

Abstract
Mechanistic understanding of how ionizing radiation induces type I interferon signaling and how to amplify this signaling module should help to maximize the efficacy of radiotherapy. In the current study, we report that inhibitors of the DNA damage response kinase ATR can significantly potentiate ionizing radiation‐induced innate immune responses. Using a series of mammalian knockout cell lines, we demonstrate that, surprisingly, both the cGAS/STING‐dependent DNA‐sensing pathway and the MAVS‐dependent RNA‐sensing pathway are responsible for type I interferon signaling induced by ionizing radiation in the presence or absence of ATR inhibitors. The relative contributions of these two pathways in type I interferon signaling depend on cell type and/or genetic background. We propose that DNA damage‐elicited double‐strand DNA breaks releases DNA fragments, which may either activate the cGAS/STING‐dependent pathway or—especially in the case of AT‐rich DNA sequences—be transcribed and initiate MAVS‐dependent RNA sensing and signaling. Together, our results suggest the involvement of two distinct pathways in type I interferon signaling upon DNA damage. Moreover, radiation plus ATR inhibition may be a promising new combination therapy against cancer.
Keywords: ATR, cGAS/STING, MAVS, radiation, type I interferon
Subject Categories: DNA Replication, Repair & Recombination; Immunology
Surprisingly, both cGAS/STING‐dependent DNA‐ and MAVS‐dependent RNA‐sensing pathways contribute to the effects of combined radiotherapy and blocked DNA damage signaling, depending on cellular context.

Introduction
Radiotherapy is widely used for cancer treatment owing to its ability to damage DNA and lead to cell death and/or senescence (Jonathan et al, 1999; Sharma et al, 2016). ATR (ataxia‐telangiectasia‐mutated (ATM) and Rad3‐related protein kinase) is a critical regulator of cellular DNA damage response (Marechal & Zou, 2013) and has become an attractive therapeutic target (Karnitz & Zou, 2015; Brown et al, 2017). AZD6738, a highly selective ATR inhibitor (ATRi) (Charrier et al, 2011), has been shown to enhance radiotherapy in a phase I clinical trial (Karnitz & Zou, 2015). However, besides the G2/M checkpoint bypass, other cellular processes influenced by ATRi, especially when combined with ionizing radiation (IR), remain poorly understood.
It is well established that the cyclic GMP‐AMP synthase (cGAS)/stimulator of IFN genes (STING) pathway is the major pathway responsible for sensing pathogenic double‐strand DNA (dsDNA) and the melanoma differentiation‐associated gene 5 (MDA5)/retinoic acid‐inducible gene I (RIG‐I)/mitochondrial antiviral‐signaling protein (MAVS) pathway is responsible for sensing pathogenic RNA (Wu & Chen, 2014). Once activated by cytosolic DNA, cGAS will catalyze the production of cyclic GMP‐AMP (cGAMP), which functions as a second messenger to activate downstream adaptor protein STING. Cytosolic RNA, once recognized by RIG‐I/MDA5, will induce a structural rearrangement to liberate their CARD domains to form oligomers; this oligomerized CARD domains of RIG‐I/MDA5 interact with and promote the polymerization of MAVS CARD, which turns on the downstream signals. These two pathways share the same downstream signaling cascade: Both MAVS and STING can recruit TANK binding kinase‐1 (TBK1) and inhibitor of kappa B kinase‐ɛ (IKKɛ) to phosphorylate the transcription factor IRF3 and promote IRF3 dimerization and accumulation in the nucleus, where it can function with IRF7 and NF‐κB to initiate the transcription of type I interferons (IFNs) and proinflammatory cytokines (Wu & Chen, 2014). Type I IFNs can be secreted, which induces the expression of IFN‐stimulated genes against pathogen infection via IFNAR/JAK/STAT signaling in an autocrine or paracrine manner (Ivashkiv & Donlin, 2014).
Recent studies demonstrated that cytosolic nucleic acid‐sensing pathways not only mediate protective immune defenses against pathogen infection but also potentiate efficient antitumor immune responses. Loss of antitumor T‐cell responses and tumor rejection were observed in STING−/− or IRF3−/− mouse models burdened with transplanted tumors (Woo et al, 2014). Similarly, the antitumor effect of radiation was greatly attenuated in IFNAR−/− mice or in the absence of a host STING pathway (Deng et al, 2014). These results strongly suggest that the cytosolic DNA‐sensing pathway in the host (mouse) plays a very important role in antitumor immunity.
Meanwhile, several studies proposed that inhibiting chromatin regulators, such as DNA methyltransferase or lysine‐specific histone demethylase 1 (LSD1), can trigger cytosolic sensing of double‐strand RNA (dsRNA) and induce type I IFN response in cancer cells. This in turn stimulates antitumor T‐cell immunity and restrains tumor growth, especially when combined with immune checkpoint blockade (Chiappinelli et al, 2015; Roulois et al, 2015; Sheng et al, 2018). These data indicate that the intrinsic RNA‐sensing pathway in tumor cells is also crucial for antitumor immunity in some circumstances. Type I IFNs induced by IR are well documented, and type I IFN‐dependent innate and adaptive immunity is essential for the efficacy of radiotherapy (Burnette et al, 2011). However, it remains unclear how radiation induces cell‐intrinsic type I IFN signaling and which cytosolic nucleic acid‐sensing pathways are involved in this process. In the current study, we sought to address this gap in knowledge by examining the pathways involved in IR‐induced type I IFN signaling to determine how best to maximize the efficacy of radiotherapy.
Results
ATR inhibition significantly potentiates IR‐induced inflammatory signals
To achieve a comprehensive understanding of cellular responses following ATRi therapy in combination with radiotherapy, we performed total RNA sequencing (RNA‐seq) using MCF10A cells treated with DMSO, IR, the ATRi AZD6738, or AZD6738 + IR. Hierarchical clustering analysis of mRNA data separated them into three clusters based on 3,463 differentially expressed genes (Fig 1A). Compared with DMSO, treatment with AZD6738 alone did not lead to any significant change in mRNA expression (Fig 1A). However, cluster 1, comprising the commonly upregulated genes in the IR and AZD6738 + IR groups, was mainly enriched in pathways including antimicrobial peptides, metal sequestration by antimicrobial proteins, and the innate immune system (Fig 1A and B). Cluster 3, comprising the commonly downregulated genes shared by the IR and AZD6738 + IR groups, was enriched in pathways involved in the cell cycle and M phase (Fig 1A and B). These data are consistent with the well‐known function of ATR in DNA damage‐induced G2/M checkpoint control. However, cluster 2, comprising the genes slightly upregulated in the IR group and possibly further amplified in the AZD6738 + IR group, was mainly enriched in the IFN and cytokine signaling pathways (Fig 1A and B), suggesting that ATR inhibition greatly enhances IFN response when combined with IR.
Figure 1. ATR inhibition significantly potentiates ionizing radiation (IR)‐induced inflammatory signals.

- Hierarchical clustering analysis of RNA sequencing data from cells treated with DMSO, 20 Gy IR, 250 nM AZD6738, or AZD6738 + IR (n = 2 biological replicates).
- Top enriched pathway analysis of differentially expressed genes in each cluster as shown in A.
- Heat map of 33 representative inflammatory genes regulated by AZD6738 + IR.
- MCF10A WT cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with 20 Gy, cells were harvested at indicated time points, and immunoblotting was performed with indicated antibodies. h, hours; pSTAT1, pSTAT1(Y701); pIRF3, pIRF3(S396); pTBK1, pTBK1(S172).
- MCF10A WT cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with indicated dose. 72 h later, cells were harvested for immunoblotting with indicated antibodies.
- Same condition as E except the irradiation dose with 20 Gy, mRNA levels of inflammatory genes as indicated were assessed by real‐time quantitative PCR, presented as mean ± standard error of the mean of three biological replicates.
We next examined the effect of AZD6738 on IR‐induced inflammatory signaling. As shown in Fig 1C, treatment with AZD6738 significantly enhanced the expression of all 33 classic inflammatory genes. Using STAT1 phosphorylation at Y701 (pSTAT1) as a surrogate marker for type I IFN signaling (Harding et al, 2017), we confirmed that AZD6738 + IR induced STAT1 activation in a time‐ and IR dose‐dependent manner (Fig 1D and E). STAT1 activation following IR alone or AZD6738 + IR normally happened after 2 days and when the IR dose was higher than 2 Gy. This activation was not only shown by pSTAT1 level but also shown by STAT1 protein level (Fig 1D and E), which is consistent with a previous study showing that prolonged IFN signal stimulation could greatly increase the protein expression of STAT1 in certain cell types (Cheon et al, 2013). The addition of type I IFN‐neutralizing antibody remarkably diminished pSTAT1 signals induced by AZD6738 + IR (Appendix Fig S1A), suggesting that pSTAT1 is a reliable readout for type I IFN signaling. Please note that STAT1 protein level positively correlates with pSTAT1 level, especially under prolonged treatment conditions, which is the consequence of a positive feedback loop resulting in STAT1 upregulation. In addition, VE‐822, another highly specific ATRi, also significantly enhanced IR‐induced STAT1 activation (Appendix Fig S1B). Moreover, inhibition of CHK1, the direct downstream target of ATR, via its specific inhibitors LY2603618 or AZD7762, also potentiated IR‐induced STAT1 activation (Appendix Fig S1C and D). Real‐time PCR results showed that the mRNA levels of multiple inflammatory genes, such as IFNB1, ISG54, CCL5, and IL6, were robustly induced by AZD6738 + IR compared with IR alone (Fig 1F), which is consistent with the RNA‐seq data (Fig 1C). Moreover, this synergistic effect was observed not only in MCF10A cells but also in another human mammary epithelial cell line (HMLE), breast cancer cell line T‐47D, and colon cancer cell line HCT 116 (Appendix Fig S1E–H). Taken together, these data indicate that inhibition of the ATR‐CHK1 axis robustly synergizes IR‐induced type I IFN signaling.
G2/M DNA damage checkpoint abrogation is required for IFN signaling induced by ATRi + IR
Cell cycle progression through mitosis has been proposed as essential step in IR‐induced type I IFN signaling (Harding et al, 2017). To examine the role of cell cycle progression in type I IFN signaling induced by AZD6738 + IR, we treated cells with the CDK1 inhibitor RO‐3306 or the CDK4/6 inhibitor PD‐0332991, which can induce cell cycle arrest in the late G2 phase or G1 phase, respectively (Appendix Fig S2). Both inhibitors significantly impaired STAT1 activation induced by AZD6738 + IR (Fig 2A and B), indicating that active cell cycle progression is required for this process.
Figure 2. G2/M DNA damage checkpoint abrogation is required for interferon signaling induced by ATR inhibition + ionizing radiation (IR).
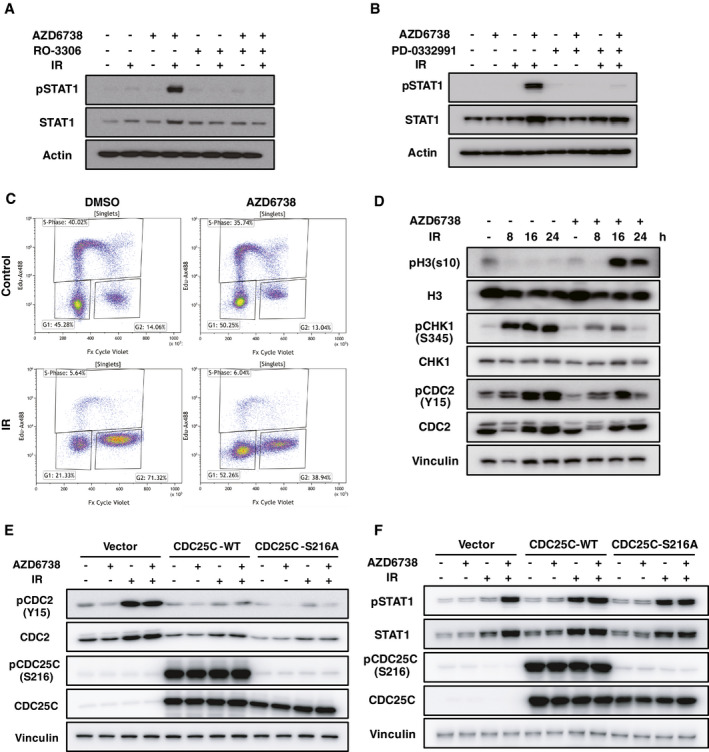
- MCF10A WT cells were pretreated with AZD6738 (250 nM) and RO‐3306 (9 μM) for 2 h, and then irradiated with 20 Gy; 3 days later, cells were harvested for immunoblotting with indicated antibodies.
- Same condition as described in A, except that RO‐3306 is replaced by PD‐0332991 (1 μM).
- MCF10A WT cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with 20 Gy; 24 h later, cell cycle distribution was measured by flow cytometry. EdU (10 μM) was added into medium 2 h before harvesting for flow cytometry.
- MCF10A WT cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with 20 Gy, and cells were harvested at indicated time points for immunoblotting.
- MCF10A control cells and CDC25C‐WT/constitutively active mutant CDC25C‐S216A overexpressed cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with 20 Gy; 24 h later, cells were harvested for immunoblotting with indicated antibodies.
- Same condition as indicated in E, except that cells were harvested 72 h later.
The ATR/CHK1/CDC25C‐dependent G2/M DNA damage checkpoint is known to often become activated after DNA damage to block the cell cycle at the G2 phase, allowing cells to repair damaged DNA (Liu et al, 2000). Abrogation of G2/M DNA damage checkpoint by AZD6738 allowed the cells to bypass IR‐induced G2 arrest, pushing more cells into mitosis and the following G1 phase, as shown by FACS and immunoblotting probed with antibodies of phos‐H3 (S10) and phos‐CDC2 (Y15) (Fig 2C and D), which are mitosis markers. Consistently, overexpression of wild‐type (WT) CDC25C or constitutively active mutant CDC25C‐S216A (Peng et al, 1997) maintained CDK1 activity even after exposure to IR (Fig 2E). As a result, overexpression of these proteins also significantly enhanced IR‐induced STAT1 activation, to a level similar to that of treatment with AZD6738 (Fig 2F). These data indicate that abrogation of the ATR/CHK1/CDC25C‐dependent G2/M DNA damage checkpoint is required for type I IFN signaling induced by ATRi + IR.
The cGAS/STING‐dependent cytosolic dsDNA‐sensing pathway is dispensable for type I IFN signaling induced by ATRi + IR in MCF10A cells
Previous studies showed that micronuclei formation and micronuclei disruption are well correlated with DNA damage‐induced STAT1 activation (Harding et al, 2017; Mackenzie et al, 2017). Those studies proposed that the dsDNA sensor cGAS could enter micronuclei after the rupture of micronuclei envelopes, sense dsDNA, and then initiate inflammatory signaling through the production of second messenger cGAMP to activate STING and downstream events (Harding et al, 2017; Mackenzie et al, 2017). We therefore generated cGAS and STING knockout (KO) clones in MCF10A cells. As anticipated, cGAMP stimulation could induce STING and IRF3 phosphorylation and STAT1 activation in WT or cGAS KO cells, but not in STING KO cells (Fig 3A). Furthermore, cGAS or STING KO could block herring testis DNA (HT‐DNA)‐induced IRF3 phosphorylation and STAT1 activation (Fig 3B). These data demonstrated that the cGAS/STING pathway is functional in MCF10A cells and is abolished in our KO cells.
Figure 3. The cGAS/STING‐dependent cytosolic dsDNA‐sensing pathway is dispensable for interferon (IFN) signaling induced by ATR inhibition + ionizing radiation (IR).

-
AMCF10A wild‐type (WT), cGAS knockout (KO), and STING KO cells were treated with cGAMP at indicated concentrations for 4 h, and cells were harvested for immunoblotting.
-
BMCF10A WT, cGAS KO, and STING KO cells were transfected with herring testis DNA (HT‐DNA, 2 μg/ml) for 6 h with Lipofectamine 3000. Immunoblotting was performed with indicated antibodies.
-
C–F(C, D, and F) MCF10A WT or two independent clones for cGAS, STING, and IRF3 KO cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with 20 Gy; 3 days later, cells were harvested for immunoblotting with indicated antibodies. cGAS, STING, and IRF3 KO were verified by immunoblotting and Sanger DNA sequencing (Appendix Fig S7). (E) Same condition as described in C, except that cells were harvested for real‐time quantitative PCR. IFNB1 mRNA level was measured and statistical analyzed by two‐way ANOVA (*** means P < 0.001). Data are presented as mean ± standard error of the mean of three biological replicates.
However, cGAS or STING KO showed only modest or undetectable effects on STAT1 activation induced by IR alone or ATRi + IR (Fig 3C and D, Appendix Fig S3A). Consistently, IFN‐β induction was statistically significantly, but very slightly decreased in cGAS or STING KO cells upon treatment with ATRi + IR (Fig 3E). In addition, knockdown of STING did not interfere with IR‐induced STAT1 activation in HCT 116 cells (Appendix Fig S3B), which are already defective in cGAS expression and IFN stimulatory DNA (ISD) sensing (Appendix Fig S3C and D). As mentioned above, ATRi significantly enhanced IR‐induced STAT1 activation in T‐47D cells, which are reported to be defective in both cGAS expression and STING expression (Zierhut et al, 2019). Furthermore, overexpression of the DNA exonuclease TREX1, which degrades cytosolic dsDNA (Grieves et al, 2015; Vanpouille‐Box et al, 2017), could not abolish STAT1 activation induced by ATRi + IR (Appendix Fig S3E). Please note that one needs to be careful when interpreting results from these overexpression experiments, since it is possible that overexpressed TREX1 may not have access and/or ability to degrade all types of DNA fragments.
Taken together, these data strongly suggest that the cGAS/STING‐dependent cytosolic dsDNA‐sensing pathway plays a very minor role in type I IFN signaling induced by IR alone or ATRi + IR in some human cells, e.g., MCF10A, HCT 116, and T‐47D cells. However, IRF3 KO blocked IFN‐β expression and STAT1 activation induced by ATRi + IR (Fig 3E and F), indicating that other pathways, and not the cGAS/STING pathway, are mainly responsible for type I IFN signaling induced by ATRi + IR, which is mediated by IRF3 activation.
MAVS‐dependent cytosolic RNA‐sensing pathway is indispensable for IFN signaling induced by ATRi + IR in MCF10A cells
As mentioned above, IRF3 activation is crucial for ATRi plus IR‐induced type I IFN signaling. Apart from the cGAS/STING‐dependent cytosolic dsDNA‐sensing pathway, the RIG‐I/MDA5/MAVS‐dependent cytosolic RNA‐sensing pathway is also important for IRF3 activation, upon sensing of cytosolic immunogenic RNAs by RIG‐I or MDA5 (Wu & Chen, 2014). To test whether the cytosolic RNA‐sensing pathway is involved in STAT1 activation induced by ATRi + IR, we generated KO cells for MAVS and its upstream RNA sensors RIG‐I and MDA5 and verified the KO using RNA Sendai virus (SeV) infection. Consistent with previous studies (Yoneyama et al, 2004; Desai et al, 2005; Kawai et al, 2005; Meylan et al, 2005; Seth et al, 2005; Xu et al, 2005), SeV‐induced IRF3 phosphorylation and STAT1 activation were significantly abrogated in RIG‐I KO, MAVS KO, and IRF3 KO cells, but not in cGAS KO, STING KO, or MDA5 KO cells (Appendix Fig S4A). MAVS KO nearly completely blocked STAT1 activation induced by IR alone or ATRi + IR (Fig 4A), as well as IFN‐β expression (Fig 4B). In addition, IRF3 activation, IFN‐β induction, and STAT1 activation were also dramatically suppressed in RIG‐I or MDA5 KO cells, but not in unrelated CDK5 KO cells (Fig 4B–E, Appendix Fig S4B and C). Meanwhile, TBK1 activation or RIG‐I upregulation induced by ATRi + IR was also abrogated in MDA5 KO cells (Fig 4D). However, MDA5 is generally believed to be activated by long dsRNA, whereas RIG‐I is believed to be activated by short dsRNA and 5′‐ppp single‐strand RNA (Wu & Chen, 2014). Thus, it is hard to imagine such robust change in single MDA5 or RIG‐I KO cells. It is possible but unlikely that one type of specific RNA needs both MDA5 and RIG‐I at the same time to initiate type I IFN signaling. Given that MDA5 and RIG‐I are well known IFN‐stimulated genes (ISGs), they are significantly induced after ATRi plus IR treatment (Fig 4C and D). We speculate that MDA5/RIG‐I upregulation could further amplify type I IFN signaling elicited by ATRi + IR, which may be the reason that even single KO of MDA5 or RIG‐I could show robust change in type I IFN signaling. To further explore this positive feedback regulation on type I IFN signaling elicited by ATRi + IR, we then knocked out another transcription factor IRF7 (a classical ISG), which function with IRF3 to modulate type I IFN expression. As anticipated, STAT1 activation was significantly diminished in IRF7 KO cells (Fig 4F). However, attenuated STAT1 activation and MDA5 induction were not as evident in TBK1 KO cells as in MAVS KO cells (Appendix Fig S4D), potentially owing to redundant functions between TBK1 and IKKɛ (Wu & Chen, 2014). In addition, overexpression of dsRNA‐specific endoribonuclease RNase III derived from E. coli drastically impaired STAT1 activation induced by ATRi + IR (Appendix Fig S4E). Consistently, knockdown of MAVS or RIG‐I also impaired IR‐induced STAT1 activation in HCT 116 cells (Appendix Fig S3B).
Figure 4. The MAVS‐dependent cytosolic RNA‐sensing pathway is indispensable for interferon (IFN) signaling induced by ATR inhibition + ionizing radiation (IR).
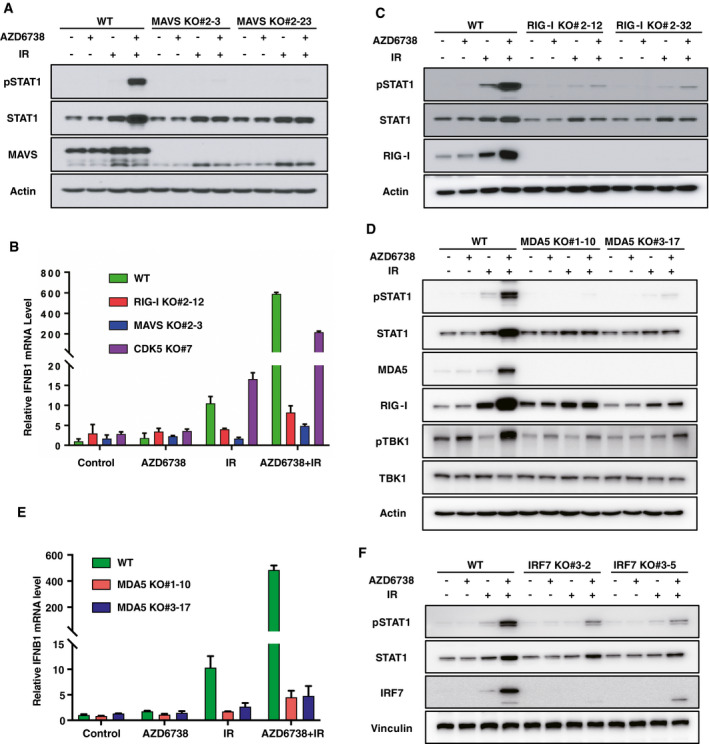
-
A–F(A, C, D, and F) MCF10A WT or two independent clones for MAVS, RIG‐I, MDA5, and IRF7 KO cells were pretreated with DMSO or AZD6738 (250 nM) for 2 h, and then irradiated with 20 Gy; 3 days later, cells were harvested for immunoblotting with indicated antibodies. MAVS, RIG‐I, MDA5, and IRF7 KO were verified by immunoblotting and Sanger DNA sequencing (Appendix Fig S7). (B and E) Same condition as described in A, except that cells were harvested for real‐time quantitative PCR instead of immunoblotting. IFNB1 mRNA level was measured by real‐time quantitative PCR. Data are presented as mean ± standard error of the mean of three biological replicates.
Source data are available online for this figure.
Taken together, these data indicate that the cytosolic RNA‐sensing pathway and positive feedback loop are required for optimized type I IFN signaling induced by ATRi + IR at least in MCF10A and HCT 116 cells.
Cytosolic nucleic acid‐sensing pathways involved in IR‐induced type I IFN signaling differ between cell lines
We sought to study the efficacy of ATRi + IR in tumor control in a syngeneic mouse tumor model. To this end, we chose a murine mammary carcinoma cell line, 4T1, and checked the synergistic effect of ATRi on IR‐induced IFN signaling in these cells. As shown in Fig 5A, ATRi significantly enhanced IR‐induced IFN‐β expression in a dose‐dependent manner.
Figure 5. Cytosolic nucleic acid‐sensing pathways involved in ionizing radiation (IR)‐induced type I interferon (IFN) signaling differ in murine and human cells.
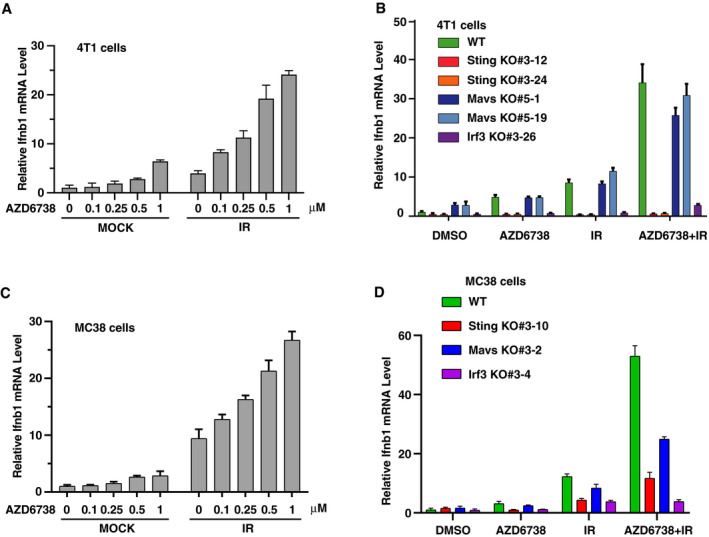
- 4T1 cells were pretreated with AZD6738 at indicated concentrations for 2 h, and then irradiated with 10 Gy. 4 days later, Ifnb1 mRNA level was measured by real‐time quantitative PCR. Data are presented as mean ± standard error of the mean of three biological replicates.
- Same condition as described in A. Ifnb1 mRNA levels were measured by real‐time quantitative PCR in WT, Sting KO, Mavs KO, and Irf3 KO 4T1 cells. Data are presented as mean ± standard error of the mean of three biological replicates. All KO cells were verified by immunoblotting and Sanger DNA sequencing (Appendix Fig S7).
- MC38 cells were pretreated with AZD6738 at indicated concentrations for 2 h, and then irradiated with 20 Gy. 3 days later, Ifnb1 mRNA level was measured by real‐time quantitative PCR. Data are presented as mean ± standard error of the mean of three biological replicates.
- Same condition as described in C. Ifnb1 mRNA levels were measured by real‐time quantitative PCR in WT, Sting KO, Mavs KO, and Irf3 KO MC38 cells. Data are presented as mean ± standard error of the mean of three biological replicates. All KO cells were verified by immunoblotting and Sanger DNA sequencing (Appendix Fig S7).
We then further generated Sting, Mavs, and Irf3 KO in 4T1 cells and verified the KO with SeV infection, HT‐DNA transfection, and murine STING agonist DMXAA treatment. SeV‐induced IRF3 phosphorylation was significantly abrogated in Mavs KO cells, but not in Sting KO cells, while HT‐DNA‐induced STING and IRF3 phosphorylation were abrogated in Sting KO cells, but not in Mavs KO cells (Appendix Fig S5A). Similarly, DMXAA induced mobility shift of STING and IRF3 phosphorylation in WT and Mavs KO cells, but not in Sting KO cells (Appendix Fig S5B). Irf3 KO was included as a control for all these experiments. However, ATRi + IR‐induced IFN‐β expression was completely diminished in Sting and Irf3 KO cells, but not in Mavs KO cells (Fig 5B). In another murine skin melanoma cell line B16‐F10, Sting KO, Mavs KO, and Irf3 KO were also generated and verified with SeV infection and HT‐DNA transfection. As anticipated, SeV‐induced IRF3 phosphorylation and STAT1 phosphorylation were significantly abrogated in Mavs KO cells, but not in Sting KO cells, while HT‐DNA‐induced IRF3 phosphorylation and STAT1 phosphorylation were abrogated in Sting KO cells, but not in Mavs KO cells (Appendix Fig S5C). Similar to 4T1 cells, ATRi + IR‐induced IFN‐β expression was significantly diminished in Sting and Irf3 KO cells, but not in Mavs KO cells (Appendix Fig S5D), although ATRi did not show a strong synergistic effect on IR‐induced IFN‐β expression (Appendix Fig S5D). In addition, we examined another widely used colon adenocarcinoma cell line, MC38. Sting KO, Mavs KO, and Irf3 KO were also generated in this cell line and verified with SeV infection and HT‐DNA transfection (Appendix Fig S5E). Similar to 4T1 cells, ATRi significantly enhanced IR‐induced IFN‐β expression in a dose‐dependent manner (Fig 5C), and IR alone or ATRi plus IR‐induced STAT1 activation was significantly compromised in Sting KO cells (Fig 5D and Appendix Fig S5F). In Mavs KO MC38 cells, IR alone or ATRi plus IR‐induced STAT1 activation was also impaired, although to a lesser extent than that in Sting KO MC38 cells (Fig 5D and Appendix Fig S5F).
These results are clearly different from those of the studies we conducted in several human cells as outlined above, which led us to question whether the difference is a genuine mouse–human difference (i.e., species difference) or a variation between cancer cells. To address this question, we further examined ten more human cancer cell lines, including four lung cancer cell lines (H358, H441, H1299, and H1355), five breast cancer cell lines (Hs 578T, MDA‐MB‐231, MDA‐MB‐468, MDA‐MB‐436, and HCC1937), one pancreatic cancer cell line PANC‐1, and one more human monocytic cell line THP‐1. We determined the expression of all core components involved in cytosolic DNA/RNA‐sensing pathway in these cell lines (Appendix Fig S5G). In comparison with other cell lines, MDA‐MB‐468 cells have extremely low cGAS expression, but these cells were reported to be cGAS‐positive (Lohard et al, 2020). We then further tested the synergistic effect of ATRi on IR‐induced inflammatory signals, with pSTAT1 level or IFNB1/CCL5 expression as readouts. Three of them, MDA‐MB‐231, H1355, and H441, showed no response to IR alone or ATRi plus IR (Appendix Fig S5H–J). In MDA‐MB‐436 cells, ATRi could not further enhance IR‐induced STAT1 activation and IFN‐β/CCL5 expression (Appendix Fig S5K and L). In the remaining seven cell lines (i.e., H358, H1299, Hs 578T, MDA‐MB‐468, HCC1937, PANC‐1, and THP‐1), ATRi enhanced IR‐induced STAT1 activation or IFN‐β/CCL5 expression to some extent (Appendix Fig S5M and S). By knocking down STING/MAVS/IRF3 or even cGAS/RIG‐I/MDA5 with highly efficient lenti‐CRISPR viruses, we demonstrated that MAVS is indispensable for ATRi plus IR‐induced STAT1 activation in MDA‐MB‐468 and PANC‐1 cells (Appendix Fig S5T and U). However, STING is indispensable for ATRi plus IR‐induced STAT1 activation in H358, H1299, and THP‐1 cells (Appendix Fig S5V and X). Furthermore, in Hs 578T, HCC1937, and MDA‐MB‐436 cells, both STING‐dependent dsDNA‐sensing pathway and MAVS‐dependent RNA‐sensing pathway contributed to IR alone or ATRi plus IR‐induced type I IFN signaling (Appendix Fig S5Y and AA), though the dependency on MAVS is more dominant than the dependency on STING in Hs 578T and MDA‐MB‐436 cells, but not in HCC1937 cells (Appendix Fig S5Y and AA).
These data led us to hypothesize that both MAVS‐ and STING‐dependent pathways are involved in IR and IR+ATRi‐induced innate immune response. However, the specific contributions of these two pathways in different cell lines may depend on their tissue origin, genetic background, and others. To provide a unified working hypothesis, we reason that DNA damage, induced by IR, could release some DNA fragments (immunogenic or not) that may be transcribed into immunogenic RNAs in different cell lines. The unknown features of these released DNA fragments, their transcription efficiency, and immunogenicity may determine the requirement of STING‐ and/or MAVS‐dependent signaling pathways for type I IFN induction.
DNA damage‐elicited AT‐rich DNA may mediates type I IFN signaling in a subset of human cells
The inconsistency among human cells and between human and murine cells led us to consider previous publications on poly(dA‐dT), which has been reported to mediate type I IFN induction in human and murine cells via divergent cytosolic nucleic acid‐sensing pathways (Ablasser et al, 2009; Chiu et al, 2009). This divergency is very similar to what we observed following IR and/or ATRi + IR treatment. We thus examined STAT1 activation induced by poly(dA‐dT) in MCF10A, 4T1, and MC38 cells. Similar to IR, poly(dA‐dT)‐induced STAT1 activation also relied on MAVS, but not cGAS or STING, in MCF10A cells (Fig 6A). In contrast, in 4T1 and MC38 cells, poly(dA‐dT)‐induced IRF3 phosphorylation and STAT1 activation were largely diminished in Sting KO but not in Mavs KO cells (Fig 6B and Appendix Fig S6A), and we included Irf3 KO as a positive control in these experiments. These data further supported the possibility that DNA damage could release some DNA fragments with similar composition as poly(dA‐dT), which may be accessed and transcribed to RNAs by RNA polymerase III and then initiate MAVS‐dependent type I IFN signaling in some human cells, as previously reported (Ablasser et al, 2009; Chiu et al, 2009).
Figure 6. DNA damage‐elicited AT‐rich DNA mediates type I interferon (IFN) signaling in human cells.
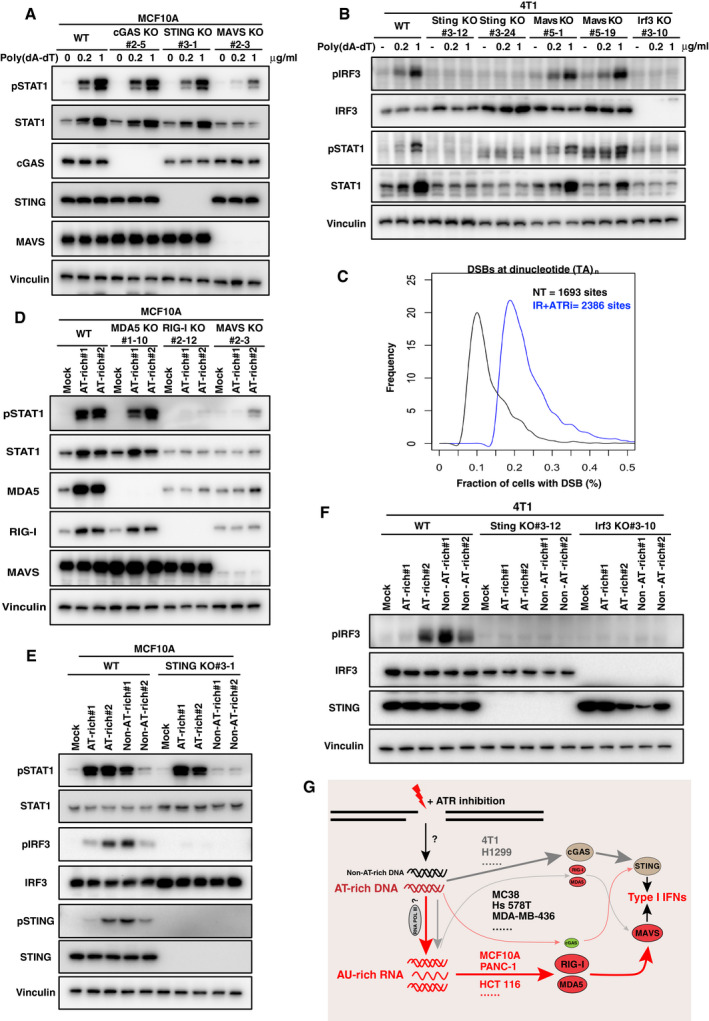
- MCF10A WT, cGAS KO, STING KO, and MAVS KO cells were treated with poly(dA‐dT)/LyoVec at indicated concentrations for 24 h. Immunoblotting was performed with indicated antibodies.
- 4T1 WT, Sting KO, Mavs KO, and Irf3 KO cells were treated with poly(dA‐dT)/LyoVec at indicated concentrations for 24 h. Immunoblotting was performed with indicated antibodies.
- END‐seq was used to map double‐strand breaks (DSBs) associated with TA dinucleotide repeats in MCF10A cells with the indicated treatments. ATR inhibitor (ATRi) AZD6738 (250 nM) was used with ionizing radiation (IR, 10 Gy) for 6 h. NT, not treated.
- MCF10A WT, MDA5 KO, RIG‐I KO, and MAVS KO cells were transfected with AT‐rich oligos (2 μg/ml) with LyoVec for 24 h. Immunoblotting was performed with indicated antibodies.
- MCF10A WT and STING KO cells were transfected with AT‐rich and non‐AT‐rich oligos (2 μg/ml) with lipofectamine 3000 for 4 h. Immunoblotting was performed with indicated antibodies.
- 4T1 WT, Sting KO, and Irf3 KO cells were transfected with AT‐rich and non‐AT‐rich oligos (2 μg/ml) with LyoVec for 24 h. Immunoblotting was performed with indicated antibodies.
- A proposed potential mechanism for the divergent cytosolic nucleic acid‐sensing pathway activated in response to radiation alone or with ATRi among different cell lines.
To test this model, we performed genome‐wide DNA double‐strand break (DSB) mapping in MCF10A cells using the END‐seq technique (Canela et al, 2016). As anticipated, END‐seq detected more frequent DSBs in cells treated with IR + ATRi compared with non‐treated cells (Appendix Fig S6B). Moreover, breakage associated with TA dinucleotide repeats (n > 25) was more common in cells treated with IR + ATRi than in non‐treated cells (Fig 6C). Consistently, a previous study demonstrated that structure‐forming repetitive DNA sequences are preferred sites for replication fork collapse caused by ATR inhibition in both murine and human cells; however, the prevailing AT‐rich feature of repeat sequences in several human cells was not identified in murine cells (Shastri et al, 2018), indicating that there may be different preferences in human versus murine cells.
We then synthesized AT‐rich and non‐AT‐rich oligonucleotides (oligos) from recurrent break sites identified in human and murine cells (Shastri et al, 2018), respectively, and tested the effect of these oligos on STAT1 activation. In MCF10A cells, transfection of AT‐rich oligos‐induced STAT1 activation was significantly diminished in RIG‐I KO and MAVS KO cells (Fig 6D), but not in STING KO cells (Fig 6E). Conversely, STING, but not MAVS, was required for IRF3 activation upon transfection with AT‐rich or non‐AT‐rich oligos in 4T1 cells (Fig 6F and Appendix Fig S6C). At the same time, STING was also important for both AT‐rich and non‐AT‐rich oligos‐induced IRF3 phosphorylation, but it is only indispensable for non‐AT‐rich oligos‐induced STAT1 activation in MCF10A cells (Fig 6E).
Furthermore, we used the RNA polymerase III‐specific inhibitor ML‐60218 (Wu et al, 2003) to inhibit RNA polymerase III activity. As anticipated, treatment with ML‐60218 significantly suppressed STAT1 activation induced by ATRi + IR (Appendix Fig S6D), supporting our working model (Fig 6G). Collectively, our data provide a potential mechanism for type I IFN signaling induced by IR alone or ATRi + IR, which may explain the preferential choice of STING‐ or MAVS‐dependent signaling in different cell lines.
Discussion
Radiotherapy, as a common treatment modality for cancer, has been used for nearly a century. Increasing evidence indicates that the efficacy of radiotherapy largely relies on type I IFN‐dependent innate and adaptive immunity, but the mechanisms of this process remain unclear. In the current study, we demonstrated that ATR inhibition significantly enhances IR‐induced type I IFN signaling in many human and murine cells. Using a series of CRISPR‐mediated isogenic KO cells, our data clearly demonstrated that besides the cGAS/STING pathway, whose frequent deficiency in a variety of cancer cell lines (Xia et al, 2016a,b; Chen et al, 2017; de Queiroz et al, 2019) indicates potentially important roles in immunosurveillance escape, the MAVS‐dependent cytosolic RNA‐sensing pathway is also crucial for type I IFN signaling induced by IR alone or ATRi + IR in some human cells.
Given that IR‐induced type I IFN showed a very similar pattern to that of poly(dA‐dT) and genome‐wide DSB mapping data upon ATR inhibition, we propose that DNA damage could release AT‐rich DNAs, which are prevalent in some human cells but may not be in other cells, such as some murine cells (Shastri et al, 2018). In murine cells, the cGAS/STING pathway may be dominant in response to DNA damage, whether the released DNA is AT‐rich or not. In contrast, in some human cells, AT‐rich DNA can be converted to immunogenic RNA via RNA polymerase III‐mediated transcription and then initiate type I IFN signaling in a MAVS‐dependent manner (Fig 6G). More specifically, we hypothesize that fragmented nuclei, other than classical micronuclei, would be produced due to mitotic catastrophe at days after high dose of radiation, which could be greatly enhanced by ATR inhibitors, because of the bypass of DNA damage‐induced G2/M arrest. Fragmented nuclear envelopes can easily rupture due to lamin B1 downregulation (Hatch et al, 2013) during this process, and as a result, immunogenic RNAs inside are accessible to cytosolic RNA sensors, which initiates type I IFN signaling (for more detailed discussion, see response to reviewers included in the Peer‐Review Process File available online with this article). Taken together, our findings suggest that ATR inhibition in combination with radiotherapy will be a promising strategy to modulate the host immune system for cancer treatment, especially with the addition of immune checkpoint blockade. Consistent with this idea, a recent study showed that AZD6738 potentiates CD8+ T‐cell‐dependent antitumor immunity following radiation, although with a different mechanism than we proposed (Vendetti et al, 2018).
Another previous study showed that poly(dA‐dT) can lead to the generation of IFN‐inducing RNA in human cells as well as in mouse cells (Chiu et al, 2009). However, it is still unclear why AT‐rich DNA sequences initiate type I IFN signaling in a MAVS‐dominant manner in some human cells and a STING‐dominant manner in some other cells, especially in murine cells. One possible explanation is that the conversion efficiency from DNA to RNA is low in these murine cells, and as a result, AT‐rich DNA cannot initiate type I IFN signaling in a MAVS‐dominant manner. Another possibility is that the process of AT‐rich DNA release is well controlled by some unknown mechanism, and genes involved may have differential expression levels or mutational status among different cell lines. Future extensive studies will be needed to determine whether any of these mechanisms can explain the differences among human cells and between human and mouse species. In addition, beyond ATR inhibitors studied here, inhibition of other DNA damage response factors, such as ATM or PARP1, has also been reported to trigger type I IFN response in a manner depending on the cGAS‐STING (ATM and PARP1) (Hartlova et al, 2015; Ding et al, 2018; Shen et al, 2019) or the RIG‐I/MAVS (PARP1) pathway (Ghosh et al, 2018). It should therefore be interesting to further explore the potential divergences among cancer cell lines and the underlying mechanism in response to different types of DNA damage. Such follow‐up work should give us a better understanding about DNA damage‐elicited interferon responses and be beneficial for radiotherapy/chemotherapy‐based cancer treatments.
MDA5 and RIG‐I, as important RNA sensors, have been implicated in the protective defense of viral infection. Aberrant activation of MDA5 could cause autoimmune and autoinflammatory diseases such as systemic lupus erythematosus and Aicardi–Goutières syndrome (Crow & Manel, 2015; Barrat et al, 2016). In addition, recent studies demonstrated that cancer treatment with inhibitors of DNA methylation or inhibitors of LSD1 results in reduced tumor growth, and this also relies on MDA5/RIG‐I/MAVS‐dependent IFN signaling (Chiappinelli et al, 2015; Roulois et al, 2015; Sheng et al, 2018). Our results suggest that MDA5 or RIG‐I expression is crucial for radiation‐induced type I IFNs, suggesting the need for systematic investigation of the relationship between genotype or expression of MDA5/RIG‐I and the response of patients to radiotherapy with or without additional agents, such as ATRi and/or immune checkpoint blockade therapies. Understanding the complexity of these responses may help us better utilize these combinatory therapies for cancer patients.
Materials and Methods
Cell culture
MCF10A cells were from American Type Culture Collection (ATCC) and were cultured in DMEM/F‐12 medium with 5% horse serum (Thermo Fisher Scientific), 20 ng/ml human EGF (Fisher Scientific), 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml recombinant human insulin (Sigma). HMLE cells were generously provided by Dr. Li Ma (MD Anderson) and were maintained in the same medium as MCF10A cells except without cholera toxin. HCT 116 cells were obtained from ATCC and cultured in McCoy 5a medium with 10% fetal bovine serum. T‐47D was obtained from ATCC and cultured in RPMI 1640 medium with 10% fetal bovine serum. MDA‐MB‐231, MDA‐MB‐468, and Hs 578T were obtained from ATCC and cultured in DMEM medium with 10% fetal bovine serum. MDA‐MB‐436 was obtained from ATCC and cultured in DMEM medium with 10% fetal bovine serum, 10 μg/ml insulin, and 16 μg/ml glutathione. HCC 1937 was cultured in RPMI 1640 medium with 10% fetal bovine serum. H358, H441, H1299, and H1355 were generously provided by Dr. Steven H. Lin (MD Anderson) and were cultured in RPMI 1640 medium with 10% fetal bovine serum. PANC‐1 was generously provided by Dr. Pawel Mazur (MD Anderson) and was cultured in RPMI 1640 medium with 10% fetal bovine serum. Mouse tumors derived 4T1 and MC38 cells were also from Dr. Steven H. Lin and were cultured in DMEM with 10% fetal bovine serum. Mouse skin melanoma cell line B16‐F10 was obtained from ATCC and cultured in DMEM medium with 10% fetal bovine serum. The cell lines were tested to verify that they were free of mycoplasma contamination.
Immunoblotting
Cells were washed once with phosphate‐buffered saline (PBS) and then directly scraped in SDS sample buffer (50 mM Tris–HCl, pH 6.8, 2% SDS, 10% Glycerol, 4% β‐mercaptoethanol, 0.025% Bromophenol blue) and denatured by heating to 99°C for 10 min. Immunoblotting was performed in SDS–polyacrylamide gel according to the standard protocol.
Irradiation
One day before treatment, cells were seeded in 6‐well plates. Irradiation was performed using a Precision X‐RAD 320 Biological Irradiator. Inhibitors were added 2 h before irradiation with indicated dose and maintained until cells were harvested at 72 h for immunoblots or reverse transcription quantitative PCR, unless otherwise stated. ATRi (AZD6738, VE‐822), CHK1 inhibitors (LY2603618, AZD7762), CDK1 inhibitor (RO‐3306, 9 μM), and CDK4/6 inhibitor (PD‐0332991, 1 μM) were obtained from Selleck Chemicals. Inhibitor concentrations were indicated in corresponding figures. For AZD6738, which was used in most experiments, the working concentration was 250 nM, unless otherwise stated. The RNA polymerase III inhibitor ML‐60218 was from Merck Millipore, and it was added into medium 2 days following irradiation and maintained for 4 or 8 h before cells were harvested for immunoblots. Human type I IFN neutralizing antibody mixture was from PBL Assay Science, and it was added into medium at the indicated dilution 2 days following irradiation and maintained for another 24 h before cells were harvested for immunoblots.
RNA extraction and reverse transcriptase quantitative PCR
WT or different KO cells with indicated treatments were washed with cold PBS, and total RNA was isolated using TRIzol reagent following the instructions of the manufacturer (Invitrogen). Next, 1 μg of RNA was reverse‐transcribed to cDNA using the High‐Capacity cDNA Reverse Transcription Kit according to the manufacturer's instructions (Thermo Fisher Scientific, 4368814). cDNA was then diluted and subjected to real‐time PCR with gene‐specific primers using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) and the 7500 real‐time PCR system (Applied Biosystems). The primer pairs used in this study were as follows: β‐actin, GCCGACAGGATGCAGAAGGAGATCA/AAGCATTTGCGGTGGACGATGGA; IFNB1, GCTTGGATTCCTACAAAGAAGCA/ATAGATGGTCAATGCGGCGTC; CCL5, CCAGCAGTCGTCTTTGTCAC/CTCTGGGTTGGCACACACTT; IFNA1, CTGAATGACTTGGAAGCCTG/ATTTCTGCTCTGACAACCTC; IL1A, TGTAAGCTATGGCCCACTCCA/AGAGACACAGATTGATCCATGCA; IL6, CACCGGGAACGAAAGAGAAG/TCATAGCTGGGCTCCTGGAG; ISG54, AAGCACCTCAAAGGGCAAAAC/TCGGCCCATGTGATAGTAGAC; β‐actin (mouse), CGCCACCAGTTCGCCATGGA/TACAGCCCGGGGAGCATCGT; Ifnb1 (mouse), TCAGAATGAGTGGTGGTTGC/GACCTTTCAAATGCAGTAGATTCA.
RNA‐seq and data analysis
MCF10A cells were treated with DMSO, AZD6738 (250 nM), IR (20 Gy), and AZD6738 + IR for 3 days (two biological replicates), and then, total RNA was extracted using the RNeasy Mini Kit (Qiagen, 74104) combined with RNase‐Free DNase (Qiagen, 79254) according to the manufacturer's instructions. The library was prepared with the Illumina TruSeq Stranded Total RNA Library Prep kit including rRNA depletion and sequencing at NextSeq 500 (Illumina) to generate 75 bp from paired‐ends.
For RNA‐seq data analysis, reads were adapter‐trimmed and preprocessed with the FastQC software for the purpose of quality control and data filtering. Genome mapping was carried out using hisat2 software (v2.0.4) and the human reference genome (UCSC hg38). The mapped reads were assembled into transcripts and genes with the human genome annotation file (genecode.v28.annotation.gtf) using StringTie software (v1.3.4d). The expression of the transcripts or genes was normalized as FPKM (fragments per kilobase of transcript per million mapped reads) for quantification. Transcripts with an FPKM score below 1 were removed, resulting in a total of 14,944 gene IDs. At 1% false discovery rate, one‐way analysis of variance, with four groups adjusted using the Benjamini–Hochberg procedure, identified 3,463 genes that were differentially expressed (downregulated or upregulated by at least 2‐fold in the three groups that were exposed to IR or the drug). Hierarchical clustering analysis separated these genes into three subgroups based on expression across four experimental groups. Pathway analysis of each subgroup was performed using the over‐representation test against KEGG or Reactome databases.
END‐seq and data analysis
MCF10A cells were treated with 10 Gy IR + ATRi (AZD6738, 250 nM) for 6 h, and then, END‐seq was used to map DSBs as previously described (Canela et al, 2016). Peak calling was performed against END‐seq reads from non‐treated cycling MCF10A cells.
CRISPR/Cas9‐mediated gene KO
gRNAs against targeting genes were designed and annealed into digested pX458 (used for cGAS/STING/IRF3 KO in MCF10A cells), LentiCRISPRv2 (used for MDA5/RIG‐I/MAVS/TBK1 KO in MCF10A cells), or modified all‐in‐one doxycycline‐inducible TLCV2 (used for IRF7 KO in MCF10A cells and Sting/Mavs/Irf3 KO in 4T1, B16‐F10, and MC38 cells) plasmids according to standard protocol. Cells were selected with FACS (pX458) or puromycin (LentiCRISPRv2, with 2 μg/ml doxycycline for TLCV2) for 3 days after transfection or lentivirus infection, and single clones were sorted and picked up for further verification by immunoblotting and Sanger DNA sequencing (Appendix Fig S7). Two independent clones for each gene deletion were used in all experiments. The gRNA sequences used in this study were as follows (m, mouse):
cGAS‐gRNA#2: AGACTCGGTGGGATCCATCG
STING‐gRNA#3: GCGGGCCGACCGCATTTGGG
STING‐gRNA#4: TACTCCCTCCCAAATGCGGT
IRF3‐gRNA#1: GCCGTAGGCCGTGCTTCCAA
IRF3‐gRNA#2: GCTGGACCTGGGGCAACTGG
MDA5‐gRNA#1: ATAGCGGAAATTCTCGTCTG
MDA5‐gRNA#3: TGGTTGGACTCGGGAATTCG
RIG‐I‐gRNA#2: GGATTATATCCGGAAGACCC
MAVS‐gRNA#2: AGTACTTCATTGCGGCACTG
TBK1‐gRNA#1: TTTGAACATCCACTGGACGA
TBK1‐gRNA#2: CATAAGCTTCCTTCGTCCAG
IRF7‐gRNA#3: CACTCTCCGAACAGCACGCG
CDK5‐gRNA: CGGCACACCCTGCATATGTG
mMavs‐gRNA#3: TTGGTGTCTCTCGTAAGCCA
mMavs‐gRNA#5: TTTGTCCTCAGGGCAGTACG
mSting‐gRNA#3: GTCCAAGTTCGTGCGAGGCT
mSting‐gRNA#4: AGCGGTGACCTCTGGGCCGT
mIrf3‐gRNA#3: ACGGGATCCTGAACCTCGTT
Poly(dA‐dT)/HT‐DNA transfection and SeV infection
Poly(dA‐dT)/LyoVec complex was purchased from InvivoGen and dissolved into 50 μg/ml, when used for cell treatment, it can be directly added into cells at indicated concentrations for 24 h unless otherwise stated. Herring testis DNA (HT‐DNA) was purchased from Sigma‐Aldrich, and the transfection of HT‐DNA into cells was performed with Lipofectamine 3000 following the standard protocol. Sendai virus was purchased from Charles River and used for cell infection at a titer of 100 HA units/ml for 4 h unless otherwise stated.
Chemicals
Murine STING ligand DMXAA was used to treat 4T1 WT or KO cells at 20 μg/ml for 1.5 h. Treatment with cGAMP was performed as described previously (Hu et al, 2016). Briefly, MCF10A cells were treated with indicated concentrations of cGAMP in digitonin permeabilization solution (50 mM HEPES, pH 7.0, 100 mM KCl, 3 mM MgCl2, 0.1 mM DTT, 85 mM sucrose, 0.2% BSA, 1 mM ATP, 0.1 mM GTP, and 10 μg/ml digitonin) at 37°C for 10 min and then changed to regular medium for 4 h before the cells were harvested for immunoblotting.
DNA oligonucleotides
The following oligonucleotides were designed based on previous study (Shastri et al, 2018) and used to stimulate cells at a concentration of 2 μg/ml with the transfection reagent LyoVec or Lipofectimine 3000 following the standard protocol:
AT‐rich#1 (from human Chr1: 101718640 ‐101721295):
ATATATATAAATATATATAATATCTATAAATATATATAAATATAAATATATATAATATCTATAAATATAGATAAATATAAATATATATAATATCTATAAATATAGATAAATATAAATATATATAACTATATATAAATATATATAACTATATATAAATATATATATAAATATATATAACTATATATATAACTATATATATA
AT‐rich#2 (from human Chr9: 44235060‐44235559):
TATATATGTATATATATGTATATATATGTATATACATATATATGTATATATATGTATATATATGTGTATATATACATATATATGTATATATATGTATATATATATGTATATATATGTATATATATACATATATATGTATATATATGTATATATATACATATATATGTATATATATGTATATATATATGTATGTATATATA
Non‐AT‐rich#1 (from mouse Chr4: 141468470‐141468919):
CAAAGTGAGTTCCAGGACAGTCAGGGCTACACAGAGAAACCCTGTCTCGGAAAAAAAAAAAAAAAAAAAAAAAAAAAACTCAAAAAAGAAAAAAAAAAAAAGATTGGTCTGTGGGGTTCCTTTTTTTGTTTTTTCGAGACAGTGTTTCTCTGTGTAGCCCTGGCTGTCCTGGAACTCACTCTGTAGACCAGGCTGGCCTC
Non‐AT‐rich#2 (from mouse Chr17: 28870500‐28870949):
GCCCTCTTCTGGTGTGTCTGAAGACAGCTACAGTGTACTTACATATAATAATAAATAAATAAATCTTTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAATTTGGTATTATCATCTTACTTGAAATACATTTTTTTTAAGATTTATTTATTAATATATACACTGTAGCTGTCTTCAGACACTCTAGAAGAGGGAG
Plasmids and lentivirus infection
Plasmids encoding CDC25C, CDC25C‐S216A, Lamin B1, TREX1, and RNase III (rnc) were purchased from Addgene. PCR‐amplified products were subcloned into pDONR201 vector and then transferred to pInducer20 vector using the Gateway system. To generate inducible expressing genes mentioned above in MCF10A cells, lentiviruses carrying the genes listed above in pInducer20 vector or pInducer20 empty vector were produced in HEK293T cells using pMD2.g and psPAX2 as packaging plasmids. The virus supernatant was filtered through a 0.45‐μm filter and used to infect targeting cells in the presence of 10 μg/ml polybrene. Stable cell pools were selected with 500 μg/ml G418 (Gemini Bio‐Products) for 4 days. Doxycycline (2 μg/ml) was added before irradiation and maintained for 3 days unless otherwise noted.
Cell cycle analysis by flow cytometry
A total of 1.5 × 106 MCF10A cells were seeded into 100‐mm plates 1 day before treatment, treated with inhibitors (AZD6738, 250 nM; RO‐3306, 9 μM; PD‐0332991, 1 μM) 2 h before irradiation (20 Gy), and cultured for another 24 h. Next, 10 μM EdU (5‐ethynyl‐2′‐deoxyuridine) was added to the culture medium for 2 h before collection. After labeling, cells were fixed with 4% paraformaldehyde for 15 min, followed by permeabilization with 0.5% Triton X‐100 in PBS for 15 min at room temperature. Cells were rinsed once with PBS and incubated with mixture reaction buffer (100 mM sodium ascorbate, 4.8 μM Alexa Fluor 488 azide, and 4 mM CuSO4) for 30 min at room temperature. After staining, cells were washed three times with 0.1% Triton X‐100 in PBS, and FxCycle Violet (1 μg/ml) was used for cell nuclei. Flow cytometry was performed on the Gallios Flow Cytometer system and analyzed using FlowJo software.
Antibodies
Antibodies for pSTAT1‐Y701 (#9167), STAT1 (#14994), pCHK1‐S345 (#2348), CHK1 (#2360), pIRF3‐S396 (#4947), IRF3 (#4302), pTBK1‐S172 (#5483), TBK1 (#3504), MDA5 (#5321), RIG‐I (#3743), MAVS (#3993, for human), MAVS (#4983, for mouse), pH3‐S10 (#9701), Histone H3 (#4499), pCDC2‐Y15 (#4539), CDC2 (#9116), pCDC25C‐S216 (#4901), CDC25C (#4688), Lamin B1 (#13435), cGAS (#15102), STING (#13647), Phospho‐STING (#85735), and IRF7 (#4920) were purchased from Cell Signaling Technology. β‐actin (A5441), HA (H3663), and Vinculin (V9264) antibodies were obtained from Sigma‐Aldrich.
Statistical analysis
All data were plotted as mean with standard error of the mean unless stated otherwise. Graphs were generated using Prism GraphPad, and statistical analysis was performed with two‐way ANOVA.
Author contributions
XF and JC conceived the project and wrote the manuscript. XF performed most of the experiments with the assistance from MT, CW, DS, HZ, ZC, LN, MH, and YX. AT, SS, and AN performed the END‐seq experiment and analyzed the data. CZ and DJ analyzed the RNA‐seq data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 4
Acknowledgements
We thank all members of the Chen laboratory for their help and constructive discussion. We thank MD Anderson's Flow Cytometry and Cellular Imaging Facility and Science Park NGS Facility for their help with the flow cytometry and RNA‐seq experiments, respectively (supported by MD Anderson's NIH Cancer Center Support Grant, P30CA016672). We thank Dr. Pawel Mazur, Li Ma and Dr. Steven H. Lin (MD Anderson Cancer Center) for providing cell lines. We thank Dr. Yi Li and Amy Tsu Ku (Baylor College of Medicine) for their help on isolating mouse primary mammary epithelial cells. This work was supported in part by institutional funds and the Pamela and Wayne Garrison Distinguished Chair in Cancer Research. J.C. also received support from CPRIT (RP160667) and NIH (CA157448, CA193124, CA210929, CA216911, and CA216437).
The EMBO Journal (2020) 39: e104036
Data availability
The RNA‐seq data from this publication have been deposited to the GEO database and assigned the identifier GSE150003 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150003).
References
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V (2009) RIG‐I‐dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III‐transcribed RNA intermediate. Nat Immunol 10: 1065–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Elkon KB, Fitzgerald KA (2016) Importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med 67: 323–336 [DOI] [PubMed] [Google Scholar]
- Brown JS, O'Carrigan B, Jackson SP, Yap TA (2017) Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov 7: 20–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL (2011) The efficacy of radiotherapy relies upon induction of type I interferon‐dependent innate and adaptive immunity. Can Res 71: 2488–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, Nussenzweig A (2016) DNA breaks and end resection measured genome‐wide by end sequencing. Mol Cell 63: 898–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, MacCormick S, Mortimore M, O'Donnell ME, Pinder JL, Reaper PM et al (2011) Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem 54: 2320–2330 [DOI] [PubMed] [Google Scholar]
- Chen YA, Shen YL, Hsia HY, Tiang YP, Sung TL, Chen LY (2017) Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS‐STING DNA sensing pathway. Nat Struct Mol Biol 24: 1124–1131 [DOI] [PubMed] [Google Scholar]
- Cheon H, Holvey‐Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, Rice CM, Jackson MW, Junk DJ, Stark GR (2013) IFNbeta‐dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J 32: 2751–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li HL, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A et al (2015) Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162: 974–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, Chen ZJ (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG‐I pathway. Cell 138: 576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N (2015) Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- Deng LF, Liang H, Xu M, Yang XM, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T et al (2014) STING‐dependent cytosolic DNA sensing promotes radiation‐induced type i interferon‐dependent antitumor immunity in immunogenic tumors. Immunity 41: 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai N, Lee J, Upadhya R, Chu Y, Moir RD, Willis IM (2005) Two steps in Maf1‐dependent repression of transcription by RNA polymerase III. J Biol Chem 280: 6455–6462 [DOI] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH et al (2018) PARP inhibition elicits STING‐dependent antitumor immunity in Brca1‐deficient ovarian cancer. Cell Rep 25: 2972–2980 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Roy S, Franco S (2018) PARP1 depletion induces RIG‐I‐dependent signaling in human cancer cells. PLoS ONE 13: e0194611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW (2015) Exonuclease TREX1 degrades double‐stranded DNA to prevent spontaneous lupus‐like inflammatory disease. Proc Natl Acad Sci USA 112: 5117–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Reenberg RAG (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA et al (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity 42: 332–343 [DOI] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW (2013) Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, Yin L, Shu HB (2016) Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45: 555–569 [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14: 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonathan EC, Bernhard EJ, McKenna WG (1999) How does radiation kill cells? Curr Opin Chem Biol 3: 77–83 [DOI] [PubMed] [Google Scholar]
- Karnitz LM, Zou L (2015) Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res 21: 4780–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Liu QH, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo GB, Carattini‐Rivera S, DeMayo F, Bradley A et al (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Gene Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Lohard S, Bourgeois N, Maillet L, Gautier F, Fetiveau A, Lasla H, Nguyen F, Vuillier C, Dumont A, Moreau‐Aubry A et al (2020) STING‐dependent paracriny shapes apoptotic priming of breast tumors in response to anti‐mitotic treatment. Nat Commun 11: 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A et al (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica‐Worms H (1997) Mitotic and G2 checkpoint control: regulation of 14‐3‐3 protein binding by phosphorylation of Cdc25C on serine‐216. Science 277: 1501–1505 [DOI] [PubMed] [Google Scholar]
- de Queiroz N, Xia T, Konno H, Barber GN (2019) Ovarian cancer cells commonly exhibit defective STING signaling which affects sensitivity to viral oncolysis. Mol Cancer Res 17: 974–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Yau HL, Singhania R, Wang YD, Danesh A, Shen SY, Han H, Liang GN, Jones PA, Pugh TJ et al (2015) DNA‐demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162: 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- Sharma RA, Plummer R, Stock JK, Greenhalgh TA, Ataman O, Kelly S, Clay R, Adams RA, Baird RD, Billingham L et al (2016) Clinical development of new drug‐radiotherapy combinations. Nat Rev Clin Oncol 13: 627–642 [DOI] [PubMed] [Google Scholar]
- Shastri N, Tsai YC, Hile S, Jordan D, Powell B, Chen J, Maloney D, Dose M, Lo Y, Anastassiadis T et al (2018) Genome‐wide identification of structure‐forming repeats as principal sites of fork collapse upon ATR inhibition. Mol Cell 72: 222–238 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G (2019) PARPi triggers the STING‐dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 79: 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng WQ, LaFleur MW, Nguyen TH, Chen SJ, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH et al (2018) LSD1 ablation stimulates anti‐tumor immunity and enables checkpoint blockade. Cell 174: 549–563.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille‐Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S (2017) DNA exonuclease Trex1 regulates radiotherapy‐induced tumour immunogenicity. Nat Commun 8: 15618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN et al (2018) ATR kinase inhibitor AZD6738 potentiates CD8 + T cell‐dependent antitumor activity following radiation. J Clin Invest 128: 3926–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA et al (2014) STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41: 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Pan J, Thoroddsen V, Wysong DR, Blackman RK, Bulawa CE, Gould AE, Ocain TD, Dick LR, Errada P et al (2003) Novel small‐molecule inhibitors of RNA polymerase III. Eukaryot Cell 2: 256–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JX, Chen ZJ (2014) Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32: 461–488 [DOI] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J, Barber GN (2016a) Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep 14: 282–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Barber GN (2016b) Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res 76: 6747–6759 [DOI] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell 19: 727–740 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H (2019) The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178: 302–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 4
Data Availability Statement
The RNA‐seq data from this publication have been deposited to the GEO database and assigned the identifier GSE150003 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150003).