

Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) emerged in the city of Wuhan, Hubei Province, China, in late 2019. Since then, the virus has spread globally and caused a pandemic. Assays that can measure the antiviral activity of antibodies or antiviral compounds are needed for SARS‐CoV‐2 vaccine and drug development. Here, we describe in detail a microneutralization assay, which can be used to assess in a quantitative manner if antibodies or drugs can block entry and/or replication of SARS‐CoV‐2 in vitro. © 2020 Wiley Periodicals LLC.
Basic Protocol 1: Microneutralization assay to test inhibition of virus by antibodies (purified antibodies or serum/plasma)
Basic Protocol 2: Screening of anti‐SARS‐CoV‐2 compounds in vitro
Support Protocol: SARS‐CoV‐2 propagation
Keywords: antivirals, COVID‐19, COVID19, medium‐throughput screening, microneutralization, neutralization, SARS‐CoV‐2
INTRODUCTION
Several “common cold” seasonal coronaviruses circulate in the human population, but in late 2019, a novel coronavirus surfaced in China that was later classified as a beta‐coronavirus and named severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; Wu et al., 2020). SARS‐CoV‐2 is closely related to SARS CoV (now re‐named SARS‐CoV‐1), which caused an epidemic from 2002‐2004 in 26 countries. As of today, millions of cases of SARS‐CoV‐2 have been reported across the globe. Currently, no vaccines and only limited therapeutics are available as treatment for COronaVIrus Disease 2019 (COVID‐19).
Both SARS‐CoV‐1 and SARS‐CoV‐2 use angiotensin‐converting enzyme 2 (ACE2) as their receptor for attachment to host cells (Letko, Marzi, & Munster, 2020). The viral protein that interacts with ACE2 is the spike protein, a large trimeric glycoprotein (Wrapp et al., 2020). The part of the spike protein that interacts with the receptor is referred to as the receptor binding domain (RBD), and this domain is one of the major targets of neutralizing antibodies (Walls et al., 2020). Several studies have already demonstrated that individuals who recover from SARS‐CoV‐2 infection seroconvert and produce high levels of antibodies against the spike protein, as measured by binding assays (Amanat et al., and Okba et al., 2020). Initial results show that the induced antibodies also neutralize the virus efficiently, but large‐scale serology studies to correlate results from binding assays and neutralization assays are needed. In addition, there is a dire need for robust assays to screen for antiviral drugs that inhibit the replication of SARS‐CoV‐2 in vitro. Several types of neutralization assays have been applied for SARS‐CoV‐2. The most common assay used is the plaque reduction neutralization test (PRNT). PRNTs require an agarose overlay, which is time‐consuming, and it is difficult to perform PRNTs at higher throughput in a Biosafety Level 3 (BSL‐3) laboratory. In addition, pseudotyped virus particle entry inhibition assays have been developed which can be used at higher throughput and at lower biosafety levels. However, they do not employ authentic virus and are, therefore, always just an approximation of “real” virus neutralization. In addition, they can only be used to test entry inhibitors, but not other types of drugs. The microneutralization assays described here are performed in a 96‐well format, which allows for medium throughput. The readout for these assays is based on staining of the virus nucleoprotein (NP), which provides quantitative assessment of inhibitory concentration and does not rely on cumbersome and subjective visual inspection of cytopathic effect. Basic Protocol 1 describes the use of the assay for testing sera, plasma, or monoclonal antibodies, while Basic Protocol 2 describes the use of the assay in the context of drug screening.
Biosafety cautions
SARS‐CoV‐2 is a BSL‐3 pathogen. Follow all appropriate guidelines for the use and handling of pathogenic microorganisms (https://www.cdc.gov/coronavirus/2019‐ncov/lab/lab‐biosafety‐guidelines.html). Also see Current Protocols article: Burnett, Lunn, & Coico (2009).
Human‐derived material such as serum and plasma samples can potentially contain infectious SARS‐CoV2, as well as other blood‐borne viral pathogens. Heat inactivation at 56°C for 1 hr is recommended prior to use. Follow all appropriate guidelines and regulations for the use and handling of human‐derived materials.
Basic Protocol 1. MICRONEUTRALIZATION ASSAY TO TEST INHIBITION OF VIRUS BY ANTIBODIES (PURIFIED ANTIBODIES OR SERUM/PLASMA)
This protocol can be used to assess the extent to which antibodies are able to neutralize SARS‐CoV‐2 in vitro. Serum or plasma samples from any species, as well as monoclonal antibodies (mAbs), can be used in this assay. This assay is similar to a PRNT, but it can be performed in a 96‐well cell culture plate and allows for a higher throughput compared to a standard PRNT. The virus that we have used in the assay is SARS‐CoV‐2 isolate USA‐WA1/2020 (BEI Resources NR‐52281), but local isolates can be used as well. The cell type described here is Vero.E6, which has been shown to be permissive for SARS‐CoV‐2, but the assay can be adapted to other cell lines (Harcourt et al., 2020). Heat inactivation of serum or plasma at 56˚C for 1 hr is recommended to minimize the effect of complement on the cells, as well as to mitigate biosafety risks. Positive and negative controls should be used at all times. For serology, naïve or pre‐pandemic sera or plasma can be used as negative controls, while convalescent patient sera or sera from immunized/infected animals can be used as positive controls. For mAbs, the negative control should be a non‐SARS‐CoV‐2 antibody of the same isotype as the tested mAbs. Any available neutralizing mAb or antiserum can be used as positive control (Amanat et al., 2020). We also preferably perform the assays with serum/plasma/mAb in the overlay at the same concentration as the initial infection, since we believe that this better reflects physiological conditions. However, the assay can be modified to have antibody only in the initial virus‐antibody incubation period or only in the overlay, to study entry inhibition only or inhibition of virus spread within the culture only, respectively.
The amount of virus used for the assay is an important variable. Virus titers can be established using different protocols (see Current Protocols article: Mendoza, Manguiat, Wood, & Drebot, 2020). In any case, a virus dose that leads to robust staining needs to be used, and approximately 10,000 TCID50/ml are used in this assay. Another crucial reagent is the staining antibody. Here, we describe an antibody against the nucleoprotein (NP), since this protein is abundant within infected cells. However, mAbs or antisera targeting other viral proteins might be used as well. Antisera raised against the whole virus might also be useful for cell staining . We perform our data analysis in GraphPad Prism, but any other similar data analysis software can be used. Preparation of the cell plates for the assay and the final staining steps may be performed outside the BSL‐3 facility. Any work with replication‐competent SARS‐CoV2 needs to be performed inside a BSL‐3 facility using appropriate personal protective equipment (PPE). Plates may only be taken out of the BSL‐3 facility once the virus has been inactivated.
Definitions
BSA = bovine serum albumin
BSL = Biosafety Level
cDMEM = complete Dulbecco's modified Eagle Medium
CPE = cytopathic effect
DMEM = Dulbecco's modified Eagle Medium
FBS = fetal bovine serum
HEPES = 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
HRP = horseradish peroxidase
ID50 = 50% inhibitory dilution
MEM = minimal essential medium
MNA = microneutralization assay
NP = nucleoprotein
OPD = o‐phenylenediamine dihydrochloride
RT = room temperature
TCID50 = 50% tissue culture infectious dose
WFI = water for injection
Materials
Vero.E6 cells (ATCC #CRL‐1586)
Complete DMEM medium (cDMEM; see recipe)
Universal mycoplasma detection kit (ATCC 30‐1012K)
2× MEM (see recipe)
Fetal bovine serum (FBS; Corning, cat. no. 35011CV)
Water for injection for cell culture (WFI; Gibco, cat. no. A1287301)
SARS‐CoV‐2 isolate USA‐WA1/2020 (BEI Resources, cat. no. NR‐52281, or similar)
Serum samples for testing
10% formaldehyde (Polysciences, cat. no. 04018‐1)
Phosphate‐buffered saline (PBS; Gibco, cat. no. 10010‐023, or equivalent)
Triton X‐100 (Fisher Bioreagents, cat. no. BP151‐100)
Non‐fat milk (AmericanBio, cat. no. AB10109‐01000, or equivalent)
Mouse anti‐SARS NP antibody, e.g., 1C7 (an in‐house mAb, provided by Dr. Thomas Moran, Thomas.Moran@mssm.edu)
Anti‐mouse IgG HRP (Rockland, cat. no. 610‐4302)
SIGMAFAST™ OPD (Sigma‐Aldrich, cat. no. P9187)
3 M hydrochloric acid (Fisher Scientific, cat. no. S25856)
Class II biological safety cabinet
96‐well cell culture plate (Corning, cat. no. 3595)
Light microscope
-
Polypropylene sterile conical tubes:
15‐ml (Denville Scientific, cat. no. C1018P or equivalent)
50‐ml (Fisher Denville Scientific, cat. no. C1060P or equivalent)
1.5‐ml microcentrifuge tubes (Denville, cat. no. C2170 or equivalent)
Pipet‐Aid
-
Sterile, serological pipettes:
5‐ml (Falcon, cat. no. 356543 or equivalent)
10‐ml (Falcon, cat. no. 357551 or equivalent)
25‐ml (Falcon, cat. no. 357535 or equivalent)
50‐ml (Falcon, cat. no. 356550 or equivalent)
Micropipettes
-
Micropipette tips:
20‐µl barrier tips (Denville Scientific, cat. no. P1121 or equivalent)
200‐µl barrier tips (Denville Scientific, cat. no. P1122 or equivalent)
200‐µl tips (USA Scientific, cat. no. 1111‐1700 or equivalent)
1000‐µl barrier tips (Denville Scientific, cat. no. P1126 or equivalent)
Sterile reservoirs (Fisher Scientific, cat. no. 07‐200‐127 or equivalent)
Biotek SynergyH1 Microplate Reader or equivalent
GraphPad Prism 7 (or equivalent software)
Determining the 50% tissue culture infectious dose (TCID50)
-
1
Maintain Vero.E6 cells in culture using cDMEM.
Cells should be mycoplasma free and should also have high viability (>95%) when used in this assay.
-
2
The day before infection, seed 20,000 cells per well in 96‐well cell culture plates.
This can be performed outside of the BSL‐3 facility, and the cells can be transferred into the facility on the day of virus infection.
-
3
The next day, prepare 10‐fold dilutions of the virus in 1× MEM supplemented with 2% FBS (1× MEM/2% FBS). 1× MEM/2% FBS is prepared by combining equal amounts of 2× MEM and WFI, plus 2% FBS.
100 μl of the original virus stock can be added to 900 μl of 1× MEM/2% FBS, and this would serve as the 10−1 dilution. A 10‐fold dilution series is then prepared from the 10−1 dilution. Dilutions up to 10−8 are usually sufficient to determine the TCID50. The dilutions of the virus are added to the plate in a horizontal manner, and row H is used as a control row with addition of only 1× MEM/2% FBS but no virus (no‐virus control). Row H is used as a reference to check CPE and assess the CPE in infected cells. Be sure to prepare enough volume of each dilution for six to eight replicates.
-
4
Check the confluency of the cells under a light microscope. Cells should be between 90% and 100% confluence. Infection efficiency may be lower if overconfluent cells are used. Remove all of the medium from the wells of the 96‐well cell culture plate and add 100 μl/well of each respective virus dilution to the plate. 100 μl per well of 1× MEM/2% FBS without virus should be added to a few rows or columns, and these wells can serve as “no‐virus” controls. Place the plate at 37°C for 1 hr.
Here and below, pipettes tips should be changed for every virus transfer, since reusing the same tips will impact the TCID50. Add the liquid to the side of the well to avoid disturbing the cell monolayer.
-
5
After 1 hr of viral absorption, remove the virus inoculum from every well. Also remove the medium from “no‐virus” control wells. Add 200 μl of 1× MEM/2% FBS to all the wells.
-
6
Incubate the cells for 3 days at 37°C.
-
7
On day 3 post infection, check the cells under the microscope for CPE (Fig. 1). Wells with the highest amount of virus should show strong CPE, and this effect should decrease as the virus is diluted further. “No‐virus” controls can be used as reference for no infection.
Figure 1.
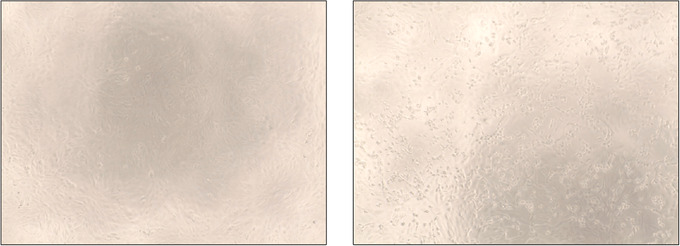
CPE induced by SARS‐CoV‐2 in Vero.E6 cells. The left panel shows healthy, uninfected Vero.E6 cells forming a monolayer. The right panel shows CPE after infection with SARS‐CoV‐2 (e.g., rounded up cells, halo around cells, etc.).
-
8
Make a note of the number of wells that are positive for each dilution and calculate the TCID50/ml using the Reed & Muench method (Ramakrishnan et al., 2016).
Performing the microneutralization assay (MNA)
-
9
Maintain Vero.E6 cells in culture using cDMEM.
Cells should be mycoplasma free and should also have high viability when used in this assay.
-
10
The day before infection, seed 20,000 cells per well in a 96‐well cell culture plate as described in step 2.
-
11
Prepare serum dilutions in empty, sterile 96‐well cell culture plates using 1× MEM/2% FBS. 1× MEM/2% FBS is prepared by adding equal amounts of 2× MEM and WFI, plus 2% FBS.
Serum/plasma samples from humans or animals infected with SARS‐CoV‐2 should be heat‐inactivated at 56°C for 1 hr prior to use. Heat‐inactivation of serum/plasma samples will ensure that any complement present in the sample is inactived, and it also increases safety since heat inactivation might inactivate infectious virus present in samples. Each sample is tested in duplicate.
-
12
Add 270 μl of 1× MEM/2% FBS to row A and add 200 μl of 1× MEM/2% FBS to row B‐G. Add 30 μl of each respective sample to row A and pipet up and down several times to mix. Once all the samples are added, transfer 100 μl from row A to B and mix 5‐10 times using the multichannel pipette. Discard tips. Load new tips on to the multi‐channel pipette and transfer 100 μl from row B to row C, then mix 5‐10 times again. Repeat this process until row G is reached. At row G, discard the 100 μl so each column has 200 μl remaining. Row H is a control row. This plate is called plate A. Refer to Figure 2 for plate layout.
Figure 2.

Plate layout for microneutralization assay. Indicated dilutions are suggestions for testing serum samples. Other dilution series or concentrations (for mAbs) might be used.
-
13
Obtain a new 96‐well cell culture plate and transfer 80 μl of each respective dilution to the new plate. Ensure that the order of the samples and dilutions is preserved in the new plate (referred to as plate B). Dilutions and preparation of plate A and plate B can all be performed in at BSL‐2.
-
14
Handle SARS‐CoV‐2 in a Biosafety Level 3 (BSL‐3) laboratory in a biosafety cabinet. Prepare 10,000 TCID50/ml of SARS‐CoV‐2 in 1× MEM/2% FBS. Add 80 μl of the virus to each well in plate B except wells H7‐H12. Add 80 μl of 1× MEM/2% FBS to rows H7‐H12. Close the lid of the 96‐well plate and tap gently with the palm of the hand to mix the serum with the virus. Each well should now have a total of 160 μl (80 μl of serum dilution plus 80 μl of virus). Incubate for 1 hr at room temperature. It is not necessary to rock the plate.
-
15
After the incubation time is over, take the Vero.E6 cells in the 96‐well cell culture plate (step 9) and remove the medium using a multi‐channel pipette or an aspirator.
The medium should be removed carefully such that the tip does not touch the cells or the cells are not dislodged in any way.
-
16
Add 120 μl of serum‐virus mixture from plate B to the cells. Ensure that the order of the serum dilutions is preserved across all plates. Tips do not need to be changed if the serum‐virus mixture is moved from the bottom of the plate to the top (i.e., starting from row H and ending at row A). Place the cells at 37°C in a humidified incubator with 5% CO2 for 1 hr.
Infection was done with only 60 μl of virus, amounting to 600 TCID50 per well. Amounts of virus can be varied depending on how long the cells are to be kept in the incubator for infection.
600 TCID50 per well has given us reasonable staining as well as reasonable sensitivity. However, the amount of virus per well can be further optimized.
-
17
After 1 hr, tilt the plate with one hand and remove all the virus‐serum mixture using a multi‐channel pipette. Ensure that all the medium is removed. Next, add 100 μl from each well of plate A to the cells (the time that the cells are without liquid should be kept as a minimum; otherwise they may dry out and die). Finally, add 100 μl per well of 1× MEM/2% FBS. Tips do not need to be changed if the wells are not touched with the tips.
-
18
Incubate the cells for 48 hr.
Plates A and plate B can be discarded.
-
19
Forty‐eight hours later, check for cytopathic effect, as cells that did not receive any virus should be healthy while wells H1‐H6 should show signs of infection. Remove all medium from each well containing cells, and add 100 μl of 10% formaldehyde.
SARS‐CoV‐2 needs to be inactivated for 24 hr at 4°C.
Staining protocol
-
20
After inactivation of virus for 24 hr, the plate can be taken out of the BSL‐3 facility and staining can be performed in a BSL‐2 facility inside a biosafety cabinet.
CAUTION: Before proceeding with this option, please discuss how inactivation needs to be tested and proven with your biosafety officer, since local regulations may differ. Some institutions may not allow bringing material out of the BSL‐3 containment facility, and staining will need to be performed within the BSL‐3 in these cases. Even after inactivation, plates should be handled at BSL‐2 in a biosafety cabinet in order to reduce residual risk.
-
21
Remove formaldehyde from the cells carefully using a multi‐channel pipette. Care should be taken not to touch the cells with the tip or dislodge the cells in any way.
Fixation with 10% formaldehyde ensures that all cells are cross‐linked onto the wells, but cells can be inspected under the microscope to ensure that they have not detached.
-
22
Wash cells by addition of 200 μl of phosphate‐buffered saline (PBS) to each well. Aspirate using a multi‐channel aspirator or remove the PBS with a multi‐channel pipette.
-
23
Prepare fresh permeabilization solution by adding 500 μl of Triton X‐100 to 500 ml of PBS (0.1% PBS/Triton). Mix vigorously. Add 150 μl per well of 0.1% PBS/Triton and incubate the cells with this solution for 15 min at RT.
-
24
Once the incubation time is over, wash the cells with 200 μl per well of PBS. Remove all the PBS and add 100 μl/well of PBS supplemented with 3% non‐fat milk. Incubate cells for 1 hr at RT.
-
25
Next, prepare primary antibody solution in PBS supplemented with 1% non‐fat milk. 1 μg/ml of 1C7 (mouse anti‐SARS NP—or equivalent) mAb was used for our assay, provided by Dr. Thomas Moran. Add 100 μl/well of primary antibody solution and incubate for 1 hr at RT.
-
26
Next, wash each well twice with 200 μl of PBS. To remove all the PBS, gently tap the plate on some paper towels to ensure that no primary antibody is left on the plate. Prepare secondary antibody solution, anti‐mouse IgG HRP, in PBS supplemented with 1% non‐fat milk at a dilution of 1:3000. Add 100 μl/well and incubate for 1 hr at RT.
-
27
Finally, wash each well twice with 200 μl/well of PBS. Dry the plate by gently tapping on paper towels. Prepare developing solution (SIGMAFAST™ OPD) according to the manufacturer instructions.
-
28
Add 100 μl of developing solution per well and wait 10‐12 min.
-
29
Stop the reaction by adding 50 μl/well of 3 M hydrochloric acid.
-
30
Measure the optical density at 490 nanometers on a Biotek SynergyH1 Microplate Reader (or equivalent) and record data. Average the “virus‐only” wells and separately average all the “no‐virus” wells. The formula to calculate percent inhibition at each well is 100 − ((X‐average of ʻno virusʼ wells)/(average of ʻvirus onlyʼ wells‐average of ʻno virusʼ wells)*100), where X is the read for each well. Non‐linear regression curve fit analysis over the dilution curve can be performed to calculate ID50. The top and bottom constraints are set at 100% and 0% (Fig. 3).
Figure 3.
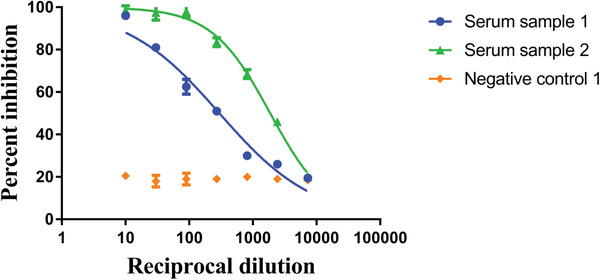
Analysis of data as reciprocal dilution of serum and percent inhibition of virus (ID50 of serum sample 1 is 1:298 and serum sample 2 is 1:1873).
Basic Protocol 2. SCREENING OF ANTI‐SARS‐CoV‐2 COMPOUNDS IN VITRO
This protocol can be used to assess the ability of a compound to inhibit SARS‐CoV‐2 infection at any stage of the viral life cycle (e.g., entry, replication, assembly, egress and spread). Repurposing of existing drugs already approved for the treatment of other diseases represents one of the most rapid strategies for the identification of compounds targeting SARS‐CoV‐2 (Gordon et al., 2020). However, any compound predicted to have antiviral activity could be tested using this assay. Here, the antiviral activity of the compound is evaluated based on its ability to inhibit SARS‐CoV‐2 replication as assessed by immunostaining for the viral NP. Specifically, Vero.E6 cells are pre‐incubated with each compound dilution for 2 hr, followed by a low‐MOI infection to allow multi‐cycle replication. 48 hr post‐infection, cells are fixed and stained prior to automated imaging and analysis. The toxicity of the test compound is determined in parallel by performing an MTT assay or any other commercial cell viability assay. The ratio of IC50 calculated from the antiviral data over the CC50 calculated from the cytotoxicity data will yield the selectivity index of a compound, indicating how selective it is for inhibiting viral replication compared to its impact on cell death. Importantly, when performing the screening, standard positive and negative controls should be included so that the results can be compared between different assays. Positive controls could be previously established—for example, SARS‐CoV‐2 inhibitors such as remdesivir, which has an expected IC50 of ∼500 nM in this assay (Bouhaddou et al., submitted). Negative controls should include diluent‐only treatment (e.g., DMSO). Of note, these conditions have been optimized for infection of Vero.E6 cells, but could be adapted to other cell lines that can support SARS‐CoV‐2 infection. In addition, time‐of‐addition and drug combination studies can also be performed with similar assay settings.
Definitions
BSA = bovine serum albumin
CC50 = 50% cytotoxic concentration
cDMEM = complete Dulbecco's modified Eagle Medium
DAPI = 4′,6‐diamidino‐2‐phenylindole
DMEM = Dulbecco's modified Eagle Medium
DMSO = dimethyl sulfoxide
FBS = fetal bovine serum
HEPES = 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
IC50 = 50% inhibitory concentration
MOI = multiplicity of infection
MTT = 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
TCID50 = 50% tissue culture infectious dose
Materials
Vero.E6 cells (ATCC #CRL‐1586)
Universal mycoplasma detection kit (ATCC 30‐1012K)
Complete DMEM medium (cDMEM; see recipe)
Viral growth medium (DMEM with 2% FBS; see recipe)
SARS‐CoV‐2 isolate USA‐WA1/2020 (BEI Resources, cat. no. NR‐52281, or similar)
Compound library
DMSO (Corning, cat. no. 25‐950‐CQC)
10% formaldehyde (Polysciences, cat. no. 04018‐1)
Anti‐SARS nucleoprotein antibody (mAb 1C7, produced in house)
Donkey anti‐mouse IgG (H+L) highly cross‐absorbed secondary antibody, Alexa Fluor Plus 488 (Invitrogen, cat. no. A32766 or equivalent)
4′,6‐Diamidine‐2′‐phenylindole (DAPI; e.g., Sigma‐Aldrich)
Phosphate‐buffered saline (PBS; Gibco, cat. no. 10010‐023, or equivalent)
Bovine serum albumin (BSA; MP Biomedicals, cat. no. 08810063)
Cell Proliferation Kit I (MTT assay, Roche, cat. no. 11465007001) or similar cytotoxicity assay.
CO2 incubator
96‐well cell culture plate (Corning, cat. no. 3595).
96‐well deep well (2‐ml) plates (Corning, cat. no. 3960, or equivalent)
Plate sealing film, adhesive (BioRad, cat. no. MSB1001)
-
Polypropylene sterile conical tubes:
15‐ml (Denville Scientific, cat. no. C1018P or equivalent)
50‐ml (Fisher Denville Scientific, cat. no. C1060P or equivalent)
Pipet‐Aid
-
Sterile, serological pipettes:
5‐ml (Falcon, cat. no. 356543 or equivalent)
10‐ml (Falcon, cat. no. 357551 or equivalent)
25‐ml (Falcon, cat. no. 357535 or equivalent)
50‐ml (Falcon, cat. no. 356550 or equivalent)
Single‐channel and multi‐channel micropipettes
-
Micropipette tips:
20‐µl barrier tips (Denville Scientific, cat. no. P1121 or equivalent)
200‐µl barrier tips (Denville Scientific, cat. no. P1122 or equivalent)
200‐µl tips (USA Scientific, cat. no. 1111‐1700 or equivalent)
1000‐µl barrier tips (Denville Scientific, cat. no. P1126 or equivalent)
Sterile reservoirs (Fisher Scientific, cat. no. 07‐200‐127 or equivalent)
1.5‐ml microcentrifuge tubes (Denville, cat. no. C2170 or equivalent)
Celigo 96‐well plates (Greiner, cat. no. 655090)
Plate cytometer (Celigo) or laser‐scanning cytometer (Acumen)
Additional reagents and equipment for TCID50 assay and immunostaining (Basic Protocol 1)
Performing the SARS‐CoV2 antiviral assay
-
1
Maintain Vero.E6 cells in culture using cDMEM.
-
2
The day before infection, seed 2000 cells per well in a 96‐well cell culture plate.
The low number of cells per well is important for the accuracy of both our immunofluorescent antiviral readout and the cytotoxicity assay used. Three compounds can be tested per 96‐well plate, and separate plates are required to test antiviral and cytotoxic activities. This and all following steps until step 6 can be performed outside of a BSL‐3 facility, and the cells and dilutions can be transferred into the facility on the day of virus infection.
-
3
Prepare compound dilutions in empty, sterile 2‐ml 96‐well deep well plates using 1× DMEM supplemented with 2% FBS (viral growth medium).
If compounds to be tested are dissolved in DMSO (common), medium supplemented with a matching percentage of DMSO should be prepared to perform dilutions.
-
4
Add 1100 μl of 1× DMEM (without DMSO) supplemented with 2% FBS to columns 1 and 7, and add 750 μl of 1× DMEM (containing DMSO if necessary) supplemented with 2% FBS to columns 2‐6 and 8‐12. Add 50% more compound than your selected maximum dose to columns 1 and 7 (2 compounds tested per row, 16 total compounds diluted per deep well plate), e.g., 3.3 μl of a 10 mM stock for concentration of 30 μM, which will dilute to a final maximum concentration of 20 μM when 50 μl of virus is added. Based on this compound example, the DMSO concentration for medium in columns 2‐6 and 8‐12 would be 0.3% (do not exceed 0.5% DMSO, due to toxicity concerns). Once all the compounds are added, pipet column 1 up and down 20 times to mix, transfer 375 μl from column 1 to column 2, and mix 5‐10 times using the multi‐channel pipette. Discard tips. Load new tips to the multi‐channel pipet, transfer 375 μl from column 2 to column 3, and mix 5‐10 times again. Repeat this process until column 6 is reached. Repeat for columns 7‐12. If the entire deep‐well plate is used, this will result 16 compounds with six 3‐fold serial dilutions of each (final concentrations of 20‐80 nM for this example).
This plate is called plate X, refer to Figure 4 for plate layout.
Figure 4.

Plate X and plate Y layout for the antiviral assay.
-
5
Take the Vero.E6 cells seeded the previous day in the two 96‐well cell culture plates (one for antiviral, one for cytotoxicity) and remove the medium using a multi‐channel pipette or an aspirator.
The medium should be removed carefully such that the tip does not touch the cells and the cells are not dislodged in any way. Transfer 100 μl of each respective dilution to the Vero.E6 plates; there should be enough volume (750 μl) in the deep‐well plate for triplicates for both antiviral and cytotoxicity testing. Using the same dilution series for both antiviral and cytotoxic assays will produce reliable matching data sets, and these should be performed at the same time. Three compounds and six DMSO control wells can be tested per 96‐well plate (plate format Y, see Fig. 4). The outer wells should be avoided due to the potential for medium evaporation to alter compound concentrations. These outer wells should be filled with medium and cells to be used as uninfected controls for viral detection through immunostaining. The cells should be incubated with compound for 2 hr prior to infection as a standard, although this time frame can be adjusted for specific experiments.
-
6
Handle SARS‐CoV‐2 in a BSL‐3 laboratory in a biosafety cabinet. Prepare 2000 TCID50/ml of authentic SARS‐CoV‐2 (SARS‐CoV‐2 isolate USA‐WA1/2020; see Basic Protocol 1, steps 1‐8, for determination of TCID50) in 1× DMEM supplemented with 2% FBS. Add 50 μl (100 TCID50 per well or 0.025 MOI) of the virus to the 100 μl of compound containing medium in each well of plate Y except the outer wells, pipet up and down gently 2‐3 times to mix each well (important for even viral infection and compound concentration across the plates). Add 50 μl of 1× DMEM with 2% FBS to the sample wells of the cytotoxicity plate to adjust compound concentrations to match the antiviral assay. Each inner well should now have a total of 150 μl. Incubate for 48 hr at 37°C in a humidified incubator with 5% CO2.
All work for testing cytotoxicity can be performed outside the BSL‐3 laboratory, e.g., at BSL‐2, since no infectious virus is involved.
-
7
After the incubation time is complete, remove the infected Vero.E6 cells in the 96‐well cell culture plates from the incubator under BSL‐3 conditions. Remove the medium using a multi‐channel pipette.
Optionally, viral titers can be quantified from the removed medium using TCID50 assay (see Basic Protocol 1) or plaque assay if desired. Fix the Vero.E6 cells by adding 200 μl of 4% formaldehyde (dilute from 10%) in PBS to each well of the 96‐well plate. Incubate in 4% formaldehyde for 24 hr at room temperature prior to removing plates from the BSL‐3 laboratory.
-
8
Once the plates have been removed from the BSL‐3 laboratory, proceed to immunostain the fixed cells with SARS‐CoV2 specific antibodies as outlined in Basic Protocol 1, steps 20‐30, with the following modifications:
Use the antibodies listed in the Basic Protocol 2 materials list for this protocol. Briefly, after three washes with 200 μl of PBS, incubate the cells with a secondary antibody solution containing anti‐mouse IgG and DAPI in PBS supplemented with 0.5% BSA at a dilution of 1:2000. Add 100 μl/well and incubate for 45 min at RT. Wash three times for 5 min in PBS with gentle shaking. A plate cytometer (Celigo) or laser scanning cytometer (Acumen) can be used to assess infected cells over total cells for highly accurate quantification of viral infection. The DMSO control should be normalized to 100% infection for comparison with compound treated wells.
-
9
Assay the uninfected Vero.E6 cell cytotoxicity controls for cell viability with the MTT assay or any other commercial cell viability assay according to the manufacturer's instructions.
Cytotoxicity measurements should be matched in incubation time with your antiviral assay.
-
10
Calculate the IC50 and CC50 for each compound using Prism software as described in Basic Protocol 1.
SARS‐CoV‐2 PROPAGATION
The SARS‐CoV2 USA‐WAS1/2020 viral strain used for the optimization of the assays described here has been propagated using the Vero.E6 cell line, as it was previously shown that the virus can replicate to high titer in this cell line (https://www.biorxiv.org/content/10.1101/2020.03.02.972935v1.full.pdf). Stocks are generated by infecting a confluent monolayer of cells at a low multiplicity of infection to avoid the generation of defective interfering particles that can lower the titer of the viral preparation. Importantly, always work with low‐passage‐number stocks that are fully sequenced, to avoid selecting mutants that are better adapted to grow in the cell line used for virus amplification.
Materials
Vero.E6 cells (ATCC CRL‐1586)
Vero.E6 growth medium: cDMEM (see recipe)
SARS‐CoV‐2 isolate USA‐WA1/2020, (BEI Resources NR‐52281 or similar)
Viral growth medium (DMEM with 2% FBS; see recipe)
Class II biological safety cabinet
75‐cm2 tissue culture flasks with filter caps (size varies according to the volume of stock needed)
Counting chamber or automatic cell counter
CO2 incubator
-
Polypropylene sterile conical tubes
15‐ml (Denville Scientific, cat. no. C1018P or equivalent)
50‐ml (Fisher Denville Scientific, cat. no. C1060P or equivalent)
Pipet‐Aid
-
Sterile, serological pipettes
5‐ml (Falcon, cat. no. 356543 or equivalent)
10‐ml (Falcon, cat. no. 357551 or equivalent)
Tabletop centrifuge, 4°C
2.0‐ml cryotubes with O‐ring
Additional reagents and equipment for TCID50 assay (Basic Protocol 1, steps 1‐8)
-
1
Maintain Vero.E6 cells in culture using cDMEM (also see Basic Protocol 1).
-
2
The day before infection seed 4 × 106 Vero.E6 cells into 75‐cm2 flasks and incubate in cDMEM for 24 hr. Make sure to prepare at least three flasks: one for virus propagation, one to be used as uninfected control, and the third one for counting the cells. Cells should be ∼90% confluent at the time of infection.
This step can be performed outside of the BSL‐3 facility, and the cells can be transferred into the facility on the day of virus infection.
-
3
On the day of the inoculation, use one of the flasks to count the cells (e.g., using a counting chamber or an automatic cell counter) and calculate the volume of viral inoculum required to infect at multiplicity of infection of 0.001. When using Vero.E6, a confluent T‐75 flask can yield about 8 × 106 cells. Remove the medium, wash once gently in PBS, and infect cells in 5 ml of viral growth medium for 1 hr at 37°C and 5% CO2, rocking the flask every 15 min to spread the viral inoculum evenly across the monolayer.
-
4
After the 1‐hr adsorption, add 5 ml of viral growth medium and incubate the flask at 37°C. Observe cell culture daily and follow the development of cytopathic effects. Typically, within 3 or 4 days, cells will become rounded‐up and will detach from the monolayer. Harvest the virus when the cytopathic effect has progressed to 80%.
-
5
Clarify the medium by centrifugation in a 50‐ml conical tube for 10 min at 1300 × g, 4°C.
-
6
Aliquot pooled supernatants into labeled 2.0‐ml O‐ring cryotubes and store samples at −80°C until further use.
-
7
Determine virus stock titer by plaque assay or TCID50 following the protocol described in Basic Protocol 1.
REAGENTS AND SOLUTIONS
Complete DMEM medium (cDMEM; used in Basic Protocol 1 and 2)
880 ml of DMEM (Gibco, cat. no. 11995‐065, or equivalent)
10 ml of penicillin‐streptomycin (Gibco, cat. no. 15140122)
10 ml of HEPES buffer (Gibco, cat. no. 15630080)
100 ml of FBS (Corning, cat. no. 35011CV)
Filter using a 0.22‐μm Stericup filter (MilliporeSigma, cat. no. S2GPU05RE)
Store up to 4 weeks at 4°C
MEM, 2× (used in Basic Protocol 1)
200 ml of 10× MEM (Gibco, cat. no 11430030)
20 ml of penicillin‐streptomycin (Gibco, cat. no. 15140122)
20 ml of HEPES buffer (Gibco, cat. no. 15630080)
20 ml of l‐glutamine (Gibco, cat. no. 25030081)
32 ml of 7.5% sodium bicarbonate (Gibco, cat. no. 25080094)
12 ml of 35% BSA (MP Biomedicals, cat. no. 08810063)
696 ml of water for injection for cell culture (WFI; Gibco, cat. no. A1287301)
Filter using a 0.22‐μm Stericup filter (MilliporeSigma, cat. no. S2GPU05RE)
-
Store up to 4 weeks at 4°C.
This medium will be used to prepare 1× MEM/2% FBS by mixing equal amounts of WFI and 2× MEM.
Viral growth medium (DMEM with 2% FBS; Basic Protocol 2)
880 ml of (DMEM Gibco, cat. no. 11995-065, or equivalent)
10 ml of penicillin‐streptomycin (Gibco, cat. no. 15140122)
10 ml of HEPES buffer (Gibco, cat. no. 15630080)
20 ml of FBS (Corning, cat. no. 35011CV)
Filter using a 0.22‐μm Stericup filter (MilliporeSigma, cat. no. S2GPU05RE)
Store up to 4 weeks at 4°C
COMMENTARY
Background Information
The microneutralization assay that is described here has been adapted from established protocols from work with other viruses such as influenza virus (Amanat, Meade, Strohmeier, & Krammer, 2019). The assay has a medium throughput, and more samples can be analyzed as compared to PRNTs. Compared to RBD‐ACE2 inhibition assays, the MNA described here will also detect neutralizing antibodies binding to epitopes outside of the RBD. Different virus isolates can be used, and the assay can likely be adapted for staining antibodies other than mAbs against NP (e.g., polyclonal sera, antibodies targeting S or M, etc.). The assay should be optimized based on the level of infection that is observed in each respective cell line used. Viral input can be varied as well. in order to make the assay more or less sensitive.
The antiviral assay described here has also been adapted from previous work with influenza virus. The immunostaining output is medium throughput and can be used to comprehensively analyze the antiviral and cytotoxic activity of many more compounds than can be done using TCID50 or plaque assay. The time course of infection, cell line used, viral strain and MOI, and antibody used can be altered to adapt this assay to answer many different questions related to the antiviral activity of a compound.
Critical Parameters and Troubleshooting
Cells that are used for the microneutralization assay in Basic Protocol 1 should be healthy, and viability should be checked periodically. The cell count should be performed with care to ensure that cells are not over‐confluent on the day of the assay. The cells should also be mycoplasma free. The TCID50 measurement of each stock should be performed with care, as each virus stock will be used numerous times for several microneutralization assays. Most importantly, the virus should be sequenced every few passages to ensure that no major mutations have occurred from the passaging of virus in cell culture. The microneutralization assay can be performed with serum/plasma or purified antibodies. If antibodies are to be used, a trial run should be performed to assess the appropriate concentrations that should be tested.
The immunostaining antiviral assay described in Basic Protocol 2 has a consistently lower sensitivity than an assay that detects infectious viral particles produced in the supernatant (TCID50/plaque assay). The IC50 determined for any particular compound in this assay will typically be 3‐ to 5‐fold higher than the aforementioned assays. Therefore, this antiviral assay is well suited for medium‐throughput screening of potential antivirals, and we suggest confirming top hits using the mentioned classical virology methods. The DAPI counterstain can be used as a proxy for toxicity in infected cells, to be paired with the MTT assay performed in uninfected cells. If the majority of DAPI staining is lost in a compound‐treated well, that compound concentration should be excluded from your final IC50 calculation.
Time Considerations
The TCID50 is only performed once for each viral stock. The TCID50 takes 3 days in total, and the microneutralization should only take 4 days, as the cells are fixed 2 days after the assay has been performed. On day 3, plates should be stained immediately, as leaving the cells in formaldehyde can negatively impact the staining. Thus, the whole microneutralization assay and data generation are completed within 4 days.
Compounds are typically added 2 hr prior to SARS‐CoV‐2 infection, although this can be adjusted based on the proposed mechanism of action of the compound or on the hypothesis being tested. The antiviral assay takes 3 days total from infection to staining, scanning, and data analysis. The compound should always be incubated on the cells for the same amount of time between antiviral and cytotoxicity assays.
Conflict of Interest Statement
Mount Sinai has licensed serological assays to commercial entities and has filed for patent protection for serological assays.
Acknowledgments
This work was partially supported by the NIAID Centers of Excellence for Influenza Research and Surveillance (CEIRS) contract HHSN272201400008C (F.K., A.G.‐S.), Collaborative Influenza Vaccine Innovation Centers (CIVIC) contract 75N93019C00051 (F.K., A.G.‐S.), DARPA project HR0011‐19‐2‐0020 (A.G.‐S.), and the generous support of the JPB foundation, the Open Philanthropy Project (#2020‐215611 to A.G.‐S.), and other philanthropic donations.
Amanat, F. , White, K. M. , Miorin, L. , Strohmeier, S. , McMahon, M. , Meade, P. , Liu, W.‐C. , Albrecht, R. A. , Simon, V. , Martinez‐Sobrido, L. , Moran, T. , García‐Sastre, A. , & Krammer, F. (2020). An in vitro microneutralization assay for SARS‐CoV‐2 serology and drug screening. Current Protocols in Microbiology, 58, e108. doi: 10.1002/cpmc.108
Literature Cited
- Amanat, F. , & Krammer, F. (2020). Sars‐Cov‐2 vaccines: Status report. Immunity, 52(4), 583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat, F. , Meade, P. , Strohmeier, S. , & Krammer, F. (2019). Cross‐reactive antibodies binding to H4 hemagglutinin protect against a lethal H4n6 influenza virus challenge in the mouse model. Emerging Microbes & Infections, 8(1), 155–168. doi: 10.1080/22221751.2018.1564369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat, F. , Stadlbauer, D. , Strohmeier, S. , Nguyen, T. H. O. , Chromikova, V. , McMahon, M. , … Krammer, F. (2020). A serological assay to detect Sars‐Cov‐2 seroconversion in humans. Nature Medicine. doi: 10.1038/s41591-020-0913-5 [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou, M. , Memon, D. , Meyer White, K. M. , Rezelj, V. V. , Marrero, M. C. , … Kroger, N. J. The global phosphorylation landscape of SARS‐CoV‐2 infection. Submitted for publication. [DOI] [PMC free article] [PubMed]
- Burnett, L. C. , Lunn, G. , & Coico, R. (2009). Biosafety: Guidelines for working with pathogenic and infectious microorganisms. Current Protocols in Microbiology, 13, 1A.1.1–1A.1.14. doi: 10.1002/9780471729259.mc01a01s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, D. E. , Jang, G. M. , Bouhaddou, M. , Xu, J. , Obernier, K. , White, K. M. , … Krogan, N. J. (2020). A Sars‐Cov‐2 protein interaction map reveals targets for drug repurposing. Nature. doi: 10.1038/s41586-020-2286-9 [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt, J. , Tamin, A. , Lu, X. , Kamili, S. , Sakthivel, S. K. , Murray, J. , … Thornburg, N. J. (2020). Isolation and characterization of Sars‐Cov‐2 from the first Us Covid‐19 patient. bioRxiv, doi: 10.1101/2020.03.02.972935. [DOI] [Google Scholar]
- Letko, M. , Marzi, A. , & Munster, V. (2020). Functional assessment of cell entry and receptor usage for Sars‐Cov‐2 and other lineage b betacoronaviruses. Nature Microbiology, 5(4), 562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza, E. J. , Manguiat, K. , Wood, H. , & Drebot, M. (2020). Two detailed plaque assay protocols for the quantification of infectious Sars‐Cov‐2. Current Protocols in Microbiology, 57(1), ecpmc105. doi: 10.1002/cpmc.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okba, N. M. A. , Muller, M. A. , Li, W. , Wang, C. , Geurts van Kessel, C. H. , Corman, V. M. , … Haagmans, B. L. (2020). Severe acute respiratory syndrome coronavirus 2‐specific antibody responses in coronavirus disease 2019 patients. Emerging Infectious Diseases. 26(7). doi: 10.3201/eid2607.200841 [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan, M. A. (2016). Determination of 50% endpoint titer using a simple formula. World Journal of Virology, 5(2), 85–86. doi: 10.5501/wjv.v5.i2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y. J. , Tortorici, M. A. , Wall, A. , McGuire, A. T. , & Veesler, D. (2020). Structure, function, and antigenicity of the Sars‐Cov‐2 spike glycoprotein. Cell, 181(2), 281–292 e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp, D. , Wang, N. , Corbett, K. S. , Goldsmith, J. A. , Hsieh, C. L. , Abiona, O. , … McLellan, J. S. (2020). Cryo‐Em structure of the 2019‐Ncov spike in the prefusion conformation. Science, 367(6483), 1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Zhao, S. , Yu, B. , Chen, Y. M. , Wang, W. , Song, Z. G. , … Zhang, Y. Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]