

Abstract
Nerve growth factor/tropomyosin receptor kinase A (NGF/TrkA) signaling plays a key role in neuronal development, function, survival, and growth. The pathway is implicated in neurodegenerative disorders including Alzheimer’s disease, chronic pain, inflammation, and cancer. NGF binds the extracellular domain of TrkA, leading to the activation of the receptor’s intracellular kinase domain. As TrkA signaling is highly dynamic, mechanistic studies would benefit from a tool with high spatial and temporal resolution. Here we present the design and evaluation of four strategies for light-inducible activation of TrkA in the absence of NGF. Our strategies involve the light-sensitive protein Arabidopsis cryptochrome 2 and its binding partner CIB1. We demonstrate successful recapitulation of native NGF/TrkA functions by optical induction of plasma membrane recruitment and homo-interaction of the intracellular domain of TrkA. This approach activates PI3K/AKT and Raf/ERK signaling pathways, promotes neurite growth in PC12 cells, and supports survival of dorsal root ganglion neurons in the absence of NGF. This ability to activate TrkA using light bestows high spatial and temporal resolution for investigating NGF/TrkA signaling.
Graphical Abstract
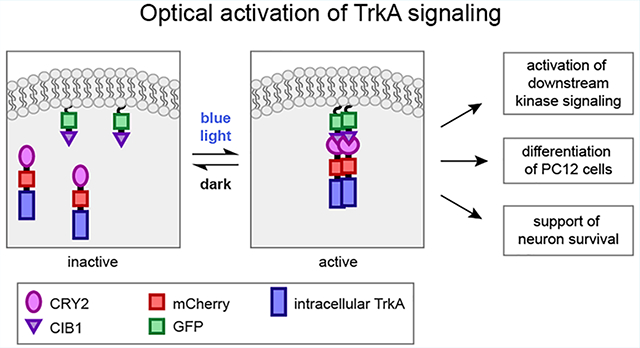
During embryonic development, nerve growth factor (NGF) plays a critical role in supporting neuronal differentiation, survival, and plasticity.1 In adults, NGF supports neural maintenance and repair, and deficits in NGF signaling have been implicated in several neurodegenerative disorders including Alzheimer’s and Parkinson’s diseases.2–4 Aberrantly elevated activity of NGF is also involved in chronic inflammatory and neuropathic pain disorders.5,6 The tropomyosin receptor kinase A (TrkA) has been shown to be the primary receptor of NGF in both neuroprotection and pain. Thus, therapeutic strategies that aim to augment NGF/TrkA signaling have been pursued for neurodegenerative diseases.7–10 For example, NGF gene therapies have been explored in clinical trials for Alzheimer’s disease,11,12 and small molecules agonists of TrkA are also being sought for potential use as therapies.13,14 On the other hand, therapeutic strategies that aim to block NGF/TrkA signaling are being explored as novel methods for pain management; Pfizer’s anti-NGF monoclonal antibody tanezumab15–17 received fast-track designation from the FDA for pain management in 2017 and has progressed to Phase 3 clinical trials.
However, the development of NGF-related drugs has been hindered partly by the incomplete understanding of NGF/TrkA signaling and its dynamics due to the lack of methods to precisely activate TrkA. It is widely accepted that NGF binds to the extracellular domain of TrkA, leading to its autophosphorylation and activation. There is less certainty regarding the identities of downstream signaling effectors or the composition of the signaling endosomes that transport TrkA after activation. TrkA signaling is dynamically regulated in space and time; the outcome of activation can differ based on subcellular environment, protein interaction partners, and the duration of activation. Thus, the ability to activate TrkA signaling with precise spatial and temporal control would be ideal for dissecting the discrete signaling events of NGF application.
The emergence of light-inducible tools presents new strategies to control intracellular activities with high spatial and temporal resolution. These optogenetic tools utilize photosensitive proteins that undergo light-gated protein–protein interactions, such as the light-oxygen-voltage (LOV) domain,18 phytochrome B,19 and cryptochrome 2 (CRY2).20 Arabidopsis CRY2 is unique in that it undergoes light-induced homo-oligomerization as well as hetero-dimerization with its binding partner CIB1.21 Optogenetic strategies based on CRY2/CIB1 hetero-dimerization have been successfully developed to control a variety of intracellular activities, such as organelle transport,22,23 gene expression,20,24 inactivation or disruption of protein function,21,25 and activation of diverse intracellular kinases.26–29 The light-mediated homo-oligomerization of CRY2 has also been used to regulate intracellular signaling of kinases such as Raf30 and transmembrane receptor kinases such as DCC.31 In particular, the use of CRY2 oligomerization-based optogenetic strategies to control the activation of TrkA has been explored in a previous work.26 This previously reported strategy appends CRY2 to the C-terminus of full length TrkA (flTrkA-CRY2), and leverages blue light to induce flTrkA-CRY2 oligomerization and activate TrkA. The light-dependent activation of flTrkA-CRY2 was assayed by the nuclear translocation of extracellular signalregulated kinase (ERK), but whether it fully recapitulates other signaling events downstream of NGF is yet to be determined.
In this work, we design and compare four different strategies for light-induced TrkA activation. We find that light-induced homo-interaction of the intracellular domain of TrkA (iTrkA) is more effective than that of full length TrkA in activating TrkA receptors and recapitulating NGF function. Particularly, recruitment and homo-oligomerization of iTrkA on the cell membrane is most efficient in activating downstream signaling pathways, inducing neurite outgrowth in PC12 cells, and supporting the survival of DRG neurons in the absence of NGF. This new method of optical activation of TrkA will be a useful tool to investigate the dynamics of NGF/TrkA signaling and to facilitate therapeutic screening of small molecule drugs that target the intracellular domain of TrkA.
The TrkA receptor consists of three domains, the extracellular domain (ECD), a transmembrane domain (TM) and the intracellular domain (iTrkA) (Figure 1A). Dimeric NGF binds the extracellular domain of TrkA and induces receptor dimerization and conformational change. The catalytic intracellular domain is then autophosphorylated and initiates downstream signal transduction.32,33 Previous work has shown that chemical-induced dimerization of plasma membrane-targeted myr-iTrkA is sufficient to activate TrkA and results in differentiation in PC12 cells.33 Inspired by this work, we hypothesized that dimerization of the iTrkA domain using light-induced protein–protein interaction may achieve optical activation of TrkA and its downstream pathways. To build the light-controlled TrkA systems, we chose to use the CRY2-CIB1 dimerization and CRY2-CRY2 homo-oligomerization.
Figure 1.

Design scheme of the light-controlled TrkA systems. We have constructed four methods of optogenetic activation of TrkA signaling by fusion to CRY2. The molecular architecture of each construct is detailed. (A) The full length TrkA receptor consists of an extracellular domain (ECD), a transmembrane domain (TM) and an intracellular domain (iTrkA). (B) Opto-full-TrkA is a fusion of CRY2 to the C-terminus of full length TrkA. (C) Opto-Lyn-iTrkA appends CRY2 to the C-terminal end of the intracellular domain of TrkA (iTrkA), and features an N-terminal Lyn membrane-targeting sequence. (D) Opto-iTrkA fuses CRY2 to the N-terminus of iTrkA. (E) Opto-iTrkA + CAAX uses CIBN-CAAX, localized to the plasma membrane, in combination with opto-iTrkA to recruit iTrkA to the membrane in a light-dependent manner.
In order to achieve light-inducible TrkA activation, we designed four different strategies using CRY2. First, CRY2 was fused to the C-terminus of full-length TrkA to make opto-full-TrkA. We expect that the fusion protein will be trafficked to the plasma membrane similar to endogenous TrkA, and that light-induced homo-oligomerization of CRY2 will initiate interaction between TrkA molecules (Figure 1B). This strategy has been explored previously.26 The second strategy fuses CRY2 to the C terminus of the truncated intracellular domain iTrkA, and is localized to the plasma membrane by an N-terminal Lyn tag (Lyn-iTrkA-CRY2, hereafter opto-Lyn-iTrkA). Light-induced CRY2 homo-interaction will induce oligomerization of iTrkA at the cell membrane (Figure 1C). Our third strategy uses a fusion of CRY2 to the N-terminus of iTrkA without any localization tag. We expect that blue light will induce the interaction of iTrkA in the cytosol (opto-iTrkA, Figure 1D). Finally, our last strategy uses coexpression of CRY2-iTrkA with CIB1-CAAX, which targets CIB1 to the plasma membrane via the CAAX motif.34 With this design, light stimulation induces both recruitment of CRY2-iTrkA to the cell membrane through CRY2-CIB1 dimerization and simultaneously induces iTrkA homo-interaction on the membrane through CRY2-CRY2 binding. We refer to the fourth strategy as opto-iTrkA + CAAX (Figure 1E). These four light-gated TrkA systems together are collectively denoted as optoTrkA systems henceforth.
In order to validate our optoTrkA systems, we examined signaling pathways downstream of TrkA. We first confirmed that our constructs could be expressed in cell culture, and that blue light stimulation induced membrane recruitment of iTrkA in cells expressing the opto-iTrkA + CAAX system (Figure S1). We next sought to assess whether blue light stimulation of optoTrkA systems would lead to the activation of pathways downstream of TrkA activation.
TrkA activation induces both the PI3K/AKT and the Raf/MEK/ERK signaling cascades. We assessed the activation of Raf/MEK/ERK by monitoring translocation of ERK2 from the cytosol to the nucleus, and probed the activation of PI3K/AKT by monitoring the translocation of the pleckstrin homology domain of AKT (PHAKT1) to the plasma membrane. NIH 3T3 cells were transfected with each of the four optoTrkA systems as well as ERK2-GFP or GFP-PHAKT1. All TrkA fusion proteins were tagged with mCherry (mCh). In the case of opto-iTrkA + CAAX, the fusion protein CIB1-CAAX was used, which contains no fluorescent reporter. mCherry fluorescence (excitation ~560 nm) was used to locate transfected cells under a microscope. Then 200 ms blue light pulses were delivered to stimulate optoTrkA systems as well as to visualize the distribution of GFP-PHAKT1 or ERK2-GFP (at 5 or 10 s intervals for 10 or 20 min, respectively).
In cells expressing opto-Lyn-iTrkA, cytosolic opto-iTrkA, or opto-iTrkA + CAAX, blue light stimulation induced visible recruitment of GFP-PHAKT1 to the plasma membrane (Figure 2A) and the nuclear translocation of ERK2-GFP (Figure 2B). On the other hand, opto-full-TrkA did not induce visible changes in the spatial distribution of either GFP-PHAKT1 or ERK2-GFP. The translocation of GFP-PHAKT1 or ERK2-GFP was quantified to show the activation of PI3K/AKT or Raf/MEK/ERK respectively. Light-induced membrane recruitment of GFP-PHAKT1 was quantified as the ratio of average intensity at the membrane to the average cytosolic intensity after light stimulation, which was then normalized to the same ratio before light stimulation. Light-induced nuclear translocation of ERK2-GFP was measured similarly by the ratio of nuclear intensity to the average cytosolic intensity (see Methods for details). Quantitative analysis shows that blue light activation of opto-full-TrkA did not induce a significant change in the membrane/cytosol ratio of GFP-PHAKT1 or the nuclear/cytosol ratio of ERK2-GFP (Figure 2C). In comparison, opto-Lyn-iTrkA, opto-iTrkA, and opto-iTrkA + CAAX demonstrated significant blue-light induced increases in GFP-PHAKT1 and ERK2-GFP translocation. To confirm that these translocation events corresponded to pathway activation, we have shown that the membrane translocation of GFP-PHAKT1 is inhibited by wortmannin, a PI3K inhibitor. Similarly, the nuclear translocation of ERK-GFP was blocked by the addition of the MEK inhibitor Trametinib (Figure S2). We also examined the kinetics of light-induced activation of AKT and ERK in cells expressing opto-iTrkA or opto-iTrkA + CAAX (Figure S3). AKT and ERK activation peaked at 2 or 5 min respectively upon light stimulation of opto-iTrkA + CAAX, comparable to kinetics reported for ligand-induced activation of TrkA in the literature.35–40 However, the activation of ERK and AKT was somewhat delayed in cells transfected with opto-iTrkA.
Figure 2.

OptoTrkA systems activate PI3K/AKT and Raf/MEK/ERK signaling pathways. (A) Opto-Lyn-iTrkA, opto-iTrkA, and opto-iTrkA + CAAX exhibit activation of the PI3K/AKT signaling pathway, assayed by the membrane translocation of PHAKT-GFP in response to blue light stimulation. Opto-full-TrkA does not induce obvious PHAKT-GFP membrane translocation (200 ms, 9.7 W/cm2, blue light pulses delivered at 5 s intervals for 10 min). (B) Opto-Lyn-iTrkA, opto-iTrkA, and opto-iTrkA + CAAX systems induce ERK activation upon blue light stimulation, assayed by the nuclear translocation of ERK-GFP. Opto-full-TrkA does not induce obvious nuclear translocation of ERK-GFP (200 ms, 9.7 W/cm2, blue light pulses delivered at 10 s intervals for 20 min). (C) Average translocation of PHAKT-GFP and ERK-GFP upon blue light stimulation for the four optoTrkA systems (for PHAKT-GFP assay, n = 10, 11, 11, 10 cells and for ERK-GFP assay, n = 11, 10, 11, 10 cells, respectively). (D) Western blot analysis of optoTrkA systems for phosphorylated ERK (Thr202 and Tyr204) and phosphorylated AKT (Ser473). Opto-iTrkA and opto-iTrkA + CAAX exhibit AKT and ERK phosphorylation upon blue light stimulation (200 μW/cm2 continuous blue light illumination for 20 min). Scale bars, 5 μm.
We further probed the light-induced activation of PI3K/AKT and Raf/MEK/ERK by immunoblot analysis of phosphorylated ERK (pERK1/2, Thr202 and Tyr204) and phosphorylated AKT (pAKT, Ser473). PC12 cells were transfected with each optoTrkA system and subjected to continuous blue light stimulation (200 μW/cm2) for 20 min. One group of cells was transfected with CIB1-GFP-CAAX alone as a negative control. A second set of samples was subjected to the same transfection and analysis without exposure to light in order to assess dark activation. Following light illumination, cells were immediately lysed and probed for pAKT, total AKT, pERK1/2, total ERK1/2, and GAPDH as a loading control. In the absence of blue light, very little phosphorylation of ERK or AKT was detected in any of the five conditions (Figure 2D). After blue light illumination, pAKT and pERK levels remain low in the CIB1-GFP-CAAX control. Cells expressing opto-Lyn-iTrkA, showed a slight increase in the levels of pAKT and pERK after blue light stimulation compared to the CAAX control. Blue light induced a significant increase in the levels of pAKT and pERK in cells expressing cytosolic opto-iTrkA, and an even greater increase in cells expressing opto-iTrkA + CAAX. In summary, our results demonstrate that three of our four optoTrkA strategies are capable of activating downstream Raf/ERK and PI3K/AKT signaling pathways, but that the extent of signaling is highly sensitive to the strategy used for activation. Specifically, blue light induced strong and robust activation of the downstream signaling in the opto-iTrkA or opto-iTrkA + CAAX systems, but much weaker activation in the opto-full-TrkA and opto-Lyn-iTrkA systems. Quantification of three independent experiments is shown in Figure S4. We also show that all of our optoTrkA systems demonstrate phosphorylation of TrkA at Tyr490 (Figure S4), though as with ERK and AKT phosphorylation, the levels are greatest in opto-iTrkA and opto-iTrkA + CAAX.
We next examined whether light-gated activation of TrkA could simulate NGF in promoting neurite growth in PC12 cells. We used a subclone of the PC12 line, NeuroScreen-1 (NS1), which is widely used to assess neurite outgrowth in response to NGF treatment.41 Though our TrkA and iTrkA constructs include the fluorescent reporter tag mCherry, this fluorescence was often too dim to be used to visualize whole-cell morphologies under low magnification (20×), especially for fine neurite structures. Thus, we made use of GFP to visualize the cell morphology. In the opto-iTrkA + CAAX system, this was achieved by using the fluorescently tagged CIB1-GFP-CAAX, and in all other systems by cotransfection with cytosolic GFP. PC12 cells transfected with CIB1-GFP-CAAX alone served as a control.
With blue light stimulation, PC12 cells expressing either the CIB1-GFP-CAAX control or opto-full-TrkA failed to grow neurites (Figure 3A,E). On the other hand, cells transfected with opto-Lyn-iTrkA, opto-iTrkA or opto-iTrkA + CAAX grew out long neurites under blue light stimulation (Figure 3B–D). Among them, cells expressing opto-iTrkA + CAAX grew out significantly longer neurites than other groups. The transfected cells kept in dark did not grow measurable neurites, except for a few cells expressing opto-Lyn-iTrkA that showed some neurite outgrowth in dark (Figure S5). To quantify the extent of PC12 cell differentiation, the percentage of cells with neurites were measured among all the transfected cells either in dark or under light illumination (Figure 3F, statistics shown in Table S1). Under blue light, 34% of opto-iTrkA and 61% of opto-iTrkA + CAAX cells grew neurites longer than the size of their cell bodies, significantly more than their dark controls (6.2% and 1.7%, respectively). 22% of opto-Lyn-iTrkA cells grew neurites with light stimulation, but this growth is not significantly greater than the corresponding dark control (13%).
Figure 3.

Light-induced activation of TrkA promotes neurite growth in PC12 cells in the absence of NGF. PC12 cells were subjected to continuous blue light illumination at 200 μW/cm2 for 24 h. (A) PC12 cells expressing opto-full-TrkA and GFP displayed no obvious neurite growth. (B) Some PC12 cells expressing opto-Lyn-iTrkA and GFP extend short neurites. (C) Some PC12 cells expressing opto-iTrkA and GFP exhibit neurite growth. (D) Most PC12 cells expressing opto-iTrkA + CAAX grow long neurites. (E) PC12 expressing only CIBN-GFP-CAAX did not show noticeable neurite growth. (F) Quantification of percentage of cells with neurite growth for different systems. (G) Quantification of the length of the longest neurite in cells bearing neurites. (H) Quantification of the average number of neurite branches in cells bearing neurites. Results are averaged from three independent sets of experiments (detailed in Supplementary Table S1) and presented as mean ± SEM. Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc test. Student’s t test was used between dark or light cells under the same transfection conditions (*P < 0.05, **P < 0.005). Scale bars, 10 μm.
We also measured the average length of the longest neurite of each cell, which was greatest in cells expressing opto-iTrkA + CAAX (118 μm) and shorter in cells transfected with opto-iTrkA (80 μm) and cells transfected with opto-Lyn-iTrkA (44 μm) (Figure 3G). Finally, the average numbers of neurites per cell were quantified to compare the morphological differences (Figure 3H). Cells expressing opto-iTrkA + CAAX had more neurite branches than other conditions. In summary, these results showed that light-induced activation of opto-iTrkA + CAAX and opto-iTrkA is sufficient to induce neurite growth in PC12 cells. In particular, opto-iTrkA + CAAX activation is the most effective in promoting neurite growth in PC12 cells, and exhibits neurites of higher complexity than other optoTrkA systems.
As NGF promotes neuronal survival through TrkA, we next tested whether light-activated TrkA signaling could mimic NGF function in supporting neuronal survival. We used embryonic DRG neuron culture as a model system, as a large proportion of these cells, known as peptidergic nociceptors, are not viable in the absence of NGF.42–44 DRG neurons were transfected with each optoTrkA system by electroporation at day 0. We used the human synapsin 1 promoter45 to express GFP or CIB1-GFP-CAAX exclusively in neurons in order to avoid high glial background expression. After transfection, neurons were plated in a 12-well plate and allowed to recover for 16 h in complete medium supplemented with 50 ng/mL NGF. NGF was then withdrawn from the culture medium and cells were treated with anti-NGF antibody to remove residual growth factor for 48 h. During this withdrawal, the cultures were subjected to blue light illumination at 200 μW/cm2 using homemade LED arrays. Another group of transfected neurons were kept in dark as controls. After 48 h in the presence or absence of blue light stimulation, the cultures were fixed and immunostained against GFP to visualize the neuron morphologies.
DRG neurons expressing opto-full-TrkA did not survive either under light illumination or in dark (Figure 4A). A small percentage of DRG neurons transfected with opto-Lyn-iTrkA (28%) survived in the presence of blue light stimulation (Figure 4B). Surviving DRG neurons had extended out long axons. On the other hand, a substantial number of DRG cells expressing opto-iTrkA (57%) or opto-iTrKA + CAAX (73%) survived after light illumination and grew very long neurites (Figure 4C and 4D). For control samples kept in dark, very few cells survived in each group of transfected DRG. To quantify the efficacy in promoting neuronal survival, the survival rates were measured for each transfection in dark or under light stimulation (Figure 4E, Table S2). Transfected DRG neurons with long neurites were identified as living cells. Quantitative analysis confirmed that opto-iTrkA + CAAX is the most effective in supporting DRG survival in the absence of NGF, followed by opto-iTrkA and opto-Lyn-iTrkA. Opto-full-TrkA did not support DRG survival. This neuron survival result corroborates the neurite growth studies in PC12 cells to show that light-induced activation of opto-iTrkA + CAAX is highly effective in mimicking endogenous NGF functions.
Figure 4.

Light-induced TrkA activation supports DRG neuron survival in the absence of NGF. After transfection, primary rat DRG neurons were allowed to recover in NGF-supplemented medium for 24 h. Then, NGF was withdrawn and replaced with anti-NGF for 48 h while the cultures were subjected to continuous blue light stimulation at 200 μW/cm2 or kept in dark for 2 days. (A) DRG neurons transfected with opto-full-TrkA and GFP did not survive under light or dark. (B) Some DRG neurons expressing opto-Lyn-iTrkA and GFP survived upon blue light stimulation, but few survived in the dark control. (C) Many DRG cells expressing opto-iTrkA survived and had extended long axons upon blue light stimulation, but not in the dark controls. (D) Most DRG neurons expressing opto-iTrkA and CIB1-GFP-CAAX were alive with long axons with blue light, but not in the dark controls. (E) Quantification of survival rate of DRG neurons for optoTrkA systems, in light and dark conditions. Results are averaged from three independent sets of experiments (see numbers of cells in each set of experiment in Table S2) and presented as means ± SEM. One-way ANOVA with Dunnett’s post hoc test was performed between cells with different transfection. t test was performed between cells in dark or in light with the same transfection (*P < 0.05, **P < 0.005). Scale bars, 50 μm.
In this work, we designed and evaluated four strategies to achieve light-induced TrkA signaling either in the cytosol or on the cell membrane. Cytosolic activation is achieved by fusing CRY2 to intracellular domain of TrkA (opto-iTrkA) so that the light-mediated homo-oligomerization of CRY2 can result in the oligomerization and autophosphorylation of iTrkA. To enable light-inducible TrkA activation at the plasma membrane, we tested three strategies: opto-full-TrkA, opto-Lyn-iTrkA, and opto-iTrkA + CAAX. We find that opto-iTrkA + CAAX is the most effective in activating downstream TrkA signaling pathways, stimulating neurite outgrowth in PC12 cells, and supporting DRG survival in the absence of NGF.
Though we focused primarily on Raf/MEK/ERK and PI3K/AKT signaling as readouts of TrkA activation, it is worth acknowledging that TrkA also activates PLC-γ and Ca2+ dependent signaling. These pathways are not independent, but rather exhibit considerable crosstalk and redundancy: it has been demonstrated that PLC-γ activity is necessary for NGF-mediated MAPK/ERK signaling, and that Ca2+ efflux impacts the extent of Akt phosphorylation.46–48 Though an in-depth study of the extent of this crosstalk in different conditions is beyond the scope of the current study, there exists precedent for the precise control of Ca2+ signaling using optogenetic methods,49 and we hope that the optogenetic tools we present will be useful in further investigation to this end.
Chang et al.’s work26 demonstrated the feasibility of using CRY2-based optogenetic protein dimerization to build light-inducible neurotrophin receptors. In this work, we sought to deliver an optoTrkA system that could reproduce more complex signaling outcomes, such as differentiation and survival. We have also demonstrated that strategies which make use of only the intracellular domain of TrkA are more effective at recapitulating native signaling than strategies that utilize the full length receptor. One possible explanation for this difference is that full length TrkA may exist as preformed yet inactive dimers prior to ligand binding.50 The extracellular domain and transmembrane domain of full-TrkA may lock the preformed dimer in a configuration that cannot be fully activated by CRY2 oligomerization. Further, the separation of the intracellular, signaling-competent domain of TrkA from its ligand-binding extracellular domain confers its own advantage, especially for use in model systems which may produce NGF endogenously. By removing the ligand-binding extracellular domain, we render the optogenetic construct inert in the absence of light induction, thus increasing confidence that any observed change is due to intentional perturbation rather than background.
Among the iTrkA systems, we observed that opto-Lyn-iTrkA is less efficient in inducing cell differentiation and supporting neuronal survival than opto-iTrkA + CAAX. opto-Lyn-iTrkA also showed higher background activities in dark than other systems, e.g., cells transfected with opto-Lyn-iTrkA had some neurite growth in dark, likely due to the enhanced homo-oligomerization of membrane-bound CRY2, which has been reported in a previous study.51
In summary, we have successfully constructed and validated two optoTrkA systems, opto-iTrkA + CAAX and opto-iTrkA, to optimally induce downstream TrkA signaling pathways. In particular, opto-iTrkA + CAAX is the most effective in mimicking the role of NGF in stimulating neurite growth in PC12 cells and supporting DRG neuronal survival. The opto-iTrkA + CAAX strategy allows spatial and temporal control of TrkA signaling to facilitate the study of mechanisms underlying NGF/TrkA signaling in a precise manner. The potential for this rapid, reversible system to be used in dissecting the neural circuitry involved in pain response in vivo is especially promising. In addition, the opto-iTrkA + CAAX strategy may prove a useful tool in screening novel therapeutics against diseases involving NGF/TrkA signaling.
METHODS
Plasmid Construction
TrkA-mCh-CRY2 (Opto-full-TrkA)
Full length TrkA was inserted into pmCherry-N1 (clontech) at XhoI and AgeI sites. TrkA template plasmids were described previously.22 CRY2 was inserted into pTrkA-mCherry with a GS N-terminal linker at BsrG1 and MfeI using InFusion (Clontech).
CRY2-mCh-iTrkA (Opto-iTrkA)
The intracellular domain of TrkA (iTrkA, aa 450–799, NP_067600.1) with GSS linker was inserted into CRY2-mCh at BsrG1 and MfeI using InFusion (Clontech).
Lyn-iTrkA-mCh-CRY2 (Opto-Lyn-iTrkA)
Lyn sequence (MGCIKSKRKDNLNDD) was directly synthesized (Integrated DNA Technology) and inserted into pmCherry-N1 at NheI and XhoI sites using ligation method (Thermo Fisher). iTrkA was then inserted at XhoI and AgeI and CRY2 was inserted at BsrG1 and MfeI using InFusion (Clontech).
hSyn-CIB1-GFP-CAAX and hSyn-GFP
Human synapsin promoter (hSyn) sequence was obtained by PCR from vector pLV-hSyn-RFP (Addgene #22909) and inserted into CIB1-GFP-CAAX7 or pEGFP-N1 (Clontech) at VspI and NheI using InFusion (Clontech).
Cell Culture and Transfection
PC12 cells (Neuro-Screen-1 subclone) were maintained in complete medium (F12K supplemented with 15% horse serum and 2.5% FBS). NIH 3T3 cells were grown in DMEM medium supplemented with 10% fetal bovine serum (FBS). All cell cultures were maintained a standard incubator at 37 °C with 5% CO2. Both PC12 and 3T3 cells were transfected with desired DNA plasmids using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. The transfected cells were allowed to recover and express the desired proteins overnight in culture medium. PC12 cells were kept in starvation medium (F12K only) overnight before light stimulation. For ERK and AKT translocation assays, 3T3 cells were starved in DMEM only for overnight before imaging.
DRG neurons were dissected from E18 embryos by standard procedure23 and maintained on poly-L-lysine (Sigma) coated coverslips (VWR) in DRG maintenance medium (Neurobasal supplemented with GlutaMax and B27, and containing 50 ng/mL NGF). For NGF withdrawal, 50 ng/mL NGF was replaced with 100 ng/mL NGF antibody (Millipore AB1526SP). The Amaxa Nucleofector was used for electroporation as previously described.52
Programmable LED Light Box
LED light boxes were constructed as previously reported for long-term light illumination.53 Briefly, a 12 well plate sized blue LED array was constructed by assembling 12 blue LEDs (LED465E, ThorLabs) on a breadboard, housed in an aluminum box, and a light diffuser film was positioned above the LED array to ensure uniform light intensity in defined area, separated by black barriers. The light intensity of each LED was controlled through a tunable resistor, and was measured by a power meter (Newport, 1931-C). The LED array was controlled by Labview through a data acquisition board (National Instrument-DAQ, PCI-6035E). The LEDs were supplied with user-defined DC voltages that were individually controllable through the Labview program.
Optical Activation in Culture
For PC12 neurite growth experiments, transfected PC12 cells were kept in dark and serum-starved for 24 h prior to blue light stimulation to minimize interference from growth factors present in serum. Cells were then illuminated with continuous blue light at 200 μW/cm2 for 24 h using a custom-built LED array housed inside a CO2 incubator. This illumination condition has been repeatedly tested and shows no obvious toxicity as demonstrated in our previous studies.23,51,53 Additional sets of transfected cells were kept in dark as controls. After 24 h, cultures were fixed and mCherry and GFP were visualized using fluorescence microscopy.
For DRG survival experiments, transfected neurons were plated in a 12-well plate and allowed to recover for 16 h in complete medium supplemented with 50 ng/mL NGF. NGF was then withdrawn from the culture medium and cells were treated with anti-NGF antibody to remove residual growth factor for 48 h. During this withdrawal, the cultures were subjected to blue light illumination at 200 μW/cm2 using homemade LED arrays. Another group of transfected neurons were kept in dark as controls. After 48 h in the presence or absence of blue light stimulation, the cultures were fixed and immunostained against GFP to visualize the neuron morphologies.
Immunoblotting
Immunoblotting experiments were carried out as previously described.54 Briefly, after desired treatment, cultured cells were lysed in PB/Triton buffer (7.74 mM Na2HPO4, 2.26 mM NaH2PO4, 5 mM EDTA, 5 mM EGTA, 100 mM NaCl, 10 mM Na4P2O7, 50 mM NaF, 1 mM Na3VO4, 1% Triton X-100), and clarified lysates were mixed with Laemmli sample buffer (Bio-Rad 1610747) and β-mercaptoethanol and boiled for 10 min. Lysed samples were subjected to electrophoresis using Bio-Rad’s Mini-PROTEAN system (1658026FC). After separation, protein was transferred to a polyvinylidene fluoride membrane (Bio-Rad 1704273), followed by standard blotting procedure. Primary antibodies were obtained from Cell Signaling Technology: anti-pTrkA (Y490) (CST 9141S), anti-pAKT (T308) (CST 9275), anti-AKT (CST 9272), anti-pERK1/2 (T202+Y204) (CST 9101), anti-ERK1/2 (CST 9102), and anti-GAPDH (CST 2118). HRP-conjugated secondary antibody (CST 7074) was used for protein band detection. Protein bands were visualized by chemiluminescence (Bio Rad 1705060).
Cell Fixation and Immunocytochemistry Staining
For PC12 neurite growth assays and DRG survival assays, cells were washed with ice-cold PBS three times before fixation in 4% paraformaldehyde in sucrose-buffered solution at room temperature for 20 min. The fixed cells were then washed with ice-cold PBS twice and proceeded to imaging or immunostaining.
DRG neurons were stained with anti-GFP (MolecularProbe, #A11122) in 5% BSA-PBS buffer at 4 °C overnight, and then probed using Alexa Fluor 488 (Thermo Fisher, R37116).
Fluorescence Imaging
Live cell imaging was performed on an epifluorescence microscope (Leica DMI6000B) equipped with an on-stage CO2 incubation chamber (Tokai Hit GM-8000) and a motorized stage (Prior). An adaptive focus control was used to actively keep the image in focus during the period of imaging. A light-emitting diode (LED) light source (Lumencor Sola) was used as the fluorescence light source. For blue-light stimulation, pulsed blue light (200 ms pulse duration at 9.7 W/cm2) was used for GFP imaging and to initiate CRY2-CIB1 and CRY2-CRY2 interactions. For mCherry imaging, pulsed green light (200 ms pulse duration) was used. Fluorescence signal from GFP was detected using a commercial GFP filter cube (Leica; excitation filter 472/30, dichroic mirror 495, emission filter 520/35); fluorescence signal from mCherry was detected using a commercial Texas red filter cube (Leica; excitation filter 560/40, dichroic mirror 595, emission filter 645/75). The images for ERK or AKT translocation were acquired with an oil-immersion 100× objective, while the images for PC12 or DRG morphologies were acquired with 10× or 40× objectives. All experiments were imaged with a sensitive CMOS camera (PCO.EDGE 5.5) (PCO).
Data Analysis
To analyze the neurite growth of PC12 cells, neurites were measured manually using the ImageJ line tool, and neurites significantly shorter than cell body were not considered. The number of cells in each measurement are included in Supplementary Table S1.
To quantitatively analyze the translocation of PHAKT1 to the plasma membrane, average intensity of membrane Imembrane was measured by selecting and measuring only membrane area. Average intensity of cytosol Icytosol was measured by selecting the whole cytosol area. Ibackground is defined as the average intensity of an image from blank areas (outside cells). The amount of PHAKT1 on membrane was indicated by ratio = (Imembrane – Ibackground)/(Icytosol – Ibackground). In Figure 2C, the normalized average intensity of PHAKT1-GFP was calculated by ratio+light/ratio−light.
Similarly, the average intensity of ERK2-GFP in the nucleus was quantified by ratio = (Inucleus – Ibackground)/(Icytosol – Ibackground). In Figure 2C, the normalized average intensity of ERK2-GFP was calculated by ratio+light/ratio−light.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Chandra Tucker (University of Colorado, Denver) for providing CIB1-GFP-Caax and CRY2-mCherry and Dr. Tobias Meyer (Stanford University) for providing PHAKT1-GFP plasmid. We also thank Zhuoluo Feng (Stanford University) for his help in constructing the controllable blue LED array. GS is a New York Stem Cell Foundation Robertson Investigator. This work was supported by the US NIH (DP2-NS082125), a Packard fellowship in Science and Engineering, a National Science Foundation Graduate Research Fellowship under Grant No. DGE-1656518 (JMH), and a seed grant from the Stanford Neurosciences Institute (BC and GS).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.8b00126.
Figures S1–S5 and Tables S1–S2 (PDF)
REFERENCES
- (1).Levi-Montalcini R, and Hamburger V (1951) Selective Growth Stimulating Effects of Mouse Sarcoma on the Sensory and Sympathetic Nervous System of the Chick Embryo. J. Exp. Zool 116 (2), 321–361. [DOI] [PubMed] [Google Scholar]
- (2).Calissano P, Matrone C, and Amadoro G (2010) Nerve Growth Factor as a Paradigm of Neurotrophins Related to Alzheimer’s Disease. Dev. Neurobiol 70 (5), 372–383. [DOI] [PubMed] [Google Scholar]
- (3).Cooper JD, Lindholm D, and Sofroniew MV (1994) Reduced Transport of [125I]nerve Growth Factor by Cholinergic Neurons and down-Regulated TrkA Expression in the Medial Septum of Aged Rats. Neuroscience 62 (3), 625–629. [DOI] [PubMed] [Google Scholar]
- (4).Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, and Mufson EJ (2004) Reduction of Cortical TrkA but Not p75(NTR) Protein in Early-Stage Alzheimer’s Disease. Ann. Neurol 56 (4), 520–531. [DOI] [PubMed] [Google Scholar]
- (5).Hirose M, Kuroda Y, and Murata E (2016) NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pract. 16 (2), 175–182. [DOI] [PubMed] [Google Scholar]
- (6).Sofroniew MV, Howe CL, and Mobley WC (2001) Nerve Growth Factor Signaling, Neuroprotection, and Neural Repair. Annu. Rev. Neurosci 24, 1217–1281. [DOI] [PubMed] [Google Scholar]
- (7).Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK, and GDNF NGF (2013) and BDNF as Therapeutic Options for Neurodegeneration. Pharmacol. Ther 138 (2), 155–175. [DOI] [PubMed] [Google Scholar]
- (8).Counts SE, and Mufson EJ (2005) The Role of Nerve Growth Factor Receptors in Cholinergic Basal Forebrain Degeneration in Prodromal Alzheimer Disease. J. Neuropathol. Exp. Neurol 64 (4), 263–272. [DOI] [PubMed] [Google Scholar]
- (9).Weissmiller AM, and Wu C (2012) Current Advances in Using Neurotrophic Factors to Treat Neurodegenerative Disorders. Transl. Neurodegener 1 (1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Aloe L, Rocco ML, Bianchi P, and Manni L (2012) Nerve Growth Factor: From the Early Discoveries to the Potential Clinical Use. J. Transl. Med 10, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tuszynski MH (2007) Nerve Growth Factor Gene Therapy in Alzheimer Disease. Alzheimer Dis. Assoc. Disord 21 (2), 179–189. [DOI] [PubMed] [Google Scholar]
- (12).Tuszynski MH, Yang JH, Barba D, U, H.-S., Bakay RAE, Pay MM, Masliah E, Conner JM, Kobalka P, Roy S, et al. (2015) Nerve Growth Factor Gene Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol. 72 (10), 1139–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jang S-W, Okada M, Sayeed I, Xiao G, Stein D, Jin P, and Ye K (2007) Gambogic Amide, a Selective Agonist for TrkA Receptor That Possesses Robust Neurotrophic Activity, Prevents Neuronal Cell Death. Proc. Natl. Acad. Sci. U. S. A 104 (41), 16329–16334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zheng X, Chen F, Zheng T, Huang F, Chen J, and Tu W (2016) Amitriptyline Activates TrkA to Aid Neuronal Growth and Attenuate Anesthesia-Induced Neurodegeneration in Rat Dorsal Root Ganglion Neurons. Medicine 95 (18), e3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ekman E, Gimbel J, Bello A, Smith M, Keller D, Annis K, Brown M, West C, and Verburg K (2011) Efficacy and Safety of Intravenous Tanezumab in Osteoarthritis Hip and Knee Pain: Comparison to Placebo and Naproxen in Two Phase III Studies (NCT00830063 & NCT00863304). J. Pain 12 (4), P55. [Google Scholar]
- (16).Sopata M, Katz N, Carey W, Smith MD, Keller D, Verburg KM, West CR, Wolfram G, and Brown MT (2015) Efficacy and Safety of Tanezumab in the Treatment of Pain from Bone Metastases. Pain 156 (9), 1703–1713. [DOI] [PubMed] [Google Scholar]
- (17).Bramson C, Herrmann D, Biton V, Carey W, Keller D, Brown M, West C, Verburg K, and Dyck P (2013) Efficacy and Safety of Subcutaneous Tanezumab in Patients with Pain Related to Diabetic Peripheral Neuropathy (NCT01087203). J. Pain 14 (4), S68. [Google Scholar]
- (18).Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, and Glotzer M (2012) TULIPs: Tunable, Light-Controlled Interacting Protein Tags for Cell Biology. Nat. Methods 9 (4), 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Levskaya A, Weiner OD, Lim WA, and Voigt CA (2009) Spatiotemporal Control of Cell Signalling Using a Light-Switchable Protein Interaction. Nature 461 (7266), 997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, and Tucker CL (2010) Rapid Blue-Light–mediated Induction of Protein Interactions in Living Cells. Nat. Methods 7 (12), 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ, and Tucker CL (2014) An Optimized Optogenetic Clustering Tool for Probing Protein Interaction and Function. Nat. Commun 5, 4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).van Bergeijk P, Adrian M, Hoogenraad CC, and Kapitein LC (2015) Optogenetic Control of Organelle Transport and Positioning. Nature 518 (7537), 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Duan L, Che D, Zhang K, Ong Q, Guo S, and Cui B (2015) Optogenetic Control of Molecular Motors and Organelle Distributions in Cells. Chem. Biol 22 (5), 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hughes RM, Bolger S, Tapadia H, and Tucker CL (2012) Light-Mediated Control of DNA Transcription in Yeast. Methods 58 (4), 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lee S, Park H, Kyung T, Kim NY, Kim S, Kim J, and Heo WD (2014) Reversible Protein Inactivation by Optogenetic Trapping in Cells. Nat. Methods 11 (6), 633–636. [DOI] [PubMed] [Google Scholar]
- (26).Chang K-Y, Woo D, Jung H, Lee S, Kim S, Won J, Kyung T, Park H, Kim N, Yang HW, et al. (2014) Light-Inducible Receptor Tyrosine Kinases That Regulate Neurotrophin Signalling Nat. Commun 5, 4057. [DOI] [PubMed] [Google Scholar]
- (27).Grusch M, Schelch K, Riedler R, Reichhart E, Differ C, Berger W, Inglés-Prieto Á, and Janovjak H (2014) Spatio-Temporally Precise Activation of Engineered Receptor Tyrosine Kinases by Light. EMBO J. 33 (15), 1713–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kim N, Kim JM, Lee M, Kim CY, Chang K-Y, and Heo WD (2014) Spatiotemporal Control of Fibroblast Growth Factor Receptor Signals by Blue Light. Chem. Biol 21 (7), 903–912. [DOI] [PubMed] [Google Scholar]
- (29).Li Y, Lee M, Kim N, Wu G, Deng D, Liu X, Heo WD, and Zi Z (2017) Spatiotemporal Control of TGF-β Signaling with Light. ACS Synth. Biol 7, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wend S, Wagner HJ, Müller K, Zurbriggen MD, Weber W, and Radziwill G (2014) Optogenetic Control of Protein Kinase Activity in Mammalian Cells. ACS Synth. Biol 3 (5), 280–285. [DOI] [PubMed] [Google Scholar]
- (31).Endo M, Hattori M, Toriyabe H, Ohno H, Kamiguchi H, Iino Y, and Ozawa T (2016) Optogenetic Activation of Axon Guidance Receptors Controls Direction of Neurite Outgrowth. Sci. Rep 6, 23976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Marlin MC, and Li G (2015) Biogenesis and Function of the NGF/TrkA Signaling Endosome. Int. Rev. Cell Mol. Biol 314, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Alfa RW, Tuszynski MH, and Blesch A (2009) A Novel Inducible Tyrosine Kinase Receptor to Regulate Signal Transduction and Neurite Outgrowth. J. Neurosci. Res 87 (12), 2624–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hancock JF, Cadwaller K, Paterson H, and Marshall CJ (1992) A CAAX or a CAAL Motif and a Second Signal Are Sufficient for Plasma Membrane Targeting of Ras Proteins. Trends Cell Biol. 2 (3), 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Burry RW (2001) p21(ras) Stimulates Pathways in Addition to ERK, p38, and Akt to Induce Elongation of Neurites in PC12 Cells. J. Neurosci. Res 63 (1), 45–53. [DOI] [PubMed] [Google Scholar]
- (36).Chen YL, Law P-Y, and Loh HH (2006) Sustained Activation of Phosphatidylinositol 3-Kinase/Akt/Nuclear Factor κB Signaling Mediates G Protein-Coupled δ-Opioid Receptor Gene Expression. J. Biol. Chem 281 (6), 3067–3074. [DOI] [PubMed] [Google Scholar]
- (37).Kim Y, Seger R, Suresh Babu CV, Hwang S-Y, and Yoo YS (2004) A Positive Role of the PI3-K/Akt Signaling Pathway in PC12 Cell Differentiation. Mol. Cells 18 (3), 353–359. [PubMed] [Google Scholar]
- (38).Pellegrino MJ, and Stork PJS (2006) Sustained Activation of Extracellular Signal-Regulated Kinase by Nerve Growth Factor Regulates c-Fos Protein Stabilization and Transactivation in PC12 Cells. J. Neurochem 99 (6), 1480–1493. [DOI] [PubMed] [Google Scholar]
- (39).Santos SDM, Verveer PJ, and Bastiaens PIH (2007) Growth Factor-Induced MAPK Network Topology Shapes Erk Response Determining PC-12 Cell Fate. Nat. Cell Biol 9 (3), 324–330. [DOI] [PubMed] [Google Scholar]
- (40).Sasagawa S, Ozaki Y-I, Fujita K, and Kuroda S (2005) Prediction and Validation of the Distinct Dynamics of Transient and Sustained ERK Activation. Nat. Cell Biol 7 (4), 365–373. [DOI] [PubMed] [Google Scholar]
- (41).Dijkmans TF, van Hooijdonk LWA, Schouten TG, Kamphorst JT, Vellinga ACA, Meerman JHN, Fitzsimons CP, de Kloet ER, and Vreugdenhil E (2008) Temporal and Functional Dynamics of the Transcriptome during Nerve Growth Factor-Induced Differentiation. J. Neurochem 105 (6), 2388–2403. [DOI] [PubMed] [Google Scholar]
- (42).Mok S-A, Lund K, and Campenot RB (2009) A Retrograde Apoptotic Signal Originating in NGF-Deprived Distal Axons of Rat Sympathetic Neurons in Compartmented Cultures. Cell Res. 19 (5), 546–560. [DOI] [PubMed] [Google Scholar]
- (43).Crowder RJ, and Freeman RS (2000) Glycogen Synthase Kinase-3 Beta Activity Is Critical for Neuronal Death Caused by Inhibiting Phosphatidylinositol 3-Kinase or Akt but Not for Death Caused by Nerve Growth Factor Withdrawal. J. Biol. Chem 275 (44), 34266–34271. [DOI] [PubMed] [Google Scholar]
- (44).Kristiansen M, Menghi F, Hughes R, Hubank M, and Ham J (2011) Global Analysis of Gene Expression in NGF-Deprived Sympathetic Neurons Identifies Molecular Pathways Associated with Cell Death. BMC Genomics 12, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nathanson JL, Yanagawa Y, Obata K, and Callaway EM (2009) Preferential Labeling of Inhibitory and Excitatory Cortical Neurons by Endogenous Tropism of Adeno-Associated Virus and Lentivirus Vectors. Neuroscience 161 (2), 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, and Kaplan DR (1994) Trk Receptors Use Redundant Signal Transduction Pathways Involving SHC and PLC-γ1 to Mediate NGF Responses. Neuron 12 (3), 691–705. [DOI] [PubMed] [Google Scholar]
- (47).Secondo A, Esposito A, Sirabella R, Boscia F, Pannaccione A, Molinaro P, Cantile M, Ciccone R, Sisalli MJ, Scorziello A, et al. (2015) Involvement of the Na+/Ca2+ Exchanger Isoform 1 (NCX1) in Neuronal Growth Factor (NGF)-Induced Neuronal Differentiation through Ca2+-Dependent Akt Phosphorylation. J. Biol. Chem 290 (3), 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rong R, Ahn J-Y, Chen P, Suh P-G, and Ye K (2003) Phospholipase Activity of Phospholipase C-γ1 Is Required for Nerve Growth Factor-Regulated MAP Kinase Signaling Cascade in PC12 Cells. J. Biol. Chem 278 (52), 52497–52503. [DOI] [PubMed] [Google Scholar]
- (49).Ma G, Wen S, He L, Huang Y, Wang Y, and Zhou Y (2017) Optogenetic Toolkit for Precise Control of Calcium Signaling. Cell Calcium 64, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shen J, and Maruyama IN (2011) Nerve Growth Factor Receptor TrkA Exists as a Preformed, yet Inactive, Dimer in Living Cells. FEBS Lett. 585 (2), 295–299. [DOI] [PubMed] [Google Scholar]
- (51).Che DL, Duan L, Zhang K, and Cui B (2015) The Dual Characteristics of Light-Induced Cryptochrome 2, Homo-Oligomerization and Heterodimerization, for Optogenetic Manipulation in Mammalian Cells. ACS Synth. Biol 4 (10), 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Guo S-L, Tan G-H, Li S, Cheng X-W, Zhou Y, Jia Y-F, Xiong H, Tao J, and Xiong Z-Q (2012) Serum Inducible Kinase Is a Positive Regulator of Cortical Dendrite Development and Is Required for BDNF-Promoted Dendritic Arborization. Cell Res. 22 (2), 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zhang K, Duan L, Ong Q, Lin Z, Varman PM, Sung K, and Cui B (2014) Light-Mediated Kinetic Control Reveals the Temporal Effect of the Raf/MEK/ERK Pathway in PC12 Cell Neurite Outgrowth. PLoS One 9 (3), e92917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Duan L, Hope J, Ong Q, Lou H-Y, Kim N, McCarthy C, Acero V, Lin MZ, and Cui B (2017) Understanding CRY2 Interactions for Optical Control of Intracellular Signaling. Nat. Commun 8 (1), 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.