

Abstract
With the worldwide spread of the novel severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) resulting in declaration of a pandemic by the World Health Organization (WHO) on March 11, 2020, the SARS‐CoV‐2‐induced coronavirus disease‐19 (COVID‐19) has become one of the main challenges of our times. The high infection rate and the severe disease course led to major safety and social restriction measures worldwide. There is an urgent need of unbiased expert knowledge guiding the development of efficient treatment and prevention strategies. This report summarizes current immunological data on mechanisms associated with the SARS‐CoV‐2 infection and COVID‐19 development and progression to the most severe forms. We characterize the differences between adequate innate and adaptive immune response in mild disease and the deep immune dysfunction in the severe multiorgan disease. The similarities of the human immune response to SARS‐CoV‐2 and the SARS‐CoV and MERS‐CoV are underlined. We also summarize known and potential SARS‐CoV‐2 receptors on epithelial barriers, immune cells, endothelium and clinically involved organs such as lung, gut, kidney, cardiovascular, and neuronal system. Finally, we discuss the known and potential mechanisms underlying the involvement of comorbidities, gender, and age in development of COVID‐19. Consequently, we highlight the knowledge gaps and urgent research requirements to provide a quick roadmap for ongoing and needed COVID‐19 studies.
Keywords: COVID‐19 comorbidity, COVID‐19 immunity, COVID‐19 multimorbidity, COVID‐19 prevention, COVID‐19 treatment, SARS, SARS‐CoV‐2 receptors
1. INTRODUCTION
Infections with the novel coronavirus SARS‐CoV‐2 resulting in COVID‐19 development represent the major medical and scientific challenges of our time. Knowledge on SARS‐CoV‐2 infection pathways and mechanisms associated with immune defense or immunopathology is growing exponentially, as it is indispensable to design the proper diagnostic and therapeutic strategies. However, there are several knowledge gaps and urgent unmet research needs in our understating of the current pandemics (Table 1). A group of experts in basic and clinical immunology has joined forces under the umbrella of the European Academy of Allergy and Clinical Immunology (EAACI) to provide a consensus report on the basic molecular and immune mechanisms associated with susceptibility, clinical presentations and severity of COVID‐19.
TABLE 1.
Summary of knowledge gaps and research needs pertaining to SARS‐CoV‐2 and COVID‐19 (as of May 20, 2020)
| Knowledge Gaps | Research Needs |
|---|---|
| Origin and evolution SARS‐CoV‐2 | To shed light on the origin of SARS‐CoV‐2 via studies of genomic epidemiology and evolutionary dynamics |
| COVID‐19 diagnosis | To develop rapid and specific point‐of‐care diagnostic test for COVID‐19 and to validate existing serological tests |
| Zoonotic transmission and exhaustive characterization of human SARS‐CoV‐2 transmission | To elucidate mechanisms of SARS‐CoV‐2 transmission from animals to humans and vice versa. To determine how demographic factors and severity of COVID‐19 patients affect SARS‐CoV‐2 transmission as well as how infectious are asymptomatic or pre‐symptomatic infected people |
| Route of SARS‐CoV‐2 transmission | To ascertain the role of fecal–oral transmission in COVID‐19 and better define the presence and duration of SARS‐CoV‐2 in oral and respiratory secretions, in fecal samples and in serum. |
| Natural history of asymptomatic and mild SARS‐CoV‐2 infection in humans | To identify SARS‐CoV‐2 a‐/oligosymptomatic carriers to track their viral loads, clinical presentations and immune response (antibody titers, immune phenotyping, etc) over time |
|
Pathogenicity of SARS‐CoV‐2 |
To investigate mechanisms of and changes in SARS‐CoV‐2 pathogenicity (as compared to SARS‐CoV) to provide the basis for the identification of novel therapeutic targets |
| Spectrum of COVID‐19 severity | To characterize the heterogeneity of COVID‐19 severity to aid in directing management and treatment of COVID‐19 patients |
| Risk factors and biomarkers associated with severe illness or mortality in COVID‐19 | To identify COVID‐19 sensitive groups and determine the causes underlying disease severity to reinforce prevention strategies and treatment of high‐risk groups |
| COVID‐19 treatment | To screen new pharmaceuticals, small molecule compounds, biologics, and other agents that have potent anti‐SARS‐CoV‐2 to empower current COVID‐19 treatments |
| Vaccine development for SARS‐CoV‐2 | To develop SARS‐CoV‐2 vaccines for prevention and ultimate eradication of SARS‐CoV‐2. Particular attention should be placed to investigate potential antibody‐dependent enhancement of viral infection in vaccine candidates |
| Pre‐clinical models for SARS‐CoV‐2 and COVID‐19 research | To develop animal models for SARS‐CoV‐2 research (mechanisms of infection, pathogenesis, treatments, etc) |
| Para‐/postinfectious syndromes in SARS‐CoV‐2 infection | To understand COVID‐19‐associated Kawasaki‐like syndrome/TSS in children and rare para‐/postinfectious symptoms in adults. |
| Long‐term sequelae of COVID‐19 | Follow‐up of COVID‐19 patients to detect potential long‐term consequences of COVID‐19 pneumonia (eg, pulmonary fibrosis, early COPD) and other manifestations (eg, renal impairment, cardiac/ vascular dysfunction, increased risk of thrombosis/ sepsis (due to endothelial dysfunction)) |
| Individual protection after SARS‐CoV‐2 infections | To understand why development of protective antibodies is not seen in all infected patients and whether this might be related to severity of SARS‐CoV‐2 infection |
2. SARS‐COV‐2 RECEPTORS: PROVEN AND POTENTIAL INTERACTION PARTNERS
On the basis of sequence homology, all human coronaviruses have animal origins: SARS‐CoV, SARS‐CoV‐2, MERS‐CoV, HCov‐NL63, and HCoV‐229E are considered to have originated from bats, 1 whereas HCoV‐ OC43 and HKU1 likely originated from rodents (Figure 1). 2 SARS‐CoV‐2 has a significant structural similarity to SARS‐CoV and MERS‐CoV and other human and animal coronaviruses. 3 , 4 It has been quickly determined that SARS‐CoV‐2, similarly to SARS‐CoV, utilizes the membrane bound form of angiotensin‐converting enzyme 2 (ACE2) to enter human cells via its spike protein (S). 5 After SARS‐CoV‐2 has bound ACE2, ACE2 will be internalized and its membrane expression decreased. Whereas ACE2 is an important regulator of bradykinin, its reduced expression in the lung environment results in local vascular leakage leading to angioedema in the affected lung tissue. 6 The host serine protease TMPRSS2 cleaves spike protein into S1 and S2 fragments, which enables fusion with the cellular membrane, entrance to the cell, and start of the replication process. 7 In addition to TMPRSS2, other proteins such as furin or human endosomal cysteine proteases are potentially capable of cleaving S, such as cathepsin L (CTSL) and cathepsin B (CTSB). 8 , 9 ACE2 is highly expressed in the lungs, small intestine, kidney, and heart, but it is not expressed on innate and adaptive immune cells. 10 , 11 , 12 , 13 As recently shown, SARS‐CoV‐2 can also use CD147 (also called basigin (BSG) or extracellular matrix metalloproteinase inducer (EMMPRIN)), to enter cells of epithelial origin, but it is not yet clear if then virus can efficiently replicate or it leads to the cell death 14 CD147 is utilized as a receptor by other viruses including SARS‐CoV and HIV‐1, as well as by malaria to enter erythrocytes. 15 , 16 , 17 It is, however, not yet clear if SARS‐CoV‐2 can replicate inside immune cells or just infects them and causes cell death. CD147 is a transmembrane immunoglobulin‐like receptor, which also exists in a secreted form. 13 , 18 At the cellular membrane, it is activated by several extracellular ligands such as cyclophilins A and B (PPIA and PPIB), S100A9 or platelet glycoprotein VI (GP6). 19 , 20 , 21 , 22 Its extracellular glycosylation sites bind to complex proteoglycans such as syndecan‐1. 23 CD147 often creates membrane complexes with CD44, one of the receptors for hyaluronan, an extracellular matrix component. 24 Coronaviruses incorporate host cyclophilins during their cellular replication cycle, which further enables them to bind to CD147. 25 , 26 CD147 is expressed in human airway and kidney epithelium, as well as in innate cells (granulocytes, macrophages, dendritic cells (DC), innate lymphoid cells (ILCs), and lymphocytes). 10 Other receptors potentially utilized by SARS‐CoV‐2 are CD26 (encoded by DPP4; a receptor for MERS‐CoV), an important T cell and also epithelial cell receptor, amino peptidase N (ANPEP; a receptor for human and porcine coronaviruses), 13 , 27 ENPEP and a glutamyl aminopeptidase, 28 as well as DC‐SIGN 29 (Figure 2).
FIGURE 1.
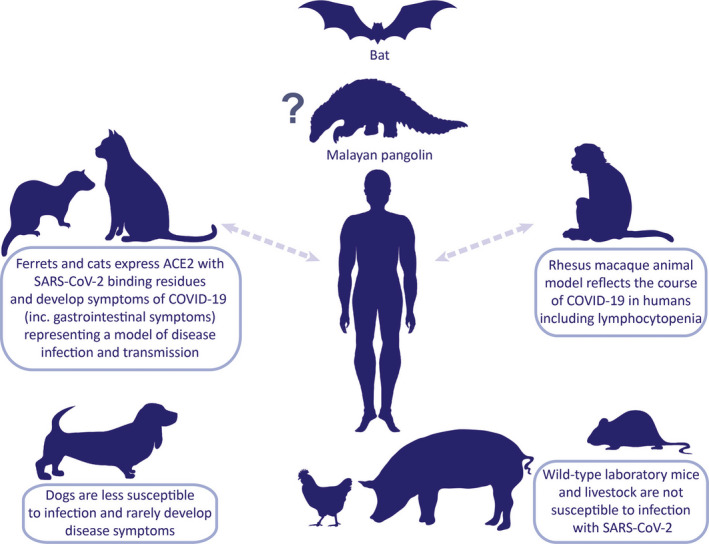
SARS‐CoV‐2 on the animal‐human interphase. Animal models that resemble clinical and pathological features of COVID‐19 are essential to investigate pathogenesis, transmission, and therapeutic strategies. SARS‐CoV‐2 shares 96.2% of its genome sequence with the bat CoV RaTG13 posing the bat as the most probable natural host of virus origin. SARS‐CoV‐2‐related coronaviruses have been identified in Malayan pangolins, which is considered as an intermediate host between bats and humans. ACE2, a critical SARS‐CoV‐2 receptor, in wild‐type mice differs from the human one; therefore, transgenic mice models with recombinant hACE2 are necessary. To this date, Rhesus macaques, with ACE2 identical to human's, have been used to study the natural course of the disease and the effectiveness of therapeutic intervention with intravenous immunoglobulins. Ferrets and cats have been shown susceptible to SARS‐CoV‐2 infection and to develop COVID‐19 symptoms including respiratory and gastrointestinal manifestations. Limited facilities and expertise in handling nonmurine species may hamper usage of the aforementioned models. Transmission between humans and animals has not been unequivocally confirmed
FIGURE 2.
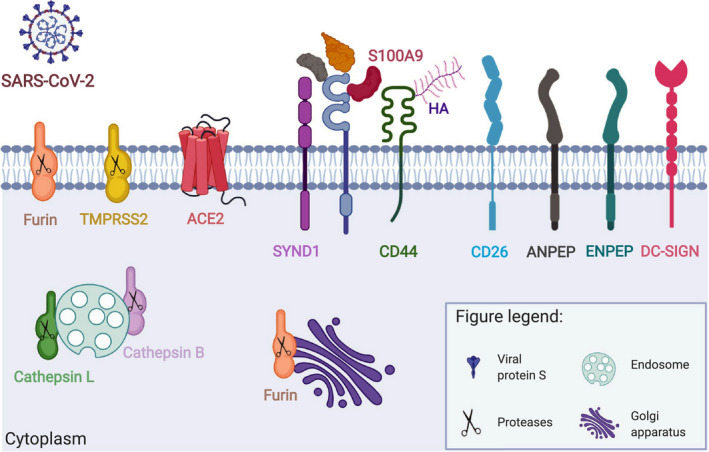
Cellular distribution of confirmed and potential SARS‐CoV‐2 receptors and interaction partners. Entry of SARS‐CoV‐2 into the host cells depends on expression of i) adequate receptors and ii) cellular proteases. The two‐step infection process is mediated by the viral Spike (S) protein. Its binding to the receptor and cleavage by proteases assures virus internalization. ACE2 and TMPRSS2 are critical complex for SARS‐CoV‐2 infection. CD147 and its extracellular (Cyclophilin A, Cyclophilin B, Platelet glycoprotein VI, S100A9, Hyaluronic acid) and transmembrane (CD44, Syndecan‐1) interaction partners can be also used for SARS‐CoV‐2 entry and/or modulation of immune responses to the virus. It has been suggested for SARS‐CoV‐2 and shown in case of other Coronavidae family members that they can also exploit other cell surface receptors (CD26, ANPEP, ENPEP, DC‐SIGN) and proteases (Furin, Cathepsin L, Cathepsin) to enter human cells. CypA, Cyclophilin A; CypB, Cyclophilin B; GPVI, Platelet glycoprotein VI; HA, Hyaluronic acid; SYND1, Syndecan‐1. This figure is modified from the original publication by Radzikowska, Ding, et al, presenting the distribution of these receptors in various human tissues and immune cells in healthy children and adults, and in patients with COVID‐19 comorbidities and risk factors (ref). Created with Biorender.com
3. ENTRANCE OF THE VIRUS THROUGH EPITHELIAL BARRIERS: COVID‐19 PATHOPHYSIOLOGY
3.1. Airway epithelium: hotspot for disease development
In the upper and lower airways ACE2 and TMPRSS2 are highly co‐expressed, 10 , 11 , 12 , 13 but there is no expression of SLC6A19, which potentially blocks the access of TMPRSS2 to ACE2 and subsequently reduces active infection. 30 , 31 In the nasal and the pharyngeal epithelium, in goblet and ciliated cell, ACE2 is expressed at high levels and co‐expressed with TMPRSS2 representing the sites of initial viral replication and a main source of infectious particles. 10 , 11 , 12 , 13 , 30 The lower airways, bronchial epithelium and type II pneumocytes (AT2 cells) highly express ACE2 and TMPRSS2, which may provide virus entrance to the lung and lead to COVID‐19 pneumonia. Moreover, CD147, CD26, ANPEP, and ENPEP are also expressed in the airway epithelium, as well as in many innate and adaptive immune cells, 10 , 13 both in bronchoalveolar lavage (BAL) and peripheral blood (Figure 3A).
FIGURE 3.
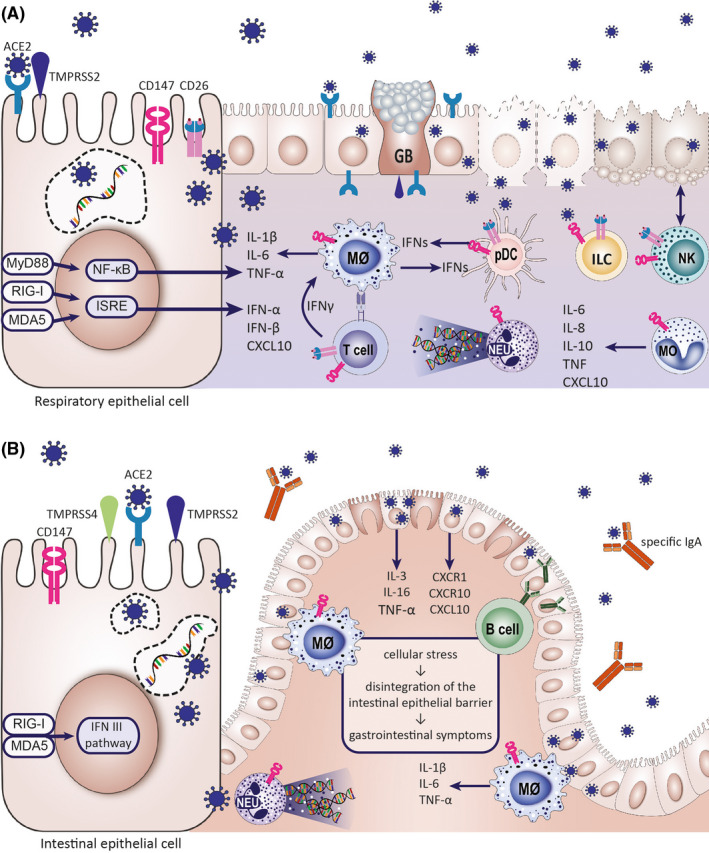
Epithelial barriers are susceptible for the SARS‐CoV‐2 infection. (A) Epithelial cells of the respiratory system are the primary site of SARS‐CoV‐2 infection. The respiratory epithelium is equipped with the receptors and other host proteins allowing viral entry: ACE2, TMPRSS2, CD147, and CD26. The highest expression of ACE2 is found in the nasopharynx. The virus was found to propagate in the lower respiratory tract as well, especially in type II alveolar cells. The effects of the virus on the respiratory epithelial barrier include cell membrane fusion and syncytium formation (which represents a mechanism of viral spread), apoptosis and virus‐mediated cell lysis leading to the loss of barrier function. Upon infection, epithelial cells release interferons, chemokines, and cytokines promoting tissue infiltration by innate immune cells, such as monocytes, NK cells, neutrophils, and, with time, inflammatory macrophages and virus‐specific lymphocytes. Immune cells express putative SARS‐CoV‐2 receptors, CD147, and CD26. (B) Gastrointestinal symptoms are seen in a substantial percentage of COVID‐19 patients. The intestinal tissue has a high expression of ACE2 receptor, TMPRSS2, and TMPRSS4 proteases. Their expression increases with intestinal epithelial cell differentiation. ACE2 expression in intestinal epithelium decreases with inflammation and shows a negative correlation with IL‐1β levels. SARS‐CoV‐2 infection results in disintegration of the intestinal epithelial barrier. Virus‐specific IgA have been found in the gastrointestinal tract. Noninfectious SARS‐CoV‐2 RNA is found in stool after negative nasal swab tests. CXCL10, C‐X‐C motif chemokine 10; CXCR1, C‐X‐C motif chemokine receptor 1; CXCR10, C‐X‐C motif chemokine receptor 10; GB, goblet cell; ILC, innate lymphoid cell; IL, interleukin; IFN, interferon; Mθ, macrophage; MO, monocyte; NEU, neutrophil; NK, natural killer cell; pDC, plasmacytoid dendritic cell; TNF, tumor necrosis factor
Once the virus enters the host cell, it releases its RNA into the cytoplasm and uses the host translation machinery to translate its polyproteins pp1a and pp1b, also known as replicases and viral essential proteases 3CLpro and PLpro. These proteases cleave polyprotein complex into several nonstructural proteins (Nsp), which together with the viral RNA‐dependent RNA polymerase form the replication complex, where the negative strand and mRNA for structural proteins (S, nucleocapsid (N), envelope (E), and membrane (M)) and accessory proteins for the virus are created. 2 , 32 , 33 After protein translation, they traffic through the ER to the Golgi apparatus, where the mature virions are assembled in budding vesicles and are exocytosed from the cell. Inside infected cells, there are several innate immune mechanisms responsible for recognizing the virus at different stages of its replication and leading to the production interferons type I (IFNα and β), type III, and pro‐inflammatory cytokines. 34 Genes encoding these interferons form the type‐1 (E1) epithelial response profile. Also, ACE2 is a typical E1 gene. 35 This response also includes mechanisms such as the expression of helicases or cytidine deaminases targeting viral RNAs (Figure 3A).
Viruses use various strategies to evade those mechanisms. 36 Yet, little is known about SARS‐CoV‐2 antiviral responses and evasion strategies of this virus, but likely much can be extrapolated from the SARS‐CoV and MERS‐CoV‐based knowledge. 34 , 37 Viral single‐stranded RNA (ssRNA), double‐stranded RNA (dsRNA), and proteins are recognized by cytosolic pattern recognition receptors (PRR), mainly RIG‐I/MDA5 and toll‐like receptors (mainly TLR7/8). This recognition leads to recruitment of MAVS, MyD88, and/or TRIF, respectively. Eventually, IRF3 and IRF7 transcription factors are activated leading to the production of type I interferons (IFN‐α and IFN‐β), whereas NF‐KB and AP‐1 transcription factors lead to the production of pro‐inflammatory cytokines such as IL‐6, IL‐8, IL‐1 β, CXCL10, and CCL2. 37
Epithelial cells produce type I and type III IFNs upon viral infection. Type I IFN act through receptors expressed in a vast number of cells. In contrast, type III IFNs seem to exert their effect mostly on epithelial cells, are less inflammatory, and are activated faster than type I IFN. 38 , 39 IFNs are one of the most potent antiviral components of the innate immune response. They work on various levels, that is, blocking viral attachment, entry, trafficking, protein production, and genome amplification and also viral assembly and egress. 38 Moreover, IFNs also activate other innate and adaptive immune responses. However, in case of COVID‐19 these responses seem to be diminished 40 or dysregulated. 41
SARS‐CoV and MERS‐CoV inhibit IFN signaling on various levels. 42 The nsp 16 mediated 2’O methylation of viral mRNA cap structure prevents coronaviruses recognition by MDA5. 43 The sequestration of viral dsRNA within double membrane vesicles (DMVs) also protects coronaviruses from detection through cytosolic PRRs. 44 Moreover, coronaviruses produce many nonstructural proteins which inhibit induction of IFNs (inhibition of IRF3 and IRF 7), and/or interferon signaling (inhibition of STAT 1 signaling). 42 A reduced antiviral response via IFN pathway inhibition, together with an ongoing pro‐inflammatory response, presumably heightened by increased viral load, may lead to excessive inflammation 41 and worsening of the disease. In an animal model of SARS‐CoV, a delayed type I interferon response resulted in accumulation of inflammatory monocytes/macrophages, leading to elevated lung cytokine/chemokine levels, vascular leakage, and impaired virus‐specific T‐cell responses. 41 A recent study in humans showed that SARS‐CoV‐2 infection induces weak IFN responses from infected pneumocytes, even weaker than in SARS‐CoV infection. 40 Interestingly, ACE2 has been recently shown to be an interferon‐stimulated gene. 12 ACE2 is also known to protect mice against acute lung injury. Therefore, it needs to be determined whether upregulation of ACE2 after the initial antiviral response is used by SARS‐CoV‐2 to enhance infection, but also if the delay of IFN responses potentially leads to impairment of ACE2‐related protection against lung injury.
3.2. From oral to rectal mucosa: What we know about the involvement of the gastrointestinal tract in COVID‐19
Based on the current knowledge of SARS‐CoV‐2 receptors’ expression on the epithelial barrier sites, the gastrointestinal tract requires special attention. Human ACE2 is homogeneously distributed on the brush border of enterocytes across the small intestine and in the lung epithelium. 45 , 46 In the oral mucosa, the basal layer of nonkeratinized, squamous epithelial cells were reported to be ACE2‐positive, while stomach epithelial cells and colon enterocytes remained negative. 45 TMPRSS2 and TMPRSS4 mediate infection of small intestinal epithelial cells. 47 These enzymes might additionally be an interesting target for therapeutic intervention, since a clinically approved protease inhibitor is available. 7 Less is known regarding the gastrointestinal distribution of CD147. 14 Enteric CD147 seems to play a role in carcinogenesis and inflammation, 48 which might shed a new light on patients’ group at risk for severe SARS‐CoV‐2 infections and needs further attention. Of note, CD26 expression was reported to be high in ileum and jejunum, low in duodenal samples and not detectable in colon epithelial cells (Figure 3B). 49
Gastrointestinal symptoms like vomiting and diarrhea in COVID‐19 are gaining attention. 50 In the previous SARS outbreak and in MERS patients, gastrointestinal complaints were found in approximately 30% of patients. In SARS‐CoV‐2 infections, diarrhea, and abdominal pain occur in 20%‐50% of COVID‐19 patients and might even precede onset of respiratory symptoms. 51 , 52 SARS‐CoV active replication was detected in small intestinal enterocytes 53 and enteroids derived from human ileum and colon in case of SARS‐CoV‐2. 47 This is highly relevant as viral excretion was detected in fecal samples and anal swabs of COVID‐19 patients. 54 While first evidence is available that human colonic fluids might rapidly inactivate SARS‐CoV‐2 in vitro, 46 MERS‐CoV was found to resist gastrointestinal fluids simulating conditions with elevated pH levels after food ingestion, while the virus rapidly lost infectivity when exposed to an acidic gastric fluid simulating fasted state. 55 These reports might explain, at least partially, the fact that even though SARS‐CoV‐2 RNA was detected in stool samples from patients, the isolates were not infective. 56 Thus, it remains unclear whether the fecal‐oral‐route might propagate disease transmission especially in reduced hygienic conditions. 57
Further understanding of the relationship between disease and the digestive tract is essential to prevent transmission and disease progression as well as to design efficient treatment of COVID‐19.
3.3. Skin barrier: more than just matter of wearing protective equipment
Recent reports indicate that in COVID‐19 the skin might also be affected. An Italian and a French study reported that 20.4% to 50% of COVID‐19 cases, respectively developed nonpruritic, erythematous rashes, urticaria or varicella‐like lesions affecting the trunk and sometimes the limbs. 58 , 59 In general, the rashes occurred 3 days after development of COVID‐19 symptoms and the median duration was 8 days. 60 In addition, acrolated ischemic, self‐healing lesions at toes, and fingers have been observed mainly in children and young adults shortly before COVID‐19 symptom appearance. 61 To put otherwise, healthy kids in quarantine upon detection of these lesions might help to prevent infection from spreading. Apparently, cutaneous manifestations of COVID‐19 are similar to skin rashes observed in other common viral infections. There is no evidence that they are related to the severity of the disease or an indication that the virus can replicate in the skin.
Unfortunately, the few studies available so far did not detect SARS‐CoV‐2 presence in skin lesions, which questions if the skin manifestations are indeed infectious or para‐infectious driven. Given that ACE2 and TMPRSS2, the receptors for SARS‐CoV‐2 entry into human cells, are absent or weakly expressed in the skin, 11 para‐infectious events seem to be more likely. Furthermore, the possibility of adverse drug reactions as causative for skin manifestations in COVID‐19 is being strongly considered in certain cases.
Some of the protective measures taken during the SARS‐CoV‐2 pandemic (use of gloves, masks or goggles) can affect the skin. Masks and goggles often induce pressure injury due to not properly fitted material, and a study in Chinese health care workers indicated that 71% suffer from skin barrier damage such as dryness, scales, papules or erythema. 62 Causative is the inevitable hand hygiene procedure—66.1% stated to wash their hands more than 10 times per day and only 22.1% used appropriate skin care products afterward. Moreover, long‐term usage of gloves over 6 hours per day is common in health care workers leading to overhydration and dysbiosis with damage to the stratum corneum and subsequent skin infection or sensitization. 63 Thus, proper education regarding the use of skin care products after the hand hygiene procedure is essential to protect the skin barrier and prevent further skin complications.
4. DEVELOPMENT AND FAILURE OF AN ADEQUATE IMMUNE RESPONSE
4.1. Innate Immunity
4.1.1. DCs and macrophages
Monocytes/macrophages and DCs play a crucial role in anti‐viral responses by linking innate and adaptive immunity. Peripheral activation and accumulation of activated pro‐inflammatory monocytes/macrophages within lungs has become one hallmark of symptomatic SARS‐CoV‐2 infection. 41 , 64 , 65 , 66 In contrast, the exact role of interactions between DCs and SARS‐CoV‐2 has not been determined yet. Previous in vitro experiments showed that different human coronaviruses display either high (229E) or poor (OC43) capacities to infect macrophages. 67 The efficiency of macrophage infection by coronaviruses was negatively correlated with IFN‐α production. 67 In COVID‐19 patients, ACE2 expression was detected on both lymph node‐associated CD68 + macrophages and tissue‐resident CD169 + macrophages. 68 It needs to be addressed, however, whether other proven and potential SARS‐CoV‐2 receptors, such as CD147 or CD209 (DC‐SIGN), both being expressed by monocytes/macrophages and DCs can facilitate viral entry to these cells. 14 , 29 , 69 , 70 Recently, SARS‐CoV‐2 particles were found in macrophages, but it remains elusive whether these findings were an effect of active cellular infection or just the consequences of physiological phagocytosis. It is tempting to speculate that SARS‐CoV‐2, similarly to HIV, can use macrophages as a Trojan horse contributing to viral spread. 68 , 71 Regardless of the exact mechanism of viral entry, both previously described coronaviruses and the new SARS‐CoV‐2 can trigger NLRP3 inflammasome activation in monocytes/macrophages, production of high levels of pro‐inflammatory mediators such as IL‐6, GM‐ CSF, IL‐1β, TNF, CXCL‐8, CCL‐3, and enhanced cell death. Subsequently, it may lead to the cytokine storm also known as cytokine release syndrome (CRS) (Figure 4). 72 Some of these cytokines (ie, IL‐6) are mainly secreted by macrophages, 66 , 73 , 74 , 75 , 76 and the evidence of macrophage activation syndrome has been reported. 77 Thus, beneficial effects of anti‐IL‐6R treatment on COVID‐19 outcomes indicate that therapies targeting macrophage‐related activities can become promising means to inhibit the inflammatory storm in the course of coronavirus disease. 78 Overloaded, activated and subsequently dying macrophages might contribute to an increase in the levels of plasma ferritin and profound dysregulation of iron metabolism. 79 , 80 High ferritin levels are common clinical findings in patients with severe COVID‐19. 81
FIGURE 4.
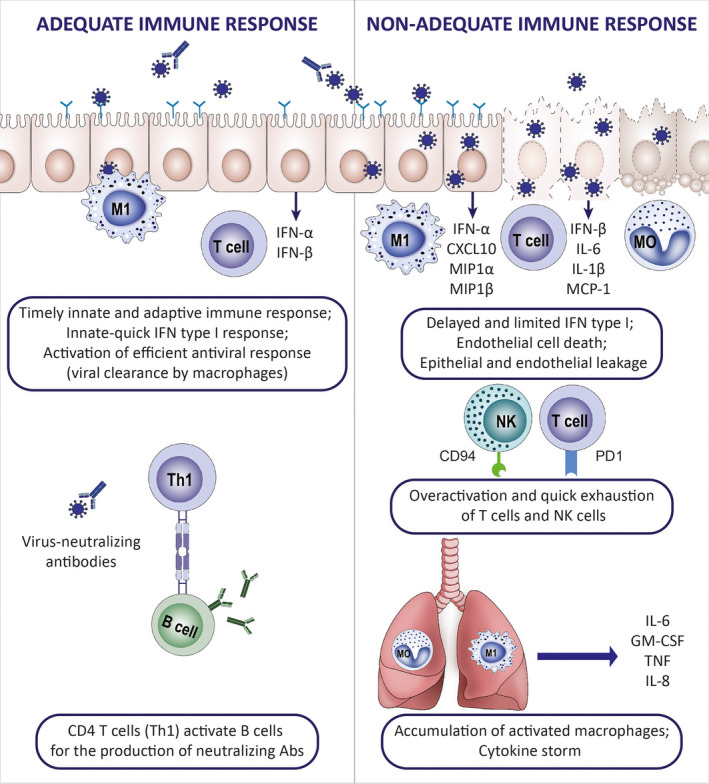
Immunology of adequate and nonadequate response to SARS‐CoV‐2 infection. The clinical course of the SARS‐CoV‐2 infection varies from an asymptomatic to a severe, life‐threatening syndrome. The number of asymptomatic carriers is unknown, and virus detection is often accidental. Data on the immune characteristics in this group are lacking. Patients who experience mild symptoms are characterized by a transient, slight decrease in lymphocyte counts and an increase in neutrophil counts in the peripheral blood. Viral clearance in this group is convergent in time with the specific antibody production. Delayed and limited IFN type I response in combination with the overactivation of pro‐inflammatory cytokine response has been suggested as a possible mechanistic explanation of hyperinflammatory syndrome in COVID‐19 patients presenting with severe clinical manifestations: respiratory insufficiency, kidney failure, thromboembolic, and other complications. Severe COVID‐19 is characterized by a systemic cytokine release syndrome (CRS), increased levels of LDH and CRP, hypoalbuminemia, deepening decrease in lymphocyte counts and immune exhaustion of T cells
4.1.2. Neutrophils and eosinophils
Neutrophils are one of the predominant lung infiltrating leukocytes in severe SARS‐CoV‐2 infection, and neutrophilia predicts poor clinical outcome. 82 Postmortem analysis of lung samples from COVID‐19 patients showed neutrophil infiltration in pulmonary capillaries and neutrophil extravasation into the alveolar space. 83 , 84 Under neutrophil‐activating conditions, such as those occurring during systemic inflammation (CRS), neutrophil extracellular traps (NETs) can be released. Although this is a way to ensnare pathogens, NET formation is linked to pulmonary diseases, particularly acute respiratory distress syndrome (ARDS). Severe COVID‐19 conditions with uncontrollable progressive inflammation presumably induce an intense crosstalk between neutrophils releasing NETs and IL‐1β secretion from macrophages, which is the driving force in further complications. 83
Although eosinophils have protective effects in different viral infections, the eosinophil response toward SARS‐CoV‐2 is incompletely understood. 85 A significant amount of COVID‐19 patients present eosinopenia, 86 , 87 although it is not reported in all cohorts. 88 The pathophysiological mechanism of eosinopenia in COVID‐19 patients is not clear but may be related to increased apoptosis, less eosinophilopoiesis, and decreased eosinophil egression from the bone marrow. 89 Increased tissue migration is unlikely because eosinophils infiltration was not found in pulmonary tissue of COVID‐19 patients, 90 but further research is needed.
4.1.3. ILC and NK cells
ILC are effector cells that respond to environmental cytokines and regulate immune responses. 91 , 92 Type 1 ILC include IFN‐γ producing subtypes and natural killer (NK) cells. Limited data are available on the role of ILC in relation to COVID‐19, but it has been shown that ILC1‐ILC3 express CD26, CD147, cyclophilins and theirs interaction partners. 10 Data on the numbers of NK cells in patients with COVID‐19 are varied. Some studies reported no differences in NK cell counts compared with healthy controls, 93 , 94 while one study found increased numbers of NK cells and suggested a role for NK cells in the CRS. 95 In contrast, other studies showed low 96 or considerably decreased numbers of NK cells in patients with SARS‐CoV‐2 infection, which was more prominent in severe cases. 97 , 98 , 99 After successful treatment, the numbers of NK cells restored to the normal levels with reduced expression of NKG2A. 99
Functional exhaustion of NK cells and CD8+ T cells was described in relation to severe SARS‐CoV‐2 infection (Figure 4). Exhausted NK and CD8+ T cells expressed CD94/NK group 2 member A (NKG2A), which functions as an inhibitory receptor, and showed diminished production of CD107a, IFN‐γ, IL‐2, granzyme B, and TNF‐α. 99 During infection, IFN‐γ induce expression of the nonclassical human leukocyte antigen E (HLA‐E). 100 HLA‐E is the ligand of NKG2A, which is expressed on epithelial cells. NKG2A blockade with monoclonal antibodies (Monalizumab) prevents the binding of HLA‐E, which may be a target for COVID‐19 therapy.
4.1.4. Complement and SARS‐CoV‐2
The complement system is engaged in both coagulation and inflammatory pathways. Histologic and immunohistochemical analysis of lung and skin have been conducted in patients with COVID‐19‐induced ARDS. The typical pulmonary findings for ARDS were accompanied with significant deposits of terminal complement components C5b‐9 (membrane attack complex), C4d, and mannose binding lectin (MBL)‐associated serine protease (MASP)2, in the microvasculature. 101 The biopsies of both damaged and normally appearing skin revealed a pauci‐inflammatory thrombogenic vasculopathy, with deposition of complement products C5b‐9 and C4d. 101 The authors conclude that a subset of sustained, severe COVID‐19 patients may be defined by a type of catastrophic microvascular injury syndrome mediated by activation of complement pathways and an associated procoagulant state. Further, C3‐deficient mice developed significantly less respiratory dysfunction despite viral loads deposited in the lung. These data indicate that SARS‐CoV‐mediated disease is largely immune driven and complement activation regulates a systemic pro‐inflammatory response to SARS‐CoV infection. 102 Most recently, the placentas from 5 healthy newborns (all negative for viral RNA and spike protein) of COVID‐19 positive mothers revealed vascular thrombosis without complement deposition, supporting COVID‐19's systemic procoagulant effects unrelated to systemic complement activation. 103
4.1.5. Trained immunity
Stimulation of innate immune cells with specific microbial antigens induces long‐lasting epigenetic and metabolic re‐programming leading to enhanced responses upon a second challenge by the same or unrelated microbial insults, a process coined as “trained innate immunity.” 104 , 105 As a consequence, trained immunity‐based vaccines (TIbV) able to induce potent responses against both specific and nonspecific antigens contained in the formulation has emerged as a novel concept in vaccinology. 105 TIbV might be especially relevant when conventional vaccines are not available, as it is the case for SARS‐CoV‐2. One of the best examples about trained immunity is the influence of BCG vaccination on unrelated pathogens. 104 BCG seems to induce nonspecific responsiveness to infections both at the level of trained immunity and prolonged heterologous Th1/Th17 responses. 106 BCG‐vaccinated infants have significantly increased production of pro‐inflammatory cytokines, increased protection against infections and reduced mortality. 107 Increased expression of PRRs in monocytes isolated from peripheral blood mononuclear cells (PBMCs) of healthy individuals 1 year after BCG vaccination has marked the importance of trained immunity. 106 Although further studies are required, lower number of cases and deaths per population during COVID‐19 pandemic seem to be reported in countries with BCG vaccination programs than those that did not have or ceased it, which could be attributed to potential BCG vaccination‐induced trained immunity effects. 108 , 109 Trials assessing the efficacy of BCG vaccination in populations at high risk of infection or with a high risk of mortality, such as hospital staff working in close contact with COVID‐19 patients or older individuals, are currently being performed in the Netherlands, Australia and Greece. 110 Future research and clinical trials are needed to demonstrate whether novel TIbV might represent a suitable strategy for the prevention and treatment of SARS‐CoV‐2 infection.
4.2. Adaptive immunity
4.2.1. T cell–related mechanisms: lymphopenia, T‐cell over‐activation and T‐cell exhaustion
Cytotoxic CD8+ T cells directly neutralize infected cells, and CD4+ T cells aid B cells to initiate humoral responses (Figure 4). 111 T cells are instrumental in developing immunological memory in the form of virus‐specific CD8+ and CD4+ T cells as shown in case of SARS‐CoV. 112 , 113 , 114 In fact, SARS‐CoV‐specific CD8+T cells have been detected in humans up to 11 years postinfection, which is longer than the specific antibodies. 114 SARS‐CoV‐2‐specific CD8+ and CD4+ T cells were also recently identified in ~70% and 100% of COVID‐19 convalescent patients, respectively. CD4+ T cells responded to spike (S) protein, which correlated with the magnitude of the anti‐SARS‐CoV‐2 IgG and IgA titers. Importantly, SARS‐CoV‐2 reactive CD4+ T cells were also detected in ~ 40%‐60% of unexposed individuals, suggesting cross‐reactive T‐cell recognition between circulating “common cold” coronaviruses and SARS‐CoV‐2, 115 which was confirmed by others. 116 Profound lymphopenia, with the subsequent shifts in the T‐cell subsets composition, is often reported in SARS‐CoV‐2 infection, similarly to SARS‐CoV and some other viruses. 94 , 98 , 117 Total numbers of CD4+ T cells and CD8+ T cells are below normal levels in most COVID‐19 patients, with the lowest numbers in the severe cases. Moreover, the number of Treg cells is also decreased, 98 whereas a recent case report of nonsevere COVID‐19 showed a progressive increase in the proportion of CD4+CXCR5+ICOS+PD‐1+ circulating follicular helper T (TFH) cells. 96 Delayed development of adaptive responses, together with prolonged virus clearance, has been reported in cases of severe SARS‐CoV‐2 infection (Figure 4). 118 Unfortunately, the mechanisms involved in the lymphocytopenia are still not known in SARS‐CoV and SARS‐CoV‐2 patients. T cells can be infected through highly expressed CD147 14 , 15 or potentially through CD26, as ACE2 expression on lymphocytes is very low, 10 except in certain tissue‐derived T cells. 119 It is yet unclear whether such infection is the reason of the death of infected T cells. Secondly, as in the case of SARS‐CoV, an alteration in the antigen‐presenting cells (APCs) function and subsequent impairment of T‐cell priming might lead to an inefficient/delayed formation of virus‐specific T cells. 120 , 121 , 122 Finally, also a high cytokine response from the infected cells might induce apoptosis of T cells. 123 The causes of lymphopenia need to be extensively studied, as it correlates with the higher risk of severe disease and increased length of hospitalization. 124 , 125 In addition to decrease in numbers, there are also other defects reported in the function of T‐cell subsets in SARS‐CoV‐2 infection. In severe pneumonia in COVID‐19 patients, it has also been shown that highly cytotoxic, activated CD8+ T cells and Th17 cells, can also participate in the CRS, together with macrophages and epithelial cells. 117 Highly activated T cells participating in viral infection often acquire an exhausted phenotype. Surviving T cells appear functionally exhausted with elevated levels of PD‐1. 126 The increased expression of PD‐1 and Tim‐3 on CD8+ T cells was found to progress with the infection. T‐cell exhaustion is a reversible process where a decrease in antigen availability, achieved either through the gradual resolution of the infection or intervention strategy, has led to exhausted T cells regaining their functions. 127
4.2.2. B cell–related mechanisms and antibody‐responses
Human SARS‐CoV‐2 infection activates mechanisms of B‐ and T‐cell immunity that result in the generation of neutralizing antibodies. 96 Initially, B cells appear to recognize SARS‐CoV‐2 through the nucleocapsid protein, which induces their activation and subsequent interaction with cognate CD4+ T cells. The antibody response is mounted 4‐8 days after the onset COVID‐19 symptoms and dominated by IgM. 128 This initial IgM‐response is followed by IgA and then IgG production (10‐18 days).
The development of mucosal IgA likely prevents SARS‐CoV‐2 re‐infection while circulatory IgA may contribute to systemic SARS‐CoV‐2 neutralization and to dampen inflammation during active infection (Figure 4). 129 The extent and quality of the IgG response to neutralize SARS‐CoV‐2 is critical. Based on previous SARS‐CoV infection reports, SARS‐CoV‐2‐neutralizing IgG antibodies should be specific for the S protein and detected in serum at 2‐3 weeks postinfection. 130 , 131 For that reason, human convalescent serum transfer has been proposed for the prevention and treatment of COVID‐19 patients. 132 In fact, and a number of clinical trials have recently reported its therapeutic value in COVID‐19. 51 , 54 , 133 , 134 However, low affinity or suboptimal IgG levels may enhance viral entry into Fcγ receptor‐expressing cells through IgG binding. This mechanism may induce the release of inflammatory cytokines and contribute to the CRS reported in some severe COVID‐19 patients. 135
In a recent pre‐print study, single‐cell RNA sequencing (scRNA‐seq) of PBMCs of 7 patients hospitalized with confirmed COVID‐19 and 6 healthy controls was performed. 136 Heterogeneous interferon‐stimulated gene (ISG) signature, HLA class II downregulation, and a novel B cell–derived granulocyte population were reported in patients with acute respiratory failure requiring mechanical ventilation. The putative contribution of this intriguing B‐cell population to COVID‐19 pathology remains to be elucidated.
A central issue of B‐cell immunity to SARS‐CoV‐2 is the duration of the antibody (IgG) response once the infection is cleared, as well as the ability for SARS‐CoV‐2 specific memory B cells to expand, or replenish, the plasma cell compartment upon re‐infection. Given that long‐lived plasma cells and high‐affinity memory B cells are thought to be germinal center‐dependent, 137 it is important to characterize the antibody and B‐cell memory repertoire (affinity, number of mutations, clonal origin, etc) of asymptomatic patients and patients that have recovered from COVID‐19. The rapid peak of viral load detected in COVID‐19 patients, 138 , 139 as compared to SARS‐CoV, may accelerate plasma cell differentiation, 140 thus limiting the germinal center phase and the degree of long‐term immunity protection.
4.3. Multiorgan involvement in severe COVID‐19
4.3.1. Lung pathologies in COVID‐19
It has yet to be determined whether SARS‐CoV‐2 shows significantly more pronounced lung tropism than other respiratory viruses. COVID‐19‐associated viral pneumonia is relatively frequently complicated by ARDS. Lung CT images of COVID‐19 pneumonia patients revealed mostly diffuse patchy ground‐glass opacities under the pleura with partial consolidation which, in clinically improving individuals, can be further absorbed and followed by formation of fibrotic tissue. 141 Postmortem analysis of COVID‐19 patients revealed extensive alveolar damage complicated by the formation of hyaline membranes, diffuse remodeling of alveolar wall and accumulation of immune cells (mostly macrophages) infiltrating air spaces. 117 , 142 Macrophages accumulating in lungs secrete type I and type III IFNs that enhance local antiviral defenses in surrounding epithelial cells. Lung‐associated macrophages contribute to development of CRS by producing IL‐6 and IL‐1β, cytokines promoting further recruitment of cytotoxic T cells and neutrophils. In consequence, activated neutrophils produce reactive oxygen species and leukotrienes that directly contribute to acute lung injury. 143 Even successful eradication of the virus does not prevent from continuous lung damage, development of frequently progressive and irreversible fibrotic consequences. 144 At this relatively early phase of the pandemic, the exact fraction of COVID‐19 patients burdened with persistent fibrotic interstitial lung disease cannot be precisely determined. Nevertheless, available and novel anti‐fibrotic therapies should also be considered as candidate strategies to manage post‐COVID‐19 long‐term lung fibrosis. 145
4.3.2. Myocardial and endothelial damage: an immunological perspective on cardiac presentation of COVID‐19
Cardiac injury is a prominent feature in COVID‐19 developed by a considerable proportion of patients and is associated with an increased mortality. 146 The pathogenesis of COVID‐19 in the cardiovascular system likely results from a combination of several mechanisms such as direct viral toxicity, systemic CRS‐mediated and stress‐related injury. These mechanisms promote cardiomyocyte and endothelial apoptosis, endothelial shedding, plaque destabilization, and increase wall shear stress, leading to myocarditis, endotheliitis, ischemia, cardiac arrhythmias, and hypercoagulability. ACE2 is highly expressed on cardiomyocytes and endothelial cells, possibly facilitating direct viral damage. However, it is unknown whether vascular derangements in COVID‐19 patients are due to endothelial cell involvement by the virus. A recent study found that endothelial cells can be infected by SARS‐Cov‐2, as postmortem analysis of kidney by electron microscopy revealed viral inclusion structures within endothelial cells. 147 The postmortem histology from patients with multiorgan failure in COVID‐19 showed endothelitis in the lung, heart, kidney, liver, and small intestine, with an accumulation of inflammatory cells associated with endothelium. 147 These findings suggest that the endotheliitis may be a combination of direct consequence of the viral involvement (ie, presence of intracellular viral bodies) and the host inflammatory response (Figure 5).
FIGURE 5.
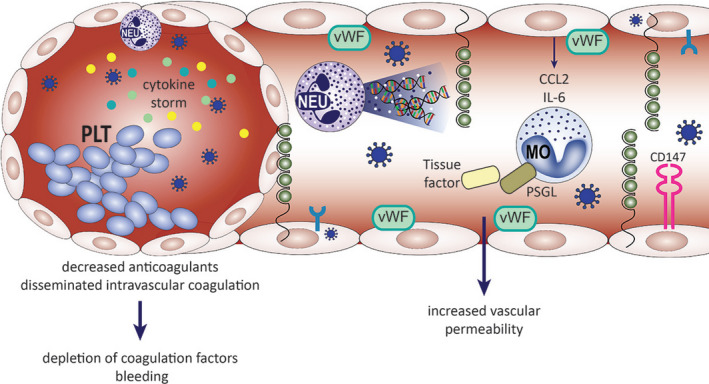
Involvement of endothelium in COVID‐19 progression. SARS‐CoV‐2 viremia is seen approximately 1 wk after the onset of illness, accompanied by an abundance of circulating pro‐inflammatory cytokines. Endothelial cells express ACE2 receptor and can be infected by the SARS‐CoV‐2. Direct viral influence on the endothelial cells, as well as systemic inflammation (depicted by activated neutrophils and extensive NET‐osis) and cytokine storm, can lead to endotheliitis, disseminated intravascular coagulation, and coagulopathy, described in severely affected COVID‐19 patients. Activated endothelial cells upregulate the expression of adhesion molecules (P‐selectin) and coagulation factors (vWF), secrete immune mediators (CCL2, IL‐6). Monocytes respond to these by releasing tissue factor and upregulate the expression of PSGL. Simultaneously, platelet activation and aggregation occurs. Increased numbers of neutrophils and monocytes in the peripheral blood correlate with severe disease course and fatalities. CCL2, CC‐chemokine ligand; IL‐6, interleukin 6; MO, monocyte; NEU, neutrophil; NET, neutrophil extracellular traps; PLT, platelets; PSGL, P‐selectin glycoprotein ligand 1; vWF von Willebrand factor.
In cardiomyocytes, SARS‐CoV‐2 appears to downregulate ACE2 and diminish its cardioprotective role, promoting left ventricular failure and hypertrophy, as well as pro‐thrombotic and pro‐oxidant pathways. 148 Few cases documented myocarditis with diffuse T‐lymphocytic inflammatory infiltrates with interstitial edema and without fibrosis, suggesting an acute inflammatory process. A recent study presenting data from autopsy series also demonstrated SARS‐CoV‐2 viral load in heart tissue. 149 , 150 , 151 Since SARS‐CoV‐2 may predispose patients to coagulopathies with clinical manifestations ranging from arterial and venous embolisms to disseminated intravascular coagulation, with very poor prognosis, early prophylactic anticoagulation in hospitalized patients is recommended. 152 Taken together, direct viral involvement, imbalanced host immune response, and systemic inflammation are proposed as important mechanisms of myocardial/endothelial injury.
4.3.3. Coagulation parameters in COVID‐19 patients
Abnormal coagulation parameters such as mild thrombocytopenia, prolonged prothrombin time, disseminated intravascular coagulation, 153 , 154 , 155 and elevated D‐dimers are seen in 36% to 43% of COVID‐19 patients. 154 , 156 In a meta‐analysis of 4 published studies, higher D‐dimers were found in patients with more severe COVID‐19. 156 Also, thrombocytopenia was reported to be associated with more severe COVID‐19 and increased risk of death (Figure 5). 155 , 157 In a trial of severe COVID patients (n = 99), anticoagulant therapy (eg, low molecular weight heparin) was associated with better prognosis. 158
Activation of endothelium, platelets, and leukocytes leads to enhanced local and systemic production of thrombin, which in turn leads to deposition of fibrin, microangiopathy, and eventual organ damage. Both pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs) initiate these processes. 159 In 16 patients with severe COVID‐19, a correlation between IL‐6 and fibrinogen levels was found, supporting a link between hyperinflammation and increased venous thromboembolism (VTE) risk. 160
Although thrombocytopenia has been implicated in patients infected with SARS‐CoV‐2, the association between platelets and the disease mortality is not clear. In COVID‐19 patients from Wuhan, China, platelet count increase was an independent risk factor reversely associated with in‐hospital mortality, as an increase of 50x109/L platelets was associated with a 40% decrease in mortality. 155 Another study of 548 patients from China found that while platelet levels were decreased when hospitalization for COVID‐19, platelet levels increased in survivors over time, but maintained lower levels or dropped significantly over time in nonsurvivors. 161 Thus, baseline platelet levels and changes over time appear to be associated with subsequent mortality and monitoring platelet levels is important in predicting prognosis of patients with SARS‐CoV‐2 infection.
4.3.4. Neurological disease presentation
Together with the choroid plexus, the blood‐brain barrier protects the brain from invading microorganisms. 162 Nevertheless, several pathogens, including viruses, are still able to traverse the barriers, 163 especially in cases of systemic inflammation 164 causing potential alterations of the central nervous system (CNS). In particular, coronaviruses might exhibit neurotropic properties, 165 and SARS‐CoV was detected in human brain. 166 , 167 Of interest, ACE2 is expressed in the human brain. 45 Based on the knowledge from animals studies, SARS‐CoV can enter the brain via the olfactory nerve leading to a rapid, transneuronal spread to connected areas of the brain. 168 As SARS‐CoV infects immune cells, the virus might penetrate the CNS also via the hematogenous route. 167
In the current COVID‐19 outbreak, a few case reports described meningitis/encephalitis or COVID‐19‐associated acute necrotizing hemorrhagic encephalopathy with or without SARS‐CoV‐2 RNA detected in the cerebrospinal fluid. 169 , 170 Moreover, autopsies of patients with COVID‐19 showed cerebral hyperemia and edema with degeneration of some neurons. 171 In a study on COVID‐19 patients from Wuhan hospitals, 78 out of 214 patients had neurological manifestations especially in severe infections. Some patients had only neurological manifestation without typical COVID‐19 symptoms. 172 In addition, olfactory and gustatory dysfunctions are often reported in patients with COVID‐19, 173 , 174 , 175 which might be due to direct effects on the nervous system.
4.3.5. COVID‐19‐related kidney failure
Recent evidence indicates that kidney injury occurring during SARS‐CoV‐2 infection can result not only from CRS and ongoing sepsis but also from direct virus‐induced impairment. 176 , 177 In fact, ACE2 is highly expressed on renal tubular cells. 178 Clinical observations of COVID‐19‐related kidney damage have been confirmed by an elegant experiment demonstrating that SARS‐CoV‐2 can directly infect human kidney organoids. 179 Moreover, this infection led to further efficient shedding of progeny viruses capable of infecting Vero E6 cells. This finding suggests that kidneys are an active player in the process of viral spread rather than only a site of virus‐induced tissue damage. The process of kidney infection by SARS‐CoV‐2 was significantly, but not completely inhibited by human recombinant soluble ACE2, which indicates that there might be other than ACE2 receptors accounting for the entry of SARS‐CoV‐2 to kidney cells. 179 The putative candidate can be CD147 being highly expressed on proximal tubular epithelium. CD147 together with one of its ligands, cyclophilin, plays a crucial role in renal inflammation and renal fibrosis. 180 Moreover, cyclophilins efficiently control the process of coronavirus replication. 181 Thus, therapeutic strategies could aim at breaking the CD147‐cyclophillins.
4.4. Multi‐morbidities as a risk factor for severe COVID‐19
Multi‐morbidities are associated with the severe course of COVID‐19. A meta‐analysis including 1,558 patients with COVID‐19 showed as independent risk factors chronic obstructive pulmonary disease (COPD) (OR: 5.97), cerebrovascular disease (OR:3.89), type 2 diabetes mellitus ((T2DM) (OR: 2.47), and hypertension (OR: 2.29). 182 , 183 In the nation‐wide report from China including 1590 patients with COVID‐19, one comorbidity was present in 25.1% and two or more comorbidities in 130 8.2% patients (Figure 6). 184
FIGURE 6.
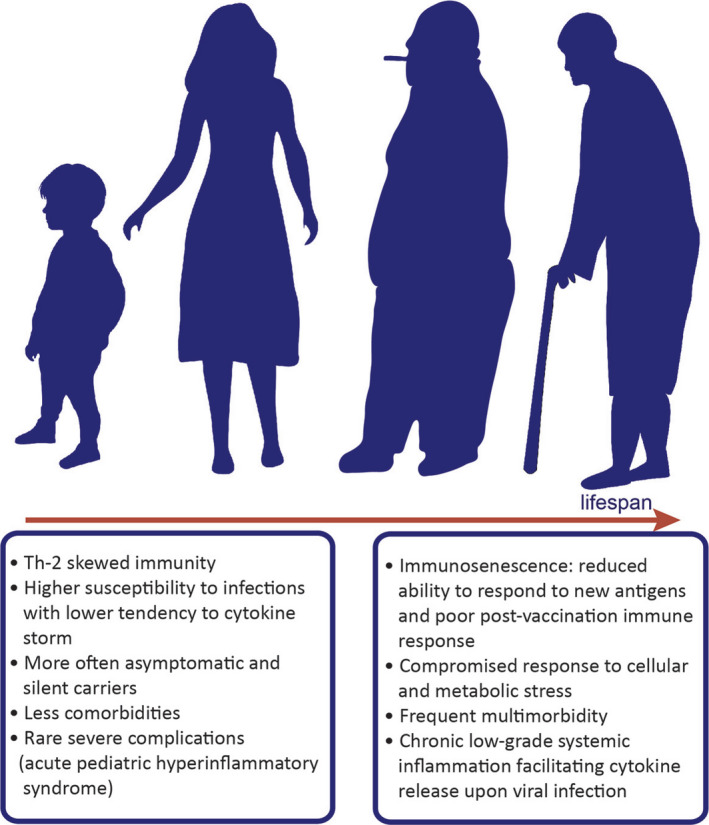
Age, gender, and comorbidities modify the onset and progression of COVID‐19. Epidemiological observations show clear differences in the course of SARS‐CoV‐2 infection between children and adults. It seems that children are less susceptible to the infection and develop less typical symptoms of the disease. Consequences of the infection on physiological development of children are unknown. Clinical data and age‐related rhesus macaque model of COVID‐19 reveal that obesity, diabetes, hypertension, smoking, chronic respiratory diseases, male gender, and older age are the most common risk factors for development of severe COVID‐19. Older age is associated with higher incidence of multimorbidity and state of low‐grade systemic inflammation. Immunosenescence could influence the adequacy of the host's response to the infection
Several mechanisms directly linked to the underlying pathological condition can contribute to the unfavorable clinical outcome. A recent study suggested that hypertension and diabetes resulted in delayed clearance of SARS‐CoV‐2. 185 The relationship between the immune dysfunction in patients with multi‐morbidities and infection with SARS‐CoV‐2 was not specifically evaluated. A transgenic diabetic mouse model expressing human CD26 had more severe disease together with a dysregulated immune response following infection. A delayed and decreased recruitment of CD4+ T cells and inflammatory monocytes and macrophages into the lung tissue and a more prominent Th17 response was oberved. 186 Interestingly, obesity is changing the expression profile of SARS‐CoV‐2 receptors. 10
Metabolic induced low‐grade systemic inflammation, as in obese patients, 187 facilitates an enhanced release of cytokines upon an acute trigger such as viral infection. As the human endocrine pancreas expresses ACE2, the coronavirus might enter islets and cause acute β‐cell dysfunction, leading to acute hyperglycemia and transient T2DM. 188 A mathematical model showed that the insulin resistance, advanced glycan end product (AGE)‐Receptor of AGE (RAGE) signaling pathway in diabetic complications and the adipocytokine signaling pathway were found in all fatal comorbidities of COVID‐19. 189 In addition, AGEs can induce monocyte CD147 expression, an effect mediated by inflammatory pathways and RAGE. 190 CD147 is highly expressed in patients with diabetic complications such as nephropathy, retinal neuropathy, and vasculopathy and was associated with chronic renal failure of other causes.
COPD and ongoing smoking contribute to COVID‐19 severity. 191 COPD and active smokers had significantly increased expression of ACE2 and its gene expression inversely related to the lung function, suggesting a dose‐dependent response. 192
Multimorbidity is also associated with elevated levels of plasminogen. Plasmin, and other proteases, may cleave a newly inserted furin site in the S protein of SARS‐CoV‐2, extracellularly, which increases its infectivity and virulence. Hyperfibrinolysis associated with plasmin leads to elevated D‐dimer in severe patients. Thus, the plasminogen system may prove a promising therapeutic target in COVID‐19. 193
4.5. Sex and aging as a risk factor for COVID‐19 course
The increased vulnerability of males compared with females to severe COVID‐19 has been reported during the pandemic. A direct endocrine link is involved as androgen receptor activity is required for the transcription of TMPRSS2 gene. 194 , 195 Male vulnerability may be further enhanced by X‐linked inheritance of genetic polymorphisms as both the androgen receptor, and the ACE2 genes loci are on chromosome X.
Old age was also associated with an increased risk of infection and worse outcome (Figure 6). Frailty is characterized by multisystem dysregulation leading to reduced physiologic reserve. Although not formally assessed in the COVID‐19 trials, frailty may be linked to infectious disease through common pathways that reduce immunity. 196 , 197 Frailty has also been shown to be associated with poor postvaccination immune response. 196 The aged immune system is characterized by a low‐grade chronic systemic inflammatory state marked by elevated inflammatory markers such as IL‐ 6 and C‐reactive protein and an increased susceptibility to infection. 198 The expression of ACE2 and TMPRSS2 genes in the type II alveolar cells of elderly and young patients is comparable. Therefore, it does not seem to be responsible for the worse outcomes observed in COVID‐19 affected elderly, but the expression of other receptors is age‐dependent. 10
4.6. Allergy‐related risk for COVID‐19
Drug hypersensitivity (11.4%) and urticaria (1.4%) were self‐reported by patients with COVID‐19. 86 In contrast, respiratory allergies and asthma were not reported as risk factors for SARS‐CoV‐2 infection. 82 , 86 , 199 , 200 , 201 , 202 However, a report from the CDC of US hospitalizations described contradicting findings in adults with asthma. Among hospitalized patients with COVID‐19, 27.3% of 18‐49 year old adults had asthma, 13.2% of 50‐64 years, and 12.9% of those of 65 years or older. 203 Currently, patients with allergic rhinitis and patients treated with allergen‐specific immunotherapy are advised to continue their therapies. 204 , 205 , 206
Another study elucidated the impact of comorbid respiratory allergy or asthma on COVID‐19 susceptibility and disease severity. 207 Children with asthma and moderate to severe allergic sensitization showed reduced ACE2 gene expression compared with children with nonatopic asthma. An additional trial including 23 patients with asthma confirmed reduced ACE2 expression in lower airway epithelial cells postallergen challenge. Finally, in vitro experiments using nasal and bronchial airway epithelium showed that IL‐13 reduced the ACE2 expression. 207 However, adult patients with asthma seem to have higher expression of TMPRSS2 and CD44, which forms a functional complex with CD147 in bronchial epithelium. 10
5. IMMUNOLOGICAL BIOMARKER PROFILING OF COVID‐19 FOR PREDICTION OF DISEASE SEVERITY
The development of serious complications and even fatal outcome in SARS‐CoV‐2 infection is strongly linked to the patients’ immune response resulting in CRS. 208 There is an urgent need for biomarkers that predict patients developing severe complications. 209 To date, there is limited information on the biomarkers associated with, or even predicting severe complications in COVID‐19. However, there is much similarity on the biomarkers that have been described before for MERS‐CoV and SARS‐CoV, also β‐coronaviruses, but also with sepsis. Many markers have been demonstrated to be increased in SARS‐CoV‐2‐infected individuals. These markers are related to innate as well as adaptive immunity, endothelial cell activation, thrombocyte activation, and leukocyte infiltration. 201 The list of markers related to severe disease, ICU, and even lethality is more limited. In ICU‐admitted COVID‐19 patients, significant increases of D‐dimer, ferritin, LDH, IL‐6, high sensitivity cardiac troponin, IL‐2, IL‐7, G‐CSF, MCP‐1, MIP‐1α, and TNF‐α were reported. 201 An even more restricted group of markers (IL‐10, MCP‐3, IL‐1ra) were increased in severe and lethal cases. 210 Differences in the biomarkers described are most probably due to the different sampling time during disease and the large heterogeneity between the patients. 201
Most likely, single biomarkers will not be predictive. On the other hand, a combination of markers (a biosignature) will help in patient stratification and may even guide patients‐tailored therapy.
6. URGENT RESEARCH NEEDS FOR MECHANISTIC, DIAGNOSTIC APPROACHES, THERAPEUTIC, AND PREVENTIVE INSIGHT
6.1. Kids versus adults: Mechanisms explaining the clinical differences
Children experience milder COVID‐19 as compared to adults, and a larger proportion of children remains asymptomatic (Figure 6). 203 Data from the USA show a strikingly low number of pediatric hospitalizations (5.7%) and very limited ICU admissions in young age. 203 Of note, children show similar chest CT results as compared to adults, with subpleural ground‐glass opacities even when having few symptoms. 211 While children may be asymptomatic, they are shedding viral particles and can therefore still be contagious with comparable virus loads. 212 ACE2 receptors are upregulated by type 1 IFNs, 12 but downregulated by IL‐13, indicating that Th1/Th2 balance may significantly influence course of SARS‐CoV‐2 infection. 207 Therefore, type 1 IFNs driving anti‐viral immunity may paradoxically promote SARS‐CoV‐2 expansion by upregulating ACE2 expression. Inflammatory responses differ throughout life, for example, pre‐existing chronic inflammation is common in elderly while absent in children. In addition, children have less potent PAMP activation, suboptimal, and Th2‐skewed cytokine production, all resulting a hypo‐inflammatory immune response. 213 This confers decreased protection against infections, but seem beneficial in preventing a CRS in SARS‐CoV‐2 infection. Hence, preferential Th2‐skewed cytokine production observed in children is presumably protective (Figure 6). Additionally, children have less often comorbidities such hypertension, diabetes, and COPD. However, in rare cases, a severe hyperinflammatory shock syndrome with features of atypical Kawasaki disease or toxic shock syndrome was reported in pediatric COVID‐19 patients, 214 which is currently intensively investigated. 215 Data are lacking on evidence surrounding transmission rates of the virus by children. The China/World Health Organization joint commission found that infected children were largely identified through contact tracing in households of adults. 216 However, no significantly different virus levels in the respiratory tract were recently reported across age groups, thus having the potential for similar transmission rates. 56 Understanding the mechanisms underlying different prognosis in children is essential for designing targeted therapies for COVID‐19.
6.2. Immunological diagnosis of SARS‐CoV‐2 infection: challenges of current approaches
PCR tests are useful for detecting SARS‐CoV‐2 RNA in an upper respiratory (preferably a nasopharyngeal) specimens. In addition, a number of diagnostic procedures to assess immunity built against SARS‐CoV‐2 are still being developed, validated, and optimized.
Antibody testing is evolving, and the market is flooded with test kits (both ELISA and rapid tests in the form of lateral flow immunoassays). However, only a small number of these kits are certified, and the results need to be interpreted with caution. Preliminary data indicate that COVID‐19 presents with a classical antibody response consisting of early induction of IgM, followed by IgA and IgG antibodies (Figure 7). 128 IgG seems to appear early in the course of clinical presentation probably due to the relatively long incubation period. However, there is not yet enough evidence with regard to the development of long‐term protective immunity. Antibody testing is so far more valuable in mapping the situation in individual populations, as planned by the WHO in the Solidarity II project. 217 Test kits for the assessment of SARS‐CoV‐2‐specific T‐cell responses for diagnostic use are currently not available.
FIGURE 7.
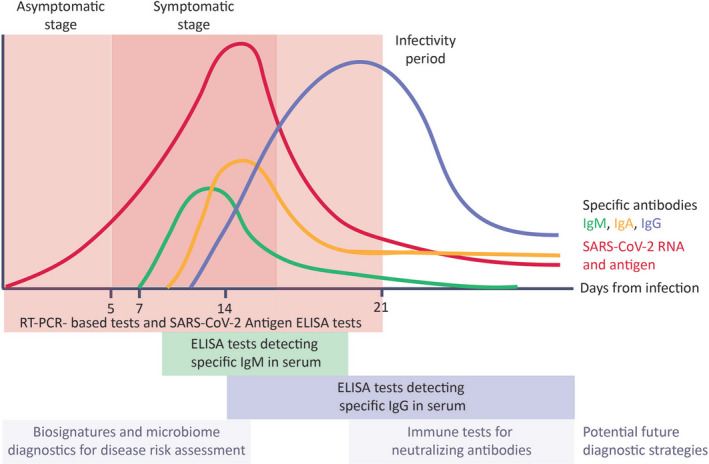
Clinical stages of COVID‐19 and their virology and immunological assessment. The success of restraining SARS‐CoV‐2 transmission depends on accurate and timely diagnostics. Asymptomatic patients transmit SARS‐CoV‐2. RT‐PCR‐based test detecting the SARS‐CoV‐2 RNA in posterior conchae nasal swabs are currently the golden standard in the initial phase of the infection. Viral antigens can be detected in patients’ blood by means of ELISA tests. ELISA tests allow for detection of virus‐specific antibodies in patients’ serum. The production of specific IgM starts after about a week from infection and IgM levels decrease with the production of specific IgG (after about 2 wks from infection). Novel diagnostic and risk‐stratification strategies could include microbiome profiling and tests detecting neutralizing antibodies
6.3. Immunological treatment approaches: biologicals, small molecules and beyond
Current evidence on the role of biologicals, small molecules, and passive immunizations in the treatment of COVID‐19 was assessed conducting a systematic literature search (see online Supporting information).
By May 15, 2020, case series and nonrandomized, small, open‐label studies report on the treatment of SARS‐CoV 2 infections were assessed. Results from controlled, randomized or placebo‐controlled, randomized trials are still lacking. Approaches include either targeting the CRS and hyperinflammatory status of lung destruction via anti‐IL‐6R antibodies, 218 , 219 , 220 , 221 , 222 , 223 , 224 , 225 , 226 , 227 , 228 , 229 , 230 , 231 , 232 , 233 , 234 , 235 , 236 , 237 IL‐1R antagonists, 238 , 239 , 240 JAK‐STAT inhibitors, 241 or inhibition of entrance by anti CD147 antibodies 237 and destruction of the virus via protective antibody delivered with convalescent plasma 51 , 54 , 66 , 133 , 134 , 242 (Table 2). Eculizumab targets complement protein C5 preventing activation of complement terminal complex, which was used off label in patients with SARS‐CoV‐2 infection and severe pneumonia or ARDS and is now evaluated in an ongoing trail (SOLID‐C19). 243 Additionally, clinical trials with type I and III interferons in COVID‐19 are currently conducted. 244 , 245 Targeting T‐cell exhaustion to reverse the dysfunctional state and restore immune responses can be achieved by anti‐PD‐1 and LAG‐3 therapies, 246 , 247 revealing novel therapeutic opportunities for persisting infections. In conclusion, prospective, randomized, and placebo‐controlled trials are needed to elucidate the clinical potential of immunomodulatory or passive immunization therapies.
TABLE 2.
Novel immunological treatment options for COVID‐19 (as of May 15, 2020)
| Treatment | Type of Drug | Mode of Action | Clinical Manifestations | Age, Gender | Intervention | Key Information (type of trial, sample size, effect) | Reference |
|---|---|---|---|---|---|---|---|
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
17/21 (severe); 4/21 (critical) 6 days postonset (median) Heterogenous clinical presentations and comorbidities |
56.8 ± 16.5 (25‐88) 18M, 3F |
400mg once iv (in addition to lopinavir) |
n = 21, prospective, open‐label, nonrandomized single‐arm, interventional trial Normalization body tempin 24h Improvement of clinical symptoms: 15/21 lower oxygen demand; 1/21 no oxygen demand improvement CT (19/21), 19/21 discharge at 13.5 d after treatment start no AE related to drug reported |
Xu et al 230 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
All critical in ICU Heterogenous clinical presentations and comorbidities |
64 (IQR 59‐72), 50‐76; 30M, 3F |
27/33 lopinivir/ritonavir; 26/33 hydroxychloroquine; 28/33 dexamethasone; 4/33 tocilizumab |
n = 4/33 tocilizumab treatment, single‐center prospective observational cohort 9/33 discharged ICU, 1 died, 23 in ICU Outcome of Tocilizumab‐treated patients not disclosed |
Piva S et al 227 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
All post–solid organ transplantation 22/90 mild, 41/90 moderate; 27/90 severe; heterogeneous clinical presentations and comorbidities All of moderate‐severe group had abnormal chest radiographs None on ECMO, 13 no oxygen |
57y (46‐68); 53M, 37F |
Heterogenous uses hydroxychloroquine (61), azithromycin (45), bolus steroids (16) Tocilizumab n = 14 N = 9 1 dose 400mg or 8mg/kg up to 800mg iv; n = 4 ‐ 2 doses; n = 1 ‐ 3 doses. |
n = 14/68 hospitalized received tocilizumab, retrospective study, 6/41 and 8/27 received tocilizumab 3/14 died, 4/14 remain ICU, 5 improvement, 2 discharged. No adverse events linked to tocilizumab |
Pereira et al 226 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade | #1 12d flu‐like symptoms, dyspnea; hypertension, respiratory distress progressing |
1; 71y/M |
D12 Lopinavir/ritonavir + hydroxychloroquine d10: 2 doses tocilizumab 12h apart |
n = 3, case report #1 d12 hospitalization, d21 deterioration and application of tocilizumab 3d fever resolution |
Di Giambenedetto et al 221 |
| #2; fever, dyspnea, chest pain | 2; 45y/M |
Day 1 Lopinavir/ritonavir + hydroxychloroquine Day 5tocilizumab: 2 doses 12h apart |
case report #2 Initial improvement, then worsening dyspnea until d5 when tocilizumab was applied d7 fever resolution, clinical improvement |
||||
| #3; flu‐like symptoms, pneumonia | 3; 53y/M |
Day 1 Lopinavir/ritonavir + hydroxychloroquine Day 3 started, 3 doses |
case report #3 respiratory symptoms worsened d2 d3 tocilizumab given progressive resolution of the symptoms day 9) |
||||
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
6/15 (severe); 7/15 (critical); 2/15 (moderate); 10 had comorbitidity |
73y (62‐70) 12M, 3F |
In 8, Tocilizumab (80 to 600mg) with methylprednisolone (20mg to 80 b.i.d.); In n = 5, 2 or more doses |
n = 15, pilot study; retrospective observational study Outcomes: 3/4 (critical receiving one dose) died, 4th persistence of CRP elevation over weeks IL‐6 decreased in 10/15 patients, increased in 4 critical and 1 severe. n = 2 disease aggravation n = 1 disease improvement n = 9 disease stabilization |
Luo et al 224 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
Kidney‐transplanted individuals Heterogenous clinical presentations and comorbidities, All presented with fever. |
59 (IQR 51‐64) 16M, 4F |
Medication adjusted according to renal function. d1‐ 7: lopinavir/ritonavir (400/100 mg bid) ‐hydroxychloroquine 200 mg bid if GFR > 30 ml/min 200 mg/day GFR > 15 ml/min and < 30 ml/min 200 mg every other day if GFR < 15 ml/min) Longer in case of deterioration Dexamethasone (20mg/d for 5 days followed by 10mg/d for 5 days) Tocilizumab (up to 2 infusions at intervals of 12 to 24 hours; 8 mg/kg (max 800 mg) |
n = 6 TCZ /20, single‐center observational study n = 1 death n = 2 discharged n = 3 inpatient Chest X‐ray improvement after 3‐11 days on tocilizumab: n = 3 no improvement n = 2 improvement n = 1 n/a |
Alberici et al 218 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade | COVID‐19‐related pneumonia and respiratory failure, not needing mechanical ventilation. Included if satisfying at least one criteria: respiratory rate ≥ 30/min, SaO2 ≤ 93% or PaO2/FiO2 300 mmHg. |
65y (IQR 54.5‐73); 64 M 21F (75%) |
Standard therapy (ST, N = 23): hydroxychloroquine (400 mg daily) and lopinavir (800 mg daily) plus ritonavir 200 mg daily Tocilizumab treatment (TCZ) started within 4 days of admission single administration, 400mg/iv |
n = 62 TCZ/Co, single‐center, retrospective, observational study Survival rate significantly higher in tocilizumab‐treated patients (60/62) as compared to controls (12/23) (HR 0.035) 92% of discharged patients recovered in Tocilizumab. Respiratory function improved in 64.8% 42% of discharged patients in control group recovered. Respiratory function worsened in all the patients; mechanical ventilation needed. |
Capra et al 219 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
SMACORE is the cohort of patients with confirmed diagnosis of COVID‐19 disease referred to the IRCCS Policlinico San Matteo Hospital of Pavia enrolled March 14th‐27th TCZ + SOC vs. standard of care (SOC) TCZ criteria: CRP > 5 mg/dl, Procalcitonin < 0.5 ng/mL, arterial partial pressure of oxygen/fractional inspired oxygen (fiO2) (PF ratio) < 300 ALT < 500 U/L. |
SOC 63.74 (IQR 16.32) 63M 28F TCZ 16.32), 62.33 (IQR 18,68) 19M 2F |
Standard of care hydroxychloroquine (200 mg bid), azithromycin (500 mg once), prophylactic dose of low weight heparin, and methylprednisolone (a tapered dose of 1 mg/ kg up to a maximum of 80 mg) for 10 days |
N = 91 SOC vs. N = 21 TCZ Retrospective registry analysis No difference regarding mortality d7 and ICU admissions (major outcomes) |
Colaneri et al 220 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade | Heterogenous clinical presentations and comorbidities; all critically ill with oxygen needed |
76.8 (52‐93) ± 11; 9M, 11F 70.7 (33‐96) ± 15; 6M, 19F |
Standard of care n = 20; antivirals, oxygen, antibiotics with occasional corticosteroids; 1 or 2 doses of TCZ average d7 from admission n = 25, same as above, no TCZ |
n = 45, retrospective case‐control study: n = 20 (TCZ, off label) to n = 25 (no TCZ) P = .002; 25% vs 72% combined death and/or ICU admission endpoint; P < .001; 0% vs 44% ICU admission for n = 20 |
Klopfen‐stein et al 223 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
Dysnpea, cough, fever with prior hypercholesterolemia Fever, cough, tachypnea with prior comorbidities Fever, dyspnea, needing Venturi; multiple comorbidities |
61y/F 57y/F 56y/M |
d2 sc. 162mg single dose; with antibiotics; previously on antivirals d6 sc. 162mg single dose; d1 antivirals d1 antivirals, d2 antibiotics for pneumonia, d8 sc. 162mg single dose |
n = 3, case series All with IL‐6 decrease and CT improvement, no adverse effects #1 fever resolved in 2 days and oxygen high flow nasal cannula progressive decrease, stopped d12 #2 worsening SpO2 lead to Venturi mask d2. d8 fever gone and oxygen stopped #3 fever resolved d8, reduction of oxygen to 4L/min from 15L/min. |
Mazzitelli et al 225 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
Hospitalized adult patients with severe COVID‐19 Inclusion criteria: a) PCR‐confirmed COVID‐19; b) Sa02 < 93% at ambient air, or tPao2/ Fio2 < 300 mm Hg c) at least 3 of the following: CRP > 10‐fold normal ferritin > 1000 ng/ml D‐dimer x 10 normal values; LDH x 2 the upper normal limits. |
62.6y (±12.5); 56M 7 F |
Tocilizumab treatment tocilizumab iv (8 mg/ kg) or s.c. (324 mg); a second administration within 24 h was given in 52/63 patients. Route of administration disposed according to the drug availability Concomitant treatment with antivirals in 63/63 patients (100%): Lopinavir/ritonavir in 45/63 patients (71.4%) Darunavir/cobicistat in 18/63 (28.6%) |
Pilot prospective open, single‐arm multicentre study. Patients were prospectively followed for, at least, 14 days after admission. Route of administration did not significantly affect mortality: 12.9% (4/31) and 10.3% (3/29) in the tocilizumab iv and s.c. groups, OR 1.16). Tocilizumab treatment within 6 days from admission was associated with an increased likelihood of survival ad d14(HR 2.2 95%CI 1.3‐6.7). |
Sciascia et al, 228 |
| Tocilizumab |
Hum. mAb IgG1 |
IL‐6R blockade |
All patients receiving noninvasive ventilation (NIV) n = 57 BCRSS = 3 or mechanical ventilation with tracheal intubation (BCRSS score > 3) n = 43 all patients: lymphopenia, high‐level inflammatory markers, |
62y (57‐71 IQR) 88 M 12 F |
Lopinavir + ritonavir 400mg bid + 100mg bid or remdesivir 100mg/d antibiotic prophylaxis hydroxychloroquine 400mg/d Dexamethasone 20mg/d NIV or Mechanical ventilation TCZ 8mg/kg (max 800mg) two infusion 12 hours apart and a third optional 24h |
N = 100 consecutive patients receiving tocilizumab; uncontrolled, single‐center observation, 58% clinical and resp. improvement within 24‐72h 37% stable; 5 worsened (4 deceased) D10: Improvement or stabilization 77%; 15% discharged 23% worsening 20% died N = 43 in ICU with mechanical ventilation, 32 (74%) improved 17 off ventilator 1 (2%) remained stable (BCRSS class 5) 10 (24%) died (all BCRSS ≥ 7 before TCZ). |
Toniati et al 229 |
| Tocilizumab or Sarilumab |
Hum. mAb IgG1 |
IL‐6R blockade | All critical disease on ECMO, heterogenous clinical presentations and comorbidities |
52.41y, ±12.49; 22M, 10F |
4/5 iv steroids, 3/5 antivirals, 2/5 tocilizumab or sarilumab, 1/5 hydroxychloroquine |
n = 3/32, report, real‐time cohort study 17 remain on ECMO, 10 died prior or shortly after decannulation, 5 extubated, 1 of the 5 discharged (9 doses remdesivir). All 5 only on v.v. ECMO 2/3 on Tocilizumab weaned from ECMO, 1 died |
Jacobs, et al ASAIO. 2020 222 |
| Convalescent Plasma | N/A | Passive immunity |
Severe disease 16.5 d (IQR 11‐19.3) since onset to CP transfusion Heterogenous clinical presentations and comorbidities Ventilation: none (n = 3) low flow nasal cannula (n = 2), high flow nasal cannula (n = 3), mechanical ventilation (n = 2) (3/10 SARS‐CoV‐2 negative at application Donor: >3w postonset, >4d postdischarge |
52.5y (IQR 45‐59.5y) 6 M 4 F |
Different co‐medication (anti‐viral 10/10; anti‐fungal 2/10; antibiotics 8/10) steroids 6/10 iv methylprednisolone. 200 ml convalescent plasma with a neutralizing antibody titers of > 1:640 |
n = 10, pilot study; open‐label prospective, nonrandomized, single‐arm study with a retrospective control group 10/10: SARS‐CoV‐2 RNA load negative serum neutralizing antibodies 9/10 1:640 (all 9 had pre‐existng neutralizing antibodies); 1/10 unavailable Compared to historic control: CP group: 3/10 discharged 7/10 improved status Control group: 3/10 deceased, 6/10 stabilized, 1/10 improved |
Duan et al 51 |
| Convalescent Plasma | N/A | Passive immunity |
Critical disease on mechanical ventilation, 1/5 ECMO 10, 19, 20, 20 and 22 days postonset of symptoms |
36‐65y 3 M 2 F |
All on Antivirals and methylprednisolone, N = 3 on interferon alpha 1b 400ml convalescent plasma with SARS‐CoV‐2 specific ELISA titer > 1:1000 and neutralizing titer > 40 |
n = 5, case series ARDS resolved in 4/5 within 12d 3 dismissed (51‐55 d postinfusion) 2 stable, on mechanical ventilation (d37 postinfusion) 5/5: SARS‐CoV‐2 RNA load negative d12 |
Shen et al 133 |
| Convalescent Plasma | N/A | Passive immunity |
N = 5 moderate‐critical disease 31‐58d postonset of symptoms CT‐chest abnormalities Critically ill with deterioration after standard treatment (arbidol) 4/5 required oxygen N = 1 asymptomatic postdischarge SARS‐CoV‐2 positive. |
56‐75; 3M, 3F |
All on Antivirals and methylprednisolone, N = 3 on interferon alpha 1b 200 ml convalescent plasma up to three times |
n = 6, case series 5/6 radiological improvement and symptom improvement 1/6 asymptomatic at enrollment |
Ye et al 242 |
| Convalescent Plasma | N/A | Passive immunity | Critically ill SARS‐Cov‐2 infected patients requiring different types of ventilation |
31‐69 2M, 2F |
All started Antiviral treatment 3/4 interferon alpha‐2b treatment 200‐400mL CP (1‐8 transfusions) transfusion from 9d −12d after admission |
n = 4, case series (including a pregnant woman) d11‐18 all 4 recovered. N = 3 discharged d24‐33 after first CP application. N = 1 discharged from ICU d35 after first CP application Not clear whether effect relates to CP or supportive care. |
Zhang et al 54 |
| Convalescent Plasma | N/A | Passive immunity |
Severe pneumonia, ARDS, ventilation CT‐chest abnormalities Critically ill with deterioration after standard treatment |
71y male; 67y female |
All on antivirals, methylprednisolone iv, and antibiotics 2 doses 250ml convalescent. plasma 12 h apart |
n = 2, case reports #1 d22 CP #2 d7 CP No adverse reaction to plasma, radiological and symptom improvement # 1 tracheostomy, off ventilation now, SARS‐CoV‐2 RNA load negative d26; #2 1 discharged d24 postinfusion; SARS‐CoV‐2 RNA load negative d20 |
Ahn JY et al 134 |
| Baricitinib | SMO | JAK1/2 pathway inhibition |
moderate (>2 of the following symptoms: fever, cough, myalgia, fatigue AND evidence of radiological pneumonia); 6 days of onset (IQR 4‐6.25) |
63.5y (IQR 57.7‐72.20y) 10M, 2F |
4mg/day for 14 days; orally in combination with lopinavir‐ritonavir) |
n = 12, pilot study; open‐label prospective, nonrandomized, single‐arm study with a retrospective control group (n = 12 lopinavir‐ritonavir, hydroxychloroquine) Outcomes: resolution of cough, dyspnea, within 14 days vs 9/12 and 8/12 in control group discharge from hospital 7/12 vs 1/12 in control group |
Cantini et al 241 |
| Meplazumab |
Hum. mAb IgG2 |
CD147 blockade |
Severity in treatment group: 4/17 (common); 6/17 (severe); 7/17 (critical); 4/11 (common); Severity in control group: 4/11 (common) 4/11 (severe); 3/11 (critical) 27/28 had fever as symptom? (16/17 in treatment group and 11/11 in control group) Comorbidities in 9 (treatment) and 4 (control) |
51y (IQR 49‐67y) 11M, 6F |
16/17 glucocorticoid, all 17 antibiotics; 7/11 glucocorticoid, 10/11 antibiotic 10mg days 1,2,5 iv for 11 patients, 6 had 2 doses only. |
n = 17, clinical trial; open‐label prospective, concurrent controlled (n = 11) for 28 days All previously treated with lopinavir/ritonavir and α‐2b. Outcomes: shorter time to discharge treatment vs control (P = .006); case severity improved (P = .021); shorter time to virus negative (P = .014). No adverse effect with treatment |
Bian et al 259 |
| Anakinra | Recombinant human anti‐IL‐1Rα | IL‐1R blockade |
Consecutive patients with SARS‐CoV‐2 pneumonia with high risk of worsening chest CT compatible with COVID‐19‐pneumonia, hospitalized in a non‐ICU, oxygen flow of ≤ 6 L/min, CRP ≥ 50 mg/L. Days since onset of symptoms 8 (median) Oxygen therapy 4.5d (median) |
46‐62y 8M, 1F |
sc. 100 mg/every 12h d1‐d3 sc. 100 mg/every 24h d4‐d10. |
N = 9 case series; prospective, observational, nonrandomized N = 1 acute resp. failure 6h after first dose (authors found no evidence of linkage to Anakinra) and stopped thereafter and ICU admission. Slow reduction of CRP over time and normalization in 5/8 at day 11 8/11 no fever d3 Reduction of affected areas in CT scan (d5‐8) 7/9 no oxygen demand d11 |
Aouba et al 238 |
| Anakinra | Recombinant human anti‐IL‐1Rα | IL‐1R blockade |
n = 8; secondary hemophagocytic lymphohistiocytosis, severe; acute respiratory failure; heterogenous clinical presentations and comorbidities n = 1 no ICU; n = 7 ICU admission |
n = 7; 51‐84y/M n = 1; 71y/F |
n = 7; iv 200mg every 8h/7 days started after ventilation with hydroxychloroquine, azithromycin n = 1; d9 300mg q.d., iv 4 days, then 100mg q.d. All on antibiotics |
n = 8, case series, nonrandomized, no placebo 3 died from refractory shock (no evidence of linkage to anakinra) n = 1, ventilation prevented, discharged d18 All with respiratory function improved, reduction of CRP, ferritin, D‐dimers, and PCT. |
Dimopoulos G. et al 240 |
| Anakinra | Recombinant human anti‐IL‐1Rα | IL‐1R blockade |
moderate to severe ARDS + hyperinflammation (CRP 100mg/L and/or Ferritin ) CPAP |
HDA: 62y (IQR 55‐71) LDA: 68y (IQR 51‐73) ST: 70y IQR (64‐78) |
N = 29 high‐dose Anakinra (HDA) 5mg/kg iv. Bid until sustained benefit then 3d low‐dose therapy + standard treatment N = 7 low‐dose Anakinra (LDA) 100mg sc. Bid until sustained benefit + standard treatment N = 16 standard treatment (ST) 200mg hydroxychloroquine 400mg lopinavir with ritonavir 100mg bid |
N = 16/29/7 (ST/LDA/HAD) Retrospective cohort analysis HDA vs ST: Survival rate d21 high‐dose Anakinra significantly higher (90% vs 56%) HDA vs ST: Mechanical ventilation free survival rate d21 (72% vs 50%; not significant) HDA: n = 7 discontinuation for adverse events d9 (IQR 8‐10) N = 4 (vs ST n = 2) Bacteriemia N = 3 (vs ST n = 5) Increased serum liver enzymes Causes of death: HDA: thromboembolism, respiratory insufficiency, and multiorgan failure (n = 1 for each). ST: respiratory insufficiency (n = 3), multiorgan failure (n = 3), and pulmonary thromboembolism (n = 1). |
Cavalli et al 239 |
6.4. Virome interactions with commensal communities: future prevention and treatment strategies for SARS‐CoV‐2 infections?
Mucosal anti‐viral immunity can be regulated by the microbiota via multiple mechanisms. The immune response to microbes is a form of host defense and entails a variety of intimate interactions with important symbiotic physiological effects on the host. 248 Specific bacterial components and specific metabolites can promote immune maturation and polarization, which ensures appropriate defense against occasional pathogens, while strongly promoting immune tolerance networks that dampen aberrant inflammatory responses. 249 , 250 The composition of the gut and lung microbiome is strongly associated with the induction of polarized immune responses within the human lung. 187 , 250 Bacterial‐derived metabolites such as short chain fatty acids (SCFAs) promote anti‐viral responses in the lungs, while also reducing inflammation. 251 , 252 Composition and metabolic activity of the gut microbiome has been associated with blood proteomic biomarkers predictive of severe COVID‐19. 253 The integration of microbial diagnostics with traditional immunological biomarkers will improve patient´s stratification and prognosis. In addition, the combination of microbial‐derived therapeutics with immune modifying drugs, such as biologicals, will enhance response to treatment and better protect from damaging inflammatory processes.
6.5. Future pandemic prevention strategies based on immunological knowledge
Apart from the well‐known measures of social distancing, washing hands, and disinfection, which have proven to limit the SARS‐CoV‐2 spread, several prevention strategies can be considered from an immunological point of view. The WHO Strategic and Technical Advisory Group for Infectious Hazards (STAG‐IH) regularly reviews and updates its risk assessment of COVID‐19 to make recommendations. 254
For the future, it is essential to define the actual prevalence of COVID‐19 in the population. Confirmation of infection at present consists of PCR for acute infection and serological tests to identify antibodies. 255 However, this may not be sufficient. The implementation of immune tests detecting neutralizing antibodies is key to define protection against SARS‐CoV‐2 (Figure 7). This can only be achieved by implementing massive testing. Moreover, multiple vaccines are under development with the aim of preventing infection, reducing disease severity, and viral shedding. A complete and continually updated list is available from the WHO. 256 , 257
Zoonotic infectious diseases have been an important concern to humankind for more than 10 000 years. Today, approximately 75% of newly emerging infectious diseases (EIDs) are zoonoses that result from various anthropogenic, genetic, ecologic, socioeconomic, and climatic factors. The COVID‐19 pandemic is an extreme reminder of the role which animal reservoirs play in public health. Also, it reinforces the urgent need for globally operationalizing a One Health approach focusing on a broad surveillance for SARS‐CoV‐2 among different animals, and the possibility of reverse zoonosis. 258 Moreover, the current pandemic highlights the essential need for a broad understanding of immunological mechanisms underlying infectious diseases to design suitable therapeutic and preventive strategies.
AUTHORS’ CONTRIBUTIONS
SM drafted the manuscript. SM, JJ, and PSI contributed to “SARS‐CoV‐2 receptors: proven virus interaction partners and structural considerations.” SM, MJ, SWCB, and SJ wrote “Airway epithelium: hotspot for disease development.” SM, SWCB, and SMH contributed to “T cell–related mechanisms: lymphopenia, T‐cells over‐activation, and T‐cell exhaustion.” LZ designed the figures and wrote the figure legends. AI and KKI wrote “Multi‐morbidities as a risk factor for COVID‐19 course,” “Sex and aging as a risk factor for COVID‐19 course” and “Allergy‐related risk for COVID‐19.” AI, SA, and vdWW contributed “Immunological diagnosis of SARS‐CoV‐2 infections: challenges of current approaches.” AI and GCC contributed to “Future pandemic prevention strategies based on immunological knowledge.” ACA and ES wrote “Skin barrier: more than just matter of wearing protective equipment.” AD contributed “Myocardial and endothelial damage: an immunological perspective on cardiological presentation of COVID‐19.” AM, SA, JSR, and vdWW wrote “B cell–related mechanisms and antibody‐responses.” BW and PF contributed “Neurological disease presentation.” BH, FW, MH, and TG wrote “Kids versus adults: mechanisms explaining the clinical differences.” ET, JM, KA, and PO contributed “Immunological treatment approaches: biologicals, small molecules and beyond.“ CO and PO wrote “Trained immunity.” EA wrote “Dendritic cells and macrophages” together with MM and PO. HSK and UE contributed “From oral to rectal mucosa: what do we know about the involvement of the gastrointestinal tract in COVID‐19.” JSR and KA provided the tables. KEF wrote “Neutrophils and eosinophils” and “Immunological biomarker profiling of COVID‐19 for prediction of disease severity.” KKI wrote “ILC and NK cells” with MH. LZ and OML wrote “Virome interactions with commensal communities: future prevention and treatment strategies for SARS‐CoV‐2 infections?”. MM wrote “Lung pathologies in COVID‐19” and “COVID‐19‐related kidney failure.” NK and PM contributed “Complement and SARS‐CoV‐2” and “Coagulation parameters in COVID‐19 patients.” SM and UE orchestrated the whole process and conducted the final editing. UE and SM wrote the Introduction and Abstract. All authors contributed to the manuscript, revised, and edited.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank Urszula Radzikowska from the Swiss Institute of Allergy and Asthma Research (SIAF) for her contribution in preparing Figure 2. The authors would like to thank the European Academy of Allergy and Clinical Immunology (EAACI) for the financial support to the sections, interest groups and working groups enabling the development of this paper. The research of SM is supported by a SNSF grant 310039_189334; JSR is funded by Ministerio de Economía y Competitividad (IJCI‐2016‐27619); KKI is supported by the FWO Post doc mandate 12W2219N; BW and PF were supported by funding from the Istituto Italiano di Tecnologia, Fondazione Telethon (project GGP19103), and Ricerca Finalizzata Giovani Ricercatori 2016 ‐ Ministero Salute Italia (project GR‐2016‐02362413); GCC is supported by a postdoctoral contract cofounded by the competitive Program “Attracting Talent,” Community of Madrid, Spain; the research of SWCB was funded by DFG (398577603), “ADAPT” EIT Health is a body of the EU receiving support from H2020 and BMBF “ESCAPE” 01KI20169A; the research of UE is supported by the H2020 grant 768641 and by the BMF grant 19056.
Dr Agache reports being Associate Editor Allergy journal. Dr Akdis reports grants from Allergopharma, Idorsia, Swiss National Science Foundation, Christine Kühne‐Center for Allergy Research and Education, European Commission's Horison's 2020 Framework Programme, Cure, Novartis Research Institutes, Astra Zeneca, Scibase, Glakso Smith‐Kline and other from Sanofi & Regeneron. Dr Eiwegger reports other from DBV , grants from Innovation fund Denmark, CIHR, other from Regeneron. He is the Co‐I or scientific lead in three investigator initiated oral immunotherapy trials including the usage of biologicals supported by the Allergy and Anaphylaxis Program Sickkids and CIHR. He serves as associate editor for Allergy. He is on the advisory board for ALK. Dr Jutel reports personal fees from ALK‐Abello, personal fees from Allergopharma , personal fees from Stallergenes, personal fees from Anergis, personal fees from Allergy Therapeutics, personal fees from Circassia, personal fees from Leti, personal fees from Biomay, personal fees from HAL, during the conduct of the study; personal fees from Astra‐Zeneka, personal fees from GSK, personal fees from Novartis, personal fees from Teva, personal fees from Vectura, personal fees from UCB, personal fees from Takeda, personal fees from Roche, personal fees from Janssen, personal fees from Medimmune, personal fees from Chiesi, outside the submitted work; Dr Nadeau reports grants and other from NIAID, other from Novartis, personal fees and other from Regeneron, grants and other from FARE, grants from EAT, other from Sanofi, other from Astellas, other from Nestle, other from BeforeBrands, other from Alladapt, other from ForTra, other from Genentech, other from AImmune Therapeutics, other from DBV Technologies, personal fees from Astrazeneca, personal fees from ImmuneWorks, personal fees from Cour Pharmaceuticals, grants from Allergenis , grants from Ukko Pharma, other from AnaptysBio, other from Adare Pharmaceuticals, other from Stallergenes‐Greer, other from NHLBI, other from NIEHS, other from EPA, other from WAO Center of Excellence, other from Iggenix, other from Probio, other from Vedanta, other from Centecor, other from Seed, from Immune Tolerance Network, from NIH, outside the submitted work; In addition, Dr Nadeau has a patent Inhibition of Allergic Reaction to Peanut Allergen using an IL‐33 Inhibitor pending, a patent Special Oral Formula for Decreasing Food Allergy Risk and Treatment for Food Allergy pending, a patent Basophil Activation Based Diagnostic Allergy Test pending, a patent Granulocyte‐based methods for detecting and monitoring immune system disorders pending, a patent Methods and Assays for Detecting and Quantifying Pure Subpopulations of White Blood Cells in Immune System Disorders pending, a patent Mixed Allergen Compositions and Methods for Using the Same pending, and a patent Microfluidic Device and Diagnostic Methods for Allergy Testing Based on Detection of Basophil Activation pending. Dr O'Mahony reports personal fees from AHL, grants from GSK, outside the submitted work; Dr Palomares reports research grants from Inmunotek SL, Novartis and MINECO (SAF2017‐84978‐R), has received fees for giving scientific lectures from: Allergy Therapeutics, Amgen, AstraZeneca, Diater, GlaxoSmithKline, S.A, Inmunotek S.L, Novartis, Sanofi‐Genzyme and Stallergenes and has participated in advisory boards from Novartis and Sanofi‐Genezyme. Prof. Schmidt‐Weber reports grants from EIT Health, body of the EU; grants from German Ministery of Education and Research (BMBF); grants from DFG, during the conduct of the study; In addition, Dr Schmidt‐Weber has a patent on drug repurposing pending Dr Schwarze reports personal fees from MYLAN, personal fees from F2F events, outside the submitted work; Industry support to educational activities of the Scottish Allergy and Respiratory Academy and of the Children's and Young people's Allergy Networtk Scotland. Industry support to EAACI, Dr Schwarze is EAACI Secretary General 2019‐2021. Dr Sokolowska reports grants from Swiss National Science Foundation, grants from GSK, outside the submitted work; all other authors have nothing to disclose.
Sokolowska M, Lukasik ZM, Agache I, et al. Immunology of COVID‐19: Mechanisms, clinical outcome, diagnostics, and perspectives—A report of the European Academy of Allergy and Clinical Immunology (EAACI). Allergy. 2020;75:2445–2476. 10.1111/all.14462
Contributor Information
Milena Sokolowska, Email: milena.sokolowska@siaf.uzh.ch.
Eva Untersmayr, Email: eva.untersmayr@meduniwien.ac.at.
REFERENCES
- 1. Li W, Shi Z, Yu M, et al. Bats are natural reservoirs of SARS‐like coronaviruses. Science. 2005;310(5748):676‐679. [DOI] [PubMed] [Google Scholar]
- 2. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang YZ, Holmes EC. A genomic perspective on the origin and emergence of SARS‐CoV‐2. Cell. 2020;181(2):223‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019‐nCoV) originating in China. Cell Host Microbe. 2020;27(3):325‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell. 2020;181(4):894‐904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van de Veerdonk FL, Netea MG, van Deuren M, et al. Kallikrein‐kinin blockade in patients with COVID‐19 to prevent acute respiratory distress syndrome. eLife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci USA. 2020;117(21):11727‐11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radzikowska U, Ding M, Tan G, et al. Distribution of ACE2, CD147, CD26 and other SARS‐CoV‐2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID‐19 risk factors. Allergy. 2020. 10.1111/all.14429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sungnak W, Huang NI, Bécavin C, et al. SARS‐CoV‐2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS‐CoV‐2 receptor ACE2 is an interferon‐stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181(5):1016–1035.e19. 10.1016/j.cell.2020.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qi F, Qian S, Zhang S, Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun. 2020;526(1):135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang K, Chen W, Zhou YS, et al. SARS‐CoV‐2 invades host cells via a novel route: CD147‐spike protein. bioRxiv. 2020;preprint:2020.2003.2014.988345. [Google Scholar]
- 15. Chen Z, Mi LI, Xu J, et al. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J Infect Dis. 2005;191(5):755‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pushkarsky T, Zybarth G, Dubrovsky L, et al. CD147 facilitates HIV‐1 infection by interacting with virus‐associated cyclophilin A. Proc Natl Acad Sci USA. 2001;98(11):6360‐6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watanabe A, Yoneda M, Ikeda F, Terao‐Muto Y, Sato H, Kai C. CD147/EMMPRIN acts as a functional entry receptor for measles virus on epithelial cells. J Virol. 2010;84(9):4183‐4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akkus MN, Ormam A, Seyis S, Baran C, Gorur A, Bilen MN. Plasma EMMPRIN levels in acute myocardial infarction and stable coronary artery disease. Clin Invest Med. 2016;39(3):E79‐87. [DOI] [PubMed] [Google Scholar]
- 19. Yurchenko V, Constant S, Eisenmesser E, Bukrinsky M. Cyclophilin‐CD147 interactions: a new target for anti‐inflammatory therapeutics. Clin Exp Immunol. 2010;160(3):305‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hibino T, Sakaguchi M, Miyamoto S, et al. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res. 2013;73(1):172‐183. [DOI] [PubMed] [Google Scholar]
- 21. Kato N, Yuzawa Y, Kosugi T, et al. The E‐selectin ligand basigin/CD147 is responsible for neutrophil recruitment in renal ischemia/reperfusion. J Am Soc Nephrol. 2009;20(7):1565‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seizer P, Borst O, Langer H, et al. EMMPRIN (CD147) is a novel receptor for platelet GPVI and mediates platelet rolling via GPVI‐EMMPRIN interaction. Thromb Haemost. 2009;101(4):682‐686. [DOI] [PubMed] [Google Scholar]
- 23. Huang W, Luo W‐J, Zhu P, et al. Modulation of CD147‐induced matrix metalloproteinase activity: role of CD147 N‐glycosylation. Biochem J. 2013;449(2):437‐448. [DOI] [PubMed] [Google Scholar]
- 24. Slomiany MG, Grass GD, Robertson AD, et al. Hyaluronan, CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 2009;69(4):1293‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. 2007;81(21):11620‐11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanaka Y, Sato Y, Sasaki T. Suppression of coronavirus replication by cyclophilin inhibitors. Viruses. 2013;5(5):1250‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whitworth KM, Rowland RRR, Petrovan V, et al. Resistance to coronavirus infection in amino peptidase N‐deficient pigs. Transgenic Res. 2019;28(1):21‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holmes RS, Spradling‐Reeves KD, Cox LA. Mammalian glutamyl aminopeptidase genes (ENPEP) and proteins: Comparative studies of a major contributor to arterial hypertension. J Data Mining Genomics Proteomics. 2017;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang Z‐Y, Huang Y, Ganesh L, et al. pH‐dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC‐SIGN. J Virol. 2004;78(11):5642‐5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu C, Zheng M. Single‐cell RNA expression profiling shows that ACE2, the putative receptor for COVID‐2019, has significant expression in nasal and mounth tissue and is co‐expressed with TMPRSS2 and not co‐expressed with SLC6A19 in the tissues. PREPRINT (Version 1) available at Research Square. 12 March 2020.
- 31. Yan R, Zhang Y, Li Y, Xia L, Zhou Q. Structure of dimeric full‐length human ACE2 in complex with B0AT1. bioRxiv. 2020;preprint:2020.2002.2017.951848. [Google Scholar]
- 32. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID‐19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 35. Zissler UM, Chaker AM, Effner R, et al. Interleukin‐4 and interferon‐γ orchestrate an epithelial polarization in the airways. Mucosal Immunol. 2016;9(4):917‐926. [DOI] [PubMed] [Google Scholar]
- 36. Finlay BB, McFadden G. Anti‐immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124(4):767‐782. [DOI] [PubMed] [Google Scholar]
- 37. DeDiego ML, Nieto‐Torres JL, Jimenez‐Guardeño JM, et al. Coronavirus virulence genes with main focus on SARS‐CoV envelope gene. Virus Res. 2014;194:124‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schoggins JW. Interferon‐stimulated genes: What do they all do? Annu Rev Virol. 2019;6(1):567‐584. [DOI] [PubMed] [Google Scholar]
- 39. Prokunina‐Olsson L, Alphonse N, Dickenson RE, et al. COVID‐19 and emerging viral infections: The case for interferon lambda. J Exp Med. 2020;217(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chu H, Chan JF‐W, Wang Y, et al. Comparative replication and immune activation profiles of SARS‐CoV‐2 and SARS‐CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID‐19. Clin Infect Dis. 2020. 10.1093/cid/ciaa410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Channappanavar R, Fehr A, Vijay R, et al. Dysregulated type I interferon and inflammatory monocyte‐macrophage responses cause lethal pneumonia in SARS‐CoV‐infected mice. Cell Host Microbe. 2016;19(2):181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kindler E, Thiel V, Weber F. Interaction of SARS and MERS coronaviruses with the antiviral interferon response. Adv Virus Res. 2016;96:219‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Menachery VD, Debbink K, Baric RS. Coronavirus non‐structural protein 16: Evasion, attenuation, and possible treatments. Virus Res. 2014;194:191‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Versteeg GA, Bredenbeek PJ, van den Worm SH, Spaan WJ. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361(1):18‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang H, Kang Z, Gong H, et al. Digestive system is a potential route of COVID‐ 19: an analysis of single‐cell coexpression pattern of key proteins in viral entry process. Gut. 2020;69(6):1010‐1018. [Google Scholar]
- 47. Zang R, Castro MFG, McCune BT, et al. TMPRSS2 and TMPRSS4 mediate SARS‐CoV‐2 infection of human small intestinal enterocytes. bioRxiv. 2020; preprint:2020.2004.2021.054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zheng H‐C, Takahashi H, Murai Y, et al. Upregulated EMMPRIN/CD147 might contribute to growth and angiogenesis of gastric carcinoma: a good marker for local invasion and prognosis. Br J Cancer. 2006;95(10):1371‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Darmoul D, Voisin T, Couvineau A, et al. Regional expression of epithelial dipeptidyl peptidase IV in the human intestines. Biochem Biophys Res Commun. 1994;203(2):1224‐1229. [DOI] [PubMed] [Google Scholar]
- 50. Pan L, Mu MI, Yang P, et al. Clinical characteristics of COVID‐19 patients with digestive symptoms in Hubei, China: A descriptive, cross‐sectional. Multicenter Study. Am J Gastroenterol. 2020;115(5):766‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duan K, Liu B, Li C, et al. Effectiveness of convalescent plasma therapy in severe COVID‐19 patients. Proc Natl Acad Sci USA. 2020;117(17):9490‐9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. D'Amico F, Baumgart DC, Danese S, Peyrin‐Biroulet L. Diarrhea during COVID‐19 infection: Pathogenesis, epidemiology, prevention and management. Clin Gastroenterol Hepatol. 2020;18(8):1663‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leung WK, To KF, Chan PK, et al. Enteric involvement of severe acute respiratory syndrome‐associated coronavirus infection. Gastroenterology. 2003;125(4):1011‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang B, Liu S, Tan T, et al. Treatment with convalescent plasma for critically Ill patients with severe acute respiratory syndrome coronavirus 2 infection. Chest. 2020;158(1):e9–e13. 10.1016/j.chest.2020.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou J, Li C, Zhao G, et al. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci Adv. 2017;3(11):eaao4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wölfel R, Corman VM, Guggemos W, et al. Virological assessment of hospitalized patients with COVID‐2019. Nature. 2020;581(7809):465‐469. [DOI] [PubMed] [Google Scholar]
- 57. Li LY, Wu W, Chen S, et al. Digestive system involvement of novel coronavirus infection: prevention and control infection from a gastroenterology perspective. J Dig Dis. 2020;21(4):199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Recalcati S. Cutaneous manifestations in COVID‐19: A first perspective. J Eur Acad Dermatol Venereol. 2020;34(5). [DOI] [PubMed] [Google Scholar]
- 59. Bouaziz JD, Duong T, Jachiet M, et al. Vascular skin symptoms in COVID‐19: A french observational study. J Eur Acad Dermatol Venereol. 2020. 10.1111/jdv.16544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marzano AV, Genovese G, Fabbrocini G, et al. Varicella‐like exanthem as a specific COVID‐19–associated skin manifestation: Multicenter case series of 22 patients. J Eur Acad Dermatol Venereol. 2020;83(1):280–285. 10.1016/j.jaad.2020.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mazzotta FT, Troccoli T. Acute acro‐ischemia in the child at the time of COVID‐19. Eur J Pediat Dermatol. 2020;30(2):71–74. [Google Scholar]
- 62. Yan Y, Chen H, Chen L, et al. Consensus of Chinese experts on protection of skin and mucous membrane barrier for health‐care workers fighting against coronavirus disease. Dermatol Ther. 2019;2020:e13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prescott SL, Larcombe D‐L, Logan AC, et al. The skin microbiome: impact of modern environments on skin ecology, barrier integrity, and systemic immune programming. World Allergy Organ J. 2017;10(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang A, Chiou J, Poirion OB, et al. Nucleus multiomic profiling reveals age‐dynamic regulation of host genes associated with SARS‐CoV‐2 infection. bioRxiv. 2020; preprint:2020.2004.2012.037580. [Google Scholar]
- 65. Blanco‐Melo D, Nilsson‐Payant BE, Liu W‐C, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036–1045.e9. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liao M, Liu Y, Yuan J, et al. The landscape of lung bronchoalveolar immune cells in COVID‐19 revealed by single‐cell RNA sequencing. medRxiv. 2020; preprint: 2020.2002.2023.20026690. [Google Scholar]
- 67. Yilla M, Harcourt BH, Hickman CJ, et al. SARS‐coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107(1):93‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Y, Feng Z, Diao B, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) directly decimates human spleens and lymph nodes . medRxiv. 2020; preprint: 2020.2003.2027.20045427. [Google Scholar]
- 69. Lozach PY, Burleigh L, Staropoli I, Amara A. The C type lectins DC‐SIGN and L‐SIGN: receptors for viral glycoproteins. Methods Mol Biol. 2007;379:51‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cai G, Cui X, Zhu X, Zhou J. A hint on the COVID‐19 Risk: population disparities in gene expression of three receptors of SARS‐CoV. In. www.preprints.org, 10.20944/preprints202002.0408.v12020 [DOI]
- 71. Park MD. Macrophages: a Trojan horse in COVID‐19? Nat Rev Immunol. 2020;20(6):351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ratajczak MZ, Kucia M. SARS‐CoV‐2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine "storm" and risk factor for damage of hematopoietic stem cells. Leukemia. 2020;1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou Y, Fu B, Zheng X, et al. Pathogenic T‐cells and inflammatory monocytes incite inflammatory storms in severe COVID‐19 patients. National Sci Rev. 2020;7(6):998–1002. 10.1093/nsr/nwaa041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. He L, Ding Y, Zhang Q, et al. Expression of elevated levels of pro‐inflammatory cytokines in SARS‐CoV‐infected ACE2+ cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J Pathol. 2006;210(3):288‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu LI, Wei Q, Lin Q, et al. Anti‐spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS‐CoV infection. JCI Insight. 2019;4(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin‐6 in COVID‐19 induced pneumonia and macrophage activation syndrome‐like disease. Autoimmun Rev. 2020;19(6):102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhou M, Zhang X, Qu J. Coronavirus disease 2019 (COVID‐19): A clinical update. Front Med. 2020;14(2):126‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nairz M, Theurl I, Swirski FK, Weiss G. "Pumping iron"‐how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017;469(3–4):397‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Soares MP, Hamza I. Macrophages and iron metabolism. Immunity. 2016;44(3):492‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Velavan TP, Meyer CG. Mild versus severe COVID‐19: Laboratory markers. Int J Infect Dis. 2020;95:304‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, et al. Targeting potential drivers of COVID‐ 19: Neutrophil extracellular traps. J Exp Med. 2020;217(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fox SE, Akmatbekov A, Harbert JL, Li G, Brown JQ, Vander Heide RS. Pulmonary and cardiac pathology in Covid‐19: The first autopsy series from new orleans. medRxiv. 2020; preprint: 2020.2004.2006.20050575. [Google Scholar]
- 85. Jesenak M, Banovcin P, Diamant Z. COVID‐19, chronic inflammatory respiratory diseases and eosinophils – Observationsfrom reported clinical case series. Allergy. 2020;75:1819‐1822. 10.1111/all.14353 [DOI] [PubMed] [Google Scholar]
- 86. Zhang J‐J, Dong X, Cao Y‐Y, et al. Clinical characteristics of 140 patients infected with SARS‐CoV‐2 in Wuhan, China. Allergy. 2020;75:1730‐1741. 10.1111/all.14238 [DOI] [PubMed] [Google Scholar]
- 87. Du Y, Tu L, Zhu P, et al. Clinical features of 85 fatal cases of COVID‐19 from Wuhan. A retrospective observational study. American Journal of Respiratory and Critical Care Medicine. 2020;201(11):1372–1379. 10.1164/rccm.202003-0543oc [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lippi G, Henry BM. Eosinophil count in severe coronavirus disease 2019. QJM: An International Journal of Medicine. 2020. 10.1093/qjmed/hcaa137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lindsley AW, Schwartz JT, Rothenberg ME. Eosinophil responses during COVID‐19 infections and coronavirus vaccination. J Allergy Clin Immuno. 2020;146(1):1–7. 10.1016/j.jaci.2020.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID‐19 Autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153(6):725‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kortekaas Krohn I, Shikhagaie MM, Golebski K, et al. Emerging roles of innate lymphoid cells in inflammatory diseases: Clinical implications. Allergy. 2018;73(4):837‐850. [DOI] [PubMed] [Google Scholar]
- 92. Morita H, Moro K, Koyasu S. Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy Clin Immunol. 2016;138(5):1253‐1264. [DOI] [PubMed] [Google Scholar]
- 93. Liu J, Li S, Liu J, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS‐CoV‐2 infected patients. EBioMedicine. 2020;55:102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang F, Nie J, Wang H, et al. Characteristics of peripheral lymphocyte subset alteration in COVID‐19 pneumonia. J Infect Dis. 2020;221(11):1762‐1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Leng Z, Zhu R, Hou W, et al. Transplantation of ACE2(‐) mesenchymal stem cells improves the outcome of patients with COVID‐19 pneumonia. Aging Dis. 2020;11(2):216‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thevarajan I, Nguyen THO, Koutsakos M, et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non‐severe COVID‐19. Nat Med. 2020;26(4):453‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992‐1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID‐19) in Wuhan, China. Clinical Infectious Diseases. 2020. 10.1093/cid/ciaa248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID‐19 patients. Cell Mol Immunol. 2020;17(5):533‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. van Montfoort N, Borst L, Korrer MJ, et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018;175(7):1744‐1755.e1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID‐19 infection: a report of five cases. Transl Res. 2020;220:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gralinski LE, Sheahan TP, Morrison TE, et al. complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio. 2018;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mulvey JJ, Magro CM, Ma LX, Nuovo GJ, Baergen RN. Analysis of complement deposition and viral RNA in placentas of COVID‐19 patients. Ann Diagn Pathol. 2020;46:151530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. 2011;9(5):355‐361. [DOI] [PubMed] [Google Scholar]
- 105. Sanchez‐Ramon S, Conejero L, Netea MG, Sancho D, Palomares O, Subiza JL. Trained immunity‐based vaccines: A new paradigm for the development of broad‐spectrum anti‐infectious formulations. Front Immunol. 2018;9:2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kleinnijenhuis J, Quintin J, Preijers F, et al. Long‐lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun. 2014;6(2):152‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jensen KJ, Larsen N, Biering‐Sørensen S, et al. Heterologous immunological effects of early BCG vaccination in low‐birth‐weight infants in Guinea‐Bissau: A randomized‐controlled trial. J Infect Dis. 2015;211(6):956‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gursel M, Gursel I. Is global BCG vaccination‐induced trained immunity relevant to the progression of SARS‐CoV‐2 pandemic?. Allergy. 2020;75:1815‐1819. 10.1111/all.14345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ozdemir C, Kucuksezer UC, Tamay ZU. Is BCG vaccination affecting the spread and severity of COVID‐19?. Allergy. 2020;75:1824‐1827. 10.1111/all.14344 [DOI] [PubMed] [Google Scholar]
- 110. O'Neill LAJ, Netea MG. BCG‐induced trained immunity: Can it offer protection against COVID‐19? Nat Rev Immunol. 2020;20(6):335‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Retamal‐Díaz A, Covián C, Pacheco GA, et al. Contribution of resident memory CD8(+) T cells to protective immunity against respiratory syncytial virus and their impact on vaccine design. Pathogens. 2019;8(3):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Murali‐Krishna K, Altman JD, Suresh M, et al. Counting antigen‐specific CD8 T cells: A reevaluation of bystander activation during viral infection. Immunity. 1998;8(2):177‐187. [DOI] [PubMed] [Google Scholar]
- 113. Libraty DH, O'Neil KM, Baker LM, Acosta LP, Olveda RM. Human CD4(+) memory T‐lymphocyte responses to SARS coronavirus infection. Virology. 2007;368(2):317‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ng O‐W, Chia A, Tan AT, et al. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post‐infection. Vaccine. 2016;34(17):2008‐2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181(7):1489–1501.e15. 10.1016/j.cell.2020.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Braun J, Loyal L, Frentsch M, et al. Presence of SARS‐CoV‐2 reactive T cells in COVID‐19 patients and healthy donors. medRxiv. 2020:2020.2004.2017.20061440. [Google Scholar]
- 117. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cameron MJ, Bermejo‐Martin JF, Danesh A, Muller MP, Kelvin DJ. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 2008;133(1):13‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID‐19 patients. Emerg Microbes Infect. 2020;9(1):761‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhao J, Zhao J, Legge K, Perlman S. Age‐related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J Clin Invest. 2011;121(12):4921‐4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yoshikawa T, Hill T, Li K, Peters CJ, Tseng CT. Severe acute respiratory syndrome (SARS) coronavirus‐induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte‐derived macrophages and dendritic cells. J Virol. 2009;83(7):3039‐3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhao J, Zhao J, Van Rooijen N, Perlman S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS‐CoV‐infected mice. PLoS Pathog. 2009;5(10):e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bahl K, Kim SK, Calcagno C, et al. IFN‐induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol. 2006;176(7):4284‐4295. [DOI] [PubMed] [Google Scholar]
- 124. Bermejo‐Martin JF, Almansa R, Menendez R, Mendez R, Kelvin DJ, Torres A. Lymphopenic community acquired pneumonia as signature of severe COVID‐19 infection. J Infect. 2020;80(5):e23‐e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). medRxiv. 2020;preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Diao B, Wang C, Tan Y, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID‐19). Frontiers Immunol. 2020;11(827). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T‐cell exhaustion, co‐stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523(7562):612‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Huang AT, Garcia‐Carreras B, Hitchings MDT, et al. A systematic review of antibody mediated immunity to coronaviruses: antibody kinetics, correlates of protection, and association of antibody responses with severity of disease. medRxiv. 2020; preprint: 2020.2004.2014.20065771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Breedveld A, van Egmond M. IgA and FcαRI: Pathological roles and therapeutic opportunities. Front Immunol. 2019;10:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tan YJ, Goh PY, Fielding BC, et al. Profiles of antibody responses against severe acute respiratory syndrome coronavirus recombinant proteins and their potential use as diagnostic markers. Clin Diagn Lab Immunol. 2004;11(2):362‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Temperton NJ, Chan PK, Simmons G, et al. Longitudinally profiling neutralizing antibody response to SARS coronavirus with pseudotypes. Emerg Infect Dis. 2005;11(3):411‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Casadevall A, Pirofski L‐A. The convalescent sera option for containing COVID‐19. J Clin Investig. 2020;130(4):1545‐1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Shen C, Wang Z, Zhao F, et al. Treatment of 5 critically Ill patients with COVID‐19 with convalescent plasma. JAMA. 2020;323(16):1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ahn JY, Sohn Y, Lee SH, et al. Use of convalescent plasma therapy in two COVID‐19 patients with acute respiratory distress syndrome in Korea. J Korean Med Sci. 2020;35(14):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID‐19. Nat Rev Immunol. 2020;20(6):339–‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wilk AJ, Rustagi A, Zhao NQ, et al. A single‐cell atlas of the peripheral immune response to severe COVID‐19. medRxiv. 2020:2020.2004.2017.20069930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Corcoran LM, Tarlinton DM. Regulation of germinal center responses, memory B cells and plasma cell formation‐an update. Curr Opin Immunol. 2016;39:59‐67. [DOI] [PubMed] [Google Scholar]
- 138. Pan Y, Zhang D, Yang P, Poon LLM, Wang Q. Viral load of SARS‐CoV‐2 in clinical samples. Lancet Infect Dis. 2020;20(4):411‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zou L, Ruan F, Huang M, et al. SARS‐CoV‐2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020;382(12):1177‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Xu Y‐H, Dong J‐H, An W‐M, et al. Clinical and computed tomographic imaging features of novel coronavirus pneumonia caused by SARS‐CoV‐2. J Infect. 2020;80(4):394‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early‐phase 2019 novel coronavirus (COVID‐19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15(5):700‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Vardhana SA, Wolchok JD. The many faces of the anti‐COVID immune response. J Exp Med. 2020;217(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID‐19: The potential role for antifibrotic therapy. Lancet Respir Med. 2020. 10.1016/s2213-2600(20)30225-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Spagnolo P, Balestro E, Aliberti S, et al. Pulmonary fibrosis secondary to COVID‐ 19: a call to arms? Lancet Respir Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Liu PP, Blet A, Smyth D, Li H. The science underlying COVID‐19: Implications for the cardiovascular system. Circulation. 2020. 10.1161/circulationaha.120.047549 [DOI] [PubMed] [Google Scholar]
- 147. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395(10234):1417‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Oudit GY, Kassiri Z, Jiang C, et al. SARS‐coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39(7):618‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sala S, Peretto G, Gramegna M, et al. Acute myocarditis presenting as a reverse Tako‐Tsubo syndrome in a patient with SARS‐CoV‐2 respiratory infection. Eur Heart J. 2020;41(19):1861‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Deng Q, Hu BO, Zhang Y, et al. Suspected myocardial injury in patients with COVID‐ 19: Evidence from front‐line clinical observation in Wuhan, China. Int J Cardiol. 2020;19:116‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Puelles VG, Lütgehetmann M, Lindenmeyer MT, et al. Multiorgan and renal tropism of SARS‐CoV‐2. N Engl J Med. 2020. 10.1056/nejmc2011400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bikdeli B, Madhavan MV, Jimenez D, et al. COVID‐19 and thrombotic or thromboembolic disease: Implications for prevention, antithrombotic therapy, and follow‐up. J Am Coll Cardiol. 2020;75(23):2950‐2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: COVID‐19, SARS‐CoV‐1, MERS‐CoV and lessons from the past. J Clin Virol. 2020;127:104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lippi G, Plebani M. Laboratory abnormalities in patients with COVID‐2019 infection. Clin Chem Lab Med. 2020;58(7):1131‐1134. [DOI] [PubMed] [Google Scholar]
- 155. Liu Y, Sun W, Guo Y, et al. Retrospective cohort study. Platelets. 2020;31(4):490‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lippi G, Favaloro EJ. D‐dimer is associated with severity of coronavirus disease 2019: A pooled analysis. Thromb Haemost. 2020;120(05):876‐ 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Lippi G, Plebani M. Procalcitonin in patients with severe coronavirus disease 2019 (COVID‐19): A meta‐analysis. Clin Chim Acta. 2020;505:190‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ito T. PAMPs and DAMPs as triggers for DIC. J Intensive Care. 2014;2(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ranucci M, Ballotta A, Di Dedda U, et al. The procoagulant pattern of patients with COVID‐19 acute respiratory distress syndrome. J Thromb Haemost. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Chen R, Sang L, Jiang M, et al. Longitudinal hematologic and immunologic variations associated with the progression of COVID‐19 patients in China. J Allergy Clin Immunol. 2020;146(1):89–100. 10.1016/j.jaci.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014;33(1):7‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Michalicová A, Bhide K, Bhide M, Kováč A. How viruses infiltrate the central nervous system. Acta Virol. 2017;61(4):393‐400. [DOI] [PubMed] [Google Scholar]
- 164. Archibald LK, Quisling RG. Central nervous system infections. In: Layon JA, Gabrielli A, Friedman WA, eds. Textbook of Neurointensive Care. London, UK: Springer; 2013:427‐517. [Google Scholar]
- 165. Wu Y, Xu X, Chen Z, et al. Nervous system involvement after infection with COVID‐19 and other coronaviruses. Brain Behav Immun. 2020;87:18‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ding Y, He LI, Zhang Q, et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS‐CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J Pathol. 2004;203(2):622‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gu J, Gong E, Zhang BO, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202(3):415‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. 2008;82(15):7264‐7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID‐19‐associated Acute Hemorrhagic Necrotizing Encephalopathy: CT and MRI Features. Radiology. 2020;201187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zhou B, She J, Wang Y, Ma X. A case of coronavirus disease 2019 with concomitant acute cerebral infarction and deep vein thrombosis. Frontiers Neurol. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. China. NHCotPsRo . Diagnosis and treatment protocol for novel coronavirus pneumonia (7th Interim Edition). 2020. China NHCOTPSRO. http://www.nhc.gov.cn/yzygj/s7653p/202003/46c9294a7dfe4cef80dc7f5912eb1989/files/ce3e6945832a438eaae415350a8ce964.pdf
- 172. Mao L, Jin H, Wang M, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Vaira LA, Salzano G, Deiana G, De Riu G. Anosmia and Ageusia: Common Findings in COVID‐19 Patients. Laryngoscope. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Giacomelli A, Pezzati L, Conti F, et al. Self‐reported olfactory and taste disorders in patients with severe acute respiratory coronavirus 2 infection: A cross‐sectional study. Clin Infect Dis. 2020. 10.1093/cid/ciaa330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Lechien JR, Chiesa‐Estomba CM, De Siati DR, et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild‐to‐moderate forms of the coronavirus disease (COVID‐19): A multicenter European study. Eur Arch Otorhinolaryngol. 2020. 10.1007/s00405-020-05965-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Pei G, Zhang Z, Peng J, et al. Renal Involvement and early prognosis in patients with COVID‐19 pneumonia. J Am Soc Nephrol. 2020;31(6):1157‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Rovin BH, Ronco P, Editors A, Kidney TEE. International and the COVID‐19 infection. Kidney Int. 2020;97(5):823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Magrone T, Magrone M, Jirillo E. Focus on receptors for coronaviruses with special reference to angiotensin‐converting enzyme 2 as a potential drug target ‐ A perspective. Endocr Metab Immune Disord Drug Targets. 2020. [DOI] [PubMed] [Google Scholar]
- 179. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181(4):905‐913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Qu X, Wang C, Zhang J, Qie G, Zhou J. The roles of CD147 and/or cyclophilin A in kidney diseases. Mediators Inflamm. 2014;2014:728673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Pfefferle S, Schöpf J, Kögl M, et al. The SARS‐coronavirus‐host interactome: Identification of cyclophilins as target for pan‐coronavirus inhibitors. PLoS Pathog. 2011;7(10):e1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Wang B, Li R, Lu Z, Huang Y. Does comorbidity increase the risk of patients with COVID‐19: Evidence from meta‐analysis. Aging (Albany NY). 2020;12(7):6049‐6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Dong X, Cao Y‐Y, Lu X‐X, et al. Eleven faces of coronavirus disease 2019. Allergy. 2020. Jul;75(7):1699‐1709. 10.1111/all.14289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Guan W‐J, Liang W‐H, Zhao Y, et al. Comorbidity and its impact on 1590 patients with COVID‐19 in China: A nationwide analysis. Eur Respir J. 2020;55(5):2000547. 10.1183/13993003.00547-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Chen X, Hu W, Ling J, et al. Hypertension and diabetes delay the viral clearance in COVID‐19 patients. medRxiv. 2020;preprint. [Google Scholar]
- 186. Kulcsar KA, Coleman CM, Beck SE, Frieman MB. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS‐CoV infection. JCI Insight. 2019;4(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Michalovich D, Rodriguez‐Perez N, Smolinska S, et al. Obesity and disease severity magnify disturbed microbiome‐immune interactions in asthma patients. Nat Commun. 2019;10(1):5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yang JK, Lin SS, Ji XJ, Guo LM. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010;47(3):193‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Chakrabarty B, Das D, Bulusu G, Roy A. Network‐based analysis of fatal comorbidities of COVID‐19 and potential therapeutics. ChemRxiv. preprint. 10.26434/chemrxiv.12136470.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Bao W, Min D, Twigg SM, et al. Monocyte CD147 is induced by advanced glycation end products and high glucose concentration: Possible role in diabetic complications. Am J Physiol Cell Physiol. 2010;299(5):C1212‐1219. [DOI] [PubMed] [Google Scholar]
- 191. Wang J, Luo Q, Chen R, Chen T, Li J. Susceptibility Analysis of COVID‐19 in Smokers Based on ACE2. Preprintsorg. 2020;preprint. [Google Scholar]
- 192. Leung JM, Yang CX, Tam A, et al. ACE‐2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID‐19. European Respiratory Journal. 2020;55(5). 10.1183/13993003.00688-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated Plasmin(ogen) as a COMMON RISK FACTOR for COVID‐19 susceptibility. Physiol Rev. 2020;100(3):1065‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Wambier CG, Goren A. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection is likely to be androgen mediated. J Am Acad Dermatol. 2020;83(1):308‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Wang LS, Williamson SR, Zhang SB, et al. Increased androgen receptor gene copy number is associated with TMPRSS2‐ERG rearrangement in prostatic small cell carcinoma. Mol Carcinogen. 2015;54(9):900‐907. [DOI] [PubMed] [Google Scholar]
- 196. Kwetkat A, Heppner HJ. Comorbidities in the elderly and their possible influence on vaccine response. Interdiscip Top Gerontol Geriatr. 2020;43:73‐85. [DOI] [PubMed] [Google Scholar]
- 197. Samson LD, Boots AMH, Verschuren WMM, Picavet HSJ, Engelfriet P, Buisman AM. Frailty is associated with elevated CRP trajectories and higher numbers of neutrophils and monocytes. Exp Gerontol. 2019;125. [DOI] [PubMed] [Google Scholar]
- 198. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: A new immune‐metabolic viewpoint for age‐related diseases. Nat Rev Endocrinol. 2018;14(10):576‐590. [DOI] [PubMed] [Google Scholar]
- 199. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet. 2020;395(10223):507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Guan W‐J, Ni Z‐Y, Hu YU, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Huang C, Wang Y, Li X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: Summary of a report of 72314 cases from the chinese center for disease control and prevention. JAMA. 2020;323(13):1239. [DOI] [PubMed] [Google Scholar]
- 203. CDC . Coronavirus Disease 2019 in Children — United States, February 12–April 2, 2020. 2020; https://www.cdc.gov/mmwr/volumes/69/wr/mm6914e4.htm. Accessed May 7, 2020 [DOI] [PMC free article] [PubMed]
- 204. Bousquet J, Akdis C, Jutel M, et al. Intranasal corticosteroids in allergic rhinitis in COVID‐19 infected patients: An ARIA‐EAACI statement. Allergy 2020;75:2511‐2515. [DOI] [PubMed] [Google Scholar]
- 205. Klimek L, Jutel M, Akdis C, et al. Handling of allergen immunotherapy in the COVID‐19 pandemic: An ARIA‐EAACI statement. Allergy. 2020;75(7):1546‐1554. 10.1111/all.14336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Johnston SL. Asthma and COVID‐19: is asthma a risk factor for severe outcomes? Allergy 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Jackson DJ, Busse WW, Bacharier LB, et al. Association of respiratory allergy, asthma and expression of the SARS‐CoV‐2 receptor, ACE2. J Allergy Clin Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Mehta P, McAuley DF, Brown M, et al. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Sotgiu G, Gerli AG, Centanni S, et al. Advanced forecasting of SARS‐CoV‐2‐related deaths in Italy, Germany, Spain, and New York State. Allergy. 2020;75:1813‐1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Yang Y, Shen C, Li J, et al. Exuberant elevation of IP‐10, MCP‐3, and IL‐1ra during SARS‐CoV‐2 infection is associated with disease severity and fatal outcomes. med Rxiv preprint. 2020. [Google Scholar]
- 211. Liu M, Song Z, Xiao K. High‐resolution computed tomography manifestations of 5 pediatric patients with 2019 novel coronavirus. J Comput Assist Tomogr. 2020;44(3):311‐ 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Jones TC, Mühlemann B, Veith T, et al. An analysis of SARS‐CoV‐2 viral load by patient age. submitted. 2020. 10.1101/2020.06.08.20125484 [DOI]
- 213. Maddux AB, Douglas IS. Is the developmentally immature immune response in paediatric sepsis a recapitulation of immune tolerance? Immunology 2015;145(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Riphagen S, Gomez X, Gonzalez‐Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID‐19 pandemic. Lancet. 2020;395(10237):1607‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. WHO . Multisystem inflammatory syndrome in children and adolescents temporally related to COVID‐19. https://www.who.int/news‐room/commentaries/detail/multisystem‐inflammatory‐syndrome‐in‐children‐and‐adolescents‐with‐covid‐19: WHO; 2020.
- 216. WHO . Report of the WHO‐China Joint Mission on Coronavirus Disease 2019 (COVID‐19). https://www.who.int/publications‐detail/report‐of‐the‐who‐china‐joint‐mission‐on‐coronavirus‐disease‐2019‐(covid‐19): WHO; 2020.
- 217. Vogel G. These are answers we need. WHO plans global study to discover true extent of coronavirus infections. Science. 2020. [Google Scholar]
- 218. Alberici F, Delbarba E, Manenti C, et al. A single center observational study of the clinical characteristics and short‐term outcome of 20 kidney transplant patients admitted for SARS‐CoV2 pneumonia. Kidney International. 2020;97(6):1083–1088. 10.1016/j.kint.2020.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Capra R, De Rossi N, Mattioli F, et al. Impact of low dose tocilizumab on mortality rate in patients with COVID‐19 related pneumonia. Eur J Intern Med. 2020;76:31‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Colaneri M, Bogliolo L, Valsecchi P, et al. Tocilizumab for treatment of severe COVID‐19 patients: Preliminary Results from SMAtteo COvid19 REgistry (SMACORE). Microorganisms. 2020;8(5):695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Di Giambenedetto S, Ciccullo A, Borghetti A, et al. Off‐label use of tocilizumab in patients with SARS‐CoV‐2 infection. J Med Virol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Jacobs JP, Stammers AH, St. Louis J, et al. Extracorporeal membrane oxygenation in the treatment of severe pulmonary and cardiac compromise in COVID‐19: Experience with 32 patients. ASAIO J. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Klopfenstein T, Zayet S, Lohse A, et al. Tocilizumab therapy reduced intensive care unit admissions and/or mortality in COVID‐19 patients. Med Mal Infect. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab treatment in COVID‐19: A single center experience. J Med Virol. 2020;92(7):814‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Mazzitelli M, Arrighi E, Serapide F, et al. Use of subcutaneous tocilizumab in patients with COVID‐19 pneumonia. J Med Virol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Pereira MR, Mohan S, Cohen DJ, et al. COVID‐19 in solid organ transplant recipients: Initial report from the US Epicenter. Am J Transplant. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Piva S, Filippini M, Turla F, et al. Clinical presentation and initial management critically ill patients with severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection in Brescia. Italy. J Crit Care. 2020;58:29‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Kuypers DRJ, de Jonge H, Naesens M, Vanrenterghem Y. A prospective, open‐label, observational clinical cohort study of the association between delayed renal allograft function, tacrolimus exposure, and CYP3A5 genotype in adult recipients. Clinical Therapeutics. 2010;32(12):2012–2023. 10.1016/j.clinthera.2010.11.010 [DOI] [PubMed] [Google Scholar]
- 229. Toniati P, Piva S, Cattalini M, et al. Tocilizumab for the treatment of severe COVID‐19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia. Italy. Autoimmun Rev. 2020:102568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Xu X, Han M, Li T, et al. Effective treatment of severe COVID‐19 Patients with tocilizumab. Proc Natl Acad Sci. 2020;117(20):10970‐10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Zhang X, Song K, Tong F, et al. First case of COVID‐19 in a patient with multiple myeloma successfully treated with tocilizumab. Blood Adv. 2020;4(7):1307‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Shi J, Wen Z, Zhong G, et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS‐coronavirus 2. Science. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Morrison AR, Johnson JM, Ramesh M, Bradley P, Jennings J, Smith ZR. Letter to the Editor: Acute hypertriglyceridemia in patients with COVID‐19 receiving tocilizumab. J Med Virol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Michot J‐M, Albiges L, Chaput N, et al. Tocilizumab, an anti‐IL6 receptor antibody, to treat Covid‐19‐related respiratory failure: A case report. Ann Oncol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Fontana F, Alfano G, Mori G, et al. Covid‐19 pneumonia in a kidney transplant recipient successfully treated with Tocilizumab and Hydroxychloroquine. Am J Transplant. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. De Luna G, Habibi A, Deux J‐F, et al. Rapid and SEvere Covid‐19 pneumonia with severe acute chest syndrome in a sickle cell patient successfully treated with tocilizumab. Am J Hematol. 2020;95(7):876‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Cellina M, Orsi M, Bombaci F, Sala M, Marino P, Oliva G. Favorable changes of CT findings in a patient with COVID‐19 pneumonia after treatment with tocilizumab. Diagn Interv Imaging. 2020;101(5):323‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Aouba A, Baldolli A, Geffray L, et al. Targeting the inflammatory cascade with anakinra in moderate to severe COVID‐19 pneumonia: Case series. Ann Rheum Dis. 2020. [DOI] [PubMed] [Google Scholar]
- 239. Cavalli G, De Luca G, Campochiaro C, et al. Interleukin‐1 blockade with high‐dose anakinra in patients with COVID‐19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2(6):e325‐e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Dimopoulos G, de Mast Q, Markou N, et al. Favorable anakinra responses in severe covid‐19 patients with secondary hemophagocytic lymphohistiocytosis. Cell Host Microbe. 2020. 10.1016/j.chom.2020.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D. Baricitinib therapy in COVID‐19: A pilot study on safety and clinical impact. J Infect. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Ye M, Fu D, Ren YI, et al. Treatment with convalescent plasma for COVID‐19 patients in Wuhan, China. J Med Virol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Diurno F, Numis FG, Porta G, et al. Eculizumab treatment in patients with COVID‐19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24(7):4040‐4047. [DOI] [PubMed] [Google Scholar]
- 244. Andreakos E, Tsiodras S. COVID‐19: lambda interferon against viral load and hyperinflammation. EMBO Mol Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer‐Smadja N. Type 1 interferons as a potential treatment against COVID‐19. Antiviral Res. 2020;178:104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682‐687. [DOI] [PubMed] [Google Scholar]
- 247. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45‐56. [DOI] [PubMed] [Google Scholar]
- 248. Lunjani N, Satitsuksanoa P, Lukasik Z, Sokolowska M, Eiwegger T, O'Mahony L. Recent developments and highlights in mechanisms of allergic diseases: Microbiome. Allergy. 2018;73(12):2314‐2327. [DOI] [PubMed] [Google Scholar]
- 249. Wypych TP, Wickramasinghe LC, Marsland BJ. The influence of the microbiome on respiratory health. Nat Immunol. 2019;20(10):1279‐1290. [DOI] [PubMed] [Google Scholar]
- 250. Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The role of lung and gut microbiota in the pathology of asthma. Immunity. 2020;52(2):241‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Roduit C, Frei R, Ferstl R, et al. High levels of butyrate and propionate in early life are associated with protection against atopy. Allergy. 2019;74(4):799‐809. [DOI] [PubMed] [Google Scholar]
- 252. Trompette A, Gollwitzer ES, Pattaroni C, et al. Dietary Fiber Confers Protection against Flu by Shaping Ly6c(‐) Patrolling Monocyte Hematopoiesis and CD8(+) T Cell Metabolism. Immunity. 2018;48(5):992‐1005.e8. [DOI] [PubMed] [Google Scholar]
- 253. Gou W, Fu Y, Yue L, et al. Gut microbiota may underlie the predisposition of healthy individuals to COVID‐19. medRxiv. 2020;preprint. [Google Scholar]
- 254. WHO . Strategic and Technical Advisory Group for Infectious Hazards (STAG‐IH). 2020; https://www.who.int/emergencies/diseases/strategic‐and‐technical‐advisory‐group‐for‐infectious‐hazards/en/. Accessed May 7, 2020
- 255. Bedford J, Enria D, Giesecke J, et al. COVID‐19: towards controlling of a pandemic. Lancet. 2020;395(10229):1015‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Lurie N, Saville M, Hatchett R, Halton J. Developing Covid‐19 vaccines at pandemic speed. N Engl J Med. 2020;382(21):1969‐1973. [DOI] [PubMed] [Google Scholar]
- 257. WHO . DRAFT landscape of COVID‐19candidate vaccines – 20 April 2020. 2020; https://www.who.int/blueprint/priority‐diseases/key‐action/novel‐coronavirus‐landscape‐ncov.pdf?ua=1
- 258. El Zowalaty ME, Jarhult JD. From SARS to COVID‐19: A previously unknown SARS‐ related coronavirus (SARS‐CoV‐2) of pandemic potential infecting humans ‐ Call for a One Health approach. One Health. 2020;9:100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Bian H, Zheng ZH, Wei D, et al. Meplazumab treats COVID‐19 pneumonia: an open‐labelled, concurrent controlled add‐on clinical trial. medRxiv. 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material