

Abstract
Introduction
Mesenchymal stromal cell (MSC) therapy mitigates lung injury and improves survival in murine models of sepsis. Precise mechanisms of therapeutic benefit remain poorly understood.
Objectives
To identify host-derived regulatory elements that may contribute to the therapeutic effects of MSCs, we profiled the microRNAome (miRNAome) and transcriptome of lungs from mice randomised to experimental polymicrobial sepsis-induced lung injury treated with either placebo or MSCs.
Methods and results
A total of 11 997 genes and 357 microRNAs (miRNAs) expressed in lungs were used to generate a statistical estimate of association between miRNAs and their putative mRNA targets; 1395 miRNA:mRNA significant association pairs were found to be differentially expressed (false discovery rate ≤0.05). MSC administration resulted in the downregulation of miR-27a-5p and upregulation of its putative target gene VAV3 (adjusted p=1.272E-161) in septic lungs. In human pulmonary microvascular endothelial cells, miR-27a-5p expression levels were increased while VAV3 was decreased following lipopolysaccharide (LPS) or tumour necrosis factor (TNF) stimulation. Transfection of miR-27a-5p mimic or inhibitor resulted in increased or decreased VAV3 message, respectively. Luciferase reporter assay demonstrated specific binding of miR-27a-5p to the 3′UTR of VAV3. miR27a-5p inhibition mitigated TNF-induced (1) delayed wound closure, increased (2) adhesion and (3) transendothelial migration but did not alter permeability. In vivo, cell infiltration was attenuated by intratracheal coinstillation of the miR-27a-5p inhibitor, but this did not protect against endotoxin-induced oedema formation.
Conclusions
Our data support involvement of miR-27a-5p and VAV3 in cellular adhesion and infiltration during acute lung injury and a potential role for miR-27a-based therapeutics for acute respiratory distress syndrome.
Keywords: ARDS, critical care, respiratory infection
Key messages.
What is the key question?
The key question of our study is whether mesenchymal stromal/stem cells (MSCs) exert their beneficial anti-inflammatory effect in septic lungs by in part modulating endogenous host-derived microRNAs and their target genes—thus amplifying their therapeutic effect by capitalising on shared endogenous post-transcriptional regulatory mechanisms.
What is the bottom line?
MSCs regulate the expression of miR-27a and its various target genes including VAV3, ACE and Nrf2 known to play critical roles in sepsis-induced lung injury and inhibition of miR-27a prevents inflammatory cell infiltration during endotoxin-induced lung injury.
Why read on?
This is the first study that links MSC regulation with microRNA alterations of various acute lung injury-related genes containing miR-27a 3′UTR regulatory sequences, suggesting that targeting this pathway may be a suitable strategy to preserve the alveolar epithelial function and to reduce inflammatory cell infiltration in acute lung injury.
Introduction
With an annual incidence of close to one million cases in North America, sepsis-related multiorgan failure is responsible for more than 200 000 deaths per year and rivals myocardial infarction as a leading cause of mortality in the developed world.1 2 The lung is the organ that fails most often dictating the need for life support, and sepsis is the highest risk factor for development and progression of acute respiratory distress syndrome (ARDS), the clinical syndrome of acute lung injury (ALI).3 In preclinical studies, mesenchymal stromal/stem cell (MSC) therapy mitigates pulmonary oedema formation, protein-rich fluid exudation and cellular infiltration.4 Network analysis of transcriptional changes following MSC administration suggests reprogramming of the immune system involving thousands of genes and multiple pathways in various organs.5 MicroRNAs (miRNAs) are centrally positioned to orchestrate this host response because of their fundamental role as modulators of biological processes and cell fate.6
miRNAs are endogenous RNA molecules that can directly bind to the 3′ untranslated region (UTR) of mRNAs to modulate gene expression at the post-transcriptional level. Binding of miRs to their seed sequences results in gene repression either by destabilising their complementary mRNAs or by preventing translation. Various studies have implicated miRNAs in the regulation of endothelial barrier integrity and demonstrated their potential as pharmacological targets for the maintenance of endothelial cell barrier function.6 Here, we hypothesise that the beneficial therapeutic effects of MSC administration in septic lungs is in part due to the modulation of host-derived endogenous miRNAs.
To identify biologically relevant, and novel, miRNAs involved in mediating the beneficial effects of MSC in preclinical models of ARDS, we capitalised on canonical binding predictions between miRs and their target mRNAs to develop a statistical algorithm that detects tightly regulated differentially expressed miRNA:mRNA pairs. Functional genomics (gain and loss of function) in vitro and in vivo studies demonstrates inhibition of host-derived miR-27a-5p in mediating the salutary effect of MSC administration in a model of preclinical lung injury.
Material and methods
All studies were approved by the animal care committee at St. Michael’s Hospital (Toronto, Ontario, Canada) in accordance with Canadian Council of Animal Care guidelines and ARRIVE criteria.7 Briefly, male C57Bl/6J mice of 10–14 weeks old (The Jackson Laboratory, Bar Harbor, Maine, USA) were randomised to caecum ligation and puncture (CLP) or sham surgery and at 6 hours post-CLP. Mice were further randomised to receive a single intrajugular injection of 2×105 bone marrow-derived MSCs or placebo (saline).4 Animals were humanely sacrificed at 28 hours and right lungs snap frozen for mRNA and miRNA profiling. Transcriptional microarray data are available in GEO (Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo GSE40180)). Detailed materials and methods are available https://thorax.bmj.com/.
MicroRNA analysis and miRNA target gene expression association
miRNA isolated from lungs (five animals per group—sham/saline, CLP/saline and CLP/MSCs) using Exiqon miRCURY LNA microRNA arrays (Exiqon, Vedbaek, Denmark). Variance-stabilising was used to normalise expression values in R/Bioconductor8 and differential expression was determined using LIMMA.9 miRNA:mRNA associated pairs were identified by matching a gene by miRNA matrix table based on in silico predictions versus empiric lung mRNA and miRNA experimental data. Predicted targets were determined using miRanda (http://mirdb.org 10 score <−0.5, conservation score >0.5) and TargetScan11 (context score <−0.3) databases with appropriate thresholds. A statistical estimate of association or correlation between miRNAs and their putative mRNA targets was determined using the equation:
where Y is the expression value of a given target gene and X is the expression of its miRNA.
We fitted a Bayes hierarchical generalised linear model12 for the equation to identify the miRNAs which significantly regulated the gene expression of the mRNA (false discovery rate, FDR set at <0.05). Adjusted p-values (Adj-p) are p-values corrected for multiple comparisons.10 Functional enrichment was performed in Enrichr.13
Cell culture, treatments and transfections
Primary human pulmonary microvascular endothelial cells (HPMECs; PromoCELL, Heidelberg, Germany), primary human distal bronchial epithelial cells (ATCC CRL-9609; BEAS2b, Toronto, Canada), RAW 264.7 (91062702; Sigma, Darmstadt, Germany) and murine MSCs (isolated from male C57Bl/6J mice, a gift from Dr Darwin Prockop, Texas A&M Health Science Center, College Station, Texas, USA) were used as previously described.4 5 All in vitro experiments were performed in triplicate. Details of in vitro experiments are presented in the online supplementary material.
thoraxjnl-2019-213561supp001.pdf (172KB, pdf)
In vivo inhibition of miR-27a-5p in murine lungs
Mice were randomised (random number generator) to receive miR-27a-5p inhibitor (INH), negative control (NC) or equal volume saline combined with transfection reagent (HiPerfect Transfection reagent). Sample size calculation was based on bronchial alveolar lavage fluid (BALf) cell count between mice treated with LPS+NC versus LPS+INH. A total sample size of six animals per group provided 80% power to detect statistically significant differences using an alpha 0.05 and beta 0.2 (https://clincalc.com/stats/samplesize.aspx). The oligonucleotide mixtures were coadministered intratracheally with LPS (10 mg/kg body weight) or equal volume saline up to a total volume of 60 μL. Dose and route of oligonucleotide administration was based on manufactures’ suggestion and pilot experiments. Animals were anaesthetised with inhaled isofluorane. LPS and oligonucleotide mixture were coadministered. The surgical area was sutured and animals were allowed to recover in a bedding free cage. All animals received subcutaneous fluid resuscitation with 50 mL/kg saline and 0.2 mg/kg buprenorphine (Buprenex) for pain. Endpoints were measured at 24 hours postinstillation. BALf total cell counts and differentials were obtained as previously described.4 14 Total protein in BALf was measured by Bradford (Bio-Rad) and IgM levels ELISA (E90-101, Bethyl BioSciences).
Statistics
Observers assessing endpoints were blinded to group assignment. Where Kolmogorov-Smirnov (K-S) test demonstrated normal distribution (based on p-value ≥0.05 for statistic (D)), data are presented as individual values (symbols), means±SD and corrected using Tukey’s test for multiple comparisons. Non-parametric data are presented as median±IQR and Dunn’s post hoc test was used to correct for multiple comparisons.
Results
Identification of significantly associated miRNA:mRNA pairs in MSC-treated septic lungs
To identify miRNA:mRNA pairs in MSC-treated versus non-treated septic lungs, we generated a gene by miRNA matrix based on in silico predictions and matched to our lung mRNA and miRNA experimental data (schematic of the experiment shown in figure 1A). Overlap between in silico (predicted) versus empiric (observed) was 72% (p≤0.01). A total of 1395 putative miRNA:mRNA associations were identified in both sham versus CLP, as well as CLP treated with placebo versus MSC comparisons (FDR ≤0.0515; online supplementary table 1). Table 1 shows top 10 differentially expressed miRNA:mRNA associations in septic lungs after treatment with MSCs. miR-27a-5p and its putative target the GEF 3 (VAV3, table 1) were selected for detailed functional genomics based on the following criteria: (1) robust statistical prediction (unbiased); (2) empirical evidence of regulation of both miRNA and its target in lungs following CLP and MSC administration; (3) novelty, that is, not known to play a role in ARDS; (4) potential for clinical relevance based on homology of both miR and gene target between human and murine orthologues; (5) biological plausibility, that is, both miR-27a and VAV3 are linked to cytoskeletal rearrangement, receptor tyrosine kinase and integrin signalling and (6) clinical relevance: involved in cell adhesion, apoptosis, phagocytosis and cell migration—processes known to play a role in ARDS.16 17 miRNA 27a-5p and its target VAV3 are regulated by MSCs in septic lungs.
Figure 1.

Identification of the significant association of miR-27a-5p-VAV3 gene expression in septic and mesenchymal stromal cell (MSC)-treated septic lungs: (A) schematic of published animal experiment.4 Mice were randomised to caecum ligation and puncture (CLP) or sham surgery (sham); in addition to antibiotics (imipenem 0.5 mg per mouse per day), fluid resuscitation and pain medication, mice were further randomised to MSCs (2.5×105 cells, 100 mL total volume, MSC) or equal volume saline (placebo). Mice were sacrificed 28 hours postsurgery and lung tissues snap frozen for analysis. (B) Principal component analysis (PCA) including genes differentially expressed in septic lungs (CLP) treated with placebo versus MSCs (n=4751, cut-off false discovery rate (FDR) 5%). Each circle represents a sample from an individual mouse. Unsupervised analysis shows clustering of treatment groups (sham=green; CLP+MSC (MSC)=blue and CLP+placebo (CLP)=red). The percentage of the total variance is described by each principal component (PC): PC1: x-axis, PC2: y-axis and PC3: z-axis. Ellipsoids represent space two SDs from the mean of the centroid for each treatment group. (C) Hierarchical clustering (Ward’s method) of 60 top miR-27a target genes differentially expressed between CLP treated with either placebo or MSCs (FDR ≤0.05). By convention, red is upregulation and blue is downregulation. (D) Ingenuity pathway analysis (IPA) showing putative network generated using predicted (dashed lines) and published interactions (solid lines) between top miR-27a-5p predicted/documented targets differentially expressed in murine septic lungs in response to MSC administration. Real-time PCR showing expression of (E) miR-27a-5p and its putative targets (F) VAV3 and (G) ACE1 in mouse lungs following sham surgery (red), CLP (green) and CLP+MSC (blue). Bar graphs are fold change (FC) over the sham control; microRNA level normalised to miR-191 and mRNA level normalised to β actin. Each symbol represents data from an individual animal. Kolmogorov-Smirnov (K-S) test of normality was applied to miRNA and mRNA expression data, statistic (D) for each analysis indicated that the data did not differ from the normal distribution. Analysis of variance shows differences in the expression between CLP and MSC treated correcting using Tukey’s test for multiple comparisons. Data are presented as individual animals, means±SDs (*p≤0.05; **p≤0.01; ***p≤0.0001). Independent in vivo experiments: mice were randomised to receive lipopolysaccharide (LPS) (10 mg/kg) or equal volume saline and 24 hours later lungs were collected for quantitative real-time (RT)-PCR. (H) Gross anatomy documenting evidence of LPS-induced lung injury at 24 hours. RT-PCR shows relative change in the expression of (I) miR-27a-3p and (J) miR-27a-5p normalised to miR-191 (n=3 per group). Bar graphs show FC over sham control normalised to miR-191; data presented as means±SEM (*p≤0.05, Mann-Whitney test).
Table 1.
Top significant miRNA:mRNA association pairs differentially expressed in lungs from septic mice treated with MSCs compared with placebo
| DE miRNA | Probe ID | DE putative target gene (Gene symbol) | Adjusted p value |
| mmu.miR.20b | ILMN_2522023 | Tusc2 | 3.7686E-229 |
| mmu.miR.93 | ILMN_2834451 | Tusc2 | 6.8076E-228 |
| mmu.miR.340.5p | ILMN_2630512 | Rsrc1 | 9.1833E-221 |
| mmu.miR.20b | ILMN_2812164 | Irak4 | 1.1008E-198 |
| mmu.miR.27a-5p | ILMN_2630512 | Rsrc1 | 9.1769E-191 |
| mmu.miR.17 | ILMN_2812164 | Irak4 | 1.746E-189 |
| mmu.miR.705 | ILMN_1222333 | Slc2a4 | 8.5175E-174 |
| mmu.miR.27a-5p | ILMN_1227993 | Vav3 | 1.2719E-161 |
| mmu.miR.339.5p | ILMN_1227610 | Rcan3 | 4.2287E-152 |
| mmu.miR.290.3p | ILMN_1242945 | Ptpn3 | 1.5424E-149 |
DE, differentially expressed; miRNA, microRNA; MSC, mesenchymal stromal cell.
thoraxjnl-2019-213561supp002.pdf (99.9KB, pdf)
Principal component analysis shows clustering of septic (CLP) versus sham-treated groups (figure 1B). MSC administration results in a shift in global transcription profiles favouring reconstitution of a ‘sham-like’ expression patterns (figure 1B). This transition is more evident from the hierarchical clustering of miR-27a putative target genes differentially expressed in septic and MSC treated lungs (figure 1C, table 2). Putative miR-27a-5p targets include VAV3 as well as other ARDS/ALI relevant targets: nuclear factor 2 (Nrf2), ACE 1 (ACE1), epidermal growth factor receptor (EGFR) and fibroblast growth factor 7 (FGF7, also known as keratinocyte growth factor (KGF)). Ingenuity pathway analysis was used to generate a putative gene interaction network using predicted (dashed lines) and documented (solid lines) interactions between miR-27a-5p targets (figure 1D). Independent experiments confirmed upregulation of miR-27a-5p and downregulation of VAV3 in CLP-placebo (figure 1E, F) and downregulation of miR-27a-5p and upregulation of VAV3 and ACE1 in the MSC-treated groups (figure 1E–G). miR-191 was used for normalisation; this miRNA was found to be statistically superior to other reference miRs.18 Additional separate mice were exposed to intratracheal LPS (10 mg/kg) to confirm miR-27a-3p and miR-27a-5 p (figure 1H–J) at 24 hours.
Table 2.
Top 30 putative miR-27a-5p targets differentially expressed in septic and MSC-treated murine lungs
| Gene symbol | P value MSC/CLP |
P value CLP/sham |
FC MSC/CLP |
FC CLP/sham |
Gene name |
| Rbfox3 | 5.85E-08 | 2.92E-01 | −2.95 | 1.21 | RNA binding fox-1 homolog 3 |
| Cldn2 | 1.45E-07 | 5.08E-10 | 3.18 | −4.19 | Claudin 2 |
| Ace1 | 7.69E-06 | 3.39E-04 | 2.24 | −2.87 | ACE I |
| Prkg2 | 8.34E-06 | 1.44E-02 | −1.88 | 1.39 | Protein kinase cGMP-dependent 2 |
| Podxl | 1.16E-05 | 7.07E-06 | −2.07 | 2.12 | Podocalyxin like |
| Npr3 | 1.71E-05 | 4.03E-06 | 1.65 | −1.72 | Natriuretic peptide receptor 3 |
| Scn3b | 2.93E-05 | 8.53E-01 | 2.22 | −1.03 | Sodium voltage-gated channel beta subunit 3 |
| Igfbp3 | 3.26E-05 | 1.54E-03 | 2.73 | −2.10 | Insulin-like growth factor-binding protein 3 |
| Stmn2 | 4.56E-05 | 7.67E-08 | −2.92 | 4.43 | Stathmin 2 |
| Egfr | 8.15E-05 | 1.20E-04 | 2.27 | −2.22 | Epidermal growth factor receptor |
| Ehd4 | 1.76E-04 | 1.10E-07 | −1.68 | 2.18 | EH domain containing 4 |
| Nrip3 | 1.76E-04 | 6.36E-05 | 2.33 | −2.49 | Nuclear receptor interacting protein 3 |
| Kcnh1 | 1.85E-04 | 4.78E-03 | 1.84 | −1.57 | K+ voltage-gated channel subfamily H member 1 |
| Vav3 | 1.95E-04 | 1.41E-06 | 2.56 | −2.82 | vav guanine nucleotide exchange factor 3 |
| Copg | 2.15E-04 | 2.78E-05 | 1.40 | −1.47 | Coatomer protein complex subunit gamma 1 |
| Gria1 | 2.17E-04 | 3.44E-05 | −1.75 | 1.89 | Glutamate ionotropic Rc AMPA type subunit 1 |
| Fgf7 | 2.58E-04 | 1.45E-02 | 1.78 | −1.45 | Fibroblast growth factor 7 |
| Pappa | 3.01E-04 | 6.99E-09 | 2.37 | −4.51 | Pappalysin 1 |
| Ppm1f | 3.04E-04 | 4.53E-05 | −1.51 | 1.61 | Protein phosphatase, Mg2+/Mn2+ dependent 1F |
| Lsm1 | 3.70E-04 | 8.45E-03 | 1.35 | −1.24 | LSM1 homologue, mRNA degradation associated |
| Hspa12b | 3.77E-04 | 4.26E-06 | −2.16 | 2.80 | Heat shock family A (Hsp70) member 12B |
| Ndrg1 | 5.57E-04 | 3.13E-03 | 2.42 | −2.11 | N-myc downstream regulated 1 |
| Cxcl12 | 6.78E-04 | 1.89E-10 | −1.96 | 4.15 | C-X-C motif chemokine ligand 12 |
| Pftk1 | 6.98E-04 | 1.70E-06 | −1.67 | 2.13 | Cyclin-dependent kinase 14 |
| Mylk | 8.14E-04 | 1.37E-05 | −1.73 | 2.09 | Myosin light chain kinase |
| Frmd6 | 8.16E-04 | 1.15E-04 | −1.54 | 1.66 | FERM domain containing 6 |
| Calcrl | 8.46E-04 | 2.26E-03 | −2.09 | 1.96 | Calcitonin receptor like receptor |
| Ykt6 | 8.85E-04 | 8.23E-03 | 1.38 | −1.29 | YKT6 v-SNARE homologue |
| Nrf2 | 8.97E-04 | 8.93E-05 | 1.34 | −1.42 | Nuclear factor, erythroid 2 like 2 |
| Slc41a2 | 9.23E-04 | 6.85E-05 | 1.71 | −1.93 | Solute carrier family 41 member 2 |
| Cxcl14 | 9.30E-04 | 1.35E-04 | 3.34 | −4.10 | C-X-C motif chemokine ligand 14 |
cGMP, cyclic guanosine monophosphate; CLP, caecum ligation and puncture; FC, fold change; MSC, mesenchymal stromal cell.
Predicted involvement of miR-27a-5p:VAV3 in inflammation and innate immunity pathways
Enrichment analysis highlighted MSC-mediated regulation of receptor tyrosine kinase signalling (Torso signalling), integrin-mediated signalling and sterol regulatory element-binding protein (table 3) in septic lungs. Canonical pathway analysis predicted alteration in: DR3 and DR4/5 death receptors, modulation by heat shock protein 70, FAS/FADD signalling and response to DNA damage (p53 pathway). Inflammation and innate immunity pathways were associated with the inhibition of cell adhesion and migration, phagocytosis, antigen presentation and cell junction function (integrin signalling, Fc gamma R-mediated phagocytosis and JAK/STAT signalling; table 4). Enrichment analysis using only miR-27a-5p predicted targets underscored involvement of this miR in regulation of cytoskeleton, chemokine and cytokine activity, focal adhesion, gap junction and leucocyte transmigration (table 5), implicating miR-27a-5p in inflammatory cell migration and barrier function.
Table 3.
Top 10 biological processes altered in septic lungs treated with MSCs
| Biological processes | FDR | Combined score |
| Torso signalling pathway (GO:0008293) | 0.0004 | 36.33 |
| Positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 0.0119 | 32.39 |
| Integrin-mediated signalling (GO:0007229) | 0.0023 | 31.64 |
| Regulation of transcription involved in cell fate commitment (GO:0060850) | 0.0009 | 30.26 |
| Regulation of transcription in anterior/posterior axis specification (GO:0044324) | 0.0012 | 29.08 |
| Positive regulation of transcription from RNA polymerase III promoter (GO:0045945) | 0.0042 | 28.45 |
| Regulation of transcription from RNA polymerase II promoter (GO:0060994) | 0.0015 | 28.35 |
| Regulation of SREBP signalling pathway (GO:2000638) | 0.0015 | 28.27 |
| Regulation of transcription from RNA polymerase II promoter (GO:1901213) | 0.0027 | 27.78 |
| Positive regulation of DNA-templated transcription, elongation (GO:0032786) | 0.0045 | 27.7 |
FDR, false discovery rate; GO, gene ontology; MSC, mesenchymal stromal cell.
Table 4.
Canonical pathways altered in septic lungs treated with MSCs
| Pathways | Combined score | Database |
| Induction of apoptosis via DR3 and DR4/5 death receptors | 9.14 | BioCarta |
| Salmonella infection | 8.42 | KEGG |
| Apoptosis modulation by HSP70 | 7.29 | Reactome |
| FAS pathway and stress induction of HSP regulation | 6.64 | Reactome |
| Glycosphingolipid biosynthesis | 6.36 | KEGG |
| Wnt signalling pathway netpath | 6.26 | Reactome |
| Adipocytokine signalling pathway | 6.25 | KEGG |
| BDNF signalling pathway | 6.14 | Reactome |
| FAS signalling pathway | 5.99 | Panther |
| Neurotrophin signalling pathway | 5.66 | KEGG |
| ER-associated degradation pathway | 5.40 | BioCarta |
| Signal transduction of S1P receptor | 5.06 | Reactome |
| FAS signalling pathway (CD95) | 5.03 | BioCarta |
| AMPK signalling pathway | 5.03 | KEGG |
| Insulin signalling | 4.72 | Reactome |
| Fc gamma R-mediated phagocytosis | 4.08 | KEGG |
| mRNA surveillance pathway | 4.02 | KEGG |
| Nicotinic acetylcholine receptor signalling pathway | 3.90 | Panther |
| Cytoskeletal regulation by Rho GTPase | 2.98 | Panther |
| Nerve growth factor pathway | 2.70 | BioCarta |
| Huntington disease | 2.42 | Panther |
| CXCR4 signalling pathway | 2.39 | BioCarta |
| Apoptotic signalling in response to DNA damage | 1.94 | BioCarta |
| EGF signalling pathway | 1.86 | BioCarta |
| JAK/STAT signalling pathway | 1.75 | Panther |
| 5HT3 type receptor-mediated signalling pathway | 1.32 | Panther |
| Muscarinic acetylcholine receptor 1 and 3 signalling pathway | 1.21 | Panther |
| 5HT2 type receptor-mediated signalling pathway | 1.13 | Panther |
| 5HT4 type receptor-mediated signalling pathway | 1.06 | Panther |
| p53 pathway | 0.51 | KEGG |
| Inflammation mediated by chemokine and cytokine signalling | 0.03 | Panther |
| Parkinson disease | −0.40 | Panther |
| Epinephrine and norepinephrine biosynthesis | −0.45 | KEGG |
| Alzheimer disease-amyloid secretase pathway | −0.62 | Panther |
EGF, epidermal growth factor ; HSP70, heat shock protein 70; MSC, mesenchymal stromal cell.
Table 5.
Top KEGG pathways regulated by top putative miR-27a-5p targets
| Name | P value | Adj p value | Combined score |
| Regulation of actin cytoskeleton | 0.00 | 0.02 | 15.46 |
| Leucocyte transendothelial migration | 0.00 | 0.03 | 12.13 |
| Chemokine signalling pathway | 0.00 | 0.05 | 10.91 |
| Focal adhesion | 0.00 | 0.05 | 10.34 |
| Gap junction | 0.00 | 0.05 | 9.99 |
| Cytokine–cytokine receptor interaction | 0.01 | 0.06 | 9.33 |
| Platelet activation | 0.01 | 0.07 | 8.4 |
miR-27a-5p and VAV3 are differentially regulated in primary human endothelial cells
Both miR-27a-5p and VAV3 target gene share 100% homology with their human orthologues (figure 2A). In HPMECs exposed to TNF, the expression of miR27a-5p is upregulated by increasing doses of TNF (10–40 ng/mL, figure 2B) and remains elevated up to 24 hours poststimulation (figure 2C). The expression of VAV3 gene (figure 2E) and protein (figure 2F) are downregulated 61% and 50%, respectively, in cells treated with TNF where the expression of miR-27a-5p and miR-27a-3p is increased (figure 2D). The expression of miR-27a-5p and miR-27a-3p is also increased by TNF in RAW 264.7 cells (murine macrophage cell line; online supplementary figure 1A) and human distal bronchial epithelial cells BEAS2b (online supplementary figure 1B). While the expression of both VAV3 and ACE1 decreased as per in silico predictions in HPMECs in response to TNF, FGF7 and EGFR increased (online supplementary figure 1B, C and online supplementary figure 2A). Protein expression of ACE1 decreased, while protein expression of EGFR and FGF7 (KGF) increased (online supplementary figure 1F, G), suggesting that in these cells these genes are not regulated as predicted.
Figure 2.
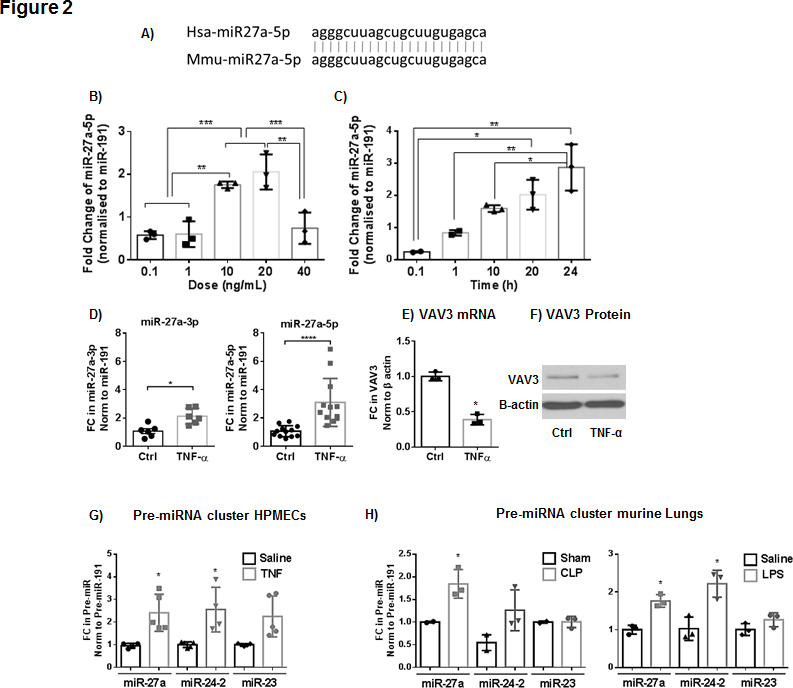
Expression of miR-27a-5p and its precursor in primary human pulmonary microvascular endothelial cells (HPMECs). (A) Sequence homology between human (hsa) and murine (mmu) miR-27a-5p. Real-time PCR relative change in the expression of miR-27a-5p in response to increasing doses (B) and time (C) of exposure to tumour necrosis factor (TNF). Bar graphs represent fold change (FC) over the sham control normalised to miR-191 (n=3). (D) Real-time PCR relative change in the expression of miR-27a-3p and miR-27a-5p in HPMECs following stimulation with TNF (10 ng/mL) for 24 hours (individual data presented and median and IQR, Mann-Whitney *p≤0.05; ****p≤0.0001) and western blot (F) and quantification (E) showing changes in VAV3 protein normalised to β-actin corresponding change in the expression of VAV3 gene (Mann-Whitney *p≤0.05). (G) Changes in the expression levels of the precursors for miR-27a, miR24-2 and miR-23 in HPMECs in response to TNF (10 ng/mL). (H) Changes in the expression of pre-miR-27a, pre-miR-24-1 and pre-miR-23 in mouse lung tissues in response to caecum ligation and puncture (CLP) (28 hours) or lipopolysaccharide (LPS) intratracheal instillation (10 mg/kg for 24 hours). Kolmogorov-Smirnov (K-S) test shows that data are normally distributed, and presented as means±SD normalised to miR-191 (*p≤0.05, analysis of variance, Tukey post hoc multiple comparisons correction). Ctrl, control.
thoraxjnl-2019-213561supp003.pdf (1.1MB, pdf)
thoraxjnl-2019-213561supp004.pdf (1.2MB, pdf)
The mirna27a gene is located on chromosome 8 in mice and chromosome 19 in humans; it lives in a cluster that encodes in both species three distinct pre-miRNAs: miR-23a, miR-24–2 and miR-27a.19 Paired miRNA:mRNA analysis did not identify other members of the cluster as differentially regulated. Notwithstanding, we found miR-24a and miR-23 were both upregulated by exposure to TNF in HPMECs (figure 2G); only miRs-27a and miRs-24 levels were upregulated by CLP and LPS in murine lungs (figure 2H).
Conditioned medium from cultured MSCs attenuated inflammation-mediated upregulation of miR-27a in vitro
Conditioned medium from cultured MSCs mitigated TNF-induced upregulation of miR-27a-5p in HPMECs and BEAS2b cells (figure 3A, B). Transfection of HPMECs with the miR-27a-5p inhibitor significantly reduced TNF-induced upregulation of miR-27a-5p (figure 3C) but did not significantly affect the expression of the miR-27a-3p fragment (figure 3D). Transfection of the miR-27a-5p mimic resulted in a 47% to 52% reduction in VAV3 expression at baseline and following TNF treatment, respectively (figure 3E). ACE1 mRNA levels were reduced following TNF and transfection of the miR-27a-5p mimic (online supplementary figure 2A) and increased following miR-27a-5p inhibitor transfection (online supplementary figure 2A). The expression of Nrf2 was not significantly modulated by the miR-27a-5p mimic or inhibitor (online supplementary figure 2B). Baseline VAV3 expression is protected from miR-27a-5p overexpression (figure 3E, F). Although TNF induced downregulation of ACE1 mRNA levels and transfection, this was significantly mitigated by miR-27a-5p inhibition. Transfection of miR-27a-5p mimic or inhibitor did not affect cell viability at 24 hours (online supplementary figure 3).
Figure 3.

VAV3 is a target of miR-27a-5p. (A) Schematic diagram of mesenchymal stromal cell (MSC)-conditioned medium (MSC-CdM) experiment. MSCs were generated from bone marrow of wild-type (WT) C57Bl6J mice. Human pulmonary microvascular endothelial cell (HPMEC) monolayers were cotreated with TNF (10 ng/mL) and 20% conditioned media from cultured MSCs or control medium (CM) for 24 hours. (B) Bar graphs showing relative fold change (FC) in the expression of miR-27a-5p and miR-27a-3p normalised to miR-191 in response to tumour necrosis factor (TNF) in the presence of CM or MSC-CdM. Data are normally distributed and presented for individual experiments as means±SD (two-way analysis of variance (ANOVA), comparison to CM alone (*p≤0.05) and comparison to CM+TNF (#p≤0.05) corrected for multiple comparisons using Tukey test). (C) HPMECs were transfected with miR-27a-5p inhibitor (INH; 25 nM) or negative control (NC; 25 nM) for 24 hours and then stimulated for 24 hours with TNF (10 ng/mL). Transfection of the miR-27a-5p inhibitor results in significant decreased levels of miR-27a expression at baseline and following TNF stimulation, but had little effect on the expression levels of the miR-3p compared with the negative control (N Ctrl) microRNA (miRNA) inhibitor (D). Transfection of HPMECs with the miR-27a-5p mimic (MIM; 25 nM) resulted in approximately a 50% reduction in the levels of expression of VAV3 at baseline and in response to TNF (E), while transfection of the miR-27a-5p inhibitor resulted in the increased expression of the levels of VAV3 following TNF stimulation compared with transfection of the microRNA NC (F). Bar graphs represent fold change (FC) over the sham control. miRNA expression was normalised to miR-191, and mRNA expression was normalised to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are normally distributed and presented for individual experiments, means±SDs (*p≤0.05, **p≤0.01, ***p≤0.0001, two-way analysis of variance (ANOVA), Tukey’s correction for multiple comparisons). (G) Genomic sequence of luciferase reporter constructs containing either the wild-type or mutated VAV3 miR-27a-5p 3′UTR binding seed sequences. Mutant 1 (Mut 1) contained a deletion of the heptameric sequence; in Mutant 2 (Mut 2) both heptameric and sextameric sequences were deleted. (I) Luciferase activity in HPMECs transfected with either the WT or mutated VAV3 3′UTR and the miR-27a-5p overexpression (MIM), miR-27a-5p inhibitor (INH) or their respective NCs (mimic (m) and inhibitor (i) Ctrl, n=3) challenged with TNF (10 ng/mL). Data are normally distributed and presented as individual experiments, means±SDs (*p≤0.05; two-way ANOVA, Tukey’s correction for multiple comparisons). 3′UTR, 3′untranslated region.
thoraxjnl-2019-213561supp005.pdf (906.3KB, pdf)
Binding of miR-27a-5p to the human 3′UTR of VAV3
Two mutation constructs containing either a deletion of the heptameric (mutant 1) or both the heptameric and sextameric (mutant 2) miR-27a-5p seed binding sequences on the 3′UTR of VAV3 were generated (figure 3G, online supplementary material and figure 4A) to demonstrate specific binding of miR-27a-5p to the 3′UTR of VAV3 (figure 3I). miR-27a-5p inhibition in TNF-treated cells rescued luciferase activity indicating that inhibition of miR-27a-5p prevents binding of miR-27a-5p to the wild-type 3′UTR and VAV3 mRNA degradation. No significant changes in luciferase activity were seen when cells were transfected with a luciferase constructs containing the 3′UTR of the housekeeping gene actinomycin B or a non-targeting control R01 (online supplementary figure 4B).
Figure 4.

Inhibition of miR-27a-5p enhances wound healing and mitigates tumour necrosis factor (TNF)-induced cell adhesion and transmigration: (A) change in fluorescent intensity showing dextran leakage across human pulmonarymicrovascular endothelial cell (HPMEC) monolayers. HPMECs were seeded onto 0.2% gelatin-coated 8-µm-pore Transwell inserts and transfected with either negative control (NC) or miR-27a-5p mimic (MIM); 24 hours later monolayers were treated with TNF (10 ng/mL) or saline for 24 hours. The following morning FITC-labelled dextran (70 KDa) was added to the upper chamber and the fluorescence of the lower chamber was measured every 30 min for 3 hours. Bar graphs show fluorescence intensity means±SDs for individual experiments normalised to NC Ctrl at time 0. Peak differences in dextran leakage between NC and TNF and MIM+TNF are seen at 1.5 hours (for normally distributed data, two-way analysis of variance (ANOVA), Tukey’s correction for multiple comparisons, **p≤0.01 and ****p≤0.0001). (B) Representative photomicrographs from experiments showing wound closure in HPMECs. HPMECs were transfected with NC, miR-27a-5p inhibitor (INH) or miR-27a-5p MIM and 24 hours later a single scratch was made straight down the middle of the monolayer and TNF (10 ng/mL) or saline was added. Micrographs from one representative experiment is shown (n=4). Distance between the leading edges was measured and bar graph show relative change in mean wound closure±SD. Scale bars are 50 mm. (C) Representative micrographs from experiments showing inhibition of THP-1 monocyte adhesion to the TNF-activated HPMECs by miR-27a-5p inhibitor (n=3). HPMECs transfected with MIM and INH and treated with TNF as described above. The following morning, THP-1 monocytes were labelled with 2 µM of CellTracker Green 5-chloromethylfluorescein diacetate and 5×105 labelled monocytes (in RPMI-1640 medium) were added to each well and allowed to attach to the monolayer for 30 min. After washing, cell adhesion was determined by measuring fluorescence (excitation/emission of 492/517 nm). Bar graph shows mean fold change in cell attachment ±SD. (D) Transendothelial migration assay. HPMECs grown in Transwell inserts as above were transfected with miR-27a-5p MIM, INH or N Ctrl and exposed to TNF as above. The following morning, HPMECs were washed, and monocyte chemoattractant protein-1 (10 ng/mL) was added to the lower chamber. Fluorescently labelled THP-1 monocytes were added to the upper chamber and cells were allowed to migrate for 2 hours. Transwells were then removed and fluorescent images of the lower chamber taken. Cells were counted for calculation of relative transmigration (n=4–5). Bar graph show relative number of transmigrated monocytes. All data are normally distributed (Kolmogorov-Smirnov test) and presented as means±SDs (*p≤0.05, **p≤0.01, two-way ANOVA, Tukey correction for multiple comparisons).
thoraxjnl-2019-213561supp006.pdf (948.5KB, pdf)
Modulation of miR-27a-5p altered wound closure, adhesion, migration and transmigration, but not barrier function
TNF administration to confluent HPMEC monolayers resulted in increased cellular leakage as measured by the amount of 70 kDa FITC-Dextran that passed through the monolayer. Transfection of miR-27a-5p mimic did not significantly increase FITC-Dextran leakage (figure 4A). Loss of cell number due to death or apoptosis was not a confounding factor (online supplementary figure 3). Wound healing scratch assay made on 24-hour serum-starved, confluent HPMECs transfected with the miR-27a-5p mimic, inhibitor or control miRNA demonstrated that exposure to TNF or/and transfection of the miR-27a-5p mimic significantly delayed wound closure (figure 4B). In contrast, miR-27a-5p inhibition enhanced wound closure at baseline and following exposure to TNF (figure 4B). Adhesion of monocytes to the HPMEC monolayer was quantified by coculturing HPMECs with fluorescently labelled monocytes THP-1 cells (described in online supplementary methods). Exposure to TNF increased monocyte adhesion compared with saline (figure 4C). Transfection with the miR-27a-5p mimic increased monocyte adhesion both at baseline and following TNF stimulation, while miR-27a-5p inhibition mitigated monocyte adhesion primarily following TNF stimulation (figure 4C). In a transwell migration assay, MiR-27a-5p inhibition attenuated transendothelial migration of THP1 cells through an HPMEC monolayer following TNF stimulation but had no effect on transendothelial migration at baseline (figure 4D). Transfection with the miR27a-5p mimic resulted in enhanced migration of monocytes across the HPMEC monolayer following TNF stimulation (figure 4D). Transfection of either 40 nM or 80 nM of an small interfering RNA (siRNA) against VAV3 resulted in significant delay in wound closure (figure 5A, B) and decrease in the expression of VAV3 protein in HPMECs (figure 5C).
Figure 5.
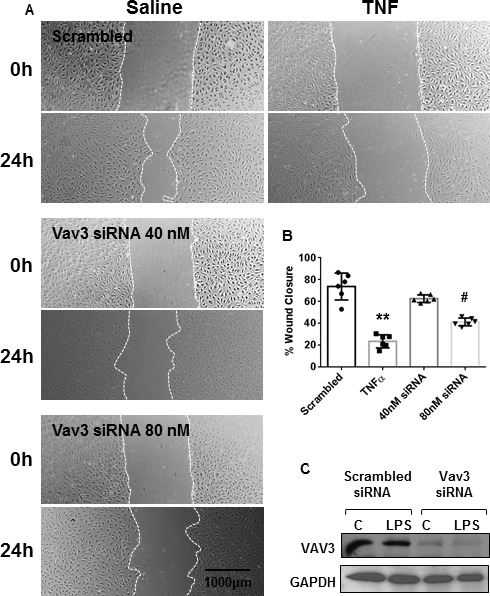
Inhibition of VAV3 delays wound closure: human pulmonarymicrovascular endothelial cells (HPMECs) were transfected with either an small interfering (siRNA) against VAV3 (40 nM or 80 nM) or scrambled control (80 nM) and 24 hours later a single scratch was made straight down the middle of the monolayer using a 100 µL pipette tip and tumour necrosis factor (TNF) (10 ng/mL) or saline added to the medium. Distance between the leading edges was measured at 0 and 24 hours. (A) Micrographs from one representative experiment is shown (n=4). (B) Bar graph of normally distributed data (Kolmogorov-Smirnov test) shows relative change in wound closure as mean % closure±SD (*p≤0.05; two-way analysis of variance, Tukey correction for multiple comparisons). (C) Western blot showing downregulation of VAV3 following transfection of siRNA against VAV3 (80 nM) or scrambled siRNA normalised to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Inhibition of miR-27a-5p in vivo attenuated pulmonary cellular infiltration but not oedema formation following LPS-induced lung injury
Intratracheal pulmonary endotoxin instillation coadministration with miR-27a-5p inhibitor results in a significant decrease in total cell count and neutrophil infiltration in mice that received the miR-27a-5p inhibitor compared with NC (figure 6A, B). Lung injury scores were calculated on the basis of the relative degree of inflammation, oedema, haemorrhage and atelectasis seen on histology (figure 6C). Coinstillation of LPS and miR-27a-5p inhibitor showed a partial decrease in total protein and IgM levels after LPS but this did not reach statistical significance (figure 6C). Endotoxin instillation resulted in both increased miR-27a-5p and decreased VAV3 protein expression (figure 6F–I). Immunohistochemistry shows decreased VAV3 antibody binding after LPS treatment (figure 6F, online supplementary material; miR-27a-5p inhibitor coinstillation attenuates VAV3 protein expression decrease (figure 6H). Expression of VAV3 also decreased following LPS instillation in mice that received the NC but was preserved in mice that received the miR-27a-5p inhibitor (figure 6I).
Figure 6.

In vivo miR-27a-5p inhibition mitigates cellular infiltration but not protein and IgM leakage into alveolar space: mice were randomised to receive vehicle (HiPerfect Transfection reagent, HPF) or lipopolysaccharide (LPS) (10 mg/kg)±miR-27a-5p inhibitor (INH, 1.6 nmol) or negative control (NCtrl, 1.6 nmol) and 24 hours later lungs were lavaged and differential cell counts were determined. (A) The photomicrograph showing a representative field of H&E stained preparations from each group of mice (magnification 100×). Kolmogorov-Smirnov test demonstrates that data are normally distributed. Bar graphs showing differences in (B) total cell count, and differential macrophages and neutrophils as mean±SD (n=4–8; *p≤0.05; two-way analysis of variance (ANOVA)) in bronchoalveolar lavage fluid (BALf). (C) Bar graphs showing changes in lung injury score; (D) total protein (mg/mL); and (E) total IgM (ng/mL) in BALf. Data are represented as means±SDs (*p≤0.05; two-way analysis of variance (ANOVA), Tukey correction for multiple comparisons). (F) Immunohistochemistry staining for VAV3 in lungs of wild-type mice 24 hours post-LPS (10 mg/kg)±INH (1.6 nmol) or NCtrl(1.6 nmol). (G) Bar graph showing fold change (FC) in the expression of miR-27a-5p normalised to miR-191. (H) Bar graph showing relative change in DAB stain showing change in VAV3 staining in lung tissues and (I) western blot showing changes in the VAV3 protein expression in lung tissues from mice 24 hours post-LPS (10 mg/kg)±INH (1.6 nmol) or NCtrl (1.6 nmol). Data represented as means±SDs (*p≤0.05; two-way ANOVA, Tukey correction for multiple comparisons). NC, negative control; Sal, saline.
Increased miR-27a-5p expression in patients who died with ARDS
Diffuse alveolar damage (DAD) is the histological correlate of severe ALI for most patients with ARDS. Absence of fresh human lung tissue from patients with ARDS is a major obstacle to further pathological, molecular and mechanistic studies in ARDS. To circumvent this limitation, we obtained extensively annotated autopsy lung tissues from patients who died with ARDS with or without biopsy proven DAD. We extracted RNA from formalin-fixed paraffin-embedded lung sections, known to contain DAD or no-DAD tissue, and determined by quantitative real-time PCR that the expression level of miR-27a-5p was significantly increased in ARDS lungs with DAD compared with no-DAD. These data support the importance of miR-27a-5p in human pathology (figure 7).
Figure 7.

Upregulation of miR-27a-5p in human acute respiratory distress syndrome (ARDS) lungs. Real-time PCR results for miR-27a-3p and miR-27a-5p expression in RNA extracted from formalin-fixed, paraffin-embedded lung autopsy samples from patients who died from ARDS with or without diffuse alveolar damage (DAD). Box plots shows fold change with respect to no-DAD cohort normalised to miR-191. Data are presented as median and IQR (n=4–8 per group; p values are shown for comparisons using Mann-Whitney test).
Discussion
Our group and others have shown that MSC administration reduces morbidity and mortality in experimental models of sepsis.4 20–23 Although these studies demonstrate the feasibility of cell-based technologies for the treatment of sepsis, mechanism(s) remain incompletely understood and are central to the development of novel therapeutics. Here, we exploited the canonical relationship between miRs and their putative targets to identify miR-27a:VAV3 pair as both modulated by MSCs and putatively involved in sepsis-induced lung injury. In vitro using a relevant cellular model of primary human pulmonary endothelial cells exposed to TNF (TNF can reproduce many of the key physiological and molecular derangements seen in sepsis), the human miR-27a-5p binds specifically to its target on the 3′UTR of VAV3 and is involved in cellular migration, adhesion and transmigration, but not convincingly in endothelial permeability. In vivo, using an independent model of ALI induced by endotoxin instillation, coadministration with the miR-27a-5p inhibitor prevents LPS-induced cellular infiltration, but not loss of barrier function. The clinical relevance of our findings is evidenced by increased miR-27a-5p expression in patients with ARDS who died with autopsy-proven DAD versus no DAD.
Using an miRNA:mRNA paired analysis approach, rather than matching miRs to putative mRNA lists post hoc, improved power and allowed us to derive important biological knowledge from the involvement of miR-27a in the regulation of the host response to injury and MSC therapy. Enrichment analysis predicted miR-27a would contribute to actin cytoskeleton, leucocyte transendothelial migration, transmigration, adhesion (focal adhesion) and gap junction regulation which we confirmed using in vitro and in vivo functional assays. In silico, VAV3 was predicted to be a key target—which we confirmed using a luciferase reporter system and siRNA against VAV3. This does not imply that VAV3 is the ‘only’ target nor that miR-27a-5p regulates the phenotype exclusively through VAV3 since other miR-27a-5p targets including ACE1, FGF7 and EGFR were also found to be regulated, both in vitro and in vivo, but the regulatory relationship to miR-27a was not exclusive, suggesting other (as of yet unknown) factors may further contribute. Importantly, inhibition of miR-27a-5p significantly reduced in vitro and in vivo evidence of endothelial cell dysfunction and ALI. Further studies will be required to elucidate the specific role(s) of VAV3 as well as other miR-27a-5p targets. In the interim, the main conclusion of our study is that miR-27a-5p plays a biologically and possibly clinically relevant role in ARDS.
How MSCs regulate miR expression levels in recipients remains highly speculative. Recent studies have highlighted the function of miRNAs in the paracrine effects of MSCs. These miRNAs may either modulate expression of proteins secreted by MSCs or be contained in microvesicles and exosomes, to exert their regulatory function in target cells. We tried to glean possible mechanisms by querying coregulation of the miR-27 cluster. miR-27a is part of the miR-23a~27a~24–2 cluster documented to have altered expression in many disease states24 involving various functions such as angiogenesis, cell differentiation, proliferation, inflammation25 and infection.26 27 All three miRNAs of this cluster are derived from a single primary transcript; transforming growth factor beta receptor 1(TGF-βR1) and SMAD transcription factors are primary regulators of the cluster, but expression pattern of different members of the cluster may vary.24 Ours is the first report underscoring the role of miR-27a-5p in acute inflammatory pulmonary injury. Previous reports implicate miR-27a in the regulation of VE-cadherin, vascular leak and recovery from ischaemic injury.28 Little is known about the role of miR-27a-5p in cell migration, transmigration and adhesion, and much of what is known has been primarily defined in malignancy. In these models, miR-27a acts as a tumour suppressor by inhibiting TGF-βRI signalling pathway.29
The Vav family of proteins (Vav1, Vav2, Vav3) are cytoplasmic guanine nucleotide exchange factors (GEFs) for Rho-family GTPases. Specific receptor signalling results in the tyrosine phosphorylation of Vav proteins and hence their activation.30 Results from mice deficient in one or more Vav proteins show critical roles in cell development, activation as well as sensing of bacterial products: LPS binding protein, CD14, Toll-like receptor 4 (TLR4) and MyD88, conveying threat signals from the cell membrane to intracellular intermediate mediators Rac1, Cdc42, ras homology gene (RhoG) and rho associated coiled-coil containing protein kinase 1 (ROCK). These processes are associated with multiple cell shape changes that rely on the reorganisation of the cytoskeleton.31 In this context, small GTPases of the RhoA family play essential regulatory roles. Vav proteins are activated by EGFR and fibroblast growth factor 2 receptor signalling.32 VAV3 also plays a key role as part of the integrin adhesome, a complex interactome involved in the transduction of integrin-related signalling.33 34 VAV3-deficient mice have hypertension and cardiac hypertrophy.35 In the immune system, deletion of Vav genes results in compromised immune responses.36
Of specific interest to ARDS, miR-27a was shown to be upregulated in microparticles derived from vascular smooth muscle cells exposed to mechanical cyclic stretch. Multipoint injection of antagomiR-27a around carotid artery decreased miR-27a expression in vivo and reversed abnormal endothelial cell proliferation suggesting a role for miR-27a in vascular remodelling.37 In studies using Townes humanised sickle cell (SS) murine models of haemolysis triggered endothelial dysfunction. In this model, haemin released during haemolysis (also happens in severe ARDS) leads to pulmonary hypertension and right ventricular hypertrophy associated with reduced lung levels of peroxisome proliferator-activated receptor γ and increased levels of miR-27a; inhibition of miR-27a reconstituted endothelin-1 levels and mitigated endothelial cell dysfunction in the SS mouse lung.38 Our study focused on the role of miR-27a in ALI, possibly longer time points would provide important clues to a role in repair and remodelling postacute injury.
While in our model the 5p mature strand of miR-27a was the primary target of regulation, our initial experiments have shown that miR-27a-3p is also upregulated in response to an inflammatory stimulus in HPMECs. Future studies involving detailed analysis of both targets will elucidate their role(s) in ARDS. Here we focused primarily on the role of miR-27a-5p in endothelial cells but given the critical role of VAV3 in immune cells, future studies using gene-specific deletion mice are warranted. Exciting results from time-series miRNA-mRNA integrated analysis identified various miRNAs whose expression differs significantly between M1 and M2 polarised macrophages and miR-27a-5p was one of the top miRs regulated during M2 polarisation.19 Furthermore, upregulation of miR-27a enhances the expression of proinflammatory cytokines in TLR4-activated macrophages via downregulation of interleukin 10 (IL-10).25 Since MSC treatment is known to induce polarisation of macrophages, possibly in an IL-10 dependent manner,20 this suggests regulation of miR-27a in myeloid derived cells might play an important role in defining myeloid endophenotypes following ALI.
In summary, although various genes were predicted to be miR-27a-5p targets (ACE1, EGFR, FGF7 and NRF2), the regulation of VAV3 by miR-27a-5p in primary human microvascular endothelial cells was the most robust in vitro. This does not preclude the coexistence of other tight regulatory relationships between miR-27a-5p and its other targets in other cells, by alternative stimuli and/or at different time points. Our in vivo experiments show proof of principle that miR-27a-5p plays an important role in early ARDS. Future studies are currently underway to demonstrate whether RNA-based therapeutics at later time points represents a viable option for treatment. From a clinical perspective, the potential pleotropic beneficial effects of miR-27a-5p inhibition suggest RNA-based therapeutics targeting this miRNA early in the course of illness may modulate the progression of ALI and ARDS.
Acknowledgments
We thank Dr Ana Paula Teixeira for her review of the manuscript and Dr Yuexin Shan for experimental technical support.
Footnotes
NY and LZ contributed equally.
Contributors: Acquisition: NY, LZ, HA, SHJM and RH; analysis: LZ, HA, NY, PH, CCdS; interpretation: CCdS, PH, WCWL, PM, DJS; approval and revising of the work: PH, SHJM, DJS, JAL, WCWL, PM, CCdS. Final approval of the version: all authors.
Funding: This work was supported by the Canadian Institutes of Health Research (Grant No: MOP-106545 to CCdS; MOP-74752 to DJS) and the Ontario Thoracic Society (OTS2010/2011/2012 to CCdS). NSERC Doctoral Canada Graduate Scholarship and Ontario Graduate Scholarship (SHJM), the Weston Foundation (JJH), the McLaughlin Centre for Molecular Medicine and a Canada Research Chair in Infectious Diseases and Inflammation (WCWL).
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available in a public, open access repository. Dataset deposited in GEO—dataset ID: GSE40180. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40180.
References
- 1. Singer M, Deutschman CS, Seymour CW, et al. The third International consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016;315:801–10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shankar-Hari M, Phillips GS, Levy ML, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the third International consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016;315:775–87. 10.1001/jama.2016.0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mikkelsen ME, Shah CV, Meyer NJ, et al. The epidemiology of acute respiratory distress syndrome in patients presenting to the emergency department with severe sepsis. Shock 2013;40:375–81. 10.1097/SHK.0b013e3182a64682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mei SHJ, Haitsma JJ, Dos Santos CC, et al. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med 2010;182:1047–57. 10.1164/rccm.201001-0010OC [DOI] [PubMed] [Google Scholar]
- 5. dos Santos CC, Murthy S, Hu P, et al. Network analysis of transcriptional responses induced by mesenchymal stem cell treatment of experimental sepsis. Am J Pathol 2012;181:1681–92. 10.1016/j.ajpath.2012.08.009 [DOI] [PubMed] [Google Scholar]
- 6. Collino F, Bruno S, Deregibus MC, et al. MicroRNAs and mesenchymal stem cells. Vitam Horm 2011;87:291–320. 10.1016/B978-0-12-386015-6.00033-0 [DOI] [PubMed] [Google Scholar]
- 7. Kilkenny C, Browne W, Cuthill IC, et al. Animal research: reporting in vivo experiments--the ARRIVE guidelines. J Cereb Blood Flow Metab 2011;31:991–3. 10.1038/jcbfm.2010.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Motakis ES, Nason GP, Fryzlewicz P, et al. Variance stabilization and normalization for one-color microarray data using a data-driven multiscale approach. Bioinformatics 2006;22:2547–53. 10.1093/bioinformatics/btl412 [DOI] [PubMed] [Google Scholar]
- 9. Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005;21:2067–75. 10.1093/bioinformatics/bti270 [DOI] [PubMed] [Google Scholar]
- 10. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 2015;43:D146–52. (Database issue). 10.1093/nar/gku1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120:15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 12. Yi N, Ma S. Hierarchical shrinkage priors and model fitting for high-dimensional generalized linear models. Stat Appl Genet Mol Biol 2012;11. 10.1515/1544-6115.1803. [Epub ahead of print: 26 Nov 2012]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shan Y, Akram A, Amatullah H, et al. ATF3 protects pulmonary resident cells from acute and ventilator-induced lung injury by preventing Nrf2 degradation. Antioxid Redox Signal 2015;22:651–68. 10.1089/ars.2014.5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–84. 10.1016/S0166-4328(01)00297-2 [DOI] [PubMed] [Google Scholar]
- 16. dos Santos CC, Slutsky AS. The contribution of biophysical lung injury to the development of biotrauma. Annu Rev Physiol 2006;68:585–618. 10.1146/annurev.physiol.68.072304.113443 [DOI] [PubMed] [Google Scholar]
- 17. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–49. 10.1056/NEJM200005043421806 [DOI] [PubMed] [Google Scholar]
- 18. Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008;14:844–52. 10.1261/rna.939908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma S, Liu M, Xu Z, et al. A double feedback loop mediated by microRNA-23a/27a/24-2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression. Oncotarget 2016;7:13502–19. 10.18632/oncotarget.6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Németh K, Leelahavanichkul A, Yuen PST, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med 2009;15:42–9. 10.1038/nm.1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lalu MM, McIntyre LL, Stewart DJ. Mesenchymal stromal cells: cautious optimism for their potential role in the treatment of acute lung injury. Crit Care Med 2012;40:1373–5. 10.1097/CCM.0b013e31824317f7 [DOI] [PubMed] [Google Scholar]
- 22. Lalu MM, Moher D, Marshall J, et al. Efficacy and safety of mesenchymal stromal cells in preclinical models of acute lung injury: a systematic review protocol. Syst Rev 2014;3:48. 10.1186/2046-4053-3-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McIntyre LA, Moher D, Fergusson DA, et al. Efficacy of mesenchymal stromal cell therapy for acute lung injury in preclinical animal models: a systematic review. PLoS One 2016;11:e0147170. 10.1371/journal.pone.0147170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chhabra R, Dubey R, Saini N. Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24-2 cluster and its implication in human diseases. Mol Cancer 2010;9:232. 10.1186/1476-4598-9-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie N, Cui H, Banerjee S, et al. miR-27a regulates inflammatory response of macrophages by targeting IL-10. J Immunol 2014;193:327–34. 10.4049/jimmunol.1400203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Z, Ruan Z, Mao Y, et al. miR-27a is up regulated and promotes inflammatory response in sepsis. Cell Immunol 2014;290:190–5. 10.1016/j.cellimm.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 27. Zhang L, Chen X, Shi Y, et al. miR-27a suppresses EV71 replication by directly targeting EGFR. Virus Genes 2014;49:373–82. 10.1007/s11262-014-1114-4 [DOI] [PubMed] [Google Scholar]
- 28. Zhao N, Sun H, Sun B, et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: an essential role for Twist-1 in HCC. Sci Rep 2016;6:23091. 10.1038/srep23091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Regis S, Caliendo F, Dondero A, et al. TGF-β1 Downregulates the Expression of CX3CR1 by Inducing miR-27a-5p in Primary Human NK Cells. Front Immunol 2017;8:868. 10.3389/fimmu.2017.00868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tybulewicz VLJ. Vav-family proteins in T-cell signalling. Curr Opin Immunol 2005;17:267–74. 10.1016/j.coi.2005.04.003 [DOI] [PubMed] [Google Scholar]
- 31. Hornstein I, Alcover A, Katzav S. Vav proteins, masters of the world of cytoskeleton organization. Cell Signal 2004;16:1–11. 10.1016/S0898-6568(03)00110-4 [DOI] [PubMed] [Google Scholar]
- 32. Moores SL, Selfors LM, Fredericks J, et al. Vav family proteins couple to diverse cell surface receptors. Mol Cell Biol 2000;20:6364–73. 10.1128/MCB.20.17.6364-6373.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gakidis MAM, Cullere X, Olson T, et al. Vav GEFs are required for beta2 integrin-dependent functions of neutrophils. J Cell Biol 2004;166:273–82. 10.1083/jcb.200404166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Faccio R, Teitelbaum SL, Fujikawa K, et al. Vav3 regulates osteoclast function and bone mass. Nat Med 2005;11:284–90. 10.1038/nm1194 [DOI] [PubMed] [Google Scholar]
- 35. Sauzeau V, Sevilla MA, Rivas-Elena JV, et al. Vav3 proto-oncogene deficiency leads to sympathetic hyperactivity and cardiovascular dysfunction. Nat Med 2006;12:841–5. 10.1038/nm1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujikawa K, Miletic AV, Alt FW, et al. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J Exp Med 2003;198:1595–608. 10.1084/jem.20030874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang L, Bao H, Wang K-X, et al. Secreted miR-27a induced by cyclic stretch modulates the proliferation of endothelial cells in hypertension via GRK6. Sci Rep 2017;7:41058. 10.1038/srep41058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang B-Y, Park K, Kleinhenz JM, et al. Peroxisome proliferator-activated receptor γ regulates the v-ets avian erythroblastosis virus E26 oncogene homolog 1/microRNA-27a axis to reduce endothelin-1 and endothelial dysfunction in the sickle cell mouse lung. Am J Respir Cell Mol Biol 2017;56:131–44. 10.1165/rcmb.2016-0166OC [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thoraxjnl-2019-213561supp001.pdf (172KB, pdf)
thoraxjnl-2019-213561supp002.pdf (99.9KB, pdf)
thoraxjnl-2019-213561supp003.pdf (1.1MB, pdf)
thoraxjnl-2019-213561supp004.pdf (1.2MB, pdf)
thoraxjnl-2019-213561supp005.pdf (906.3KB, pdf)
thoraxjnl-2019-213561supp006.pdf (948.5KB, pdf)