

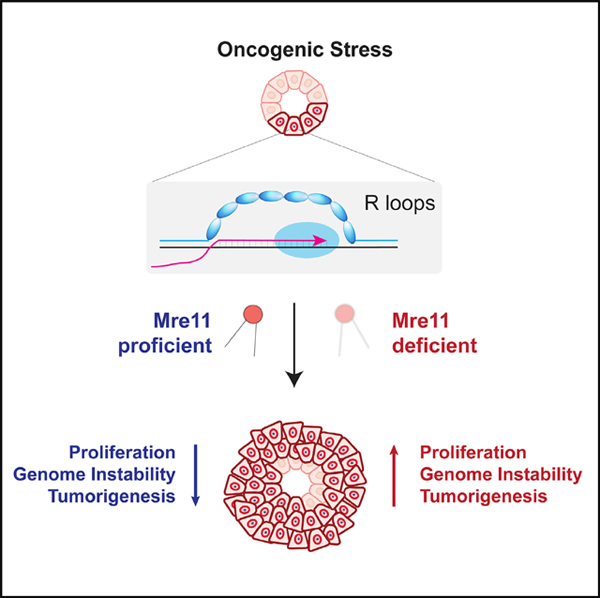
SUMMARY
The Mre11-Rad50-Nbs1 complex is a DNA doublestrand break sensor that mediates a tumor-suppressive DNA damage response (DDR) in cells undergoing oncogenic stress, yet the mechanisms underlying this effect are poorly understood. Using a genetically inducible primary mammary epithelial cell model, we demonstrate that Mre11 suppresses proliferation and DNA damage induced by diverse oncogenic drivers through a p53-independent mechanism. Breast tumorigenesis models engineered to express a hypomorphic Mre11 allele exhibit increased levels of oncogene-induced DNA damage, R-loop accumulation, and chromosomal instability with a characteristic copy number loss phenotype. Mre11 complex dysfunction is identified in a subset of human triple-negative breast cancers and is associated with increased sensitivity to DNA-damaging therapy and inhibitors of ataxia telangiectasia and Rad3 related (ATR) and poly (ADP-ribose) polymerase (PARP). Thus, deficiencies in the Mre11-dependent DDR drive proliferation and genome instability patterns in p53-deficient breast cancers and represent an opportunity for therapeutic exploitation.
In Brief
The origins of genome instability in cancer remain poorly understood. Fagan-Solis et al. reveal a p53-independent genome integrity checkpoint pathway mediated by Mre11 that protects against genome instability in breast cancer. Mre11 dysfunction in breast cancer models induces a genomic loss signature and vulnerability to PARP and ATR inhibitors.
Graphical Abstract
INTRODUCTION
Structural chromosomal instability (CIN) is a frequent hallmark of clinically aggressive cancers, such as triple-negative (estrogen receptor, progesterone receptor, and HER2-negative) breast cancer (TNBC) and high-grade serous ovarian cancer (HGSOC), yet its etiology remains poorly understood (Cancer Genome Atlas, N. and Cancer Genome Atlas Network, 2012; Jiang et al., 2010; Jonkers et al., 2001). Cancers with high levels of structural CIN are characterized by nearly universal disruption of the p53 pathway and frequent genetic aberrations that drive a hyper-proliferation phenotype (e.g., amplification of c-Myc or Cyclin E and/or Rb1 deletion). Cancers with high levels of CIN also have frequent perturbation of DNA damage response (DDR) pathway genes (Kniinenburg et al., 2018), although their relevance to genome instability and therapeutic sensitivity in p53-deficient cancers remains unclear.
Oncogene-induced hyper-proliferation stimulates DNA replication stress (Bartkova et al., 2005, 2006; Di Micco et al., 2006; Halazonetis et al., 2008), resulting in accumulation of single- and double-strand breaks (DSBs) during S phase (Gaillard et al., 2015; Hills and Diffley, 2014; Macheret and Halazonetis, 2015) and activation of the DDR. The etiology of oncogene-induced replication stress has been extensively studied. Proposed mechanisms include nucleotide depletion, oxidative stress, misregulated replication origin firing, re-replication, perturbed replication fork kinetics, and under-replicated genomic DNA (Gaillard et al., 2015; Hills and Diffley, 2014; Kotsantis et al., 2018). Recent evidence suggests that oncogene expression stimulates genome-wide activation of ectopic intragenic origins, which results in replication stress due to a higher rate of transcription replication conflicts (TRCs) (Macheret and Halazonetis, 2018). Furthermore, depletion of RNA-DNA hybrids (i.e., R-loops) by RNase H overexpression reduces the burden of oncogene-induced DSBs (Kotsantis et al., 2016). Despite these advances, the relevance of pathways that regulate R-loop-mediated genome instability in tumorigenesis models remains poorly understood.
The Mre11-Rad50-Nbs1 complex is a DSB sensor that lies at the nexus between DNA repair and DDRs. The Mre11 complex is critical for ataxia telangiectasia mutated (ATM) activation at DSBs and downstream activation of G2/M and p53-dependent G1/S cell cycle checkpoints (Oh and Symington, 2018; Stracker and Petrini, 2011; Syed and Tainer, 2018). The nuclease and structural functions of the Mre11 complex promote the resection of DSBs to generate 3′ overhangs, which are a prerequisite for homologous recombination (HR)-mediated repair and replication fork stability (Hashimoto et al., 2011; Lemacon et al., 2017; Trenz et al., 2006). Although complete deficiency is lethal, hypomorphic alleles of Mre11 complex genes are causative for ataxia-telangiectasia-like disorder and Nijmegan breakage syndrome and have helped to establish critical roles for this pathway in suppression of replication-associated DSBs and response to exogenous clastogens. Recent findings also implicate the Mre11 complex and its closely associated nuclease, Sae2/CtIP, in the resolution of R-loops in mammals (Chang et al., 2018; Makharashvili et al., 2018).
Accumulating evidence supports a tumor-suppressive function for the Mre11 complex. Individuals with rare germline variants in Mre11 complex genes are at elevated risk of developing breast cancer (Damiola et al., 2014; Heikkinen et al., 2006). Although somatic mutations in Mre11 complex genes are infrequent in cancer (~3%; Zehir et al., 2017), aberrantly reduced protein expression has been identified in subsets of bladder, colorectal, breast, and ovarian cancers (Bartkova et al., 2008; Brandt et al., 2017; Choudhury et al., 2010; Situ et al., 2019; Zaki et al., 2014). Mice expressing the hypomorphic Mre11ATLD1 allele also exhibit accelerated tumorigenesis driven by p53 mutation (Theunissen et al., 2003), Chek2 deficiency (Stracker et al., 2008), and mammary-specific Her2 expression (Gupta et al., 2013). Her2-driven breast cancers engineered to express a hypomorphic Mre11 complex exhibit increased genome instability and greater metastatic potential (Gupta et al., 2013). The mechanisms for these diverse tumor-suppressive effects have not been elucidated and may entail ATM- and p53-independent pathways. Indeed, ATM-independent tumor suppression was recently demonstrated using mice with knockin of a hypomorphic Nbs1 allele (Nbs1ΔB) (Balestrini et al., 2016). Here, Mre11 complex dysfunction was associated with common fragile site instability and dramatically increased genomic instability in the induced lymphomas. Collectively, these observations highlight an important role for the Mre11 complex in tumor suppression and protection against genome instability. A greater understanding of these Mre11-dependent effects may have important implications for classification of human tumors and identification of potential therapeutic vulnerabilities.
The goal of this study was to characterize the effects of Mre11 complex dysfunction on oncogenic proliferation, DNA damage, and genome instability in p53-proficient and p53-deficient inducible breast cancer models. We use single-cell, whole-genome sequencing to characterize the effect of Mre11 dysfunction on the landscape of Her2-induced genomic instability in mammary epithelial hyperplasia. We further establish p53-independent effects of Mre11 in suppressing oncogene-induced cell proliferation, DNA damage, and R-loops induced by diverse oncogenic drivers. We characterize a murine model of Rb1−/−Trp53−/− breast cancers with Mre11 hypomorphism that reveals abundant expression of R-loops and a marked increase in genomic deletions relative to other types of chromosomal rearrangements. Finally, drug sensitivity analyses of engineered p53-deficient breast tumors with or without Mre11 dysfunction identify therapeutic vulnerabilities that may be clinically exploitable.
RESULTS
Early Induction of Chromosomal Instability by Oncogenic Stress in Primary Mammary Epithelial Cells
We previously demonstrated that breast cancers initiated by Her2/Neu expression (abbreviated here as “Her2”) develop with shorter latency and increased levels of CIN in mice expressing a hypomorphic allele of Mre11 (Mre11ATLD1/ATLD1; abbreviated Mre11ATLD; Gupta et al., 2013). To investigate whether Mre11 hypomorphism alters CIN patterns during early stages of oncogenic hyperplasia, we established a system for propagating primary murine mammary epithelial cells (pMMECs) on lethally irradiated LA7 mammary stem cell feeder cells (Ehmann et al., 1984; Jechlinger et al., 2009). Consistent with our previously reported in vivo findings, we observed significantly greater Her2-induced proliferation in Mre11ATLD pMMECs relative to wild-type (WT) pMMECs (Figure 1A). Because oncogene-induced CIN during preneoplasia is stochastic and precedes clonal expansion, analysis of CIN patterns requires a single cell-based approach. Accordingly, we performed low-depth (~0.2x), single-cell, whole-genome sequencing (scWGS) in WT and Mre11ATLD pMMECs 14 days after lentiviral transduction with EGFP alone or EGFP+Her2 (Figure 1A). Copy number alterations (CNAs) were detected at single-cell resolution by pooling mapped reads into genomic bins with average size of 25 kb, using a previously described analysis pipeline (Garvin et al., 2015; Martelotto et al., 2017). The CNA profiles of 24 single cells from each genotype (WT+EGFP, WT+Her2, Mre11ATLD+EGFP, and Mre11ATLD+Her2) are depicted as a clustered heatmap, with the total fraction of altered genome for each cell shown on the right as a bar graph (Figure 1B). Significantly, WT pMMECs expressing EGFP had very few CNAs, corroborating the low level of background signal associated with the scWGS methodology and analysis pipeline employed in this study. In contrast, there is a substantial increase in the fraction of genome altered per cell after both Her2 expression and Mre11 perturbation (Figure 1B, bar graph on right). Unsupervised clustering was also performed but did not reveal any clonally related individual cells (Figure 1B), which is consistent with the relatively short time period after transduction when the cells were analyzed. Although the majority of the observed CNAs are non-recurrent and thus are indicative of a sporadic etiology, there are some focal CNAs that were recurrently seen in multiple cells within the same genotype. An interesting example is a focal region in chromosome 1 (boxed region in Figure S1) that has increased ploidy in 4/24 WT+Her2 pMMECs and 5/24 Mre11ATLD+Her2 pMMECs, but not in the other genotypes analyzed. This minimal region of CNA overlap contains Parp1, which is instrumental for single-strand break repair (Ray Chaudhuri and Nussenzweig, 2017) and is frequently overexpressed in human HER2+ breast cancers (Stanley et al., 2015). Furthermore, a recent study suggests that Parp1 activity reduces the rate of replication fork progression (Maya-Mendoza et al., 2018). Consistent with these prior studies, our findings are suggestive that Parpl copy number gains may confer a growth advantage in the setting of Her2-induced mammary preneoplasia.
Figure 1. Oncogene Expression Rapidly Induces Copy Number Aberrations in Individual pMMECs.
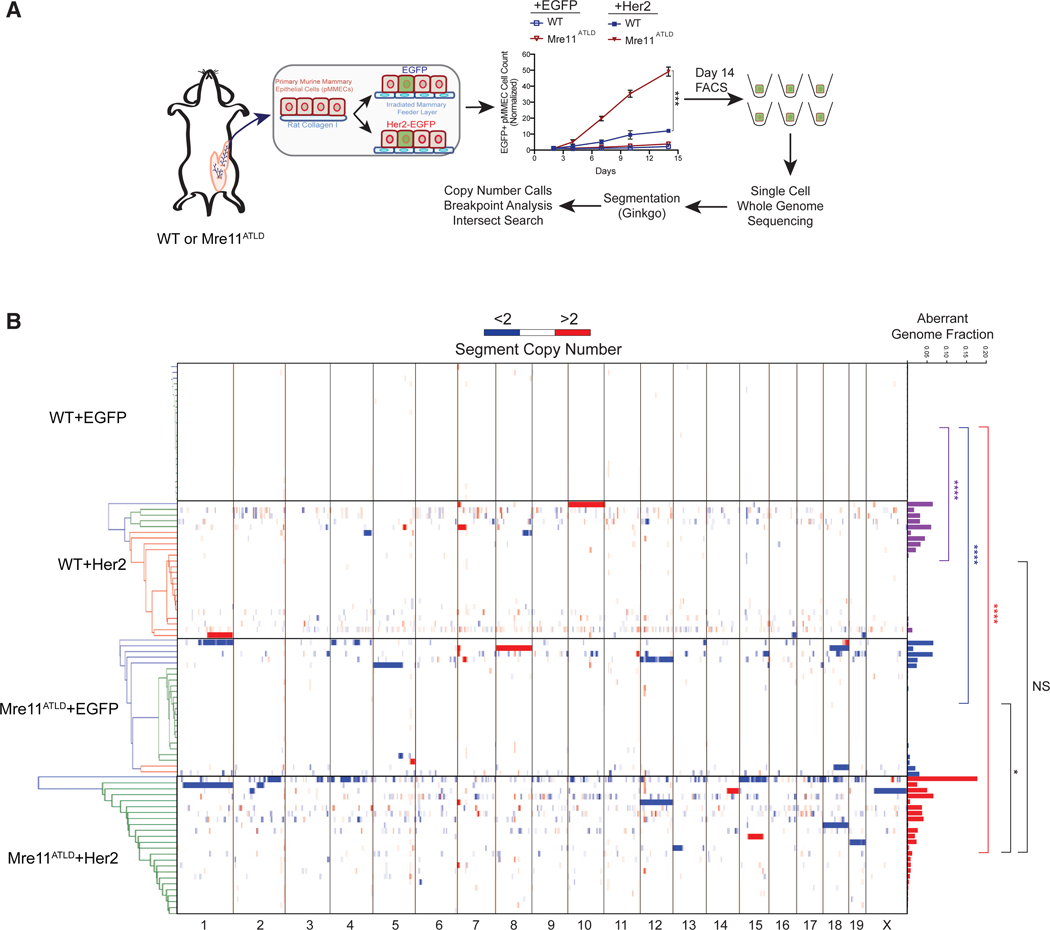
(A) Schematic representation of the single-cell, whole-genome sequencing process. WT or Mre11ATLD pMMECs were transduced with EGFP or Her2-EGFP. Growth curves on an irradiated feeder layer are shown. The mean of three biological replicates is shown with error bars depicting the standard error of the mean (SEM). ***p < 0.001, calculated using a two-tailed t test on log transformed day 14 data. On day 14, fluorescence-activated cell sorting (FACS) was performed to sort single EGFP+ cells into a 96-well microtiter plate. These cells were then processed for sequencing as described in the methods.
(B) Heatmap showing copy number of individual segments for each cell. The individual cells are clustered by geometric distance, which does not reveal any clonal relationships. The histograms at the right show the total fraction of aberrant genome per cell. p values were calculated by two-tailed Mann-Whitney test. ****p < 0.0001; *p < 0.05.
See also Figure S1.
Mre11 Dysfunction Results in a Copy Number Loss Phenotype
We observed a significant increase in CNAs per cell after Her2 expression in both WT and Mre11ATLD pMMECs (Figure 2A), indicating that oncogenic stress stimulates CIN early in neoplasia and does not require DDR deficiency. In fact, we did not observe an increase in CNAs per cell in Mre11ATLD pMMECs expressing Her2, suggesting that oncogene expression is the major driver of CNA development in this model. Consistent with a role for Mre11 in suppressing spontaneous genomic instability, Mre11ATLD pMMECs expressing EGFP also had a modest increase in CNAs compared to the respective WT control (Figure 2A). We next evaluated the size distribution of observed CNAs in the different genotypes. Notably, the CNA size distributions in both EGFP and Her2-expressing Mre11ATLD pMMECs were significantly larger than the CNA size distribution observed in WT+Her2 pMMECs (Figure 2B). This difference was even more striking when evaluating CNAs that give rise to genomic gains versus losses. Although the size distribution of CNA gains and losses (≤5,000 kb) was equivalent in the WT pMMEC genotypes, we observed a highly significant (p < 0.0001) enrichment for larger size genomic loss CNAs in both EGFP and Her2-expressing Mre11ATLD pMMECs (Figure 2C). The enrichment for genomic loss CNAs in Mre11ATLD pMMECs is also visually evident in the scWGS heatmaps shown in Figure 1B. To better characterize the relationship between CNA size and the observed enrichment for genomic losses, we plotted the cumulative frequency distribution of CNA gains and losses according to individual aberration size (Figure 2D). This analysis demonstrated that the enrichment for genomic loss was evident for CNAs larger than 1,000 kb in Mre11ATLD+EGFP cells and for CNAs greater than 500 kb in Mre11ATLD+Her2 cells. In contrast, no enrichment for CNA losses was observed in WT+Her2 pMMECs. The enrichment for genomic losses in the setting of Mre11 hypomorphism is reminiscent of copy number losses that result from accumulation of under-replicated DNA (UR-DNA) in settings of elevated replication fork stress in other model organisms (Salim et al., 2017; Yarosh and Spradling, 2014). Thus, our findings are consistent with replication fork instability caused by Mre11 dysfunction in the setting of oncogenic stress, resulting in an accumulation of UR-DNA. Furthermore, because copy number losses are one mechanism by which loss of heterozygosity (LOH) in cancer may arise, an enrichment for larger sized deletions in Mre11ATLD pMMECs may be consistent with the high levels of LOH that are observed in human breast and ovarian cancers with homologous recombination deficiency (Wang et al., 2012).
Figure 2. Mre11 Dysfunction Alters the Profile of Copy Number Aberrations to Favor Genomic Loss.
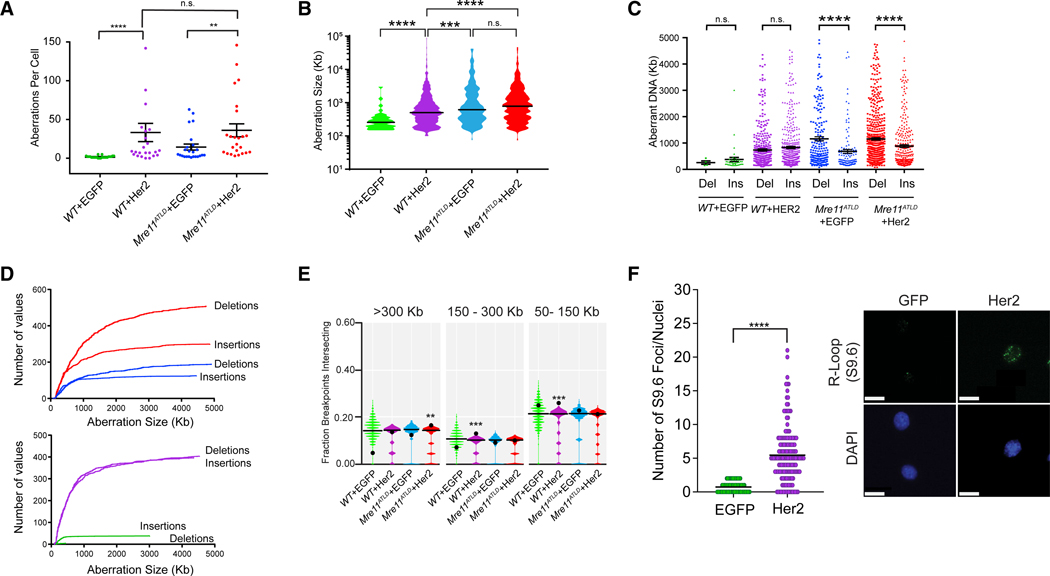
(A) Scatterplot showing total aberrations per cell in the different genotypes of pMMECs (mean ± SEM). **p < 0.01; ****p < 0.0001 bytwo-tailed Mann-Whitney test.
(B) Violin plots depicting the size distribution of aberrant copy number regions in each cell type. The line represents the median value. ***p < 0.001; ****p < 0.0001 by two-tailed Mann-Whitney test.
(C) Scatterplot CNA segment size for deletions and insertions, demonstrating a bias toward genomic deletions in Mre11ATLD genotypes. Error bars are median ± 95% confidence level. ****p < 0.0001 by two-tailed Mann-Whitney test.
(D) Cumulative size distribution CNA deletions and insertions for WT+EGFP (green), WT+Her2 (purple), Mre11ATLD+EGFP (blue), and Mre11ATLD+Her2 (red) pMMECs.
(E) Breakpoint regions in Mre11ATLD+Her2 cells are associated with the largest genes (>300 kb). Violin plots show an empirical null distribution of overlaps generated by shuffle permutation of the observed CNA data, as described in the methods. Black line in violin plots is the median value, and the black dot is the observed value. p values are approximated from the empirical null distribution. *p = 0.03–0.05; **p = 0.01–0.03; ***p < 0.01.
(F) Expression ofHer2 increases the number of R-loop DNA-RNA hybrids, detected by S9.6 immunofluorescence staining of WT pMMECs transduced with EGFP control or Her2. Scatterplot of the number of S9.6 foci per nucleus in control and Her2-expressing cells is shown. On the right are representative images ofS9.6 nuclear foci used to generate the scatterplot. ****p < 0.0001 using a two-tailed t test. Scale bar represents 5 μm.
See also Figure S2.
Chromosomal Aberrations Are Associated with Large Genes
Next, we investigated whether there were any distinguishing features of the genomic regions where the oncogene-induced CNAs were observed. The sparseness of our scWGS data precluded single-base-pair resolution of CNA breakpoints. In contrast, we approximated the genomic region of the chromosomal aberration breakpoint as contained within the two genomic bins that span the transition in copy number (Figure S2A; STAR Methods). We next evaluated whether these breakpoint-containing regions were enriched in previously annotated genomic features and used a shuffle permutation of the observed CNA profiles for each of the genotypes as a means of establishing statistical significance of the enrichment (Supplemental Methods). We did not observe any significant enrichment or depletion of satellite, simple repeat, long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), or CpG island sequences among the breakpoint regions for any of the genotypes (Figure S2B). In contrast, there was significant enrichment of breakpoint-containing regions with genes. Genes were further subdivided according to size into four bins of <50 kb, 50–150 kb, 150–300 kb, and >300 kb. There was a significant enrichment of chromosomal breakpoint regions within large genes (>300 kb) relative to random chance for Her2-expressing Mre11ATLD pMMECs (Figure 2E). In contrast, chromosomal breakpoint regions in WT+Her2 pMMECs were relatively enriched in moderately sized genes between 50 kb and 300 kb. Large genes are known to take longer than one cell cycle to transcribe and have been associated with R-loops, chromosomal fragile sites, and recurrent deletions in many cancers (Glover et al., 2017; Helmrich et al., 2006, 2011; Le Tallec et al., 2013; Wilson et al., 2015). Recent work has also demonstrated an increase in transcription-replication conflicts (TRCs) upon oncogene expression (Macheret and Halazonetis, 2018), which may be due to greater transcriptional activity and/or persistence of R-loops (Kotsantis et al., 2016). We thereby assessed global R-loop levels in our pMMEC model using immunofluorescence with the monoclonal S9.6 antibody and observed a significant increase in R-loops 3 days after transduction with lentivirus expressing Her2-EGFP in pMMECs, relative to control pMMECs transduced with EGFP alone (Figure 2F). Collectively, these findings indicate that oncogenic stress in early mammary neoplasia stimulates R-loop expression and CNAs enriched in large genes, which in the setting of Mre11 dysfunction becomes strongly enriched in a copy number loss phenotype.
Mre11 Suppresses Oncogenic Proliferation Independently of p53 and ATM
We next investigated the effect of Mre11 hypomorphism in transgenic mammary hyperplasia models induced by c-Myc (Myc) overexpression, Rb1 deletion, and/or Trp53 deficiency–genetic aberrations that are prevalent in human TNBCs and basal-like breast cancer (Cancer Genome Atlas, N. and Cancer Genome Atlas Network, 2012). Because the Mre11ATLD allele is a premature stop codon in the C-terminal region of Mre11, we hypothesized that a comparable hypomorphic allele could be generated using CRISPR/Cas9 gene targeting in pMMECs, as has previously been demonstrated in murine embryonic fibroblast cell lines (Wyatt et al., 2016). pMMECs isolated from Rosa26LSL-Cas9-EaFP (abbreviated R26Cas9) mice (Platt et al., 2014) were transduced with lentivirus expressing Cre recombinase and single guide RNA (sgRNA) targeting the C-terminal region of Mre11 (Cre-sgMre11) or a control non-coding region on chromosome 2 (Cre-sgControl; Figures 3A and 3B). Effective target site mutagenesis was confirmed by Sanger sequencing of 10 cloned amplicons each from R26Cas9 pMMECs transduced with Cre-sgControl or Cre-sgMre11 (Figure S3A). CRISPR-directed mutations corresponding to the expressed sgRNA were identified, which resulted in an Mre11 frameshift mutation in all 10 clones analyzed from sgMre11-transduced pMMECs (Figure S3A). Furthermore, pMMECs transduced with Cre-sgMre11 had reduced expression of Mre11 (Figure S3B), phenocopying the destabilizing effect of the Mre11ATLD allele on Mre11 complex protein stability (Theunissen et al., 2003) These findings validated the pMMEC platform to evaluate phenotypes associated with oncogene expression and/or Mre11 hypomorphism.
Figure 3. Mre11 Suppresses Oncogenic Proliferation in pMMECs Independently of Trp53 and ATM.
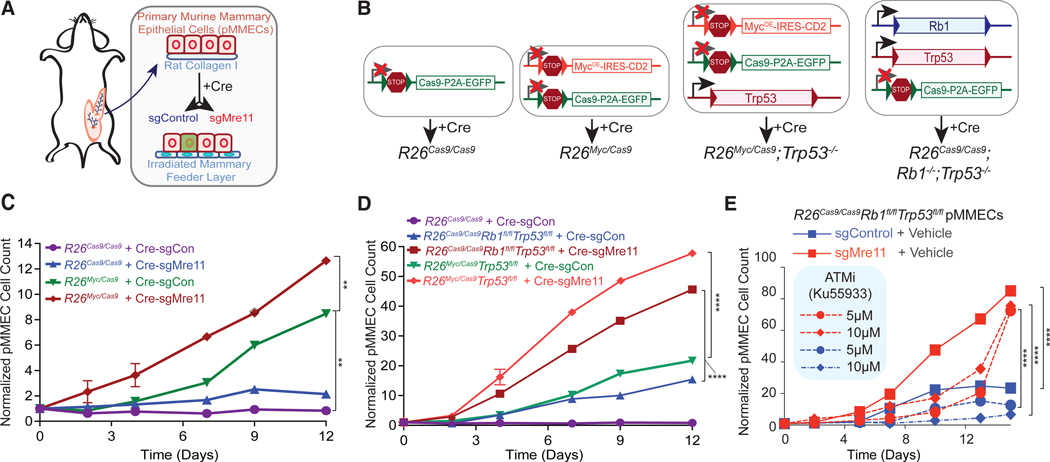
(A) Graphic representing how pMMECs are harvested from the mice and manipulated in vitro to assess growth rates.
(B) Description of compound transgenic mice used for pMMEC experiments and resulting genotypes after introduction of Cre recombinase.
(C) pMMEC growth curves examining the effect of Myc overexpression and/or Mre11 hypomorphic mutation. Cell counts are normalized to their respective day 0 counts.
(D) Significant effect of Mre11 mutation on oncogenic growth induced by Myc overexpression or Rb1 deletion in p53-deficient pMMECs. Cell counts are normalized to their respective day 0 counts.
(E) ATM inhibitor Ku55933 does not phenocopy the growth-stimulating effects of Mre11 mutation in Rb1−/−Trp53−/− pMMECs. Statistical significance in (C)–(E) was determined by two-tailed t test on log-transformed data comparing day 12. The p values were adjusted for multiple comparisons by the method of Holm-Sidak. Data are represented as mean ± SEM.
See also Figure S3.
We next interbred the R26Cas9 mice with transgenic mice containing a Cre-inducible Myc overexpression cassette also at the Rosa26 locus (Rosa26LSL-MycOE-hCD2 or R26Myc) to generate combination transgenic R26Myc/Cas9 mice (Figures 3A and 3B). pMMECs derived from these mice were transduced with lentivirus expressing Cre recombinase and either sgControl or sgMre11, revealing greater Myc-induced proliferation in cells with a hypomorphic Mre11 complex (Figure 3C). Notably, expression of Cre-sgMre11 in R26Cas9/Cas9 pMMECs did not result in a substantial increase in proliferation, indicating that this anti-proliferative effect of Mre11 is oncogene specific.
To investigate whether suppression of oncogene-induced proliferation by the Mre11 complex is p53 dependent, we generated R26Myc/Cas9; Trp53fl/fl mice. Cre recombinase expression in pMMECs isolated from these mice induces deletion of Trp53 exon 2 in conjunction with activated expression of Cas9 and Myc (Figures 3B and S3). Mre11 complex hypomorphism induced by expressing Cre recombinase and sgMre11 resulted in significantly increased proliferation relative to the R26Myc/Cas9; Trp53fm pMMECs transduced with Cre and sgControl (Figure 3D). We also generated and analyzed R26Cas9Trp53fl/flRb1fl/fl mice, representing another model of human TNBC (Figure 3B; Cancer Genome Atlas, N. and Cancer Genome Atlas Network, 2012; Jiang et al., 2010; Jonkers et al., 2001). Once again, R26Cas9Trp53mflRb1ml pMMECs transduced with Cre recombinase and sgMre11 demonstrated significantly increased proliferation relative to cells expressing sgControl (Figure 3D). These findings strongly argue that Mre11-mediated suppression of oncogenic proliferation is p53 independent. Given that the Mre11 complex has both ATM-dependent and independent effects in the response to DNA damage and replication stress (Rein and Stracker, 2014), we sought to determine the ATM dependency of Mre11-mediated suppression of oncogenic proliferation. R26Cas9Trp53flmRb1fl/fl pMMECs were transduced with either sgControl or sgMre11 and then treated with ATM inhibitor Ku55933 or vehicle control. ATM inhibition (Figure S3D) did not increase proliferation of Rb1−/−Trp53−/− pMMECs (Figure 3E). A possible explanation for these observations is that growth suppression by ATM is p53 dependent, consistent with emerging clinical evidence that ATM and TP53 may be epistatic in breast cancer (Weigelt et al., 2018). Conversely, Mre11 hypomorphism stimulated oncogenic proliferation irrespective of ATM inhibition (Figure 3E). These observations collectively indicate that Mre11-mediated suppression of oncogenic proliferation is both p53 and ATM independent.
Mre11 Suppresses Oncogene-Induced R-Loops and DNA Damage
The Mre11 complex suppresses the accumulation of spontaneous, replication-associated DSBs (Oh and Symington, 2018). In contrast, some studies have demonstrated that Mre11 nuclease activity can also promote destabilization of stalled replication forks (Malacaria et al., 2019; Schlacher et al., 2011). To determine whether Mre11 promotes or suppresses oncogene-induced DSBs in pMMECs, we measured γH2AX foci, 53BP1 foci, and neutral COMET tails (Figures 4A, 4B, and S4A). Mre11 hypomorphism induced by CRISPR/Cas9-mediated targeting of Mre11 resulted in significantly higher levels of nuclear DSBs in both p53-proficient and p53-deficient pMMEC models. Similarly, Mre11 hypomorphism increased the levels of single-stranded DNA damage, as measured by pRPA2 immunofluorescence and alkaline COMET assay (Figures 4B and S4B). The increase in pRPA2 foci was most significant in cells that were no longer in S phase (Figure S4C), indicative of a deficiency in resolution/repair of replication stress in Mre11 mutant pMMECs. These findings argue that Mre11 suppresses the accumulation of oncogenic DNA damage (single-stranded breaks and DSBs) in both p53-proficient and p53-deficient models of mammary preneoplasia.
Figure 4. Mre11 Suppresses Oncogene-Induced DNA Damage and R-Loops in p53-Proficient and p53-Deficient Models.
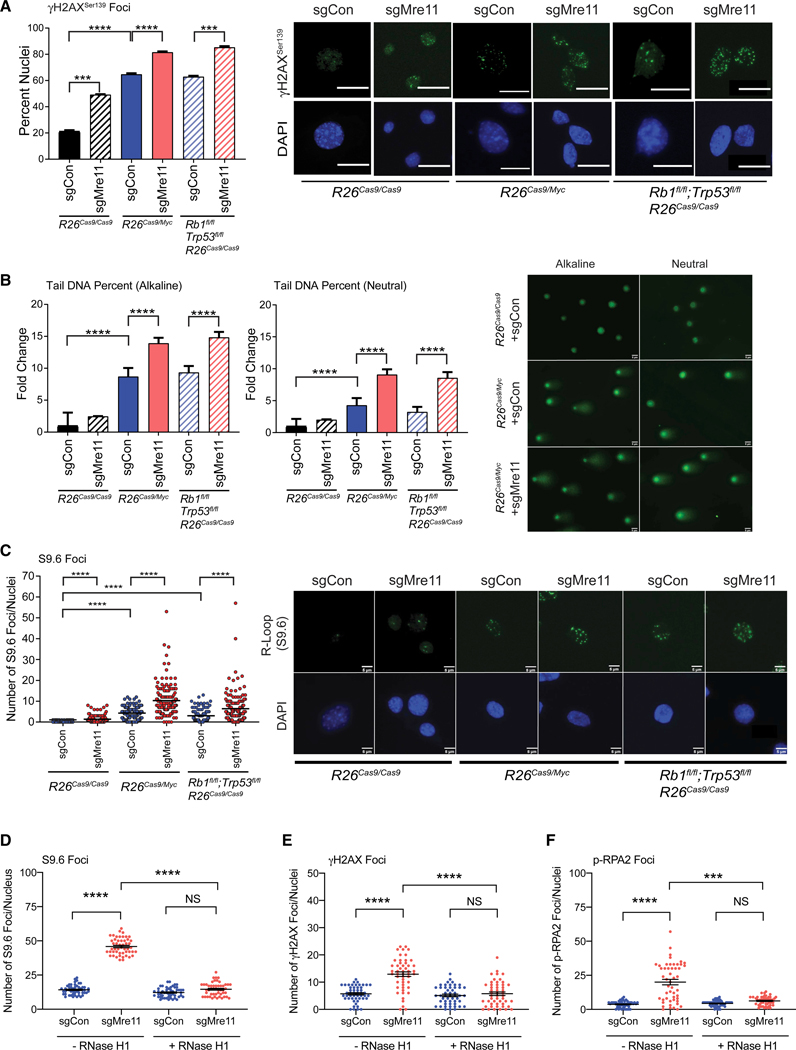
(A) Mre11 suppresses oncogene-induced γH2AX foci formation in both p53-proficient and p53-deficient pMMECs. Bar graphs show quantification of the percent of nuclei containing ≥5 γH2AXfoci in the different genotypes. Representative images (right) of the nuclei containing γH2AX foci are shown. White bar indicates 5 μm.
(B) Bar graphs depicting the fold change in tail DNA percent for both alkaline (left) and neutral (right) COMET assays in pMMECs with the genotypes shown post-Cre-sgRNA transduction. Representative images of alkaline and neutral COMETs in R26Cas9+sgControl, R26Cas9/Myc+sgControl, and R26Cas9/Myc+sgMre11 pMMECs are shown. Data are represented as mean ± SEM.
(C) Mre11 suppresses oncogene-induced R-loop formation independently of Trp53. Scatterplot shows a quantification of the nuclear S9.6foci in pMMECs from the genetic backgrounds shown after transduction with Cre-sgControl versus Cre-sgMre11. Representative images (right) of the nuclei containing S9.6 foci are shown. White bar indicates 5 μm.
(D) S9.6 (R-loop) foci after RNase H1 overexpression in R26CasaRb1fl/flTrp53fl/fl pMMECs transduced with Cre-sgControl or Cre-sgMre11.
(E and F) Additionally, RNase H1 overexpression counteracts the increase in (E) γH2AX and (F) p-RPA2 foci seen in Mre11 hypomorphic R26CasaRb1fl/flTrp53fl/fl pMMECs.
***p < 0.001; ****p < 0.0001. p values are calculated using a two-tailed Mann-Whitney test.
See also Figure S4.
Due to our prior finding that oncogenic stress in the setting of Mre11 hypomorphism induces copy number aberrations in genomic regions known to have high levels of R-loops (see Figure 2E), we assessed global R-loop levels using S9.6 immunofluorescence. As previously demonstrated with Her2 expression (see Figure 2F), we again found that oncogenic stress stimulates R-loop foci in both R26Myc and R26Cas9Rbr−/−Trp53−/− pMMECs (Figure 4C). Significantly, we also found R-loop levels increased substantially further upon Mre11 dysfunction, in both p53-proficient and p53-deficient models (Figure 4C). These observations are consistent with a recent study implicating the Mre11 complex as a critical mediator of transcription-replication conflicts in yeast and mammals (Chang et al., 2019). To establish whether the increase in R-loops mediates the increased levels of oncogenic DNA damage in pMMECs with hypomorphic Mre11, we modified our Cre-sgControl/sgMre11 lentiviral construct to also co-overexpress RNase H1 (Figure S4D). As expected, co-overexpression of RNase H1 abolished the increase in R-loops observed in the setting of hypomorphic Mre11 (Figure 4D). RNase H1 expression also rescued the increase in oncogenic DSBs and single-stranded DNA breaks induced by Mre11 dysfunction (Figures 4E and 4F). These findings were also confirmed by neutral and alkaline COMET assays (Figures S4E and S4F). Thus, an aberrant accumulation of R-loops in pMMECs with Mre11 dysfunction is necessary for the observed increase in oncogenic DNA damage. Collectively, these findings support a model wherein Mre11 mitigates DNA damage incurred at sites of oncogene-induced transcription-replication conflicts (TRCs) (see Figure 7).
Figure 7. Model Depicting Genome Stabilizing Functions of Mre11 at Oncogene-Induced Transcription-Replication Conflicts.
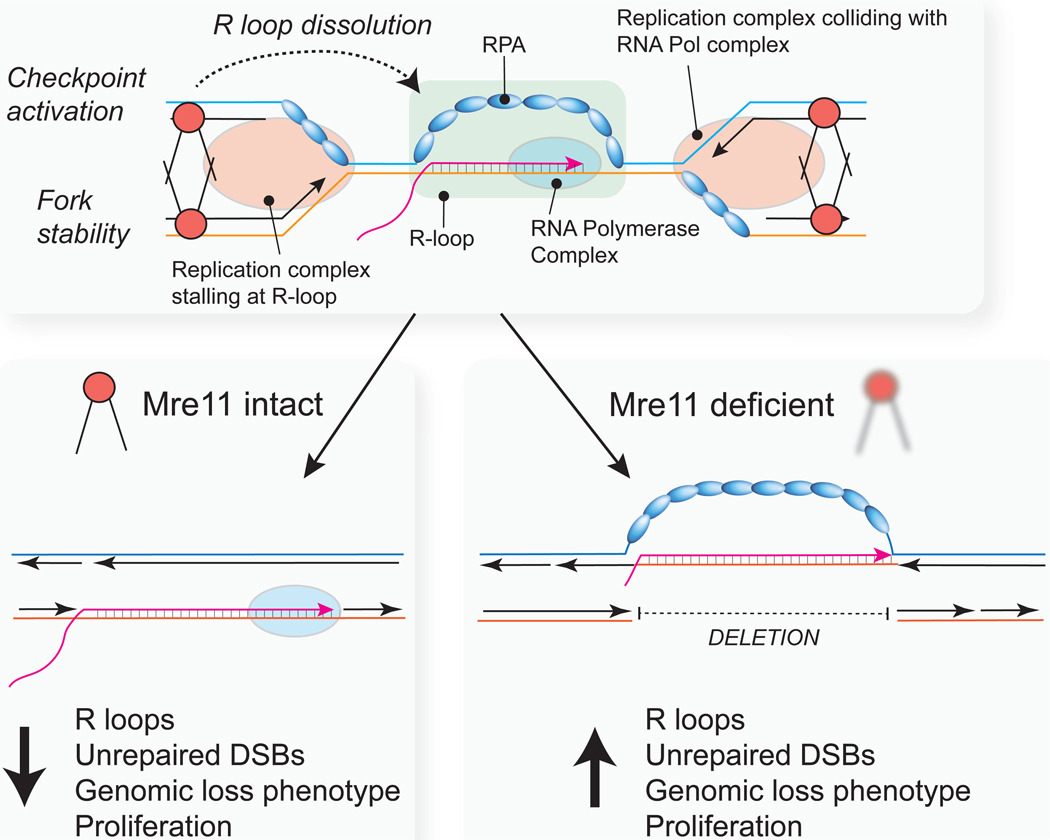
Mre11 dysfunction during oncogenic breast neoplasia results in accumulation of R-loops, replication-associated DSBs, under-repllcated DNA, an enrichment of genomic deletions, and uncontrolled proliferation. See the Discussion for further details.
Mre11-Deficient Mammary Tumors Have Increased R-Loops and a Genomic Loss Phenotype
To extend and validate these results in vivo, R26Cas9Rb1fl/flTrp53fl/fl and R26Myc/Cas9 female mice were administered intraductal injections of lentivirus expressing Cre-sgControl or Cre-sgMre11 into mammary gland 4. Two weeks after injection, the mice were euthanized and gland 4 (injected) and gland 5 (uninjected control) were processed for histopathology. There was a qualitative (Figure 5A) and quantitative (Figure 5B) increase in mammary epithelial hyperplasia after intraductal injection with Cre-sgMre11, relative to glands injected with Cre-sgControl. Thus, Mre11 suppresses oncogenic hyperplasia in vivo in response to diverse oncogenic drivers and in the setting of induced p53 deficiency.
Figure 5. Cell Proliferation, R-Loops,and Genomic LossSignatureAre Elevated inRb1−/−Trp53−/− MammaryTumors Engineeredwith Mre11 Hypomorphism.
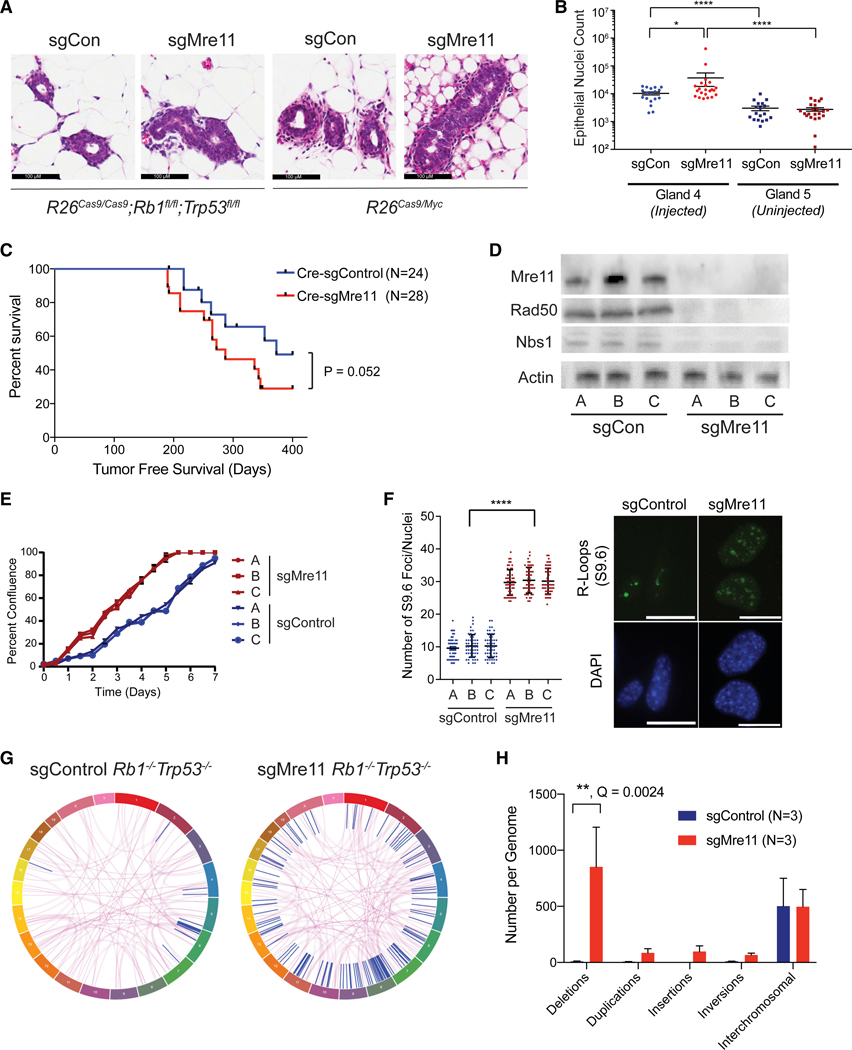
(A) Representative images of mammary hyperplasia 2 weeks after intraductal injection of R26Cas9Rb1fl/flTrp53fl/fl or R26Cas9/Myc mice with Cre-sgControl or Cre-sgMrell-expressing lentivirus.
(B) Quantification of mammary hyperplasia in R26Cas9Rb1fl/flTrp53fl/fl mice as shown in(A). The fifth, non-injected, mammary gland serves as a negative control. Data are represented as mean ± SEM. Significance was determined using two-tailed Mann-Whitney test.
(C) Kaplan-Meier tumor-free survival plot of R26Cas9Rb1fl/flTrp53fl/fl mice after mammary intraductal injection of either Cre-sgControl or Cre-sgMre11 lentivirus. p value was calculated using a two-tailed Gehan-Breslow-Wilcoxon test.
(D) Mre11, Rad50, and Nbs1 protein levels in tumorsfrom mice in (C), validating protein destabilization induced by CRISPR/Cas9-mediated Mre11 mutagenesis.
(E) Cellsfrom the sgMre11 tumors growfaster in vitro than cellsfrom the sgControl tumors. Three independenttumor lines are shown for each genotype. Growth curves were acquired using the Incucyte Live Cell Analysis system. Data are represented as mean ± SEM.
(F) Cellsfrom sgMre11-derived tumors have more R-loops than cells from sgControl tumors. Three independent tumor lines of each genotype were evaluated by immunofluorescence using the S9.6 antibody. Data are represented as mean ± standard deviation. Significance was determined using a two-tailed Mann-Whitney test.
(G) Representative circos plot of structural variations identified by whole-genome sequencing of WT or Mre11 hypomorphic mammary tumors derived from the R26Cas9Rb1fl/flTrp53fl/fl model. Purple lines indicate translocations between different chromosomes. Blue lines indicate intra-chromosomal rearrangements.
(H) Analysis of structural aberrations identified from tumor-normal WGS from three WT and three Mre11 hypomorphic mammary tumors from the R26Cas9Rb1fl/flTrp53fl/fl model. Deletions (>50 bp) are highly over-represented in Mrell hypomorphic Rb1−/−Trp53−/− mammary tumors. Data are represented as mean ± SEM. Q value was estimated using a two-tailed t test with false discovery rate correction using Graphpad Prism v8. All other comparisons were not statistically significant (i.e., Q < 0.05).
See also Figure S5.
Cohorts of Cre-sgControl and Cre-sgMre11-injected R26Cas9Rb1fl/flTrp53fl/fl mice were monitored for mammary tumor development. Tumor initiation frequency was higher in glands injected with Cre-sgMre11 (~70%) versus Cre-sgControl (~50%), and there was a trend toward a shorter latency period (Figure 5C). Tumor-free survival rates, however, did not achieve statistical significance (p = 0.052), suggesting that tumor latency in the R26Cas9Rb1fl/flTrp53fl/fl model may also be dependent on the stochastic acquisition of secondary genetic events. Mammary tumor lines were established from the induced tumors, and as expected, expression of the entire Mre11 complex (Mre11, Rad50, and Nbs1) was substantially reduced in tumors derived from mice injected with Cre-sgMre11 (Figure 5D). Consistent with our observations with pMMECs, sgMre11 mammary tumor lines had a higher proliferation rate (Figure 5E) and significantly higher levels of R-loops (Figure 5F) relative to sgControl mammary tumor lines.
We also performed matched tumor-normal whole-genome sequencing (~25x mean depth of coverage) of three Cre-sgControl and three Cre-sgMre11-induced mammary tumors in R26Cas9Rb1fl/flTrp53fl/fl mice. Site-specific mutagenesis of the Mre11 locus was confirmed in all of the Cre-sgMre11 mammary tumors (data not shown). Single-nucleotide variant mutation signatures were not statistically different between the Cre-sgControl and Cre-sgMre11 tumors (Figure S5). Notably, there was no increase in COSMIC signature 3, which has been associated with homologous-recombination-deficient cancers (Alexandrov et al., 2013; Riaz et al., 2017; Rosenthal et al., 2016). A similar lack of COSMIC signature 3 enrichment has been observed in human breast cancers with ATM deficiency (Weigelt et al., 2018). Structural variants (SVs) were observed in both sgControl and Cre-sgMre11 Rb1−/−Trp53−/− mammary tumors (Figure 5G). Significantly, the observed SVs in Mre11 hypomorphic tumors were highly enriched for deletions relative to control tumors (Figures 5G and 5H). Thus, the genomic loss phenotype observed in early oncogenic hyperplasia induced in the setting of Mre11 hypomorphism (see Figures 2C and 2D) is also preserved upon tumorigenesis in a distinct p53-deficient breast cancer model. These observations indicate that Mre11 complex dysfunction is causative for a genomic scar phenotype consisting of an enrichment for copy number losses/genomic deletions.
Breast Cancers with Mre11 Dysfunction Are Hypersensitive to DNA-Damaging Therapy and Inhibitors of ATR and PARP
Our finding that the Mre11 complex has p53-independent, tumor-suppressive functions leads to a hypothesis that Mre11 complex dysfunction may be selected for in p53-deficient breast cancers. Indeed, a prior study has demonstrated downregulation of Mre11 complex proteins in TNBC (Bartkova et al., 2008), 90% of which harbor p53 pathway deficiency (Cancer Genome Atlas, N. and Cancer Genome Atlas Network, 2012). To validate these findings, we performed immunohistochemistry for Mre11 and Nbs1 on a tissue microarray of 254 early-stage TNBCs that were treated at Memorial Sloan Kettering Cancer Center. Ninety-three percent of the patients received adjuvant chemotherapy. We identified 23 samples (9%) with less than 10% of tumor nuclei staining for Mre11 or Nbs1 protein, while infiltrating stromal cells retained normal expression (Figure 6A). Notably, we found that all tumors with low Mre11 expression also had low Nbs1 expression, consistent with underexpression or destabilization of the Mre11 complex. The lack of immunoreactivity for Mre11 and Nbs1 was also confirmed on whole tissue sections from the original tumor blocks. These “Mre11 complex low” cancers presented with similar T- and N- stage at presentation relative to cancers with normal Mre11 complex expression (Figure S6). Notably, patients with Mre11 complex low TNBC had a significantly better breast-cancer-specific survival compared to the remainder of TNBCs with normal Mre11 complex expression (Figure 6B).
Figure 6. Favorable Clinical Outcomes and Therapeutic Vulnerabilities of Mre11 Hypomorphic p53-Deficient Breast Cancers.
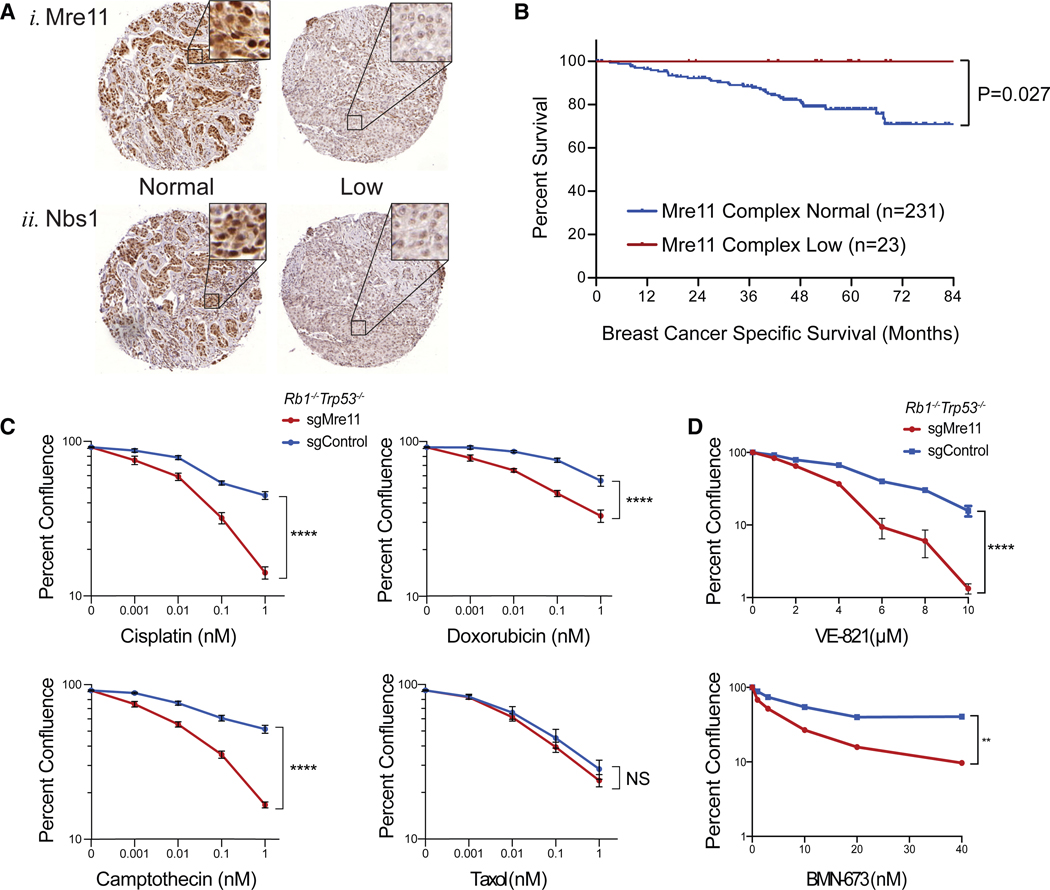
(A) Immunohistochemistry for(1)Mre11 and (2) Nbs1 performed on tissue microarrays of primary triple-negative breast cancers (n = 254). In both cases (1 and 2), the left panel represents an example of a “normal” expressor and the right case represents an example of a “low” expressing tumor.
(B) Kaplan-Meier breast-cancer-specific survival of patients with TNBC that had normal expression of Mre11 and Nbs1 or patients with TNBC that had low expression of both Mre11 and Nbs1 (i.e., Mre11 complex low). p value was calculated by a two-tailed log rank test.
(C) In vitro sensitivity of sgControl and sgMre11 Rb1−/−Trp53−/− mammary tumor lines treated with the indicated doses of cisplatin, doxorubicin, camptothecin, and Taxol. Percent confluence was measured using an Incucyte Live-Cell Analysis System 7 days after drug exposure.
(D) Similar drug sensitivity assays using the ATR inhibitorVE-821 and PARP inhibitorBMN-673. The data shown represent the average of at least two independent cell lines for each genotype with three replicates each. Significance was determined using a two-tailed t test at the highest drug dosage. Data are represented as mean ± SEM.
See also Figure S6.
Because disease-free survival of patients with TNBC is highly associated with improved chemotherapeutic responses (Cortazar et al., 2014), we hypothesized that Mre11 hypomorphism may promote hypersensitivity to DNA-damaging therapeutics in breast cancer. To directly address this possibility, we used breast tumor lines derived from R26Cas9Trp53fl/flRb1fl/fl mice injected with Cre-sgControl or Cre-sgMre11 to evaluate the effect of Mre11 hypomorphism on sensitivity to DNA-damaging therapeutics. We found that breast cancer cells expressing hypomorphic Mre11 were hypersensitive to several DNA-damaging therapeutics commonly used to treat breast cancer, but not to the anti-microtubule agent Taxol (Figure 6C). We also evaluated the sensitivity of these mammary tumor lines to inhibitors of ATR (VE-821) and PARP (BMN-673), particularly because both of these pathways are components of the replication stress response and the ATR pathway has been implicated in resolution of R-loops (Cristini et al., 2018; Hodroj et al., 2017; Nguyen et al., 2018). We observed significantly greater hypersensitivity of Mre11 hypomorphic Rb1−/−Trp53−/− breast tumor lines to both VE-821 and BMN-673, relative to control Rb1−/−Trp53−/− breast tumor lines (Figure 6D). Collectively, these findings indicate that Mre11 complex dysfunction is evident in a subset of TNBC patients and associated with improved clinical outcomes after DNA-directed cancer therapy.
DISCUSSION
Seminal studies (Bartkova et al., 2005, 2006; Di Micco et al., 2006; Gupta et al., 2013; Halazonetis et al., 2008) have described an Mre11-dependent DDR activated by oncogene-induced replication stress in preneoplasia that mediates physiologically significant tumor suppression. Some of these studies have claimed that the oncogene-induced DDR exerts these effects primarily through p53 activation. To our knowledge, ours is the first study to directly assess the effects of DDR perturbation in p53-deficient models of oncogenic preneoplasia. We demonstrate that Mre11-mediated suppression of oncogenic proliferation, DNA damage, and genome instability are p53 independent. Thus, our findings challenge current models of the oncogene-induced DDR and its relation to p53: rather than being epistatic to p53, the Mre11-dependent DDR mediates p53-independent effects that regulate oncogenic phenotypes, including proliferation, genome instability, and therapeutic sensitivity.
Single-cell, whole-genome sequencing of premalignant mammary epithelial cells revealed important clues into the critical functions of the Mre11-dependent DDR in response to oncogenic stress. We found that oncogene expression was sufficient to rapidly induce widespread CNAs in otherwise completely normal primary mammary epithelial cells, independently of Mre11 status. Recent genomic analyses of human breast preneoplasia have also suggested an early origin of chromosomal instability, which does not seem to require DDR deficiency (Martelotto et al., 2017; Rane et al., 2015). In the setting of Mre11 dysfunction, we observed a striking enrichment in genomic losses rather than gains, which was further exacerbated by oncogene expression. A similar enrichment in genomic deletions was seen in Rb1−/−Trp53−/− mammary tumors engineered to express hypomorphic Mre11 alleles, indicating that this genomic scar signature of Mre11 dysfunction persists during tumorigenesis. A possible explanation for the observed enrichment in genomic losses may be the accumulation of UR-DNA in oncogene-expressing cells with Mre11 dysfunction. Coupled with the observation that Mre11 dysfunction results in higher levels of unrepaired oncogene-induced DSBs, the increased levels of UR-DNA in this setting may be due to a higher rate of replication fork collapse (Figure 7).
The location of oncogene-induced chromosomal aberrations in the setting of Mre11 dysfunction were non-randomly distributed and significantly enriched in large genes (>300 kb). These genomic regions are known to be occupied by R-loops, depleted in replication origins, and associated with chromosomal fragility (Helmrich et al., 2011). Thus, our findings reinforce prior reports of fragile site instability induced by oncogenic stress (Miron et al., 2015) and complement the recent demonstration of oncogene-induced TRCs that stimulate intragenic dormant origin firing (Macheret and Halazonetis, 2018). Our results suggest that Mre11 promotes replication fork stability at oncogene-induced TRCs (Figure 7). R-loops can either be persistent structures that are stabilized by DNA secondary structure or transient intermediates of gene transcription. Further work to determine whether one of these types of R-loops is a greater contributor to oncogene-induced CIN is warranted. We observed a striking increase in R-loop expression in mammary hyperplasias and tumors with Mre11 dysfunction. Mitigation of R-loops by RNase H1 expression in Mre11 hypomorphic cells diminished oncogenic DNA damage. A causative role for R-loops in the etiology of oncogenic DNA damage can be explained in the setting of replication fork collisions, which can give rise to fork uncoupling and/or collapse. Thus, our findings support a model wherein the Mre11 complex has critical functions in mitigating DNA damage at TRCs in oncogene-expressing cells (Figure 7). Similar conclusions were drawn in a recent analysis of yeast and human cell line models (Chang et al., 2019). A more detailed examination of how the Mre11 complex promotes genome integrity at TRCs—particularly in consideration of head-on versus co-directional collisions (Hamperl et al., 2017)—may provide additional clues regarding the etiology of genome instability during tumorigenesis.
The observation that Mre11 mutant cells have increased proliferation despite elevated levels of unrepaired DNA damage points to an oncogene-induced DNA damage checkpoint that is mediated by Mre11. The nature of the Mre11-mediated checkpoint in response to oncogenic stress remains to be further elucidated. Our finding that this checkpoint remains operative in p53-deficient cells suggests a possible role for an Mre11-mediated G2/M checkpoint in response to oncogenic stress. We found that ATM inhibition, however, did not phenocopy the effect of Mre11 hypomorphism in promoting oncogenic proliferation, suggesting that non-canonical mechanisms may also be involved.
Reduced expression of Mre11 complex proteins has been identified in a variety of human malignancies (Bartkova et al., 2008; Brandt et al., 2017). Our data provide direct evidence supporting a role for Mre11 in mediating resistance to DNA-damaging therapy as well as PARP and ATR inhibitors in TNBCs. Indeed, low expression of Mre11 and Nbs1 was associated with improved clinical outcomes in a cohort of TNBC patients who were treated with adjuvant chemotherapy. The finding that these cancers may also be hypersensitive to targeted DDR pathway inhibitors represents opportunities for targeted therapy that may reduce the need for highly toxic combination chemotherapeutic regimens in a subset of TNBC patients.
The Mre11-dependent DNA damage response is evolutionary more primitive than p53 and functions as a mechanism to preserve genome integrity in response to endogenous and exogenous genotoxic stresses. The observation that DDR gene perturbations are prevalent across many human cancer types (Knijnenburg et al., 2018) suggests that aspects of the DDR are also tumor suppressive. Our findings reveal a p53-independent checkpoint function of the Mre11 complex that suppresses genome instability and uncontrolled proliferation induced by oncogenic mutations. Disruption of this genome integrity checkpoint may drive the catastrophic loss of chromosomal stability that is observed in a variety of clinically aggressive cancers. Elucidating additional mediators of this tumor-suppressive pathway may reveal new opportunities for personalized therapy of human cancers, including those with p53 deficiency.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gaorav Gupta (gaorav_gupta@med.unc.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293T/17 cells were obtained from American Type Culture Collection (ATCC, CRL-11268) and were cultured according to manufactures’ specifications. LA-7 cells obtained from American Type Culture Collection (ATCC, CRL-2283). LA-7 cells were maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum, 20 mM HEPES, and 10 ug/ml Insulin (“LA-7 medium”). Prior to use as feeder cells, LA-7’s were lethally irradiated with 70 Gy ionizing radiation using a Rad Source RS2000 irradiator.
Primary Murine Mammary Epithelial Cells (pMMECs)
pMMECs were derived by harvesting the 4th and 5th mammary glands from 6–12-week-old female transgenic mice with the desired genotype. Glands were incubated in Liberase digestion medium (EpiCult-B Mouse Medium Kit (Stem Cell Technologies, 285 Units CollagenaseType 3 (Worthington), 20mM HEPES (GIBCO), 20 ug/mL Liberase Blendzyme 2 (Roche) and shaken (vertically) at 37°C overnight. The resulting digestion was spun down and resuspended in 3 mls trypsin with EDTA and 1000U DNase and incubated at 37°C for 5 min. LA-7 medium (DMEM-F12 media, 10% FBS, 20 mM HEPES, 10 μg/mL Insulin, 1XL-glutamine, 1X Penicillin-Streptomycin) was added to neutralize the trypsin. Cells were spun down and resuspended in 10U Dispase (Stem Cell Technologies) and 1000U DNase I (Worthington Biochemical) and incubated at 37°C for 5 min. Cells were washed twice with LA-7 medium and the resulting cells were resuspended in EpiCult-B Mouse Medium Kit (Stem Cell Technologies) and seeded onto Cultrex3D-Culture Matrix (Trevigen) coated 6 well plates. For longer term cell growth experiments, pMMECs were seeded on lethally irradiated LA-7 cells and cultured in LA-7 medium. All cells were cultured to 80% confluence then passaged by trypsinization. Cells were tested monthly for mycoplasma using PlasmoTest Kit.
Transgenic Mouse Models
Mice used in this study were housed in the Division of Comparative Medicine at the University of North Carolina at Chapel Hill, a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). R26LSL-Cas9 (jaX#024857) and R26LSL-MycOE (JAX#020458) transgenic mouse strains were obtained from the Jackson Laboratory. Rb1fl/fl and Trp53fl/fl mouse strains were generously provided by the Perou laboratory, and originally obtained from the Frederick National Laboratory for Cancer Research (Strains #01XC1 and #01XC2). A subset of interbred mouse strains used in this study were analyzed by the Mouse Universal Genotyping Array (MUGA) from Neogen Genomics, and determined to be > 90% FVB with a minor contribution from C57BL/6J. For mammary tumor induction studies, six to twelve-week-old female R26Cas9/Cas9; Rb1fl/fl; Trp53fl/fl mice received bilateral intraductal injections, into the fourth mammary gland, containing 5 × 105 transduction units (TU) of either LentiCRISPR-Cre-V2-sgControl (Cre-sgControl) or LentiCRISPR-Cre-V2-sgControl (Cre-sgMre11) lentivirus. Mouse cohorts were palpated for the development of mammary tumors twice weekly, and three times weekly after mammary tumors had formed. Mice were euthanized using humane experimental endpoints in accordance with UNC Institutional Animal Care and Use Committee (IACUC) guidelines. At necropsy, mammary tumors were harvested and sectioned into four pieces. Two pieces were immediately flash frozen for RNA and DNA extraction. One piece with any remaining glands (4th and 5th) were fixed in 4% paraformaldehyde and processed for paraffin embedding and H&E staining (Histoserv Inc.). One piece was taken for creation of tumor lines. Briefly, tumor pieces were incubated in digestion medium (DMEM, 10% FBS, 1mg/ml Collagenase Type 3,1mg/ml Hyaluronidase) and shaken (horizontally) at 37°C for four hours. The resulting digestion was spun down and resuspended in trypsin with DNase and incubated at 37°C for 5min. LA-7 medium was added to neutralize the trypsin. Cells were spun down and resuspended in Dispase and deoxyribonuclease and incubated at 37°C for 5 min. Cells were washed twice with LA-7 medium and passed through a 70 μm filter. The resulting cells were resuspended in LA-7 media and seeded into co-culture with irradiated LA-7 feeder cells. The cells were cultured over 10 passages and analyzed via flow for GFP expression to monitor tumor cell outgrowth. Tumor lines were then utilized for downstream assays when GFP expression reached < 90%.
METHOD DETAILS
Cloning
LentiCRISPR-Cre-V2-sgRNA LumiFluor plasmid
This plasmid was created by using restriction enzymes (XbaI and BglII) to cut the Cre sequence from the pLV-Cre_LKO1 plasmid and swapping it for the Cas9 sequence in lentiCRISPR V2 using restriction digest and T4 ligation. In order to get rid of the BsmbI site within Cre, Gibson cloning was used (HiFi DNA Assembly Master Mix; NEB) to change the sequence of a Valine residue from GTC to GTA, thus removing the site while preserving the protein sequence. Using the remaining BsmB1 sites, the sequences for sgControl (53bp1 intron sequence) and sgMre11 were inserted into the sgRNA scaffolding region.
LentiCRISPR-Cre-V2-sgRNA-RNasH1
This plasmid was created via Gibson cloning of the RNaseH1 gene from the pEGFP-RNASEH1 plasmid (Addgene #108699) to replace the LumiFluor gene in the LentiCRISPR-Cre-V2-sgControl LumiFluor and LentiCRISPR-Cre-V2-sgMre11 LumiFluor plasmids.
Lentiviral_pRRL-EF1a-NeuT-LumiFluor
This plasmid was created via Gibson cloning of the Lentiviral_pRRL-EF1a-GpNLuc plasmid to insert the NeuT gene (constitutively active truncation mutant of Neu, the rodent ortholog of Her2) and an autocleavage P2A sequence upstream of the luciferase gene. NeuT-P2A was synthesized as a gene block (IDT), using pSV2-NeuT (Addgene #10919) as the reference sequence. Because NeuT is commonly used to recapitulate Her2-initiated tumorigenesis, we refer to this construct as “Her2” in the text and figures of the manuscript.
All plasmids created were confirmed by Sanger sequencing (Eton Bioscience Inc.). LentiCRISPR-Cre-V2-sgControl-Lumifluor, LentiCRISPR-Cre-V2-sgMre11-Lumifluor, LentiCRISPR-Cre-V2-sgControl-RNaseH1, LentiCRISPR-Cre-V2-sgMre11-RNaseH1, Lentiviral_pRRL-EF1a-GpNLuc, and Lentiviral_pRRL-EF1a-NeuT-LumiFluor will be made available by the Lead Contact.
Topo cloning
pMMECs were infected twice with either Cre-sgControl or Cre-sgMre11. Upon confirmation of at least 80% viral efficiency, Topo Cloning was performed per manufactures protocol. Sequencing was performed by Eton Biosciences and analysis performed using Bioedit software.
Immunofluorescence
pMMECs were infected twice with either Cre-sgControl, Cre-sgMre11, GFP, or NeuT virus. Upon confirmation of at least 80% viral efficiency, cells were seeded onto 3D-matrix coated coverslips, were treated with EdU for 10 min, and subsequently fixed by cold Methanol:Acetone (1:1) incubation at −20Cfor 10 mins. Cells then underwent EdU detection using the EdU detection kit (Baseclick) in accordance with kit instructions. Cells were then blocked in PBS + 5% FBS for 1 hour, followed by incubation in the appropriate primary antibody for 1 hour (p-γH2AX; 53bp1; p-RPA2; or S9.6), secondary antibody for 30 min, then DAPI for 1 min. Coverslips were then mounted onto slides with Prolong Gold mounting medium, cured for 2 hours and stored at 4C in the dark until imaging. Coverslips were examined on an Olympus BX61 upright wide field microscope. Resulting foci were analyzed using Fiji software (Schindelin etal., 2012).
Comet assay
pMMECs were infected twice with either Cre-sgControl, Cre-sgMre11, GFP, or NeuT virus and confirmation of at least 80% viral efficiency was determined by Flow Cytometry (Attune NxT) for GFP expression. The presence of SSBs and DSBs were analyzed via Alkaline (SSBs/DSBs) and Neutral (DSBs) comet assay using the Trevigen comet assay kit according to manufacturer’s protocol. Comet images were captured by fluorescence microscopy using Olympus BX61 upright wide field microscope. The tail DNA percent was quantified using the ImageJ software with OpenComet plug-in (Gyori et al., 2014).
Growth assays
pMMECs were infected twice with either Cre-sgControl or Cre-sgMre11 virus then seeded into 12 well plates at a density of 3 × 104 cells/well onto LA-7 feeder cells. Duplicate samples were harvested every 2–3 days for 15 days. Total cells/well were counted, cells were fixed in 3% PFA and subjected to flow analysis (Attune NxT) for the presence of GFP. Prior to ATMi growth assays, the dose of ATMi that resulted in 50% and 100% inhibition of the ATM pathway was determined. Wild-type mouse embryonic fibroblasts (MEFs) were treated with 0,1,3,5, 7,10,15, or 20 uM of the ATM inhibitor Ku55933for30 min. They were then subjected to 10 Gy, incubated under normal culture conditions and then cell lysate was collected for western blotting analysis of the phosphorylated form of the ATM downstream protein, Kap1. For ATMi growth assays pMMECs were treated as above but with the additional presence of either 5 or 10 uM of ATMi.
Viral production and infection
HEK293T/17 cells were transfected, using Polyethylenimine (PEI), with viral packaging plasmids, psPax2 and pMD2.G, and either LentiCRISPR-Cre-V2-sgControl-Lumifluor, LentiCRISPR-Cre-V2-sgMre11-Lumifluor, Lentiviral_pRRL-EF1a-GpNLuc, or Lentiviral_pRRL-EF1a-NeuT-LumiFluor, LentiCRISPR-Cre-V2-sgControl-RNaseH1, or LentiCRISPR-Cre-V2-sgMre11-RNaseH1 plasmids. Twenty-four hours post transfection, cells were washed and refed with fresh medium. Viral containing media was collected for 3 days. Collected media was filtered (0.45 um) then spun down for 2 hours at 16C at 21,000 rpm. Virus containing pellet was resuspended in PBS and incubated at 4°C for 24 hours then aliquoted and stored at −80C. For lentiviral infections, cells were transduced with the appropriate virus combined with 4μg/ml Polybrene overnight. Cells were refed with fresh viral containing medium and incubated overnight two to three times (2–3 back to back infections). Following the last infections cells were washed three times with PBS and cultured with MEGM. For testing viral efficacy, a small sample of cells were fixed with 3% Paraformaldehyde (PFA) and were assessed via flow cytometry (Attune NxT) for the presence of GFP indicating Cas9 expression or were stained with anti-CD2-PE indicating Myc expression.
Chemotherapeutic and inhibitor sensitivity assays
Tumor cells were seeded at a density of 1500 cells per well of a 96 well dish and allowed to attach overnight. After the cells were attached, various drugs or DMSO control were added to the media to the final concentrations shown in the figures. Each treatment was done in triplicate. Cells underwent IncuCyte live cell imaging (S’artorius), scanned every two hours to determine cell confluence and growth rates over seven days.
Automated analysis of mouse mammary tissues
The process of quantitative image analysis begins with the acquisition of high-resolution digital slides. FFPE sections of mouse mammary tissue stained with hematoxylin and eosin were scanned on an Aperio ScanScope XT (Leica Biosystems). Images were then uploaded to eSlide Manager and visualized with ImageScope 12.3 (Leica Biosystems). Separate tissue sections on each image were annotated by KFS. Mouse mammary glands four and five were differentiated based on the location of the lymph node which separates the glands. The annotated images were then imported to Definiens Architect XD 2.7.0 Build 60765 ×64 for analysis with Tissue Studio version 4.4.2. Using the Tissue Studio portal, the annotated images were preselected for region-of-interest (ROI) detection. The Definiens Composer algorithm was used to segment the tissue into different ROIs: Brown Adipose, Epithelium, RBCs, Stroma, Glass, and White Adipose. This algorithm was trained on representative regions to classify all the tissue within the ROIs in the final analysis. The program then calculated the total tissue area and the area percentages for each of the ROIs. In addition, the program was configured to detect and score nuclei within each of the ROIs, based on a hematoxylin stain threshold and average size set by the analyst. All nuclei were counted and classified as small (<20 μm2), medium (20–40 μm2), or large (> 40 μm2). These values were used to calculate a histological score equal to (1 × % nuclei small) + (2 × % nuclei medium) + (3 × % nuclei large). The analysis output included all quantitative results as well as screen captures of the ROI detection plus overlays and the cellular analysis (nuclei). Slide scanning and tissue quantification was performed by the Bentley R. Midkiff at the Translational Pathology Lab at UNC.
Human breast cancer tissue microarrays
Tissue microarrays were constructed from 271 patients with non-metastatic TNBC (ER/PR <1%; HER2 0/1+, or HER2 2+/FISH not amplified) who underwent surgical resection of their primary tumor at our institution between 2002 and 2007. Exclusion criteria were < 1 cm primary tumor size, prior breast radiation, inflammatory breast cancer, and neoadjuvant chemotherapy. Immunohistochemistry (IHC) was performed using rabbit polyclonal anti-sera generated against human Mre11 (1:3000) and human Nbs1 (1:3000). Tumors were classified as low-expressors of the Mre11 complex if < 10% of cancer cells had detectable nuclear protein expression of Mre11 and/or Nbs1 relative to background staining levels, and determined by two independent reviewers. 254 out of 271 cases were deemed evaluable. Lack of immunoreactivity was confirmed by repeating the IHC using whole tissue sections in a subset of Mre11 complex low-expressors.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell whole genome sequencing
Mammary epithelial cells for single cell sequencing were derived from littermate WT and Mre11ATLD1/ATLD1 female mice. Lentivirus expressing EGFP or EGFP+HER2 was added to the cells for 24 hours. Following this infection, the cells were transferred to 10 cm tissue culture dishes containing irradiated LA7 cell feeder layers. The cells were maintained in culture for 2 weeks at which time they were dissociated into a single cell suspension and sorted for EGFP positive cells into a 96 well plate. These were used for the whole genome single cell sequencing described below.
Single cell sequencing libraries were prepared using the WGA4 kit (Sigma-Aldrich) as previously described (Garvin et al., 2015; Martelotto et al., 2017). Half of the individual cell libraries (48/96) were pooled into each of two library pools. Each pool was run on a single lane for 100-cycle single end sequencing (HiSeq2500, Illumina). The resulting FASTQ files and a bed file defining the indices for the pool, were used as input into Volur (https://github.com/pkMyt1/Volur). The pipeline trimmed 27 nucleotides from the end of each read, aligned the reads to the GRCm38 reference with BowTie2 (Langmead and Salzberg, 2012), converted the SAM files to BAM with SamTools (Li et al., 2009), and output gzipped, demultiplexed BED files suitable for use with Ginkgo (Garvin et al., 2015). Ginkgo was run using a variable 25 Kb segment size, 76 base pair bowtie simulated reads, and global (sample with lowest LOD) segmentation. All other parameters were left as the default. Once Ginkgo completed the segment copy data file was downloaded. This file was modified by removing the chromosome-Y elements, masking alignment errors by setting the copy number in those regions to 2, and normalizing any cell called triploid to diploid. Chromosome Y was removed because these were female mice. These mice are not the same genetic background as the GRCm38 reference. This resulted in regions of copy gain or loss common to all samples.
Quantification and statistical analysis
Since we did not provide a known diploid sample for Ginkgo to use as a reference, any cell that was scored as being triploid by Ginkgo was adjusted by subtracting 1 from the copy value of each segment. Segments observed in the Mre11 ATLD1 cells that were likely mapping errors because of the mouse strain used were masked by setting those regions to a copy number of 2 creating a no triploid, Mre11ATLD1 masked segment copy bed file. Ginkgo defines the copy number based on how many reads mapped to each predefined segment. The mapped reads for this determination may cross the segment boundaries. Because of this the copy number transition cannot be assigned to the segment boundaries. To account for this uncertainty, we use a breakpoint for our analysis. We define a breakpoint region as the start coordinate of the preceding segment through the stop coordinate of the segment containing the copy number change. Using the masked segment copy file, the number and location of each breakpoint was derived for each cell type. The unique breakpoints were then intersected with bed files of interest using BedTools (Quinlan and Hall, 2010) via the Python pybedtools API within Völur.
Statistical significance of breakpoint region intersects
The determination that an observed set of breakpoint regions is enriched for overlap with annotated genome regions (e.g., genes, obtained from http://www.Ensembl.org//useast.ensembl.org/?redirectsrc=//www.ensembl.org%2F) was based on generation of an empirical null distribution dataset by randomly shuffling the genomic locations of the individual breakpoint regions for each cell and then determining the frequency of intersects with the genome feature of interest. This process was repeated 10,000 times for each intersect set. The observed frequency of intersection value was considered significantly different than the randomly shuffled dataset if it was < 5% or > 95% of this empirical null distribution. Specific association with a target set was defined as being those target sets that were not enriched for the control cells but were enriched for the oncogene expressing or Mre11ATLD1 cells.
Tumor whole genome sequencing
Whole genome sequencing services were provided by Novogene. Reads were trimmed using SeqPurge (Sturm et al., 2016), aligned to the mm10 mouse reference using bwa-mem (Li, 2013) and subsequently realigned with ABRA2 (Mose et al., 2019). The resultant BAM files were sorted and duplicate marked using biobambam2 (Tischler and Leonard, 2014). Small variants were called using Strelka2, Mutect2 and Cadabra (Cibulskis et al., 2013; Kim et al., 2018; Mose et al., 2019). Mutect2 calls were filtered using the GATK’s best practices whereas the Strelka2 and Cadabra default filters were applied. Quality thresholds were used to filter calls using values of QSS_NT > = 70 and QSI_NT > = 40 for Strelka2, SNV TLOD > = 9 and Indel TLOD > = 10 for Mutect2, and QUAL > = 15 for Cadabra. Variants were additionally filtered using a panel of normals constructed using the Mutect2-GATK best practices. Structural variants were called using Manta (Chen et al., 2016) and copy number variants were called with CNVKit (Talevich et al., 2016). Mutation signature analysis was performed using deconstructSigs (Rosenthal et al., 2016). Circos plots were created with Circa (http://omgenomics.com/circa).
Statistical Analyses
Two-tailed statistical tests for experimental data were conducted using Graphpad Prism version 8. The specific test used in each analysis is indicated in the corresponding figure legend.
DATA AND CODE AVAILABILITY
The custom algorithms developed for this study are available at https://github.com/pkMyt1/Volur. Whole genome sequencing reads for murine breast cancers with matched normal liver have been uploaded to NCBI Sequence Read Archive (SRA) with accession number PRJNA595908. Single cell whole genome sequencing BAM files and copy number segment files have not been uploaded to SRA due to data format incompatibility, and can be requested from the Lead Contact.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, (1:10,000 for IF) | Thermo Fisher Scientific | Cat# A-21200, RRID:AB_2535786 |
| Chicken anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, (1:10,000 for IF) | Thermo Fisher Scientific | Cat# A-21441, RRID:AB_2535859 |
| Mouse Anti-beta-Actin Monoclonal Antibody, Unconjugated, Clone AC-15 (1:10,000 for WB) | Sigma-Aldrich | Cat# A1978, RRID:AB_476692 |
| Rabbit Anti-Phospho-p53 (Ser15) Antibody (1:1000 for WB) | Cell Signaling Technology | Cat# 9284, RRID:AB_331464 |
| Mouse Anti-DNA-RNA Hybrid [S9.6] Antibody (1:500 for IF) | Kerafast | Cat# ENH001, RRID:AB_2687463 |
| Mouse Anti-Human CD2 Monoclonal Antibody, Clone LT2 (1:10 for Flow) | Miltenyi Biotec | Cat# 130-091-115, RRID:AB_244321 |
| Rabbit Anti-RPA2, (phospho Ser4, Ser8) Polyclonal, Unconjugated antibody (1:500 for IF) | Novus | Cat# NBP1-23017, RRID:AB_1726226 |
| Rabbit Anti-53BP1 Polyclonal Antibody (1:500 for IF) | Bethyl | Cat# A300-272A, RRID:AB_185520 |
| Goat anti-Hamster IgG Secondary Antibody, HRP (1:10,000 for WB) | Thermo Fisher Scientific | Cat# PA1-29626, RRID:AB_10985385 |
| Goat Anti-Rabbit IgG, IRDye® 680LT Conjugated antibody (1:10,000 for WB) | LI-COR Biosciences | Cat# 926-68021, RRID:AB_10706309 |
| Goat Anti-Mouse IgG, IRDye® 800CW Conjugated antibody (1:10,000 for WB) | LI-COR Biosciences | Cat# 926-32210, RRID:AB_621842 |
| Mouse Anti-p53 (1C12) mAb Antibody (1:1000 for WB) | Cell Signaling Technology | Cat# 2524, RRID:AB_331743 |
| Rabbit Anti-phosphorylated Histone H2AX (g-H2AX) Polyclonal Antibody (1:500 for IF) | Trevigen | Cat# 4418-APC-100 |
| Rabbit Anti-KAP1 Polyclonal Antibody, Unconjugated (1:500 for WB) | Abcam | Cat# ab10484, RRID:AB_297223 |
| Rabbit Anti-KAP1 (phospho S824) antibody, (1:500 for WB) | Abcam | Cat# ab70369, RRID:AB_1209417 |
| Mouse Anti-NBS1 Monoclonal Antibody, Unconjugated (1:10,000 for WB) | Novus | Cat# NB100-221, RRID:AB_10001212 |
| Rabbit Anti-RAD50 Polyclonal Antibody, Unconjugated (1:10,000 for WB) | Novus | Cat# NBP2-20054 |
| Armenian Hamster Anti-Mre11 Monoclonal Antibody, Unconjugated (1:500 for WB) | Novus | Cat# NBP2-59677 |
| Mouse Anti-RNaseH1 (H-4) Monoclonal Antibody, Unconjugated (1:1000 for WB) | Santa Cruz Biotechnology | Cat# sc-376326, RRID:AB_10987730 |
| Bacterial and Virus Strains | ||
| Endura DUOs Electrocompetent Cells | Lucigen | 60242-2 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Liberase Blendzyme 2 | Roche | 11988425001 |
| Cultrex 3D-Culture Matrix | Trevigen | 3447-020-01 |
| EpiCult-B Mouse Medium Kit | Stem Cell Technologies | 05610 |
| HuMEC Ready Medium (1X) | GIBCO | 12752010 |
| Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 | GIBCO | 11320082 |
| HEPES (1M) | GIBCO | 15630080 |
| Insulin | Sigma-Aldrich | 407709-50MG |
| Penicillin-Streptomycin (10,000 U/mL) | GIBCO | 15140122 |
| DNase I | Worthington | LS002060 |
| Dispase | Stem Cell Technologies | 07913 |
| Trypsin EDTA | GIBCO | 25200-056 |
| Polyethylenimine, Linear (MW 25,000) | Polysciences | 23966 |
| Bovine Serum Albumin | Fisher Scientific | BP9706-160 |
| Carbenicillin | Fisher Scientific | BP26481 |
| Ampicillin | Fisher Scientific | B1760-25 |
| Talazoparib (BMN-673) | Selleck Chemicals | S7048 |
| KU-55933 (ATM Kinase Inhibitor) | Selleck Chemicals | S1092 |
| VE-821 (ATR Kinase Inhibitor) | Sigma-Aldrich | SML1415 |
| Mechlorethamine hydrochloride (HN2) | Sigma-Aldrich | 122564 |
| Paclitaxel | Sigma-Aldrich | T7402 |
| cis-Diammineplatinum(II) dichloride | Sigma-Aldrich | P4394 |
| Doxorubicin hydrochloride | Sigma-Aldrich | D1515 |
| (S)-(+)-Camptothecin | Sigma-Aldrich | C9911 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | 107689 |
| Corning® Cell-Tak and Tissue Adhesive | Corning | 354240 |
| Critical Commercial Assays | ||
| PlasmoTest | Invivogen | REP-PT1 |
| RNAeasy Plus Mini Kit | QIAGEN | 74136 |
| Comet Assay Kit | Trevigen | 4250-050-K |
| Q5® Hot Start High-Fidelity 2X Master Mix | New England Biolabs | M0494S |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | E2621L |
| EdU-Click 594 | Baseclick | BCK-Edu594 |
| TOPO® TA Cloning® Kit for Sequencing | Invitrogen | 450030 |
| T4 DNA Ligase | New England Biolabs | M0202S |
| Experimental Models: Cell Lines | ||
| HEK293T/17 | ATCC | CRL-11268 |
| LA-7 | ATCC | CRL-2283 |
| Experimental Models: Organisms/Strains | ||
| FVB;129-Rb1tm2Brn/Nci Referred to in this manuscript as “Rb1FL” | Frederick National Laboratory for Cancer Research | 01XC1 |
| FVB.129P2-Trp53tm1Brn/Nci Referred to in this manuscript as “Trp53FL” | Frederick National Laboratory for Cancer Research | 01XC2 |
| C57BL/6N-Gt(ROSA) 26Sortm13(CAG-MYC,-CD2*,)Rsky/J Referred to in this manuscript as “R26MycOE” | The Jackson Laboratory | 020458 |
| B6;129-Gt(ROSA) 26Sortm1(CAG-cas9*,-EGFP)Fezh/J Referred to in this manuscript as “R26Cas9” | The Jackson Laboratory | 024857 |
| Oligonucleotides | ||
| sgControl: CTGATTTGAATAATGATGCC | Generated in this study | N/A |
| sgMre11: TGGAGATCACTACTCGAGGC | Generated in this study | N/A |
| pLV-Cre_LKO1 | Addgene | 12106 |
| LentiCRISPR V2 | Addgene | 52961 |
| psPAX2 | Addgene | 12260 |
| pMD2.G | Addgene | 12259 |
| pEGFP-RNASEH1 | Addgene | 108699 |
| LentiCRISPR-Cre-V2-sgControl-LumiFluor | Generated in this study | N/A |
| LentiCRISPR-Cre-V2-sgMre11-LumiFluor | Generated in this study | N/A |
| Lentiviral_pRRL-EF1a-GpNLuc | Gift from Antonio Amelio, Ph.D. | N/A |
| Lentiviral_pRRL-EF1a-NeuT-LumiFluor | Generated in this study | N/A |
| LentiCRISPR-Cre-V2-sgControl-RNaseH1 | Generated in this study | N/A |
| LentiCRISPR-Cre-V2-sgMre11-RNaseH1 | Generated in this study | N/A |
| Software and Algorithms | ||
| BowTie2 v2.3.4.1 | Langmead and Salzberg, 2012 | https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.3.4.1 |
| Samtools v1.6.0 | Li et al., 2009 | http://www.htslib.org/download/ |
| BedTools v2.26.0 | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| Python R v3.5 | https://www.python.org/ | N/A |
| Ginkgo | Garvin et al., 2015 | http://qb.cshl.edu/ginkgo/?q= |
| Volur algorithm | Generated in this study | https://github.com/pkMyt1/Volur |
| GNU Gzip v1.5 | N.A. | https://www.gnu.org/software/gzip/ |
| Graphpad Prism v8 | N.A. | https://www.graphpad.com/ |
| Python-Levenshtein Library v0.12.0 | N.A. | https://github.com/ztane/python-Levenshtein |
| Fiji | Schindelin et al., 2012 | https://imagej.net/FijitDownloads |
| SnapGene software v4.3.4 | GSL Biotech | https://www.snapgene.com |
| Open Comet v1.3.1 | Gyori et al., 2014 | http://www.cometbio.org |
| Other | ||
| Mouse Reference Sequence GRCm38 | http://www.Ensembl.org//useast.ensembl.org/?redirectsrc=//www.ensembl.org%2F | http://www.ensembl.org//useast.ensembl.org/Mus_musculus/Info/Index?redirectsrc=//www.ensembl.org%2FMus_musculus%2FInfo%2FIndex |
| Microsatellites, CpG Islands, Simple Repeats, SINE Elements, LINE Elements, LTR | UCSC Genome Browser | https://genome.ucsc.edu/cgi-bin/hgTables |
| Genome annotation tables | http://www.Ensembl.org//useast.ensembl.org/?redirectsrc=//www.ensembl.org%2F | Ensembl v91 |
Highlights.
Mre11-mediated tumor suppression is p53 independent
Mre11 suppresses DNA damage at oncogene-induced R loops
Breast cancers with Mre11 dysfunction exhibit a genomic loss signature
Mre11 dysfunction induces sensitivity to PARP and ATR inhibitors
ACKNOWLEDGMENTS
We thank Christine E. Foster, Bentley R. Midkiff, Muzaffar Akram, and Brian Cooley and the UNC Animal Surgery Core Lab for their expert technical assistance and members of the Gupta laboratory for advice and support. G.P.G. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. This work was supported by Susan G. Komen CCR16377075 (G.P.G.), NCI R37 CA227837 (G.P.G.), NIH GM59413 and NCI CA087497 (J.H.J.P.), the UNC-Lineberger Cancer Center core grant (P30 CA016086), the MSK Cancer Center core grant (P30 CA008748), and the University Cancer Research Fund. J.S.R.-F. is funded in part by the Breast Cancer Research Foundation. The UNC Translational Pathology Laboratory is supported in part by grants from the NCI (5P30CA016086-42), NIH (U54-CA156733), NIEHS (5 P30 ES010126-17), UCRF, and NCBT(2015-IDG-1007). K.D.F.-S. is funded by Ruth L. Kirschstein National Research Service Award (NRSA) Individual Postdoctoral Fellowship (Parent F32) (F32 CA 206345). The UNC Flow Cytometry Core Facility is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center, by the North Carolina Biotech Center Institutional Support Grant 2017-IDG-1025, and by the National Institutes of Health 1UM2AI30836-01. The Microscopy Services Laboratory is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The UNC Translational Pathology Laboratory is supported in part by grants from the NCI (5P30CA016086-42), NIH (U54-CA156733), NIEHS (5 P30 ES010126-17), UCRF, and NCBT (2015-IDG-1007).
Footnotes
DECLARATION OF INTERESTS
G.P.G. has ownership interest (including patents) in and is a consultant/advisory board member for Naveris, Inc., outside the scope of the present study. J.S.R.-F. reports personal/consultancy fees from VolitionRx, Page.AI, Goldman Sachs, Grail, Ventana Medical Systems, Invicro, Roche Diagnostics, and Genentech, outside the scope of the present study. J.H.J.P. is a consultant for Ideaya Biosciences, Novus Biologicals, and Atropos Therapeutics, outside the scope of the present study.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.020.
REFERENCES
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. ; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrini A, Nicolas L, Yang-Lott K, Guryanova OA, Levine RL, Bassing CH, Chaudhuri J, and Petrini JH (2016). Defining ATM-independent functions of the Mre11 complex with a novel mouse model. Mol. Cancer Res. 14, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, Mistrik M, Aittomäki K, Blomqvist C, Heikkilä P, et al. (2008). Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2, 296–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt S, Samartzis EP, Zimmermann AK, Fink D, Moch H, Noske A, and Dedes KJ (2017). Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer 17, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N; Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EY, Wells JP, Tsai S-H, Coulombe Y, Chan YA, Zhu YD, Fournier L-A, Hieter P, Masson J-Y, and Stirling PC (2018). MRE11-RAD50-NBS1 activates Fanconi Anemia R-loop suppression at transcription-replication conflicts. bioRxiv. 10.1101/472654 [DOI] [Google Scholar]
- Chang EY, Tsai S, Aristizabal MJ, Wells JP, Coulombe Y, Busatto FF, Chan YA, Kumar A, Dan Zhu Y, Wang AY, et al. (2019). MRE11RAD50-NBS1 promotes Fanconi anemia R-loop suppression at transcription-replication conflicts. Nat. Commun. 10, 4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Choudhury A, Nelson LD, Teo MT, Chilka S, Bhattarai S, Johnston CF, Elliott F, Lowery J, Taylor CF, Churchman M, et al. (2010). MRE11 expression is predictive of cause-specific survival following radical radiotherapy for muscle-invasive bladder cancer. Cancer Res. 70, 7017–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, Bonnefoi H, Cameron D, Gianni L, Valagussa P, et al. (2014). Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172. [DOI] [PubMed] [Google Scholar]
- Cristini A, Groh M, Kristiansen MS, and Gromak N (2018). RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 23, 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiola F, Pertesi M, Oliver J, Le Calvez-Kelm F, Voegele C, Young EL, Robinot N, Forey N, Durand G, Vallee MP, et al. (2014). Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 16, R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG, Bensimon A, et al. (2006). Oncogeneinduced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642. [DOI] [PubMed] [Google Scholar]
- Ehmann UK, Peterson WD Jr., and Misfeldt DS (1984). To grow mouse mammary epithelial cells in culture. J. Cell Biol. 98, 1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard H, García-Muse T, and Aguilera A (2015). Replication stress and cancer. Nat. Rev. Cancer 15, 276–289. [DOI] [PubMed] [Google Scholar]
- Garvin T, Aboukhalil R, Kendall J, Baslan T, Atwal GS, Hicks J, Wigler M, and Schatz MC (2015). Interactive analysisand assessment ofsingle-cell copy-number variations. Nat. Methods 12, 1058–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Wilson TE, and Arlt MF (2017). Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Vanness K, Barlas A, Manova-Todorova KO, Wen YH, and Petrini JH (2013). The Mre11 complex suppresses oncogene-driven breast tumorigenesis and metastasis. Mol. Cell 52, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, and Bartek J (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Puddu F, and Costanzo V (2011). RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 19, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen K, Rapakko K, Karppinen SM, Erkko H, Knuutila S, Lundán T, Mannermaa A, Børresen-Dale AL, Borg A, Barkardottir RB, et al. (2006). RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 27, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Stout-Weider K, Hermann K, Schrock E, and Heiden T (2006). Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 16,1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, and Tora L (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. [DOI] [PubMed] [Google Scholar]
- Hills SA, and Diffley JF (2014). DNA replication and oncogene-induced replicative stress. Curr. Biol. 24, R435–R444. [DOI] [PubMed] [Google Scholar]
- Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, and Maiorano D (2017). An ATR-dependent function forthe Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 36, 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jechlinger M, Podsypanina K, and Varmus H (2009). Regulation of transgenes in three-dimensional cultures of primary mouse mammary cells demonstrates oncogene dependence and identifies cells that survive deinduction. Genes Dev. 23, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, Weigman VJ,Tsao MS, Lane TF, Perou CM, and Zacksenhaus E (2010). Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Invest. 120, 3296–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, and Berns A (2001). Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 29, 418–425. [DOI] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, and Saunders CT (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, and Petermann E (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 7, 13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Petermann E, and Boulton SJ (2018). Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 8, 537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, and Debatisse M (2013). Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancerdeletions lie infragile sites hosting large genes. Cell Rep. 4, 420–428. [DOI] [PubMed] [Google Scholar]
- Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, and Vindigni A (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequencesand assemblycontigs with BWA-MEM. arXiv, arXiv:1303.3997, https://arxiv.org/abs/1303.3997. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 70, 425–448. [DOI] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makharashvili N, Arora S, Yin Y, Fu Q, Wen X, Lee JH, Kao CH, Leung JW, Miller KM, and Paull TT (2018). Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. eLife 7, e42733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malacaria E, Pugliese GM, Honda M, Marabitti V, Aiello FA, Spies M, Franchitto A, and Pichierri P (2019). Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 70, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelotto LG, Baslan T, Kendall J, Geyer FC, Burke KA, Spraggon L, Piscuoglio S, Chadalavada K, Nanjangud G, Ng CK, et al. (2017). Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat. Med. 23, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, and Bartek J (2018). High speed offork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- Miron K, Golan-Lev T, Dvir R, Ben-David E, and Kerem B (2015). Oncogenes create a unique landscape of fragile sites. Nat. Commun. 6, 7094. [DOI] [PubMed] [Google Scholar]
- Mose LE, Perou CM, and Parker JS (2019). Improved indel detection in DNA and RNA via realignment with ABRA2. Bioinformatics 35, 2966–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, Zou L, and Graubert TA (2018). Spliceosome mutations induce R-loop-associated sensitivitytoATR inhibition in myelodysplastic syndromes. Cancer Res. 78, 5363–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, and Symington LS (2018). Role of the Mre11 complex in preserving genome integrity. Genes (Basel) 9, E589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 759, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SU, Mirza H, Grigoriadis A, and Pinder SE (2015). Selection and evolution in the genomic landscape of copy number alterations in ductal carcinoma in situ (DCIS) and its progression to invasive carcinoma of ductal/no special type: a meta-analysis. Breast Cancer Res. Treat. 753, 101–121. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, and Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein K, and Stracker TH (2014). The MRE11 complex: an important source of stress relief. Exp. Cell Res. 329, 162–169. [DOI] [PubMed] [Google Scholar]
- Riaz N, Blecua P, Lim RS, Shen R, Higginson DS,Weinhold N, Norton L,Weigelt B, Powell SN, and Reis-Filho JS (2017). Pan-canceranalysisof bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 8, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, and Swanton C (2016). DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim D, Bradford WD, Freeland A, Cady G, Wang J, Pruitt SC, and Gerton JL (2017). DNA replication stress restricts ribosomal DNA copy number. PLoS Genet. 13, e1007006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9,676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Situ Y, Chung L, Lee CS, and Ho V (2019). MRN (MRE11-RAD50-NBS1) complexin human cancerand prognostic implications in colorectal cancer. Int. J. Mol.Sci. 20, E816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley J, Klepczyk L, Keene K, Wei S, Li Y, Forero A, Grizzle W, Wielgos M, Brazelton J, LoBuglio AF, and Yang ES (2015). PARP1 and phospho-p65 protein expression is increased in human HER2-positive breast cancers. Breast Cancer Res. Treat. 150, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, and Petrini JH (2011). The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Couto SS, Cordon-Cardo C, Matos T, and Petrini JH (2008). Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol. Cell 31, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm M, Schroeder C, and Bauer P (2016). SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinformatics 17, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A, and Tainer JA (2018). The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 87, 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, and Petrini JH (2003). Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol. Cell 12, 1511–1523. [DOI] [PubMed] [Google Scholar]
- Tischler G, and Leonard S (2014). biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 9, 13. [Google Scholar]
- Trenz K, Smith E, Smith S, and Costanzo V (2006). ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 25, 1764–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZC, Birkbak NJ, Culhane AC, Drapkin R, Fatima A, Tian R, Schwede M, Alsop K, Daniels KE, Piao H, et al. ; Australian Ovarian Cancer Study Group (2012). Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 18, 5806–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, Pareja F, James PA, Couch FJ, Eccles DM, et al. ; kConFab Investigators (2018). The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J. Natl. Cancer Inst. 110, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Arlt MF, Park SH, Rajendran S, Paulsen M, Ljungman M, and Glover TW (2015). Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 25, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Miecz-kowski P, Wood RD, Gupta GP, and Ramsden DA (2016). Essential roles for polymerase θ-mediated end joining in the repair of chromosome breaks. Mol. Cell 63, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh W, and Spradling AC (2014). Incomplete replication generates somatic DNA alterations within Drosophila polytene salivary gland cells. Genes Dev. 28, 1840–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki BI, Suriawinata AA, Eastman AR, Garner KM, and Bakhoum SF (2014). Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer 120, 1733–1742. [DOI] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The custom algorithms developed for this study are available at https://github.com/pkMyt1/Volur. Whole genome sequencing reads for murine breast cancers with matched normal liver have been uploaded to NCBI Sequence Read Archive (SRA) with accession number PRJNA595908. Single cell whole genome sequencing BAM files and copy number segment files have not been uploaded to SRA due to data format incompatibility, and can be requested from the Lead Contact.