

Abstract
Abstract: By providing long‐term protection against infectious diseases, vaccinations have significantly reduced death and morbidity worldwide. In the 21st century, (bio)technological advances have paved the way for developing prophylactic vaccines that are safer and more effective as well as enabling the use of vaccines as therapeutics to treat human diseases. Here, we provide a focused review of the utility of genetic code expansion as an emerging tool for the development of vaccines. Specifically, we discuss how the incorporation of immunogenic noncanonical amino acids can aid in eliciting immune responses against adverse self‐proteins and highlight the potential of an expanded genetic code for the construction of replication‐incompetent viruses. We close the review by discussing the future prospects and remaining challenges for the application of these approaches in the development of both prophylactic and therapeutic vaccines in the near future.
Keywords: genetic code expansion, immunochemistry, protein modifications, vaccine development, virus attenuation
Vaccine variations: This article reviews the utility of genetic code expansion as an emerging tool for the development of vaccines. It highlights how the incorporation of immunogenic noncanonical amino acids (ncAA) can aid in eliciting immune responses against adverse self‐proteins and highlights the potential of an expanded genetic code for the construction of live‐attenuated virus vaccines.
1. Introduction
The routine vaccination of large populations was the single most effective medical intervention in reducing death and morbidity in the 20th century. [1] For example, pioneering work on vaccines resulted in the eradication of small pox [2] and the reduction of polio cases from 350.000 in 1988 to 66 in 2019. [3] Today, routine vaccinations continue to protect millions each year against a growing number of infectious diseases. Nevertheless, recent and ongoing pandemics such as those caused by H1N1 influenza A virus, [4] human immunodeficiency virus (HIV), [5] and severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) [6] serve as a stark reminder that advances in vaccine development remain highly relevant. Such efforts will also be crucial for creating more efficient vaccines that protect against infectious diseases, such as malaria or tuberculosis, [7] and aid efforts in which vaccines are not used prophylactically but as the primary form of treatment for a disease (=immunotherapy). [8]
Scientific and (bio)technological innovations have repeatedly enabled the development of effective vaccines. [9] Many of the techniques for vaccine production and development that have been in use for decades (e. g., serial passaging in vitro or through abnormal hosts) [10] continue to remain relevant today. However, new approaches based on technologies that have become available over the past decades are being explored for the production of safe and effective vaccines. [11] Herein, we focus on genetic code expansion [12] as an innovative tool for the development of prophylactic and therapeutic vaccines. With a broad chemical biology audience in mind, we first provide accessible introductions to immunology and relevant genetic code expansion strategies, before discussing protein‐based vaccines, live‐attenuated viruses and providing a general outlook on the utility of an expanded genetic code for the development of vaccines in the future.
1.1. A hitchhiker's guide to vaccine immunology
Prophylactic vaccinations aim to provide long‐term protection against pathogens (i. e., immunological memory) through activation of the adaptive immune system. [13] Thus, vaccines function by inducing the production of effector cells or molecules that are capable of neutralizing pathogens by controlling their replication or inactivating their toxic components. Depending on the type of vaccine, mode of delivery and additives used, the exact mechanisms by which protection is achieved will differ. [14] Here we focus on the induction of a T cell‐dependent humoral immune response, that is the secretion of specific antibodies and the formation of memory B cells against pathogen‐specific antigens upon immunization. [15] For an in‐depth review of more complex pathways that result in the secretion of antibodies in mucosal membranes (mucosal immunity) or the production of cytotoxic T lymphocytes (cell‐mediated immunity), the reader is referred to specialized literature. [16]
Overall, the main steps (numbered 1–15 in Figure 1) toward acquiring vaccine‐induced humoral immunity are: stimulation of the innate immune system, leading to local inflammation and transport of antigens to the draining lymph nodes (dLNs; 1–4); activation of antigen specific T and B cells, upon which the latter undergoes affinity maturation and differentiation into antibody‐producing plasma B cells (5–12); and antibody secretion in the blood (and tissue) and build‐up of immunological memory (13–15).[ 13 , 15 ]
Figure 1.
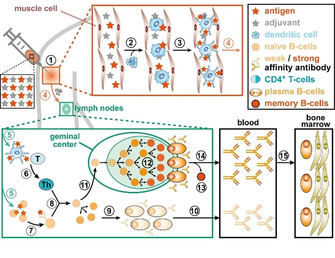
Schematic representation of the T cell‐dependent humoral immune response.
Stimulation of the innate immune system is triggered by (1) injecting a vaccine that contains a mixture of weakened pathogens or pathogen‐specific antigen(s) and immune response‐boosting additives (=adjuvants). The resulting local inflammation leads, amongst others, to recruitment of dendritic cells, which take up the antigens and are subsequently activated (2). The activated dendritic cells undergo changes in their morphology and surface receptors, facilitating the presentation of antigens (3) and inducing their migration to the dLNs (4). In response to these antigen‐presenting cells entering the dLNs, naïve antigen‐specific CD4+ T cells undergo activation and differentiate to helper T cells (5, 6). In parallel, activation takes place of naïve B cells that feature surface antibodies with (weak) affinity for antigens drained from the site of infection, triggering a multistep process that results in the display of antigens on the surface of activated B cells (7). Subsequently, the interaction between antigen‐presenting activated B cells and antigen‐specific helper T cells (8) induces B‐cell proliferation and their differentiation into plasma B cells (9). The low‐affinity antibodies secreted by these plasma cells typically appear in the bloodstream within a few days after the immunization (10). In a slower process, the interaction between activated helper T and B cells also triggers the generation of a geminal center (11), where B cells undergo massive clonal expansion and affinity maturation (12). In this fine‐tuning process, B cells displaying high affinity antibodies are selected and subsequently differentiate either into memory B cells (13) or to long lived plasma cells. The latter enter the bloodstream and tissues where they secrete large amounts of high affinity antibodies (14). Lastly, a few of these plasma cells migrate toward survival niches (15), which are mostly located in the bone marrow, from which they provide long‐term production of antibodies.
Depending on the type of vaccine and the immunological memory generated, prevention against infectious diseases can be short‐term or last lifelong. [17] Among the different types of vaccines available, weakened live pathogens often elicit a robust immune response, although safety concerns can limit their applicability (e. g., for immunocompromised patients). [18] Conversely, vaccines based on inactivated pathogens are safer, but often require repeated administration of booster doses to prolong protection. Similar trade‐offs between safety and efficiency are encountered for well‐defined vaccines based on subunits of pathogens (i. e., proteins or polysaccharides) [19] or those applied for therapeutic applications in immunotherapy. [8] As mentioned before, biotechnological innovations play a key role in overcoming these limitations, with this review focusing on the utility of genetic code expansion strategies for the development of improved prophylactic and therapeutic vaccines.
1.2. The ins and outs of genetic code expansion
“Genetic code expansion” is an umbrella term for strategies that enable the ribosomal incorporation of noncanonical amino acids (ncAAs) into peptides or proteins of interest. These strategies facilitate ncAA incorporation either through the global reassignment of sense codons or the suppression of nonsense codons (i. e., stop or quadruplet codons).[ 12 , 20 ] The latter strategy has proven particularly powerful, as it allows for site‐selective incorporation of the non‐native building block. Faithful and efficient expansion of the genetic code by suppression of in‐frame nonsense codons in vivo requires three principal components: 1) a metabolically stable, nontoxic ncAA that is readily taken up by the target organism (=bioavailability); 2) a reassigned nonsense codon in the gene of interest, with the amber stop codon (UAG) or quadruplet codons being the preferred choices; and 3) an orthogonal translation system (OTS), consisting of an aminoacyl tRNA synthetase (aaRS)/tRNA pair, which exclusively recognizes the ncAA. In this context, orthogonality refers to the fact that none of the OTS components interact with endogenous amino acids, aaRSs or tRNAs and vice versa.
Engineering OTSs for expanding the genetic code of prokaryotes and eukaryotes typically starts from existing aaRS/tRNA pairs from archaea, which are poorly recognized by prokaryotic and eukaryotic translation components, thereby providing an initial level of orthogonality. [21] Next, the tRNA anticodon is mutated to the desired sequence (e. g., CUA for the reverse‐complement of the UAG amber stop codon) and, if necessary, its orthogonality can be improved by consecutive rounds of positive (and negative) selection. Lastly, the substrate binding pocket of the aaRS is subjected to site‐directed mutagenesis and variants are selected that allow the incorporation of the ncAA of interest. These engineering efforts typically result in selective and efficient OTSs that load a ncAA onto an orthogonal tRNA, with the charged tRNAs subsequently being recruited to the ribosome, where in‐frame UAG stop codons in mRNAs can be suppressed (Figure 2).
Figure 2.
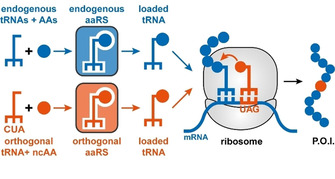
Schematic depiction of genetic code expansion by suppression of nonsense codons (P.O.I.=protein of interest).
Although low read‐through efficiencies typically result in limited protein yields, the suppression of nonsense codons has enabled the incorporation of more than 150 ncAAs into proteins of interest in a variety of organisms. [12] For the majority, these efforts have focused on introducing ncAAs harboring functional groups that enable site‐selective protein modification and/or aid in elucidating, altering or regulating protein function. [22] More recently, ncAAs featuring uniquely reactive side chains have also been employed to select cyclic peptides with novel ring architectures, design enzymes with new‐to‐nature reactivities and create organisms whose lives are dependent on these non‐native building blocks (=synthetic auxotrophy). [23] In this minireview, we add to this growing list of applications that repurpose OTSs by highlighting their utility for vaccine development.
2. Protein‐Based Vaccines
The immune system of mammals is finely tuned to discriminate between endogenous (self) proteins and those from potentially pathogenic foreign entities (nonself). [24] Tolerance to self‐proteins is primarily achieved through the inactivation of self‐reactive B and T cells during their development, a process that is crucial to prevent misguided attacks of the immune system. [25] The consequences of inadvertently breaking this self‐tolerance is readily apparent by the adverse effects of >80 known autoimmune diseases. [26] Nevertheless, breaking self‐tolerance can also be desirable, for example for cancer immunotherapy, [8] or for treating diseases caused the by adverse actions of self‐proteins, such as chronic inflammations and osteoporosis. However, eliciting strong effector mechanisms (e. g., neutralizing antibodies) against adverse self‐proteins remains challenging. Intentionally breaking self‐tolerance typically relies on immunization with modified self‐proteins to elicit antibodies that are selective for both the modified and the parent self‐protein. [27] Although such cross‐reactive immune responses to adverse self‐proteins have been induced by introducing foreign T‐helper cell epitopes into chimeric antigens or by extensive chemical derivatization of self‐antigens, the design of effective immunotherapeutics remains a slow process.
In contrast to nonselective chemical modification or the introduction of epitopes which can disrupt protein folding, genetic code expansion offers a controlled means to modify therapeutically relevant proteins. For breaking self‐tolerance, nitrated or sulfonated ncAAs are of particular interest. In addition of these oxidative stress‐induced modifications in self‐proteins being associated with the onset of a number of autoimmune diseases, [28] nitroaryl groups have also been used as highly immunogenic hapten‐molecules that induce a strong immune response when attached to proteins. [29] Although not fully understood, the molecular basis for recognition of nitroaryl groups by the immune system is typically attributed to the interactions of the electron‐deficient π systems with tryptophan and tyrosine residues that are common in B‐ or T‐cell receptors. Thus, the ncAAs p‐nitro‐l‐phenylalanine (pNO2Phe), 3‐nitro‐l‐tyrosine (3NO2Tyr) and p‐sulfo‐l‐phenylalanine (SO3Tyr) can be considered immunogenic amino acids (Figure 3A). Indeed, injection of proteins of interests featuring these immunogenic ncAAs has proven a surprisingly straightforward strategy to produce high‐titer of antibodies that were cross‐reactive to the wild‐type protein (Figure 3B). Here, we will focus on studies performed on the murine tumor necrosis factor‐α (mTNF‐α) and the murine receptor activator of nuclear factor‐κ B ligand (mRANKL).
Figure 3.
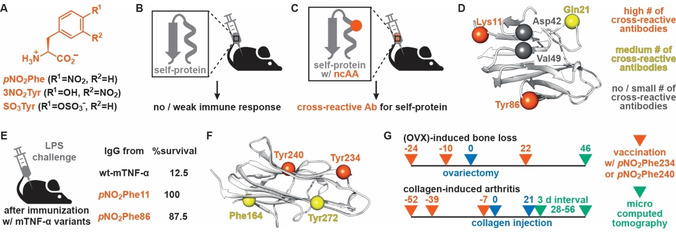
A) Structures of immunogenic ncAAs. B), C) Principles of breaking self‐tolerance via site‐selective incorporation of immunogenic ncAAs. B) While injection of a self‐protein in mice does not elicit a strong immune response, C) immunization with an immunogenic ncAA‐bearing self‐protein results in the generation of antibodies that show cross‐reactivity for the self‐protein. D) Structural representation of mTNF‐α (PDB ID : 2TNF). α‐Carbons of solvent‐exposed residues that were targeted for ncAA incorporation are shown as spheres with color representing the effectiveness of these mutations to elicit cross‐reactive antibodies. E) Summary of survival rates of mice challenged by LPS injection after immunization with relevant mTNF‐α variants. F) Structural representation of mRANKL (PDB ID : 1JTZ). α‐Carbons of solvent‐exposed residues that were targeted for ncAA incorporation are shown as spheres with color representing the effectiveness of these mutations to elicit cross‐reactive antibodies (color code as in (D)). G) Representative timeline for immunization of mice against OVX‐induced bone loss and CIA.
2.1. Breaking mTNF‐α self‐tolerance in mice
TNF‐α is a multifunctional cytokine that plays an important role in the acute phase of inflammation or infection and has been identified as a clinical target for treatment of multiple inflammatory diseases, such as Crohn's disease and rheumatoid arthritis. [30] To elucidate its role and function in these processes, studying murine TNF‐α in mouse models has proven particularly useful. For example, mTNF‐α‐deficient mice are viable and do not show an apparent phenotype, thus indicating that immunization against this self‐protein would not have adverse effects. [31] To test the protective effect of eliciting immunological memory against mTNF‐α, mice are typically subjected to a lipopolysaccharide (LPS) challenge. The injection of bacterial LPSs triggers the production and subsequent release of high‐levels of mTNF‐α, which induces a potentially lethal septic shock. [32] However, the presence of cross‐reactive antibodies against the self‐protein in challenged mice results in the breakdown of released mTNF‐α, thereby decreasing the severity of the induced shock. [33]
To test the utility of genetic code expansion for eliciting antibodies against self‐proteins, Schultz and co‐workers produced a series of mTNF‐α variants, in which surface‐exposed residues were mutated to immunogenic amino acids (Figure 3D). [34] Immunization of mice with these variants yielded antibodies against the mutant protein, while – in accordance with self‐tolerance – attempts at immunization with wild‐type mTNF‐α or with variants featuring somatic mutations failed to elicit a significant immune response. Notably, the ability of ncAA‐bearing mTNF‐α variants to generate cross‐reactive antibodies against the self‐protein upon immunization was dependent on the site of mutation (Figure 3D). Specifically, variants featuring immunogenic ncAAs at positions Lys11 or Tyr86 gave rise to high levels of antibodies cross‐reactive to wild‐type mTNF‐α. Encouragingly, the antibody titers in mice were found to be sustained at >80 % for at least 40 weeks after immunization, signifying the potential for inducing persistent immunity against the self‐protein. Lastly, the protective potential of immunization with ncAA‐bearing mTNF‐α variants was evaluated by the previously outlined LPS challenge model. Whereas mice that received sham injections or vaccines containing wild‐type protein succumbed quickly after the challenge (survival rate of 12.5 % after 3 days), immunization with pNO2Phe11‐ or pNO2Phe86‐mTNF‐α provided substantial levels of protection (Figure 3E). A survival rate of >87.5 % for these variants attested on the ability to elicit cross‐reactive antibodies that are effective in vivo.
A follow‐up study by Ramirez‐Montagut and co‐workers aimed to shed light on the mechanism by which incorporation of immunogenic ncAAs into mTNF‐α results in the formation of antibodies with cross‐reactivity for wild‐type proteins. [35] One key insight this study revealed was that mice with different genetic backgrounds responded differently to the immunization with modified proteins. For example, while pNO2Phe11mTNF‐α was immunogenic in C57BL/6 mice (see above), the same mutant protein failed to elicit a significant antibody response in FVB/N mice. One marked difference with respect to immune response between these two mouse strains is located in genes that encode for major histocompatibility complex (MHC) class II molecules. In the humoral immune response (see Introduction), these molecules are responsible for antigen‐presentation by dendritic or B cells, thus facilitating interaction of those cells with naïve and antigen‐specific CD4+ T cells.[ 13 , 15 , 36 ] Class II MHC genes are known to be highly polymorphic and certain genetic variations in these genes are linked to predisposition for autoimmune disease and responsiveness to immunization. [37] Critically, vaccination with pNO2Phe11mTNF‐α proved only effective in mice that featured the H‐2b haplotype, while mice with a different haplotype or featuring MHC class II molecules with only three amino acids mutations responded weakly or not at all to the same immunization regime. Isolated CD4+ T cells from H‐2b mice immunized with pNO2Phe11mTNF‐α proved to undergo activation when presented with peptide antigens that featured pNO2Phe but failed to do so in response to the wild‐type peptide epitope. In comparison CD4+ T cells isolated from mice featuring a different haplotype or mutated MHC‐class II molecules did not undergo activation in presence of either the wild‐type or pNO2Phe‐containing peptide epitope. Together, these results are consistent with 1) the negative selection of autoreactive T‐cells in the thymus [38] and 2) ncAA‐incorporation generating modified (neo‐)epitopes that can activate T cells, thus promoting a T cell‐dependent immune response. Therefore, a plausible mechanism for immunization with proteins featuring immunogenic ncAAs involves the activation of CD4+ T cells by these neo‐epitopes being presented on MHC class II molecules on B cells, triggering clonal proliferation of antigen‐specific B cells and the subsequent production of antibodies that are cross‐reactive to the wild‐type protein.
Lastly, immunization with mTNF‐α variants featuring 3NO2Tyr or SO3Phe in position Lys11 proved to follow the same mechanism. This observation is notable as these ncAAs are naturally occurring post‐translational modifications, which have been linked to the onset of a number of inflammatory diseases, autoimmune disorders and are believed to contribute to oncogenic signaling. [28] Using genetic code expansion strategies to produce homogeneous proteins featuring these post‐translational modifications could therefore prove valuable in efforts aimed at deciphering the biological consequences of formation of 3NO2Tyr or SO3Phe‐bearing neo‐epitopes.
2.2. Preventing RANKL‐induced bone loss in mice
The TNF cytokine family member RANKL is another self‐protein for which raising cross‐reactive antibodies is of clinical interest. Existing in both membrane‐bound and soluble form, RANKL is an essential factor in the activation of osteoclasts, which are cells responsible for breaking down the tissue in bones (=bone resorption). [39] As such RANKL is a therapeutic target for diseases in which patients suffer from bone erosion caused by an imbalance in bone remodeling, such as in osteoporosis (OP) and rheumatoid arthritis (RA). [40] Indeed, the recombinant monoclonal antibody denosumab, which prevents the activation of osteoclasts by binding to RANKL, is currently employed for the treatment of these diseases. [41] Although effective, the repeated injection of denosumab is also costly, preventing a more widespread use.
In principle, eliciting cross‐reactive antibodies against RANKL by immunization with ncAA‐bearing variants could provide a cheaper alternative for the treatment of OP and RA. [42] To enable such developments, Tao et al. produced pNO2Phe‐containig variants of murine RANKL (85 % sequence identity to human RANKL) and tested their ability to elicit cross‐reactive antibodies for the self‐protein following injection into mice. [43] As it was the case for mTNF‐α, the immune response generated was dependent on the site of mutation (Figure 3F). From the four positions evaluated, mutations of Tyr234 and Tyr240 gave rise to high titers of cross‐reactive antibodies that persisted in mice for at least 24 weeks.
The protective potential of pNO2Phe234‐mRANKL injection was evaluated in mouse models for OP and RA. While bone loss induced in female mice by ovariectomy (OVX) is a common model for OP, [44] injection of chicken type II collagen results in collagen‐induced arthritis (CIA), which shares several pathological features with RA. [45] As anticipated, repeated injection of pNO2Phe234‐mRANKL (Figure 3G) reduced the severity of symptoms both in OVX‐induced OP and CIA mice, while immunization with the self‐protein did not have a significant effect on the progression of either disease.[ 43 , 46 ] Specifically, OVX mice immunized with the mutant protein showed significantly reduced bone loss in micro‐computed tomography analysis. Similarly, in the CIA model, immunization with the pNO2Phe234‐mRANKL did not delay the onset of disease, but the severity of clinical symptoms and joint damage was significantly decreased when compared to nonimmunized mice. Together, these results provide proof‐of‐concept for the potential of applying immunization with pNO2Phe‐bearing proteins as a potential alternative treatment for RANKL‐induced and related diseases in humans.
3. Live‐Attenuated Virus Vaccines
Traditionally, viral vaccines have come in either of two forms: as inactivated whole virus particles or live‐attenuated viruses (LAVs). [13] Inactivation of viruses is straightforward by heat or the use of formaldehyde and essentially renders replicating viruses into inert antigens able to elicit an immune response. LAVs, on the other hand, contain viruses that remain infective and able to undergo limited replication upon injection, but have been sufficiently weakened to not cause damage. [47] Given that LAVs retain some degree of infectivity, they typically elicit a stronger immune response than fully inactivated viruses particles. As such, LAV‐based vaccines often provide long‐term protection without booster doses and in some cases can be administered orally. [17] However, as LAVs maintain their ability to replicate, they have a low chance to revert to virulence, and as a result cause the disease they are meant to prevent. [48]
Attenuation of viruses has long been (and often remains) an empirical process in which an infectious virus is serially passaged through cell cultures, abnormal hosts and/or below normal human body temperature (i. e., cold‐adapted attenuation). [9a] The virus gradually weakens as a result of adapting to these new environments, thereby lowering its virulence and ability to infect and spread in the intended host. With the advent of molecular biology, more rational approaches toward virus attenuation have become available. [49] For example, genes encoding for proteins involved in replication or assembly can be deleted from the viral genome or rendered inactive by mutation. While resulting viruses are, per se, replication‐incompetent their production remains possible in cell‐lines that express genes encoding for the missing or dysfunctional proteins. More recently, viral replication has also been controlled by installing rare codons into viral genomes to slow the translation of viral proteins (=codon deoptimization) and by the introduction of microRNAs and zinc finger nucleases that selectively break down viral RNA.[ 11 , 19 , 29a ] Nevertheless, LAVs created by these means share some of the safety concerns associated with those based on viruses attenuated through empirical approaches. Additionally, they often elicit a weaker immune response than their progenitors and their applicability can be limited to certain classes of viruses (e. g., RNA viruses or nonintegrating DNA viruses).
In principle, making viral replication dependent on the incorporation of ncAAs is a promising strategy for creating LAV‐based vaccines. Instead of deleting or inactivating viral genes, nonsense codons are introduced as premature termination‐codons (PTCs) at permissive sites throughout the viral genome (Figure 4A). [23] Introduction of PTCs results in incomplete translation of viral proteins, thus rendering the virus replication‐incompetent. However, these viruses can be produced in cell lines which express an OTS that permits readthrough of the PTC, when its cognate ncAA is supplied. The advantages of this strategy are threefold: 1) PTC viruses produced in these transgenic cell lines feature minimal structural deviations when compared to their progenitor viruses, which is desired for attaining a high level of infectivity and a strong immune response; 2) safety of the PTC virus can be enhanced by incorporating multiple nonsense codons throughout the viral genome, making reversion to a replication‐competent state by mutation less likely; and 3) the introduction of stop codons to generate LAVs is possible on the DNA and RNA level and therefore widely applicable to different classes of viruses. [50] In the following sections, we highlight the utility of PTC‐harboring viruses for controlling the replication of HIV‐1 and for creating prophylactic and therapeutic vaccines for influenza A.
Figure 4.
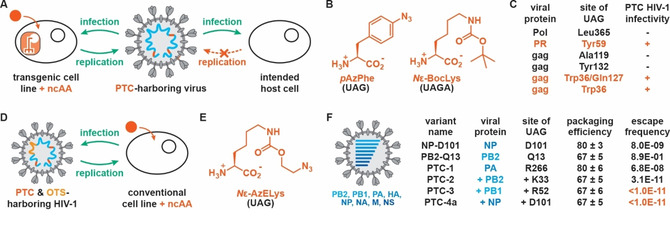
A) PTC‐harboring viruses can infect and replicate upon addition of a ncAA in transgenic cell lines that feature an OTS specific for this ncAA. Conversely, PTC viruses remain the infective for the intended host cells but cannot propagate in the absence of the OTS and ncAA. B) Structures of ncAAs used for the construction of PTC‐harboring HIV‐1. C) Infectivity of PTC‐harboring HIV‐1 viruses created by incorporating ncAAs at different sites in viral proteins. D) Replication of PTC‐harboring HIV‐1 can be controlled by the addition of ncAAs if an OTS is introduced at a permissive site in the HIV genome. E) Structure of Nϵ‐AzELys, which was used to create PTC‐harboring influenza A viruses. F) Schematic representation of nucleic acids found inside the influenza A capsid and their effect on packaging efficiency and escape frequency of PTC‐harboring influenza A viruses featuring Nϵ‐AzELys at different positions in the viral genome.
3.1. Controlling the replication of live‐attenuated HIV‐1
Since HIV was identified as the causal agent for AIDS in 1983, [51] major progress in the prevention and treatment of HIV/AIDS has been made. However, despite continuous efforts, it is estimated that as of 2018 more than 37 million people are living with the virus, with 1.7 million people being newly infected that year. [52] While the development of a successful vaccine is presumably the only means to control this ongoing pandemic, this task has proven challenging. [53] Experimental vaccines that have been developed either do not provide sufficient levels of protection (e. g., around 30 % in the RV144 efficacy trial) [54] or are not safe enough to be tested in clinical trials. For example, while deleting the nef gene in simian immunodeficiency virus resulted in a highly effective LAV vaccine (>95 % protection in macaques), [55] it also showed pathogenic potential in animal models. [56] As such, applying new technologies for the development of safe and effective HIV vaccines continues to be highly relevant.
Toward this end, Guo and co‐workers were the first to apply genetic code expansion for the construction of replication‐incompetent, PTC‐harboring HIV‐1. [50a] In a trial‐and‐error approach they installed UAG nonsense codons at permissive sites into a number of HIV‐1 genes and tested viral assembly and infectivity of the resulting PTC viruses. Three structurally similar tyrosine analogues were tested for the production of PTC‐containing HIV‐1 in engineered cell lines featuring matching OTSs. While p‐iodo‐l‐phenylalanine was not effectively incorporated in viral proteins and p‐acetyl‐l‐phenylalanine yielded noninfectious variants, only p‐azido‐l‐phenylalanine (pAzPhe, Figure 4B) displayed the desired high suppression efficiency and gave rise to infectious viruses. In addition of PTC‐virus production varying with the choice of ncAA, the viral protein targeted and the site of mutation proved also crucial, which in part could be ascribed to the complex nature of HIV‐1 gene splicing and protein processing. [57] While PTC mutations at various sites in the HIV‐1 genome resulted in the pAzPhe‐dependent production of mature HIV‐1 in transgenic cells, the resulting virions often lacked the ability to infect potential host cells (Figure 4C). Encouragingly though, variants featuring either one stop codon at position Tyr59 in the gene coding for the HIV‐1 protease (PR) or two PTC mutations at positions Trp36 and Gln127 in the group‐specific antigen (gag) protein yielded infectious viruses in presence of pAzPhe in transgenic cells. While this study provided proof‐of‐concept for the creation of a live PTC viruses, the chance of these HIV‐1 strains to regain functional replication by mutation of the installed nonsense codon(s) to sense codon(s) raises considerable safety concerns for their potential application as LAV vaccines.
To decrease the likelihood of PTC HIV‐1 variants to revert to a virulent state, the use of a nonsense quadruplet codon to encode for a ncAA was explored. [58] For this, an OTS adapted to decode UAGA codons with N ϵ‐(tert‐butoxycarbonyl)‐l‐lysine (N ϵ‐BocLys, Figure 4B) in Escherichia coli could be repurposed for the incorporation of this ncAA in mammalian (HEK 293T) cells. [59] To avoid mistranslation of five in‐frame UAG‐A sequences in the HIV‐1 genome, which could interfere with viral assembly, Guo and co‐workers created an HIV‐1 variant that lacked these sequences and displayed replication and infectivity levels comparable to the parent strain. Introducing N ϵ‐BocLys by suppression of UAGA codons at the previously identified positions Tyr59 in PR or Trp36 in gag gave rise to infectious HIV‐1 virions, although at significantly lower levels than the parent variant. A theoretically lower escape frequency of these HIV‐1 variants results from the fact that reversion of the +1 frameshift caused by the incorporation of the quadruplet codon requires a deletion event, which is rare compared to simple point mutations that can revert UAG‐harboring PTC viruses. [60]
Lastly, another point of concern for replication‐incompetent PTC‐harboring viruses is that immunization with a single infection cycle HIV virus would likely result in an insufficient immune response. [61] To clear this hurdle, Guo et al. generated a PTC‐harboring HIV‐1 mutant, for which genes encoding for the OTS for decoding UAG with N ϵ‐BocLys were inserted at a permissive site in the viral genome. [62] Consequently, the replication of this mutant could be controlled in conventional cell lines by simply supplementing N ϵ‐BocLys into the media (Figure 4D). Unfortunately, the infectivity of the resulting PTC‐harboring virus was significantly lower than for the parent HIV‐1, presumably due to the low expression levels of the OTS components. Nevertheless, this type of PTC‐harboring virus could potentially allow for controlling HIV‐1 replication by co‐administering the ncAA, thus keeping the virus infective until a sufficiently strong immune response is elicited.
3.2. Toward prophylactic and therapeutic influenza A vaccines
Yearly, worldwide influenza outbreaks result on average in about 3–5 million cases of severe illness and >300.000 deaths. [63] Rendering the influenza viruses responsible for this loss of life into avirulent vaccines while maintaining sufficient infectivity remains challenging. [64] Attenuated influenza viruses do in principle match these criteria, but immune escape due to antigenic drift and shift adds to the challenges associated with creating effective vaccines against seasonal influenza viruses. [65] With the aim of providing a more robust alternative to available attenuated vaccines, Zhuo and co‐workers set out to create PTC‐harboring influenza A strains that could reproduce exclusively in transgenic cell lines and subsequently be used for prophylactic and therapeutic vaccination. [50b]
To allow for the production of PTC‐harboring influenza A viruses, a HEK293T cell line was constructed with an OTS for N ϵ‐2‐azidoethyloxycarbonyl‐l‐lysine (N ϵ‐AzELys, Figure 4E). Transfection of this packaging cell line with the genome of an influenza strain featuring a UAG codon at a randomly chosen site (Asp101 in the nucleoprotein, NP) indeed resulted in the reproduction and correct packaging of influenza A virions (Figure 4F). Critically, no virus production was observed in a conventional cell line upon infection with the progeny virus, which contrasts results obtained for cold‐adapted live attenuated influenza vaccine (CAIV) and codon‐deoptimized influenza A viruses which both showed residual infectivity and reproductivity. [66] Moreover, introduction of PTCs proved a general strategy, as for eight different influenza proteins at least one site for mutation was identified that resulted in high packaging and propagation efficiency. Aware of mutation of the UAG codon being a straightforward escape mechanism for PTC‐harboring viruses, influenza A strains harboring 1 to 8 nonsense codons in different viral proteins were constructed (Figure 4F). These viruses retained good packaging efficiencies (at least 67 % compared to the wild‐type), while showing escape frequencies over 20 passages ranging from 10−2 to 10−9 for variants featuring one nonsense codon to <10−11 (below the detection limit in the assay) for those harboring more than three stop codons.
To evaluate the in vivo safety of PTC‐harboring influenza A strains, PTC‐4a was selected as none of its four mutations were in envelope genes, thus providing wild‐type‐like representation of surface antigens. Following injection of PTC‐4a at 100.000 times the median lethal dose (LD50) of the parent influenza strain, no mice succumbed or showed any symptoms associated with influenza infection (i. e., body weight loss or other health issues). Moreover, lower viral titers were elicited after injection of PTC‐4a not only compared to the wild‐type virus, but also to a CAIV, further underling its overall safety in vivo. Similarly, when guinea pigs inoculated with PTC‐4a were caged together with non‐infected ones, no transmission was detected, which again contrasts results obtained for wild‐type viruses or CAIV, where transmission was readily observed. Thus, these experiments demonstrate the overall safety of PTC‐harboring influenza viruses.
Besides showing high levels of safety, PTC‐4a was also able to elicit a strong immune response when injected into mice, ferrets or guinea pigs. In fact, this replication‐incompetent influenza A virus not only gave rise to robust antibodies in serum (humoral immunity), but also to high level of secretory immunoglobulin A in the lungs (mucosal immunity) and cytotoxic T lymphocytes (cell‐mediated immunity). Notably, the levels for all three elicited immune effectors were boosted significantly upon a second injection of PTC‐4a and overall comparable to those levels elicited by CAIV. Furthermore, the protective efficacy of PTC‐4 A virus was high: when challenged with 50 times the LD50 of the wild‐type virus, all mice survived after immunization even with just one dose of PTC‐4a, with injection of two doses preventing any symptoms associated with influenza infection. The strong immune response elicited provided also significant protection, when mice were challenged with antigenically distant influenza strains. Such heterologous protection was ascribed to surface and internal antigens that are highly conserved among distant influenza strains and to a broad protective effect of influenza‐specific of cytotoxic T‐lymphocytes. [67]
Lastly, somewhat counterintuitively, co‐injection of PTC‐4a and wild‐type influenza A into mice resulted in reduction of viral titers when measured over the following days. This inhibition of the wild‐type virus by PTC‐4a cannot be ascribed to acquired immunity, but results from attenuation by genetic reassortment between the PTC‐harboring virus and the wild‐type.[ 65 , 68 ] Upon infecting the same host cell, genes are being exchanged between the two strains and progeny viruses can become replication incompetent when they are packaged with at least one PTC‐harboring gene (Figure 5). Sequencing analysis of propagated viruses confirmed the creation of an assortment of replication‐incompetent PTC‐harboring influenza strains. Notably, the attenuation effect was directly proportional to the number of PTCs introduced, with a variant featuring four or more UAG stop codons essentially preventing proliferation of wild‐type influenza in vitro. Combined, these experiments indicate that viruses harboring multiple PTCs could not only find use in the prophylactic prevention of viral diseases, but also applied as therapeutic vaccines for the neutralization of replicating viruses.
Figure 5.
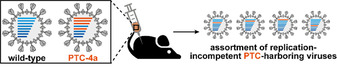
Principles of genetic reassortment. Co‐injection of wild‐type and PTC‐4a slows viral replication due to the formation of an assortment of replication‐incompetent PTC‐harboring influenza viruses.
4. Challenges and Future Prospects
The proof‐of‐concept studies highlighted in this review attest on the future applicability of genetic code expansion strategies for the development of protein‐ and LAV‐based vaccines. For the former, the incorporation of ncAAs such as pNO2Phe into self‐proteins provides a straightforward means to break self‐tolerance and elicit a robust humoral immune response. Similarly, the production of LAVs by introduction of PTCs has proven effective for containing viral replication in vitro and in vivo.
The precise control over the site and number of introduced ncAAs into nonimmunogenic proteins makes suppression of nonsense codons a promising strategy for creating protein‐based vaccines. Their potential for inducing immunity against self‐proteins makes the technique a promising tool for creating sought‐after therapeutic vaccines against inflammatory diseases and different types of cancer. [69] In addition to the development of vaccines against self‐proteins, this strategy could potentially find application for eliciting immune response against pathogen‐associated proteins that are not or weakly immunogenic. While the results obtained for self‐proteins are promising, the fact that the formation of cross‐reactive antibodies was dependent on the genetic background of mice in genes encoding for MHC class‐II molecules is problematic. The genetic variability of MHC genes in the population [70] and the difficulty of understanding and predicting T‐cell specificity for particular peptides [71] are important hurdles to overcome for these protein‐based vaccines to become applicable for treating human diseases. Therefore, more systematic studies on reliably identifying possible site(s) of modification in proteins and the development of OTSs for more immunogenic ncAAs, such as N ϵ‐dinitrophenylacetyl‐l‐lysine) [72] and three halotyrosine residues [73] could prove valuable for furthering the development of immunogenic self‐proteins. Lastly, the ability to generate homogenous self‐proteins that feature naturally occurring, stress‐induced post‐translational modifications could provide useful insights for elucidating mechanisms of activation and progression of autoimmune disorders as well as cancer. This information could further unveil which modifications generate an immunogenic epitope and therefore expand the applicability of incorporating ncAAs for the development of vaccines.
Creating LAVs by making viral replication dependent on the presence of an OTS and a matching ncAA could be widely applicable to develop vaccines for different viruses. Especially for viruses for which attenuation using conventional methods is challenging (e. g., herpes simplex virus), [74] a straightforward means to create replication‐incompetent variants should be of particular interest. Thus far however, approaches for identifying suitable incorporation sites for ncAAs have largely been trial‐and‐error and their effect on virus assembly and infectivity was not always straightforward to interpret. Complex processing of viral genomes and proteins leading up to assembly of full virions (e. g., for retroviruses) complicates finding permissive sites for PTC incorporation. [75] Thus, other virus classes likely provide better targets for development of PTC‐harboring LAVs, as demonstrated for influenza A. In addition to further exploring the consequences of incorporating PTCs into viral genomes for the generation of infective virions, the resulting LAVs must also undergo a rigorous validation with respect to their safety in vivo. Intriguingly, the incorporation of PTCs into genomes might also be applicable for the development of vaccines against persistent infectious diseases caused by bacterial pathogens. Specifically, multiple studies have recently succeeded to make bacterial survival strictly dependent on the presence of a ncAA and a matching OTS (synthetic biocontainment).[ 23 , 76 ] Adapting these biocontainment strategies to bacterial pathogens – note that the ncAA incorporation in Mycobacterium tuberculosis has previously been reported [77] – could pave the way toward the creation of infective pathogens that can only replicate in engineered cell lines. Lastly, to prevent reversion to replicating pathogens in host by mutation of introduced nonsense codons, essential enzymes of the pathogens could be engineered to make their function depend on the incorporation a ncAA. [78]
Overall, we are confident that future studies will continue to highlight and further progress the utility of genetic code expansion for vaccine development.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
C.M. thanks Dr. Manuela Bersellini for her careful reading of this manuscript and for many helpful suggestions. C.M. also acknowledges the Netherlands Organisation for Scientific Research (NWO, Veni grant 722.017.007).
J. A. Fok, C. Mayer, ChemBioChem 2020, 21, 3291.
References
- 1. Greenwood B., Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Behbehani A. M., Microbiol. Rev. 1983, 47, 455–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moffett D. B., Llewellyn A., Singh H., Saxentoff E., Partridge J., Iakovenko M., Roesel S., Asghar H., Baig N., Grabovac V., et al., Morb. Mortal. Wkly. Rep. 2019, 68, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Girard M. P., Tam J. S., Assossou O. M., Kieny M. P., Vaccine 2010, 28, 4895–4902. [DOI] [PubMed] [Google Scholar]
- 5. Sharp P. M., Hahn B. H., CSH Perspect. Med. 2011, 1, a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., et al., Nature 2020, 579, 265–269; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., et al., Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rappuoli R., Aderem A., Nature 2011, 473, 463–469. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Mellman I., Coukos G., Dranoff G., Nature 2011, 480, 480–489; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Waldmann T. A., Nat. Med. 2003, 9, 269–277. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Plotkin S. A., Plotkin S. L., Nat. Rev. Microbiol. 2011, 9, 889–893; [DOI] [PubMed] [Google Scholar]
- 9b. Plotkin S., Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enders J. F., Weller T. H., Robbins F. C., Science 1949, 109, 85–87. [DOI] [PubMed] [Google Scholar]
- 11. Kanekiyo M., Ellis D., King N. P., J. Infect. Dis. 2019, 219, S88-S96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Dumas A., Lercher L., Spicer C. D., Davis B. G., Chem. Sci. 2015, 6, 50–69; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Chin J. W., Nature 2017, 550, 53–60; [DOI] [PubMed] [Google Scholar]
- 12c. Young D. D., Schultz P. G., ACS Chem. Biol. 2018, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Bloom B. R., Lambert P. H., The Vaccine Book , 2nd ed., Elsevier, Amsterdam, 2016; [Google Scholar]
- 13b. Plotkin S. A., Orenstein W. A., Offit P. A., Plotkin's Vaccines, 7th ed., Elsevier, Philadelphia, 2018.29424838 [Google Scholar]
- 14. Pulendran B., Ahmed R., Nat. Immunol. 2011, 12, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Slifka M. K., Antia R., Whitmire J. K., Ahmed R., Immunity 1998, 8, 363–372. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Holmgren J., Czerkinsky C., Nat. Med. 2005, 11, S45–53; [DOI] [PubMed] [Google Scholar]
- 16b. Kaech S. M., Wherry E. J., Ahmed R., Nat. Rev. Immunol. 2002, 2, 251–262. [DOI] [PubMed] [Google Scholar]
- 17. Vetter V., Denizer G., Friedland L. R., Krishnan J., Shapiro M., Ann. Med. 2018, 50, 110–120. [DOI] [PubMed] [Google Scholar]
- 18. Eibl M. M., Wolf H. M., Immunotherapy 2015, 7, 1273–1292. [DOI] [PubMed] [Google Scholar]
- 19. Delany I., Rappuoli R., De Gregorio E., EMBO Mol. Med. 2014, 6, 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Agostini F., Voller J. S., Koksch B., Acevedo-Rocha C. G., Kubyshkin V., et al., Angew. Chem. Int. Ed. 2017, 56, 9680–9703; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9810–9835; [Google Scholar]
- 20b. Tsiamantas C., Rogers J. M., Suga H., Chem. Commun. 2020, 56, 4265–4272. [DOI] [PubMed] [Google Scholar]
- 21. Liu C. C., Schultz P. G., Annu. Rev. Biochem. 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Davis L., Chin J. W., Nat. Rev. Mol. Cell Biol. 2012, 13, 168–182; [DOI] [PubMed] [Google Scholar]
- 22b. Huang Y., Liu T., Synth. Syst. Biotechnol. 2018, 3, 150–158; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Neumann H., FEBS Lett. 2012, 586, 2057–2064. [DOI] [PubMed] [Google Scholar]
- 23. Mayer C., ChemBioChem 2019, 20, 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matzinger P., Science 2002, 296, 301–305. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Van Parijs L., Abbas A. K., Science 1998, 280, 243–248; [DOI] [PubMed] [Google Scholar]
- 25b. Wing K., Sakaguchi S., Nat. Immunol. 2010, 11, 7–13. [DOI] [PubMed] [Google Scholar]
- 26. Sakaguchi S., Powrie F., Ransohoff R. M., Nat. Med. 2012, 18, 54–58. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Kalliolias G. D., Ivashkiv L. B., Nat. Rev. Rheumatol. 2016, 12, 49–62; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Cappellano G., Orilieri E., Woldetsadik A. D., Boggio E., Soluri M. F., et al., Am. J. Immunol. 2012, 1, 136–146; [PMC free article] [PubMed] [Google Scholar]
- 27c. Dalum I., Butler D. M., Jensen M. R., Hindersson P., Steinaa L., et al., Nat. Biotechnol. 1999, 17, 666–669. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Doyle H. A., Mamula M. J., Curr. Opin. Immunol. 2012, 24, 112–118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Ryan B. J., Nissim A., Winyard P. G., Redox Biology 2014, 2, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Jones L. H., Nat. Chem. 2015, 7, 952–960; [DOI] [PubMed] [Google Scholar]
- 29b. Palm N. W., Medzhitov R., Proc. Natl. Acad. Sci. USA 2009, 106, 4782–4787; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29c. Berd D., Expert Rev. Vaccines 2004, 3, 521–527. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Bradshaw R. A., Dennis E. A., Handbook of Cell Signaling, Academic Press, Amsterdam, 2004; [Google Scholar]
- 30b. Bradley J. R., J. Pathol. 2008, 214, 149–160; [DOI] [PubMed] [Google Scholar]
- 30c. Steeland S., Libert C., Vandenbroucke R. E., Int. J. Mol. Sci. 2018, 19, 1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pasparakis M., Alexopoulou L., Episkopou V., Kollias G., J. Exp. Med. 1996, 184, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niessen F., Schaffner F., Furlan-Freguia C., Pawlinski R., Bhattacharjee G., et al., Nature 2008, 452, 654–658. [DOI] [PubMed] [Google Scholar]
- 33. Silva A. T., Bayston K. F., Cohen J., J. Infect. Dis. 1990, 162, 421–427. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Grunewald J., Hunt G. S., Dong L., Niessen F., Wen B. G., et al., Proc. Natl. Acad. Sci. USA 2009, 106, 4337–4342; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b. Grunewald J., Tsao M. L., Perera R., Dong L., Niessen F., et al., Proc. Natl. Acad. Sci. USA 2008, 105, 11276–11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gauba V., Grunewald J., Gorney V., Deaton L. M., Kang M., et al., Proc. Natl. Acad. Sci. USA 2011, 108, 12821–12826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blum J. S., Wearsch P. A., Cresswell P., Annu. Rev. Immunol. 2013, 31, 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.
- 37a. Tsai S., Santamaria P., Front. Immunol. 2013, 4, 321; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37b. Yucesoy B., Talzhanov Y., Johnson V. J., Wilson N. W., Biagini R. E., et al., Vaccine 2013, 31, 5381–5391; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37c. Luckey D., Weaver E. A., Osborne D. G., Billadeau D. D., Taneja V., Sci. Rep. 2019, 9, 19061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takaba H., Takayanagi H., Trends Immunol. 2017, 38, 805–816. [DOI] [PubMed] [Google Scholar]
- 39. Geusens P., Ther. Adv. Musculoskelet. Dis. 2012, 4, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.
- 40a. Guo Q., Wang Y., Xu D., Nossent J., Pavlos N. J., et al., Bone 2018, 6, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40b. Ukon Y., Makino T., Kodama J., Tsukazaki H., Tateiwa D., et al., Int. J. Mol. Sci. 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chiu Y. G., Ritchlin C. T., Expert Opin. Biol. Ther. 2017, 17, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ono T., Hayashi M., Sasaki F., Nakashima T., Inflammation Regener. 2020, 40, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li F., Li H., Zhai Q., Li F., Wu T., et al., Biochem. Biophys. Res. Commun. 2018, 499, 648–654. [DOI] [PubMed] [Google Scholar]
- 44. Roberts B. C., Giorgi M., Oliviero S., Wang N., Boudiffa M., et al., Bone 2019, 127, 260–270. [DOI] [PubMed] [Google Scholar]
- 45. Brand D. D., Latham K. A., Rosloniec E. F., Nat. Protoc. 2007, 2, 1269–1275. [DOI] [PubMed] [Google Scholar]
- 46. Wu T., Li F., Sha X., Li F., Zhang B., et al., Int. Immunopharmacol. 2018, 64, 326–332. [DOI] [PubMed] [Google Scholar]
- 47. Minor P. D., Virology 2015, 479–480, 379–392. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Galen J. E., R. Curtiss III , Vaccine 2014, 32, 4376–4385; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48b. Bull J. J., Virus Evol. 2015, 1, vev005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lauring A. S., Jones J. O., Andino R., Nat. Biotechnol. 2010, 28, 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.
- 50a. Wang N., Li Y., Niu W., Sun M., Cerny R., et al., Angew. Chem. Int. Ed. 2014, 53, 4867–4871; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4967–4971; [Google Scholar]
- 50b. Si L., Xu H., Zhou X., Zhang Z., Tian Z., et al., Science 2016, 354, 1170–1173. [DOI] [PubMed] [Google Scholar]
- 51.
- 51a. Gallo R. C., Sarin P. S., Gelmann E. P., Robert-Guroff M., Richardson E., et al., Science 1983, 220, 865–867; [DOI] [PubMed] [Google Scholar]
- 51b. Barre-Sinoussi F., Chermann J. C., Rey F., Nugeyre M. T., Chamaret S., et al., Science 1983, 220, 868–871. [DOI] [PubMed] [Google Scholar]
- 52. Walter E. Robert, Anthony S. F., Emerging Infect. Dis. 2018, 24, 413. [Google Scholar]
- 53. Dieffenbach C. W., Fauci A. S., Ann. Intern. Med. 2011, 154, 766–771. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Hsu D. C., O′Connell R. J., Hum. Vaccines 2017, 13, 1018–1030; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54b. Kim J. H., Excler J. L., Michael N. L., Annu. Rev. Med. 2015, 66, 423–437. [DOI] [PubMed] [Google Scholar]
- 55. Daniel M. D., Kirchhoff F., Czajak S. C., Sehgal P. K., Desrosiers R. C., Science 1992, 258, 1938. [DOI] [PubMed] [Google Scholar]
- 56.
- 56a. Baba T. W., Liska V., Khimani A. H., Ray N. B., Dailey P. J., et al., Nat. Med. 1999, 5, 194–203; [DOI] [PubMed] [Google Scholar]
- 56b. Koff W. C., Johnson P. R., Watkins D. I., Burton D. R., Lifson J. D., et al., Nat. Immunol. 2006, 7, 19–23. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Bell N. M., Lever A. M. L., Trends Microbiol. 2013, 21, 136–144; [DOI] [PubMed] [Google Scholar]
- 57b. Hryckiewicz K., Bura M., Kowala-Piaskowska A., Bolewska B., Mozer-Lisewska I., HIV AIDS Rev. 2011, 10, 61–64. [Google Scholar]
- 58. Chen Y., Wan Y., Wang N., Yuan Z., Niu W., et al., ACS Synth. Biol. 2018, 7, 1612–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Niu W., Schultz P. G., Guo J., ACS Chem. Biol. 2013, 8, 1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cuevas J. M., Geller R., Garijo R., Lopez-Aldeguer J., Sanjuan R., PLoS Biol. 2015, 13, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wyand M. S., Manson K. H., Garcia-Moll M., Montefiori D., Desrosiers R. C., J. Virol. 1996, 70, 3724–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yuan Z., Wang N., Kang G., Niu W., Li Q., et al., ACS Synth. Biol. 2017, 6, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lambert L. C., Fauci A. S., N. Engl. J. Med. 2010, 363, 2036–2044. [DOI] [PubMed] [Google Scholar]
- 64. Jang Y. H., Seong B. L., Clin. Exp. Vaccine Res. 2012, 1, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tong S., Li Y., Rivailler P., Conrardy C., Castillo D. A., et al., Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.
- 66a. Nogales A., Baker S. F., Ortiz-Riano E., Dewhurst S., Topham D. J., et al., J. Virol. 2014, 88, 10525–10540; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66b. Mueller S., Coleman J. R., Papamichail D., Ward C. B., Nimnual A., et al., Nat. Biotechnol. 2010, 28, 723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.
- 67a. Impagliazzo A., Milder F., Kuipers H., Wagner M. V., Zhu X., et al., Science 2015, 349, 1301–1306; [DOI] [PubMed] [Google Scholar]
- 67b. Burton D. R., Poignard P., Stanfield R. L., Wilson I. A., Science 2012, 337, 183–186; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67c. Stanekova Z., Vareckova E., Virol. J. 2010, 7, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou N. N., Senne D. A., Landgraf J. S., Swenson S. L., Erickson G., et al., J. Virol. 1999, 73, 8851–8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wraith D. C., Front. Immunol. 2017, 8, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Buhler S., Sanchez-Mazas A., PLoS One 2011, 6, e14643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Singh N. K., Riley T. P., Baker S. C. B., Borrman T., Weng Z., et al., J. Immunol. 2017, 199, 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ren W., Ji A., Wang M. X., Ai H. W., ChemBioChem 2015, 16, 2007–2010. [DOI] [PubMed] [Google Scholar]
- 73. Jang H. S., Gu X., Cooley R. B., Porter J. J., Henson R. L., et al., ACS Chem. Biol. 2020, 15, 562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dudek T., Knipe D. M., Virology 2006, 344, 230–239. [DOI] [PubMed] [Google Scholar]
- 75. Painter M. M., Collins K. L., in Encyclopedia of Microbiology 4th ed. (Ed.: Schmidt T. M.), Academic Press, Oxford, 2019, pp. 613–628. [Google Scholar]
- 76. Simon A. J., Ellington A. D., F1000Research 2016, 5, (F1000 Faculty Rev):2118. [Google Scholar]
- 77. Wang F., Robbins S., Guo J., Shen W., Schultz P. G., PLoS One 2010, 5, e9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.
- 78a. Koh M., Yao A., Gleason P. R., Mills J. H., Schultz P. G., J. Am. Chem. Soc. 2019, 141, 16213–16216; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78b. Tack D. S., Ellefson J. W., Thyer R., Wang B., Gollihar J., et al., Nat. Chem. Biol. 2016, 12, 138–140. [DOI] [PubMed] [Google Scholar]