to detect autophagosomes and autolysosomes after PFKP-knocked down VSMCs were treated with Ang II and SC-79. Scale bars, 50 μm. (J) RFP-GFP-LC3B double-labeled adeno-associated virus was used to detect autophagosomes and autolysosomes after PFKP-knocked down VSMCs were treated with Ang II and MHY1485. Scale bars, 50 μm. (H) Levels of IL-1β, IL-6, and TNF-α in the culture medium were examined by ELISA after PFKP-knocked down VSMCs were treated with Ang II and SC-79. ∗p < 0.05 versus si-Ctl+Ang II; #p < 0.05 versus si-PFKP+Ang II. (K) Levels of IL-1β, IL-6, and TNF-α in the culture medium were examined by ELISA after PFKP-knocked down VSMCs were treated with Ang II and MHY1485. ∗p < 0.05 versus si-Ctl+Ang II; #p < 0.05 versus si-PFKP+Ang II.
Among signaling pathways that regulate autophagy and inflammatory response, the PI3K-Akt-mTOR pathway is one of the most important signaling pathways.28 Thus, we knocked down PFKP in VSMCs and detected the activity of Akt/mTOR signaling. The results showed that knockdown of PFKP significantly decreased the phosphorylation of Akt and mTOR, but had no significant effect on extracellular signal-regulated kinase 1/2 (ERK1/2) (Figures 7F, 7I, and S4C). To further examine whether Akt/mTOR signaling is required for PFKP downregulation-induced autophagy, we used Akt/mTOR signaling activators SC-79 (10 μM) and MHY1485 (10 μM) to pretreat the PFKP-knocked down VSMCs for 2 h, and we found that activation of Akt/mTOR signaling by SC-79 or MHY1485 partially abolished VSMC autophagy induced by knockdown of PFKP, as evidenced by decreased Beclin1 and Atg5 expression and conversion of LC3BI to LC3BII as well as by reduced formation of autolysosomes (Figures 7F, 7G, 7I, and 7J), with corresponding changes in the expression of IL-1β, IL-6, and TNF-α (Figures 7H and 7K).
Altogether, these results indicate that Sal-miR-58 downregulates PFKP protein level through modulating KLF3 and then NEDD4L expression, which in turn decreases activation of Akt/mTOR signaling and thus facilitates VSMC autophagy induced by Sal-miR-58 in the context of chronic Ang II stimulation and aneurysm formation (Figure 8).
Figure 8.

Proposed Model for Sal-miR-58-Induced Autophagy and Anti-inflammation in VSMCs
Sal-miR-58 enters VSMCs after exogenous administration and downregulates KLF3 expression through direct binding to the 3′ UTR of KLF3 mRNA, which in turn relieves KLF3 repression of NEDD4L expression, rendering upregulated NEDD4L expression. Increased NEDD4L facilitates the ubiquitination and degradation of PFKP, leading to a decrease in the activation of Akt/mTOR signaling and thus resulting in induction of VSMC autophagy.
Discussion
Polyphenols, most prominently flavonoids derived from medicinal plants, have been shown to exert anti-inflammatory, antioxidant, anti-neurodegenerative, and cytoprotective actions.10,29 The active ingredients of Salvia miltiorrhiza, such as phenolic acids and diterpene quinones, have anti-inflammatory and anti-oxidative effects.12,13,30 Although plant-derived miRNAs are well known to have cross-kingdom regulatory effects on mammalian target genes,12,31,32 it remains unknown whether Salvia miltiorrhiza-derived miRNAs may regulate autophagy and inflammation in mammalian VSMCs. The important findings of the present study are that (1) Sal-miR-58 specifically present in Salvia miltiorrhiza inhibits the formation of AAA by inducing VSMC autophagy and suppressing the inflammatory response in a mouse AAA model; (2) Sal-miR-58 reduces KLF3 expression via direct binding to the 3′ UTR of KLF3 mRNA and thus relieves KLF3 repression of NEDD4L transcription; (3) NEDD4L upregulation elicited by KLF3 deficiency in VSMCs increases the ubiquitination and degradation of PFKP by NEDD4L; and (4) decreased activation of Akt/mTOR signaling by PFKP facilitates VSMC autophagy induced by Sal-miR-58.
A previous study demonstrated that tanshinone IIA, a main active ingredient of Salvia miltiorrhiza, inhibited the development of elastase-induced experimental AAAs by suppressing proteolysis, inflammation, and oxidative stress, thus preserving VSMCs.30 Moreover, tanshinone IIA attenuated elastase-induced AAA in rats via the inhibition of myeloid differentiating factor 88 (MyD88)-dependent TLR-4 signaling.33 In this study, we screened and identified Sal-miR-58 from Salvia miltiorrhiza as a novel effective component. We found that exogenous administration of Sal-miR-58 to Ang II-induced mouse AAA models could effectively inhibit the formation of AAAs. These findings suggest that multiple ingredients in Salvia miltiorrhiza may act on different targets, which in turn generates a range of actions that prevent the development of AAAs through multiple effects. Overall, the present study further extended the active component of Salvia miltiorrhiza. Sal-miR-58 may represent a novel series of universal modulators that play a vital role in mediating plant-animal interactions at the molecular level.
It is well known that miRNAs regulate gene expression at the post-transcriptional level by promoting mRNA degradation or by inhibiting mRNA translation. However, some miRNAs can also regulate gene expression post-transcriptionally only by inhibiting mRNA translation. In this study, we found that KLF3 expression was regulated by Sal-miR-58 only via inhibiting translation, which reduces the protein level of KLF3, but does not affect its mRNA level. It has been shown that KLF3 serves as a transcriptional repressor through its interaction with the co-repressor carboxyl-terminal binding protein (CtBP),34 and it plays diverse and important roles in cardiovascular development and function in mice.35 In cultured VSMCs, we confirmed that KLF3 was a direct target of Sal-miR-58 and was negatively regulated by Sal-miR-58, as evidenced by a luciferase reporter assay, western blotting, and immunofluorescence staining. Treating VSMCs with Sal-miR-58 induced autophagy by downregulating KLF3 expression, which may be the basis of cardiovascular protection by Sal-miR-58. Previous studies showed that transcriptional activators KLF4 and KLF5 participated in the development and progression of AAAs. For example, Salmon et al.36 reported that KLF4 plays a critical role in aortic aneurysm formation by regulating VSMC phenotypic switching and could be a potential therapeutic target for AAA disease. Our recent study indicated that myeloid-specific KLF5 knockout resulted in decreased aortic aneurysm formation by inhibition of macrophage infiltration and migration.5 To our knowledge, this is the first study to demonstrate that transcriptional repressor KLF3 was involved in AAA formation, showing that downregulation of KLF3 by Sal-miR-58 is directly correlated with the inhibitory effect of Sal-miR-58 on Ang II-induced inflammation. KLF family members mainly bind to CACCC box sequences in enhancers or promoters of target genes and regulate the transcription of multiple genes.37 In the present study, we found that the proximal promoter region of NEDD4L contains putative KLF3 binding sites (CACCC motif). Because NEDD4L is an important E3 ubiquitin ligase involved in autophagic activation,38 we investigated whether KLF3 regulates NEDD4L expression. ChIP with KLF3 antibodies followed by qPCR indicated that KLF3 directly bound to upstream sequences of the NEDD4L promoter region, and repressed the NEDD4L gene expression at the transcription level. Thus, we identified KLF3 as a novel regulator of NEDD4L expression.
NEDD4L belongs to the NEDD4 family of E3 ubiquitin ligases and is the closest homolog to NEDD4 and is widely expressed in many tissues.39 Previous studies showed that NEDD4-1 expression is regulated at the translational level by mTOR complex 1 (mTORC1),40 and is transcriptionally upregulated by the forkhead box protein M1B (FoxM1B).41 In this study, we showed that Sal-miR-58 could significantly upregulate the expression of NEDD4L by relieving KLF3 repression of NEDD4L expression, thus leading to the induction of VSMC autophagy. Consistent with our finding, Wang et al.26 reported that knockdown of NEDD4L in mouse embryonic fibroblasts suppressed both basal autophagy and endoplasmic reticulum (ER) stress-induced autophagy, whereas overexpression of NEDD4L induced autophagy. Moreover, deletion of NEDD4L in lung epithelia caused massive inflammation and cystic fibrosis-like lung disease.42 Taken together, these results suggest that NEDD4L plays an indispensable role in autophagy and inflammation in a cell type-dependent manner. Sal-miR-58 induced VSMC autophagy through upregulating NEDD4L expression, which is repressed by KLF3 in the absence of Sal-miR-58.
NEDD4L negatively regulates the stability of its target proteins at the post-translational level by the ubiquitin-mediated proteasomal degradation pathway. For example, NEDD4-1 can ubiquitinate PTEN and thus target PTEN for proteasomal degradation.43 NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy.38 In this study, we used quantitative ubiquitylation analysis to identify substrates regulated by NEDD4L in Ang II-stimulated VSMCs. Using coIP experiments and proteasome inhibitor, we showed that NEDD4L interacted with and ubiquitylated PFKP, leading to NEDD4L-mediated proteasomal degradation of PFKP. PFKP is a rate-controlling enzyme of the glycolytic pathway.44 Previous studies showed that PFKP is significantly upregulated in tumor tissues,45,46 and KLF4 plays an essential role in maintenance of high glycolytic metabolism by transcriptional activation of the PFKP gene in breast cancer cells.47 Moreover, stabilization of PFKP by Akt promotes tumorigenesis in human glioblastoma development.48 Remarkably, a recent study reported that the knockdown of PFKP in oral squamous cell carcinoma dramatically inhibited the expression of LC3-II and Atg5, as well as decreased starvation-mediated glycolysis and autophagy, indicating that PFKP is necessary for starvation-mediated autophagy and glycolysis.49 Differently, in this study, we found that transfecting VSMCs with Sal-miR-58 significantly attenuated the level of PFKP protein, and downregulation of PFKP induced VSMC autophagy. This discrepancy in the effect of PFKP on autophagy between the studies might be due to differences in the context of autophagy between cancer cells and VSMCs used in the experiments.
Considering that phosphorylated (phospho-)PFKP can activate PI3K/Akt signaling,50 and the PI3K/Akt/mTOR signaling pathway participates in the process of autophagy triggered by different stimuli,51,52 as well as that all kinds of active ingredients in Salvia miltiorrhiza, such as cryptotanshinone, tanshinol, and tanshinone IIA, inhibit the growth and invasion of cancer cells by modulating the PI3K/Akt/mTOR signaling,53,54 we hypothesized that Sal-miR-58 might induce VSMC autophagy via blocking the PFKP-mediated PI3K/Akt/mTOR regulatory pathway. Consistent with our hypothesis, we observed that knockdown of PFKP significantly reduced the phosphorylation level of Akt and mTOR, and activation of Akt/mTOR signaling partially abolished autophagy induced by knockdown of PFKP. These results suggest that PFKP mediates Sal-miR-58-induced autophagy and anti-inflammatory effects by inhibiting the Akt/mTOR signaling pathway. However, how PFKP regulates the Akt/mTOR signaling needs to be addressed in future studies.
In summary, our studies identified Salvia miltiorrhiza-derived Sal-miR-58 as a novel vasoprotective agent, and this is the first study to demonstrate that Sal-miR-58 induces autophagy and attenuates inflammation in VSMCs through cross-species modulation of the KLF3/NEDD4L/PFKP regulatory pathway.
Materials and Methods
Total RNA Isolation from Salvia miltiorrhiza
Salvia miltiorrhiza total RNA was isolated with a plant RNA kit (Omega Bio-tek, R6827-01) according to the manufacturer’s instructions. The eluted RNA was stored at −80°C. RNA contamination and degradation were detected on 1% agarose gels, and RNA purity was examined using a NanoDrop 2000 spectrophotometer from Thermo Scientific. Reverse transcription and qRT-PCR were run on an ABI 7500 Fast system (Life Technologies) using a DNeasy plant mini kit (QIAGEN, 69106). The reference gene U6 was used as an internal control according to the manufacturer’s instructions and calculated using the 2−ΔΔCt method as previously described.55 All primer sequences are listed in Table S1.
High-Throughput Sequencing and Bioinformatics Approaches
High-throughput sequencing and bioinformatics for Salvia miltiorrhiza were performed in Novogene in Beijing, China. A total amount of 3 μg of total RNA was used for input material for the plant miRNA database. A sequencing database was generated using the NEBNext multiplex small RNA library prep set for Illumina (NEB, USA) following the manufacturer’s recommendations, and index codes were added to attribute sequences of each sample. The small RNA tags were mapped to the reference sequence by Bowtie without mismatch to analyze their expression, and the characteristics of hairpin structure of miRNA precursor can be used to predict novel miRNAs. At the same time, custom scripts were used to obtain the identified miRNA counts as well as base bias on the first position with certain length and on each position of all identified miRNAs, respectively. High-throughput sequencing and bioinformatics for Salvia miltiorrhiza were independently repeated and verified by qRT-PCR.
Animals
All animal studies were approved by the Ethics Review Committee for Animal Experimentation at HeBei Medical University (approval ID: HebMU 20080026). ApoE−/− mice were purchased from the Department of Laboratory Animal Science, Vital River, Beijing, China. Genotyping was identified by qRT-PCR.
Ang II-Induced Mouse AAA Model
The Ang II-induced AAA model in ApoE−/− mice was generated as described previously.56 Briefly, Ang II (1,000 ng/kg/min) was administered subcutaneously via an Alzet osmotic minipump (Alzet Scientific Products, model 2004) implanted into ApoE−/− mice of 8–10 weeks of age under anesthesia with 1.5% isoflurane. The Alzet osmotic minipump was placed into the subcutaneous space of the mice through a small incision for 28 d (five mice per group). Sal-miR-58 (10 mg/kg) was administered by intraperitoneal injection to ApoE−/− mice treated with or without Ang II 3 times/week for 4 weeks (five mice per group). At the endpoint of the experiments, all experimental mice had survived. Following 4 weeks, the abdominal aortas were examined by ultrasound to measure the maximum external diameters. Blood samples of mice were collected, and the abdominal aortas were harvested for analysis of RNA, protein, histology, and immunofluorescence staining.
Cell Culture and Treatment
Mouse VSMCs (ATCC, no. CRL-2797TM) were cultured in low-glucose Dulbecco’s modified Eagle’s medium (DMEM, Gibco, 31600-034) containing 10% fetal bovine serum (FBS) (Gemini, 900-108), 100 U/mL penicillin, an 100 μg/mL streptomycin, in a humidified incubator at 37°C and 5% CO2. The cells at passages 3–5 were used in all of the experiments. For the functional tests of Sal-miR-58 in vitro, 1 × 106 cells/well were transiently transfected with Sal-miR-58. 2 × 10−7 M Ang II (MedChemExpress, 30553) was used to stimulate VSMCs for 24 h. Before stimulation with Ang II, 1 × 106 cells/well VSMCs were incubated in serum-free medium for 24 h. Human embryonic kidney 293A cells were obtained from ATCC and cultured in high-glucose DMEM with 10% FBS.
RNA Extraction from the Tissues, Serum, or Cells and PCR
Total RNA was extracted from mouse tissues or cells with TRIzol reagent (QIAGEN, 10296-028) according to the manufacturer’s instructions, and a miRNeasy mini kit was use to extract RNA of tissues or cells, and a miRNeasy serum/plasma kit was used to extract RNA of serum. The quality of the RNA was measured using a NanoDrop 2000 spectrophotometer from Thermo Scientific. Reverse transcription and qRT-PCR were run on an ABI 7500 Fast system (Life Technologies) using miRNA detection kits by GenePharma (Shanghai, China) or a DNeasy plant mini kit (QIAGEN, 69106). The reference gene U6 was used as an internal control according to the manufacturer’s instructions and calculated using the 2−ΔΔCt method as previously described.55 All primer sequences are listed in Table S1.
Oxidation of Small RNAs with Periodate
Periodate oxidation was performed as previously described with slight modifications.16 Briefly, total RNA was extracted from mouse serum, Salvia miltiorrhiza, or mouse tissues using TRIzol reagent according to the manufacturer’s instructions. A 100-μL mixture consisting of 20 μg of small RNA fraction and 10 mM NaIO4 was incubated at 0°C for 40 min in the dark. The oxidized RNA was precipitated twice by ethanol, rinsed once with 80% ethanol, aired dried, dissolved in double-distilled H2O (ddH2O), and then subjected to Solexa sequencing or a qRT-PCR assay. The qRT-PCR assay was conducted using the miScript PCR system (QIAGEN, 218161) according to the manufacturer’s instructions.
Western Blot Analysis
Protein was extracted from abdominal aortas and cultured VSMCs as previously described.57 Equal amounts of protein were electrophoresed on SDS-PAGE and electrotransferred to a polyvinylidene fluoride membrane (Millipore). The membranes were blocked with 5% milk in Tris-buffered saline with Tween 20 (TBST) for 2 h at room temperature and incubated overnight at 4°C with the following primary antibodies: anti-Atg5 (1:1,000, Abcam, ab108327), anti-Beclin1 (1:1,000, Abcam, ab207612), anti-NF-κB p50 (1:1,000, Abcam, ab32360), anti-NF-κB p65 (1:1,000, Abcam, ab16502), anti-LC3B (1:1,000, Abcam, ab48394), anti-mTOR (1:1,000, Abcam, ab2732), anti-phospho-mTOR (1:1,000, Abcam, ab109268), anti-KLF3 (1:1,000, Santa Cruz Biotechnology, sc-514500), anti-β-tubulin (1:1,000, Abcam, ab6046), anti-NEDD4L (1:1,000, Abcam, ab46521), anti-PFKP (1:1,000, Abcam, ab204131), anti-β-actin (1:1,000, Santa Cruz Biotechnology, sc-47778), anti-eEF1A1 (1:1,000, Abcam, ab157455), anti-MYH9 (1:1,000, Abcam, ab75590), anti-VDAC3 (1:1,000, Proteintech, 14451-1-AP), anti-integrin β1 (1:1,000, Abcam, ab179471), anti-HA tag (1:1,000, Abcam, ab9110), anti-Flag tag (1:1,000, Proteintech, 20543-1-AP), anti-Erk1/2 (1:1,000, Abcam, ab17942), anti-phospho-Erk1/2 (Thr202/Tyr204) (1:1,000, Cell Signaling Technology, 4370), anti-Akt (1:1,000, Cell Signaling Technology, 2920), and anti-phospho-Akt (Ser473) (1:1,000, Cell Signaling Technology, 4060). Then, membranes were washed, and after incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (1:5,000, Rockland Immunochemicals, KOA0136) for 1 h at room temperature, protein blots were treated with the Immobilon western chemiluminescent HRP substrate (Millipore) and detected by ECL (enhanced chemiluminescence) Fusion Fx (Vilber Lourmat). Images were captured and processed by FusionCapt Advance Fx5 software (Vilber Lourmat). All experiments were replicated three times.
ELISA
The concentrations of IL-1β, IL-6, and TNF-α were determined in the culture supernatants from 1 × 107 cells/well VSMCs treated with the different stimuli using a commercial ELISA kit (Proteintech, KE1002, KE1003, KE1007) according to the manufacturer’s instructions. The absorbance at 450 nm was detected with a Multiskan Ascent (SPECTRAFluor Plus, Tecan).
Immunofluorescence Staining
Immunofluorescence staining was performed with 4-μm paraffin cross-sections from mouse abdominal aorta as well as with paraformaldehyde-fixed cells. After deparaffinization with xylene and rehydration, the slides were blocked by pre-incubation with 10% normal goat serum (KPL, 710027) for 1 h. Paraformaldehyde-fixed VSMCs were permeabilized by incubation with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 30 min. Then, the tissue section or cells were incubated overnight at 4°C with the following primary antibodies: anti-α-SMA (1:50, Santa Cruz Biotechnology, sc-130617), anti-Beclin1 (1:100, Abcam, ab207612), anti-LC3B (1:100, Abcam, ab48394), anti-KLF3 (1:50, Santa Cruz Biotechnology, sc-514500), anti-NEDD4L (1:100, Abcam, ab46521), and anti-PFKP (1:100, Abcam, ab204131). Secondary antibodies were rhodamine-labeled antibody to rabbit immunoglobulin G (IgG) (1:50, KPL, 031506) and fluorescein-labeled antibody to mouse IgG (1:50, KPL, 021815), or rhodamine-labeled antibody to mouse IgG (1:50, KPL, 031806) and fluorescein-labeled antibody to rabbit IgG (1:50, KPL, 021516), and 4′,6-diamidino-2-phenylindole (DAPI) (1:50, MP Biomedicals, 157574) was used to stain nuclei in each experiment. Images were captured by confocal microscopy (DM6000 CFS, Leica Microsystems) and processed by LAS AF software.
CoIP Assays
coIP was carried out as described previously.58 Briefly, the proteins from 1 × 107 cells/well VSMCs were first immunoprecipitated with 3 μg of anti-NEDD4L, anti-PFKP, and anti-ubiquitin antibodies, respectively, for 1 h at 4°C, followed by incubation with protein A-agarose overnight at 4°C. The complexes of protein A-agarose-antigen-antibody were pelleted by centrifugation at 12,000 rpm for 2 min at 4°C. Then, the collections were washed with 600 μL of IPH buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40 [NP-40], and 0.1 mM phenylmethylsulfonyl fluoride) for 20 min at 4°C for five times. The bound proteins were resolved by SDS-PAGE, followed by western blotting with antibodies against PFKP, NEDD4L, Flag, HA, and Ubi.
ChIP Assays
The ChIP assay was carried out as described previously.58 In brief, 1% formaldehyde was used to treat VSMCs for 10 min to cross-link proteins with DNA. The cross-linked chromatin was then prepared and sonicated to an average size of 400–600 bp. The DNA fragments were immunoprecipitated overnight at 4°C with the antibodies against KLF3 (Santa Cruz Biotechnology, sc-514500) and anti-IgG (as a negative control). After reversal of cross-linking, KLF3 occupancy on the NEDD4L gene promoter (the region between –380 and +34 bp) was examined by PCR with the primers listed in Table S2 for ChIP, and a negative control region upstream of the NEDD4L promoter (the region between –1880 and –1040 bp) was also amplified. All experiments were replicated at least three times.
Luciferase Assays
Human embryonic kidney 293A cells were maintained in high-glucose DMEM supplemented with 10% FBS as previously described.59 For luciferase assays, 3 × 104 cells/well were seeded in a 24-well plate and grown for 24 h prior to co-transfection with a Sal-miR-58 (GenePharma) or negative control (NC) mimic combined with 100 ng of wild-type KLF3 3′ UTR reporter plasmids or mutant KLF3 3′ UTR reporter plasmids. 293 cells were also transfected with a luciferase-harboring NEDD4L promotor (KLF3 binding site), pcDNA3.1-KLF3, empty vector, and the pTK-RL plasmid using Lipofectamine 2000 reagent (Invitrogen, 11668019) according to the manufacturer’s instructions. Luciferase assays were performed using a Dual-Glo luciferase assay system (Promega, Madison, WI, USA) 24 h after transfection. Specific promoter activity was expressed as the relative activity ratio of firefly luciferase to Renilla luciferase. All transfected constructs were evaluated in at least three separate wells per experiment.
Analysis of Autophagic Flux in VSMCs Infected with mRFP-GFP-LC3 Double-Labeled AAV
Analysis of autophagic flux in VSMCs infected with mRFP-GFP-LC3 was performed as previously described.60 Briefly, 2 × 104 cells/well were infected with Adv-mRFP-GFP-LC3 (Hanbio Biotechnology) at an MOI of 20. After the culture media containing adenoviral particles were replaced with fresh media, the cells were cultured for another 24 h. The collected cells were washed with PBS, fixed with 4% paraformaldehyde, and images were captured by confocal microscopy (DM6000 CFS, Leica Microsystems) and processed by LAS AF software.
Protein Extraction and Label-Free Quantitative Analysis of Ubiquitylome
VSMCs were sonicated three times on ice using a high-intensity ultrasonic processor (Scientz Biotechnology) in lysis buffer (8 M urea, 1% protease inhibitor cocktail). The remaining debris was removed by centrifugation at 12,000 × g at 4°C for 10 min. Finally, the supernatant was collected and the protein concentration was determined with a bicinchoninic acid (BCA) kit according to the manufacturer’s instructions. Ubiquitylome analysis by label-free quantification was performed in PMT Biolabs in Hangzhou, China. For liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, the NEDD4L-modified peptides were desalted with C18 ZipTips (Millipore) according to the manufacturer’s instructions. The resulting MS/MS data were processed using a MaxQuant search engine (v1.5.2.8). Tandem mass spectra were searched against the database concatenated with the reverse decoy database. Ubiquitylome analysis (label-free) was independently repeated and verified by western blot analysis.
siRNA Transfection
siRNAs targeting mouse KLF3 (si-KLF3), PFKP (si-PFKP), and NEDD4L (si-NEDD4L) were designed and synthesized by GenePharma (Shanghai, China). Non-specific siRNA (si-Ctl) was purchased from Santa Cruz Biotechnology. Transfection was performed using Lipofectamine 2000 reagent according to the manufacturer’s instructions. Twenty hours following transfection, VSMCs were treated with Ang II (10−7 M). Cells were then harvested and lysed for western blotting. All the sequences of siRNAs are listed as Table S3.
Statistical Analysis of Experimental Data
All statistical analyses were performed using GraphPad Prism 5 software (GraphPad, La Jolla, CA, USA). The data presented as bar graphs are the means ± SEM of at least three independent experiments. Differences between two groups were assessed using analysis of variance followed by a Student’s t test. For multiple comparisons or repeated measurements, ANOVA or repeated ANOVA followed by a Tukey’s post hoc test was used. A p value of <0.05 was considered statistically significant.
Author Contributions
J.-k.W. and B.Z. engaged in study design and coordination, material support for obtained funding, and supervised the study. Y.Q. performed most of the experiments and statistical analyses and wrote the manuscript. G.-s.Y., H.-j.Y., and J.Z. performed parts of the experiments. Z.Y. analyzed the data. X.-h.Z., H.-y.Z. and J.-h,S. reviewed the data and helped in the preparation of the manuscript. All authors reviewed and approved the final manuscript.
Conflict of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (nos. 31671182, 31871152, and 81770285); the Hebei Scientific Research Project of High-Level Talents (no. GCC2014026); the Foundation Project of the Affiliated Hospital of Hebei University (no. 2019Q008); and from the Hebei Scientific Research Project of Traditional Chinese Medicine (nos. 2018151 and 2018147).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.06.015.
Supplemental Information
References
- 1.Netea-Maier R.T., Plantinga T.S., van de Veerdonk F.L., Smit J.W., Netea M.G. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12:245–260. doi: 10.1080/15548627.2015.1071759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osonoi Y., Mita T., Azuma K., Nakajima K., Masuyama A., Goto H., Nishida Y., Miyatsuka T., Fujitani Y., Koike M. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy. 2018;14:1991–2006. doi: 10.1080/15548627.2018.1501132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tai S., Hu X.Q., Peng D.Q., Zhou S.H., Zheng X.L. The roles of autophagy in vascular smooth muscle cells. Int. J. Cardiol. 2016;211:1–6. doi: 10.1016/j.ijcard.2016.02.128. [DOI] [PubMed] [Google Scholar]
- 4.Ackers-Johnson M., Talasila A., Sage A.P., Long X., Bot I., Morrell N.W., Bennett M.R., Miano J.M., Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler. Thromb. Vasc. Biol. 2015;35:817–828. doi: 10.1161/ATVBAHA.114.305218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma D., Zheng B., Suzuki T., Zhang R., Jiang C., Bai D., Yin W., Yang Z., Zhang X., Hou L. Inhibition of KLF5-Myo9b-RhoA pathway-mediated podosome formation in macrophages ameliorates abdominal aortic aneurysm. Circ. Res. 2017;120:799–815. doi: 10.1161/CIRCRESAHA.116.310367. [DOI] [PubMed] [Google Scholar]
- 6.Martinet W., De Meyer G.R. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ. Res. 2009;104:304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 7.Berman A.Y., Motechin R.A., Wiesenfeld M.Y., Holz M.K. The therapeutic potential of resveratrol: a review of clinical trials. NPJ Precis. Oncol. 2017;1:35. doi: 10.1038/s41698-017-0038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonkowski M.S., Sinclair D.A. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016;17:679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Cabo R., Carmona-Gutierrez D., Bernier M., Hall M.N., Madeo F. The search for antiaging interventions: from elixirs to fasting regimens. Cell. 2014;157:1515–1526. doi: 10.1016/j.cell.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmona-Gutierrez D., Zimmermann A., Kainz K., Pietrocola F., Chen G., Maglioni S., Schiavi A., Nah J., Mertel S., Beuschel C.B. The flavonoid 4,4′-dimethoxychalcone promotes autophagy-dependent longevity across species. Nat. Commun. 2019;10:651. doi: 10.1038/s41467-019-08555-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z.M., Xu S.W., Liu P.Q. Salvia miltiorrhiza Burge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 2018;39:802–824. doi: 10.1038/aps.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang W., Song J.K., Zhang X., Zhou Q.M., He G.R., Xu X.N., Rong Y., Zhou W.X., Du G.H. Salvianolic acid A attenuates ischemia reperfusion induced rat brain damage by protecting the blood brain barrier through MMP-9 inhibition and anti-inflammation. Chin. J. Nat. Med. 2018;16:184–193. doi: 10.1016/S1875-5364(18)30046-3. [DOI] [PubMed] [Google Scholar]
- 13.MEIm X.-D., Cao Y.-F., Che Y.-Y., Li J., Shang Z.-P., Zhao W.-J., Qiao Y.-J., Zhang J.-Y. Danshen: a phytochemical and pharmacological overview. Chin. J. Nat. Med. 2019;17:59–80. doi: 10.1016/S1875-5364(19)30010-X. [DOI] [PubMed] [Google Scholar]
- 14.Guo X., Niu J., Cao X. Heterologous expression of Salvia miltiorrhiza microRNA408 enhances tolerance to salt stress in Nicotiana benthamiana. Int. J. Mol. Sci. 2018;19:E3985. doi: 10.3390/ijms19123985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Hou D., Chen X., Li D., Zhu L., Zhang Y., Li J., Bian Z., Liang X., Cai X. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res. 2012;22:107–126. doi: 10.1038/cr.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu B., Yang Z., Li J., Minakhina S., Yang M., Padgett R.W., Steward R., Chen X. Methylation as a crucial step in plant microRNA biogenesis. Science. 2005;307:932–935. doi: 10.1126/science.1107130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Au K.Y., Pong J.C., Ling W.L., Li J.C. miR-1303 regulates mycobacteria induced autophagy by targeting Atg2B. PLoS ONE. 2016;11:e0146770. doi: 10.1371/journal.pone.0146770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pozuelo-Rubio M. 14-3-3 Proteins are regulators of autophagy. Cells. 2012;1:754–773. doi: 10.3390/cells1040754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X., Yu J., Zhong B., Lu J., Lu J.J., Li S., Lu Y. Pharmacological activities of dihydrotanshinone I, a natural product from Salvia miltiorrhiza Bunge. Pharmacol. Res. 2019;145:104254. doi: 10.1016/j.phrs.2019.104254. [DOI] [PubMed] [Google Scholar]
- 20.Wortmann M., Skorubskaya E., Peters A.S., Hakimi M., Böckler D., Dihlmann S. Necrotic cell debris induces a NF-κB-driven inflammasome response in vascular smooth muscle cells derived from abdominal aortic aneurysms (AAA-SMC) Biochem. Biophys. Res. Commun. 2019;511:343–349. doi: 10.1016/j.bbrc.2019.02.051. [DOI] [PubMed] [Google Scholar]
- 21.Priyadarshini L., Aggarwal A. Astaxanthin inhibits cytokines production and inflammatory gene expression by suppressing IκB kinase-dependent nuclear factor κB activation in pre and postpartum Murrah buffaloes during different seasons. Vet. World. 2018;11:782–788. doi: 10.14202/vetworld.2018.782-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Somade O.T., Ajayi B.O., Tajudeen N.O., Atunlute E.M., James A.S., Kehinde S.A. Camphor elicits up-regulation of hepatic and pulmonary pro-inflammatory cytokines and chemokines via activation of NF-kB in rats. Pathophysiology. 2019;26:305–313. doi: 10.1016/j.pathophys.2019.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Roedig H., Nastase M.V., Wygrecka M., Schaefer L. Breaking down chronic inflammatory diseases: the role of biglycan in promoting a switch between inflammation and autophagy. FEBS J. 2019;286:2965–2979. doi: 10.1111/febs.14791. [DOI] [PubMed] [Google Scholar]
- 24.Mehto S., Chauhan S., Jena K.K., Chauhan N.R., Nath P., Sahu R., Dhar K., Das S.K., Chauhan S. IRGM restrains NLRP3 inflammasome activation by mediating its SQSTM1/p62-dependent selective autophagy. Autophagy. 2019;15:1645–1647. doi: 10.1080/15548627.2019.1628544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahid S., Kim G., Johnson N.R., Wafula E., Wang F., Coruh C., Bernal-Galeano V., Phifer T., dePamphilis C.W., Westwood J.H., Axtell M.J. MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature. 2018;553:82–85. doi: 10.1038/nature25027. [DOI] [PubMed] [Google Scholar]
- 26.Wang H., Sun R.Q., Camera D., Zeng X.Y., Jo E., Chan S.M., Herbert T.P., Molero J.C., Ye J.M. Endoplasmic reticulum stress up-regulates Nedd4-2 to induce autophagy. FASEB J. 2016;30:2549–2556. doi: 10.1096/fj.201500119. [DOI] [PubMed] [Google Scholar]
- 27.Nazio F., Cecconi F. Autophagy up and down by outsmarting the incredible ULK. Autophagy. 2017;13:967–968. doi: 10.1080/15548627.2017.1285473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heras-Sandoval D., Pérez-Rojas J.M., Hernández-Damián J., Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014;26:2694–2701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Singh P., Anand A., Kumar V. Recent developments in biological activities of chalcones: a mini review. Eur. J. Med. Chem. 2014;85:758–777. doi: 10.1016/j.ejmech.2014.08.033. [DOI] [PubMed] [Google Scholar]
- 30.Shang T., Liu Z., Zhou M., Zarins C.K., Xu C., Liu C.J. Inhibition of experimental abdominal aortic aneurysm in a rat model by way of tanshinone IIA. J. Surg. Res. 2012;178:1029–1037. doi: 10.1016/j.jss.2012.04.068. [DOI] [PubMed] [Google Scholar]
- 31.Fei Y.X., Wang S.Q., Yang L.J., Qiu Y.Y., Li Y.Z., Liu W.Y., Xi T., Fang W.R., Li Y.M. Salvia miltiorrhiza Bunge (Danshen) extract attenuates permanent cerebral ischemia through inhibiting platelet activation in rats. J. Ethnopharmacol. 2017;207:57–66. doi: 10.1016/j.jep.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z., Wang Y., Tan W., Wang S., Liu J., Liu X., Wang X., Gao X. A Review of Danshen combined with clopidogrel in the treatment of coronary heart disease. Evid. Based Complement. Alternat. Med. 2019;2019:2721413. doi: 10.1155/2019/2721413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shang T., Ran F., Qiao Q., Liu Z., Liu C.J. Tanshinone IIA attenuates elastase-induced AAA in rats via inhibition of MyD88-dependent TLR-4 signaling. Vasa. 2014;43:39–46. doi: 10.1024/0301-1526/a000326. [DOI] [PubMed] [Google Scholar]
- 34.McConnell B.B., Yang V.W. Mammalian Krüppel-like factors in health and diseases. Physiol. Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelsey L., Flenniken A.M., Qu D., Funnell A.P., Pearson R., Zhou Y.Q., Voronina I., Berberovic Z., Wood G., Newbigging S. ENU-induced mutation in the DNA-binding domain of KLF3 reveals important roles for KLF3 in cardiovascular development and function in mice. PLoS Genet. 2013;9:e1003612. doi: 10.1371/journal.pgen.1003612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salmon M., Johnston W.F., Woo A., Pope N.H., Su G., Upchurch G.R., Jr., Owens G.K., Ailawadi G. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation. 2013;128(11, Suppl 1):S163–S174. doi: 10.1161/CIRCULATIONAHA.112.000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearson R.C., Funnell A.P., Crossley M. The mammalian zinc finger transcription factor Krüppel-like factor 3 (KLF3/BKLF) IUBMB Life. 2011;63:86–93. doi: 10.1002/iub.422. [DOI] [PubMed] [Google Scholar]
- 38.Lin Q., Dai Q., Meng H., Sun A., Wei J., Peng K., Childress C., Chen M., Shao G., Yang W. The HECT E3 ubiquitin ligase NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy. J. Cell Sci. 2017;130:3839–3850. doi: 10.1242/jcs.207068. [DOI] [PubMed] [Google Scholar]
- 39.Goel P., Manning J.A., Kumar S. NEDD4-2 (NEDD4L): the ubiquitin ligase for multiple membrane proteins. Gene. 2015;557:1–10. doi: 10.1016/j.gene.2014.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsia H.E., Kawabe H. Translational regulation of the expression of the ubiquitin E3 ligase Nedd4-1 by mTORC1-dependent signaling. Commun. Integr. Biol. 2015;8:e1017161. doi: 10.1080/19420889.2015.1017161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovacevic Z., Chikhani S., Lui G.Y., Sivagurunathan S., Richardson D.R. The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid. Redox Signal. 2013;18:874–887. doi: 10.1089/ars.2011.4273. [DOI] [PubMed] [Google Scholar]
- 42.Kimura T., Kawabe H., Jiang C., Zhang W., Xiang Y.Y., Lu C., Salter M.W., Brose N., Lu W.Y., Rotin D. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc. Natl. Acad. Sci. USA. 2011;108:3216–3221. doi: 10.1073/pnas.1010334108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strathe A.V., Bruun T.S., Tauson A.H., Theil P.K., Hansen C.F. Increased dietary protein for lactating sows affects body composition, blood metabolites and milk production. Animal. 2020;14:285–294. doi: 10.1017/S1751731119001678. [DOI] [PubMed] [Google Scholar]
- 44.Wang J., Zhang P., Zhong J., Tan M., Ge J., Tao L., Li Y., Zhu Y., Wu L., Qiu J., Tong X. The platelet isoform of phosphofructokinase contributes to metabolic reprogramming and maintains cell proliferation in clear cell renal cell carcinoma. Oncotarget. 2016;7:27142–27157. doi: 10.18632/oncotarget.8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minton D.R., Fu L., Chen Q., Robinson B.D., Gross S.S., Nanus D.M., Gudas L.J. Analyses of the transcriptome and metabolome demonstrate that HIF1α mediates altered tumor metabolism in clear cell renal cell carcinoma. PLoS ONE. 2015;10:e0120649. doi: 10.1371/journal.pone.0120649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanders E., Diehl S. Analysis and interpretation of transcriptomic data obtained from extended Warburg effect genes in patients with clear cell renal cell carcinoma. Oncoscience. 2015;2:151–186. doi: 10.18632/oncoscience.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moon J.S., Kim H.E., Koh E., Park S.H., Jin W.J., Park B.W., Park S.W., Kim K.S. Krüppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. J. Biol. Chem. 2011;286:23808–23816. doi: 10.1074/jbc.M111.236737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J.H., Liu R., Li J., Zhang C., Wang Y., Cai Q., Qian X., Xia Y., Zheng Y., Piao Y. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017;8:949. doi: 10.1038/s41467-017-00906-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen G., Liu H., Zhang Y., Liang J., Zhu Y., Zhang M., Yu D., Wang C., Hou J. Silencing PFKP inhibits starvation-induced autophagy, glycolysis, and epithelial mesenchymal transition in oral squamous cell carcinoma. Exp. Cell Res. 2018;370:46–57. doi: 10.1016/j.yexcr.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 50.Lee J.H., Liu R., Li J., Wang Y., Tan L., Li X.J., Qian X., Zhang C., Xia Y., Xu D. EGFR-phosphorylated platelet isoform of phosphofructokinase 1 promotes PI3K activation. Mol. Cell. 2018;70:197–210.e7. doi: 10.1016/j.molcel.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bi Y., Wang G., Liu X., Wei M., Zhang Q. Low-after-high glucose down-regulated Cx43 in H9c2 cells by autophagy activation via cross-regulation by the PI3K/Akt/mTOR and MEK/ERK1/2 signal pathways. Endocrine. 2017;56:336–345. doi: 10.1007/s12020-017-1251-3. [DOI] [PubMed] [Google Scholar]
- 52.Chang H., Li X., Cai Q., Li C., Tian L., Chen J., Xing X., Gan Y., Ouyang W., Yang Z. The PI3K/Akt/mTOR pathway is involved in CVB3-induced autophagy of HeLa cells. Int. J. Mol. Med. 2017;40:182–192. doi: 10.3892/ijmm.2017.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L., Chen C., Duanmu J., Wu Y., Tao J., Yang A., Yin X., Xiong B., Gu J., Li C., Liu Z. Cryptotanshinone inhibits the growth and invasion of colon cancer by suppressing inflammation and tumor angiogenesis through modulating MMP/TIMP system, PI3K/Akt/mTOR signaling and HIF-1α nuclear translocation. Int. Immunopharmacol. 2018;65:429–437. doi: 10.1016/j.intimp.2018.10.035. [DOI] [PubMed] [Google Scholar]
- 54.Su C.C. Tanshinone IIA can inhibit MiaPaCa-2 human pancreatic cancer cells by dual blockade of the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR pathways. Oncol. Rep. 2018;40:3102–3111. doi: 10.3892/or.2018.6670. [DOI] [PubMed] [Google Scholar]
- 55.Wang C., Han M., Zhao X.M., Wen J.K. Krüppel-like factor 4 is required for the expression of vascular smooth muscle cell differentiation marker genes induced by all-trans retinoic acid. J. Biochem. 2008;144:313–321. doi: 10.1093/jb/mvn068. [DOI] [PubMed] [Google Scholar]
- 56.Daugherty A., Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor −/− mice. Ann. N Y Acad. Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhang X.H., Zheng B., Han M., Miao S.B., Wen J.K. Synthetic retinoid Am80 inhibits interaction of KLF5 with RARα through inducing KLF5 dephosphorylation mediated by the PI3K/Akt signaling in vascular smooth muscle cells. FEBS Lett. 2009;583:1231–1236. doi: 10.1016/j.febslet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 58.Meng F., Han M., Zheng B., Wang C., Zhang R., Zhang X.H., Wen J.K. All-trans retinoic acid increases KLF4 acetylation by inducing HDAC2 phosphorylation and its dissociation from KLF4 in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2009;387:13–18. doi: 10.1016/j.bbrc.2009.05.112. [DOI] [PubMed] [Google Scholar]
- 59.Li H.X., Han M., Bernier M., Zheng B., Sun S.G., Su M., Zhang R., Fu J.R., Wen J.K. Krüppel-like factor 4 promotes differentiation by transforming growth factor-β receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J. Biol. Chem. 2010;285:17846–17856. doi: 10.1074/jbc.M109.076992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hua F., Li K., Yu J.J., Lv X.X., Yan J., Zhang X.W., Sun W., Lin H., Shang S., Wang F. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat. Commun. 2015;6:7951. doi: 10.1038/ncomms8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.