

Key Points
Using CRISPR screens, we identify mechanisms increasing expression of the immunotherapy target BCMA and antibody-drug conjugate efficacy.
We also identify antigen-independent mechanisms regulating response of cancer cells to BCMA CAR-T cells.
Abstract
Cancer cells commonly develop resistance to immunotherapy by loss of antigen expression. Combinatorial treatments that increase levels of the target antigen on the surface of cancer cells have the potential to restore efficacy to immunotherapy. Here, we use our CRISPR interference– and CRISPR activation–based functional genomics platform to systematically identify pathways controlling cell surface expression of the multiple myeloma immunotherapy antigen B-cell maturation antigen (BCMA). We discovered that pharmacologic inhibition of HDAC7 and the Sec61 complex increased cell surface BCMA, including in primary patient cells. Pharmacologic Sec61 inhibition enhanced the antimyeloma efficacy of a BCMA-targeted antibody-drug conjugate. A CRISPR interference chimeric antigen receptor T cells (CAR-T cells) coculture screen enabled us to identify both antigen-dependent and antigen-independent mechanisms controlling response of myeloma cells to BCMA-targeted CAR-T cells. Thus, our study shows the potential of CRISPR screens to uncover mechanisms controlling response of cancer cells to immunotherapy and to suggest potential combination therapies.
Visual Abstract

Introduction
Immunotherapy has transformed the treatment of many types of cancer, including multiple myeloma (MM). B-cell maturation antigen (BCMA) is currently being evaluated in numerous clinical trials as an immunotherapy target in MM.1 BCMA-targeted immunotherapy agents have shown improved responses in patients with relapsed and refractory disease.2,3 However, as with other MM therapies, resistance and relapse to BCMA-targeted therapies have emerged as significant challenges and present an unmet need.4,5
An important mechanism by which cancer cells can become resistant to different forms of immunotherapy in the clinic is the downregulation or loss of the targeted antigen,6,7 also termed “antigen escape.”6,8,9 Ongoing clinical trials using BCMA-targeted chimeric antigen receptor T cells (CAR-T cells) have reported antigen loss in some patients experiencing relapse,4,5 indicating that reduced cell surface levels of BCMA may be an important mechanism of therapy resistance. However, the underlying cellular mechanisms remain to be understood. CRISPR-based genetic screens are a powerful research tool for defining mechanisms of treatment resistance in cancer cells to different immunotherapies,10-12 designing strategies to overcome resistance,11,13 identifying novel immunotherapy target antigens,14 and better understanding immune checkpoint regulation.13
The current study used a CRISPR-interference/CRISPR-activation (CRISPRi/CRISPRa) functional genomics platform15,16 to systematically elucidate the mechanisms by which the cell surface expression of BCMA is controlled in MM cells and to test whether some of these mechanisms would be potential targets for combination therapy to enhance BCMA-directed immunotherapy. We also conducted a CRISPRi screen for genes controlling sensitivity of MM cells to BCMA-directed CAR-T cells. To our knowledge, this study is the first genetic screen for genes in MM controlling response to CAR-T cells directed against a clinically relevant target. The results show the potential of CRISPR screens to elucidate mechanisms controlling the response of cancer cells to immunotherapy and the identification of potential pharmacologic strategies to enhance immunotherapy.
Materials and methods
CRISPRi and CRISPRa flow cytometry screen
CRISPRi and CRISPRa cell lines were generated as detailed in the supplemental Methods. For transduction of each sublibrary, AMO1 cells expressing the CRISPRi or CRISPRa machinery were spin-infected with the virus at 700g for 2 hours at 32°C. Forty-eight hours later, the cells were analyzed for percentage of infection by using flow cytometry and were treated with 1 μg/mL of puromycin to obtain a pure population of single guide RNA (sgRNA)–expressing cells. On day 12 and day 5 postinfection with the CRISPRi and CRISPRa sublibraries, cells were stained for cell surface BCMA and flow-sorted to enrich for populations of cells expressing low or high cell surface levels of BCMA. Briefly, for each sublibrary, cells were resuspended in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline containing 0.5% fetal bovine serum) at a concentration of 10 × 106 cells/mL. The cells were blocked by using Human BD Fc Block (#564220; BD Biosciences), stained with PE/CY7-BCMA (19F2) (#357508; BioLegend) antibody, and resuspended in FACS buffer for flow sorting. The top and bottom 30% of cells expressing BCMA as determined from PE/Cy7-BCMA histogram were flow-sorted by using FACSAria II (BD Biosciences). The various cell populations were then processed for next-generation sequencing as previously described15,17 and sequenced on a HiSeq-4000 (Illumina). To identify significant hit genes, sequencing reads were analyzed by using the MAGeCK-iNC pipeline as previously described.18
CRISPRi validation screen
sgRNAs targeting the selected 41 top hits identified from the primary CRISPRi screen were cloned into a custom library of 90 sgRNAs, including 2 sgRNAs per gene and 8 nontargeting control sgRNAs. The sgRNAs were transduced into a panel of CRISPRi-MM cell lines (KMS11, AMO1, RPMI8226, OPM2, and KMS12-PE). The validation screen was performed similar to the primary screen in which cells were stained by using PE/Cy7-BCMA or FITC-CD38 (#303504; BioLegend). Knockdown phenotypes for both BCMA and CD38 were hierarchically clustered based on Pearson correlation using Cluster 3.019 and Java TreeView 3.0 (http://jtreeview.sourceforge.net/).20
Antibody-drug conjugate dose response assays
A BCMA-targeted antibody-drug conjugate (ADC), HDP-101, was produced as previously described.21 For drug combination studies, cells were treated with either dimethyl sulfoxide (DMSO) or an indicated concentration of drugs before seeding. Increasing concentrations of HDP-101 were then added to cells to obtain a dose–response curve. Ninety-six hours posttreatment, cell viability was measured by using CellTiter-Glo 2.0 reagent (#G9241; Promega) following the manufacturer’s protocol. Raw luminescence signals were collected by using a SpectraMax M5 Plate Reader (Molecular Devices). All measurements were taken in triplicate, and raw counts were normalized as the percentage of signal relative to untreated cells or percent maximum signal when comparing >1 drug. Sigmoidal dose–response curve fitting for calculation of 50% inhibitory concentration was performed by using Prism version 7 (GraphPad Software).
CAR-T cell cytotoxicity assay
Generation of CAR-T cells is detailed in the supplemental Materials and methods. For T-cell cytotoxicity assays, green fluorescent protein (GFP)/CAR-T cells22 were cocultured in the presence of blue fluorescent protein–CRISPRi MM cells stained with eFluor-670 (#65-0840-85; Thermo Fisher Scientific) at a ratio of 1:1 for 24 hours. Baseline target cell death was measured by incubating CRISPRi MM cells without any CAR-T cells over the same time period. Cells were then stained with BV786-CD69 (FN50) (#310932; BioLegend) to determine T-cell activation status and propidium iodide to assess overall cell death.
CRISPRi-CAR survival screen
AMO1 CRISPRi cells expressing an sgRNA library targeting 12 838 genes (including kinases, phosphatases, cancer drug targets, apoptosis genes, proteostasis genes, and mitochondrial genes) were cocultured in the presence or absence of GFP/BCMA CAR-T cells at a ratio of 1:1 for 24 hours. Surviving cells were then harvested, and the different cell populations were processed for next-generation sequencing as previously described.15,17 Sequencing reads were analyzed by using the MAGeCK-iNC pipeline developed in our laboratory to identify significant hit genes as described.18
De-identified primary MM bone marrow aspirates were obtained from the University of California, San Francisco (UCSF), hematologic malignancy tissue bank in compliance with the UCSF Institutional Review Board protocols.
Results
Genome-wide CRISPR screens to identify genes controlling cell surface BCMA expression
To identify novel genes or pathways regulating cell surface expression of BCMA in MM cell line, genome-wide CRISPRi and CRISPRa screens were performed in an MM cell line (Figure 1A). Performing parallel CRISPRi knockdown and CRISPRa overexpression screens can yield complementary insights.15,23 Knockdown of individual genes by CRISPRi can inhibit the function or activity of an entire pathway or protein complexes for which the individual gene is required, whereas overexpression of an individual gene does not in general enhance the function of an entire pathway or protein complex. However, whereas CRISPRi screens can only interrogate the function of genes expressed in the cell line used in the screen, CRISPRa can uncover consequences of inducing a gene that is not normally expressed.
Figure 1.
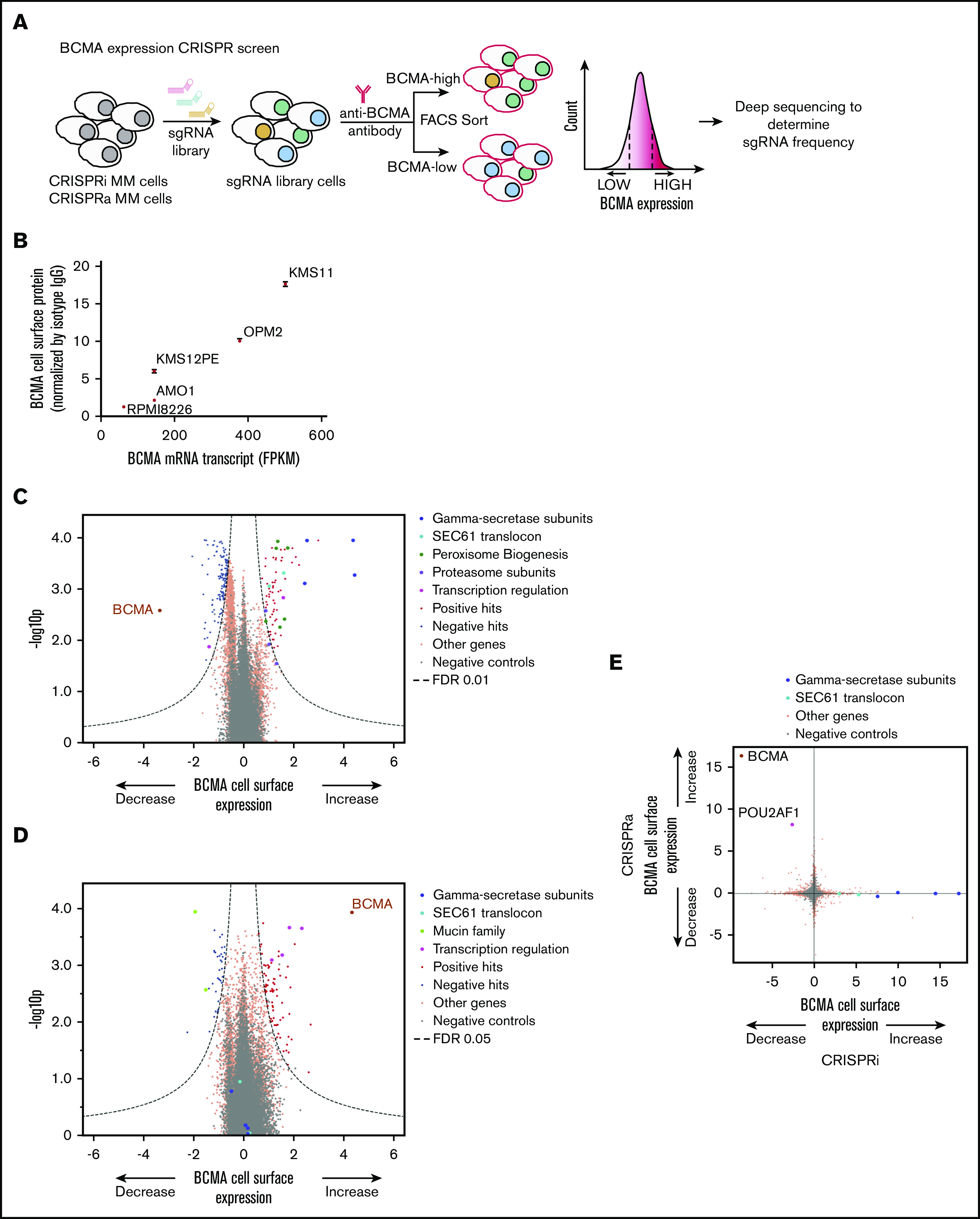
Genome-wide CRISPRi/CRISPRa screens to identify genes regulating cell surface expression of BCMA. (A) Schematic representation of our genome-wide CRISPRi and CRISPRa screens to identify modulators of BCMA expression. AMO1 cells constitutively expressing the CRISPRi or CRISPRa machinery were transduced with a genome-wide lentiviral sgRNA library. After transduction, cells were stained for cell surface levels of BCMA and sorted by FACS to enrich for populations with low or high levels of cell surface BCMA. Frequencies of cells expressing a given sgRNA were determined in each population by using next-generation sequencing. (B) BCMA expression levels in a panel of MM cell lines. BCMA transcript FPKM levels obtained from the Keats Laboratory database (https://www.keatslab.org) were plotted against cell surface expression levels of BCMA quantified according to flow cytometry. The flow cytometry data are the means of 3 biological replicates, and error bars denote standard deviations. Note that some error bars are not visible because values are small. Volcano plots indicating the BCMA expression phenotype and statistical significance for knockdown (CRISPRi) (C) or overexpression (CRISPRa) (D) of human genes (orange dots) and quasi-genes generated from negative control sgRNA (gray dots). Hit genes corresponding to functional categories are color-coded as labeled in the panel. (E) Comparison of phenotypes from the CRISPRi and CRISPRa screens. Selected hit genes are color-coded. IgG, immunoglobulin G; mRNA, messenger RNA.
To select a suitable cell line for these primary screens, levels of BCMA expression on the RNA (www.keatslab.org) and cell surface protein levels for a panel of MM cell lines (Figure 1B) were compared. We found that AMO1 cells expressed moderate levels of cell surface BCMA. This line was therefore selected for the primary screen as we reasoned it would enable us to identify modifiers that either increase or decrease BCMA surface levels.
AMO1 cells were lentivirally transduced to express the CRISPRi and CRISPRa machinery, and CRISPR functionality was tested by using sgRNAs targeted toward CD38 and CXCR4, respectively (supplemental Figure 1). The genome-wide screen was conducted as shown in Figure 1A. Briefly, AMO1 cells expressing the CRISPRi and CRISPRa machinery were transduced with the pooled genome-wide sgRNA library. The cells were then stained for cell surface BCMA by using fluorescent tagged antibody and subjected to flow sorting into low and high BCMA populations. Frequencies of cells expressing each sgRNA were identified by using next-generation sequencing. The genome-wide CRISPRi screen identified several genes regulating cell surface expression of BCMA (Figure 1C; supplemental Table 1). Knocking down BCMA itself or its transcription factor POU2AF124 resulted in significant downregulation of cell surface BCMA expression, thus validating the screen. Furthermore, all of the subunits of the γ secretase complex were among the top hits, and their knockdown resulted in a significant increase in cell surface BCMA. This finding was consistent with previous reports showing that γ secretase complex cleaves membrane-bound BCMA to a soluble form (sBCMA).9,25 Moreover, several functional categories of genes regulating expression levels of BCMA were identified, including subunits of the Sec61 translocon complex, peroxisome biogenesis, proteasome subunits, and regulators of transcription.
Our CRISPRa genome-wide screen (Figure 1D; supplemental Table 2) identified genes in the Mucin family (MUC1 and MUC21) and several genes involved in transcriptional regulation (POU2AF1, CBFA2T3, MAML2, and RUNX3) that regulated surface BCMA. Systematic comparison of the parallel CRISPRi and CRISPRa screens (Figure 1E) revealed that overexpression and knockdown of some genes had opposing effects on BCMA surface levels, including BCMA itself and POU2AF1. However, other genes were hits in only the CRISPRi or CRISPRa screens, notably the subunits of the γ secretase complex and the Sec61 complex; this finding highlights the fact that CRISPRi and CRISPRa screens can uncover complementary results, as previously described.15,23
Validation of hit genes in a panel of MM cell lines
To test the generality of the findings, we decided to validate hit genes from the primary screen performed on the AMO1 cells in a panel of CRISPRi-MM lines, in which we confirmed CRISPRi activity (supplemental Figure 1). Forty-one hits were chosen, based both on strength of the phenotype from the CRISPRi screen and on their potential as therapeutic targets. The secondary screen was performed similarly to the genome-wide screen to validate changes in BCMA. The screen was conducted in parallel for a different cell surface protein expressed in all MM lines, CD38, to investigate whether hit genes selectively affected BCMA expression.
As observed in the primary CRISPRi screen, knockdown of the subunits of γ secretase subunits significantly increased cell surface BCMA (Figure 2A). This effect was found to be selective for BCMA, as γ secretase knockdown had no effect on CD38 expression in a panel of MM cell lines. This finding was further validated by using a pharmacologic inhibitor of γ secretase complex, RO4929097.26 MM cell lines treated with the indicated concentration of RO4929097 exhibited an increase in both BCMA cell surface expression according to flow cytometry (Figure 2B) and in total protein levels of BCMA according to immunoblotting (Figure 2C). Thus, we were able to successfully validate genes from our primary screen in a panel of MM cell lines, using both genetic and pharmacologic perturbations.
Figure 2.
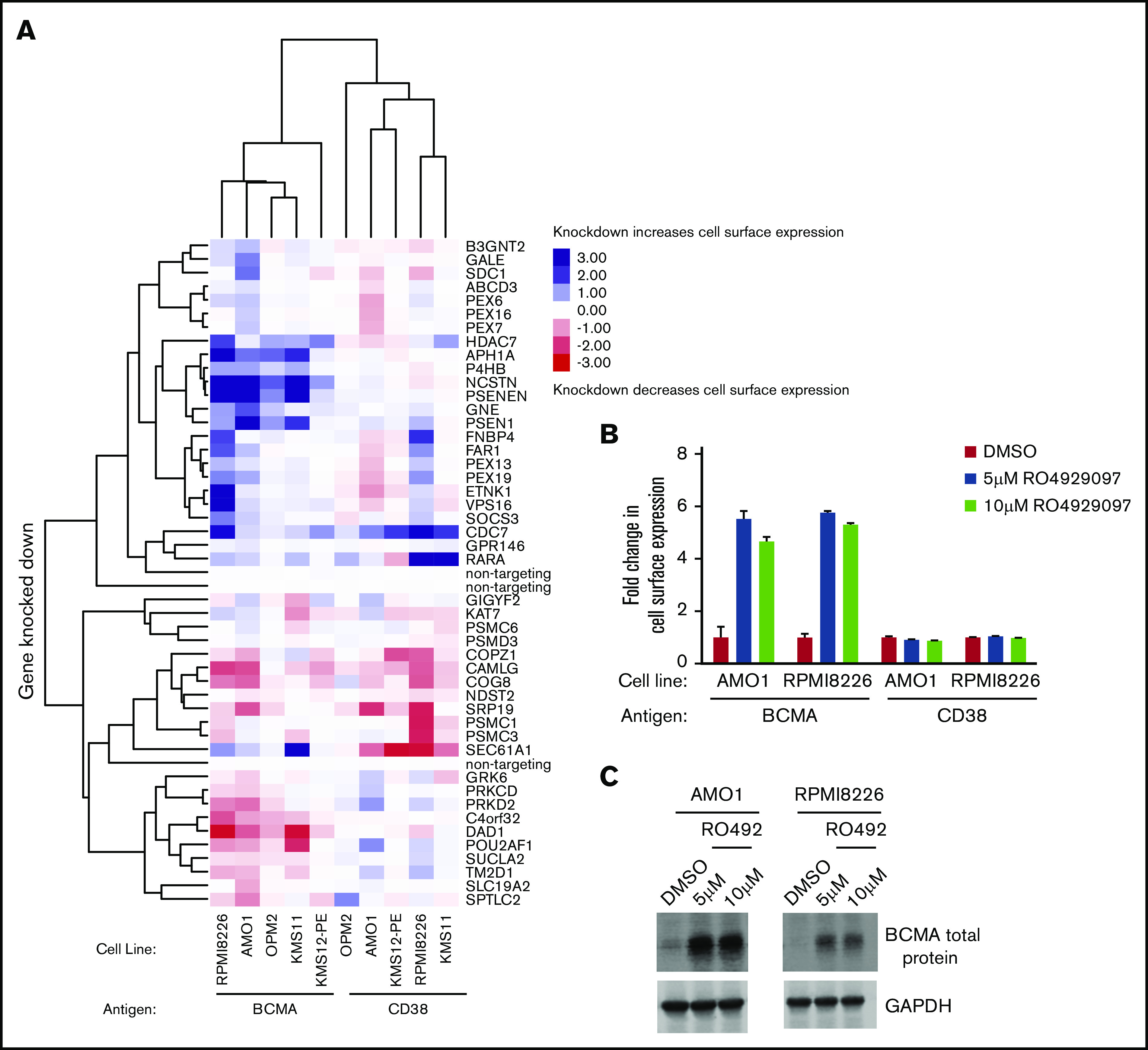
Validation of hit genes from the primary screen. (A) Heat map representation of knockdown phenotype scores from CRISPRi validation screens in a panel of MM cell lines for cell surface levels of BCMA and CD38. Both genes and screens were hierarchically clustered based on Pearson correlation. AMO1 and RPMI8226 cells treated with indicated concentrations of the γ-secretase inhibitor RO4929097 or DMSO for 48 hours were analyzed by using flow cytometry (B) and immunoblotting (C) for changes in BCMA levels. Fold changes in BCMA cell surface levels as shown in panel B were determined by normalizing to DMSO-treated cells. Data points are means of three biological replicates, and error bars denote standard deviations. Western blot of endogenous BCMA is shown in panel C. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a loading control.
Several of the novel factors from our primary screen were also validated in the larger panel of MM cells (Figure 2A; supplemental Tables 3 and 4), most notably the transcriptional regulator HDAC7 and SEC61A1, a subunit of the SEC61 translocon. Knockdown of both factors increased cell surface levels of BCMA but not CD38.
Class IIa HDAC inhibition upregulates BCMA transcription
Our primary CRISPRi screen showed that knockdown of HDAC7, but not any other HDAC gene, upregulates BCMA expression (Figure 3A). HDAC7 is a member of the histone deacetylase (HDAC) family, which has emerged as crucial transcriptional corepressors.27 HDAC7 belongs to the class II family of HDACs, which are subdivided into class IIa including HDAC4, HDAC5, HDAC7, and HDAC9 and class IIb including HDAC6 and HDAC10.27 We validated our screen findings by using TMP269, a pharmacologic inhibitor targeting class IIa HDACs.28 Treatment of RPMI8226 cells with increasing concentrations of TMP269 showed a twofold increase in BCMA protein levels (Figure 3B-D). Because HDACs have been established to regulate transcription of several genes, quantitative polymerase chain reaction was performed to analyze for changes in transcript levels of BCMA. The results showed a twofold increase in BCMA transcript levels (Figure 3E), indicating that class II HDAC inhibition regulates transcription of BCMA. No significant change was observed in the CD38 protein or transcript levels (Figure 3B,E). Furthermore, treatment of RPMI8226 cells with a combination of TMP269 and the γ-secretase inhibitor RO4929097 revealed a further increase in BCMA surface expression levels, supporting the notion that they act through different mechanisms (Figure 3F).
Figure 3.
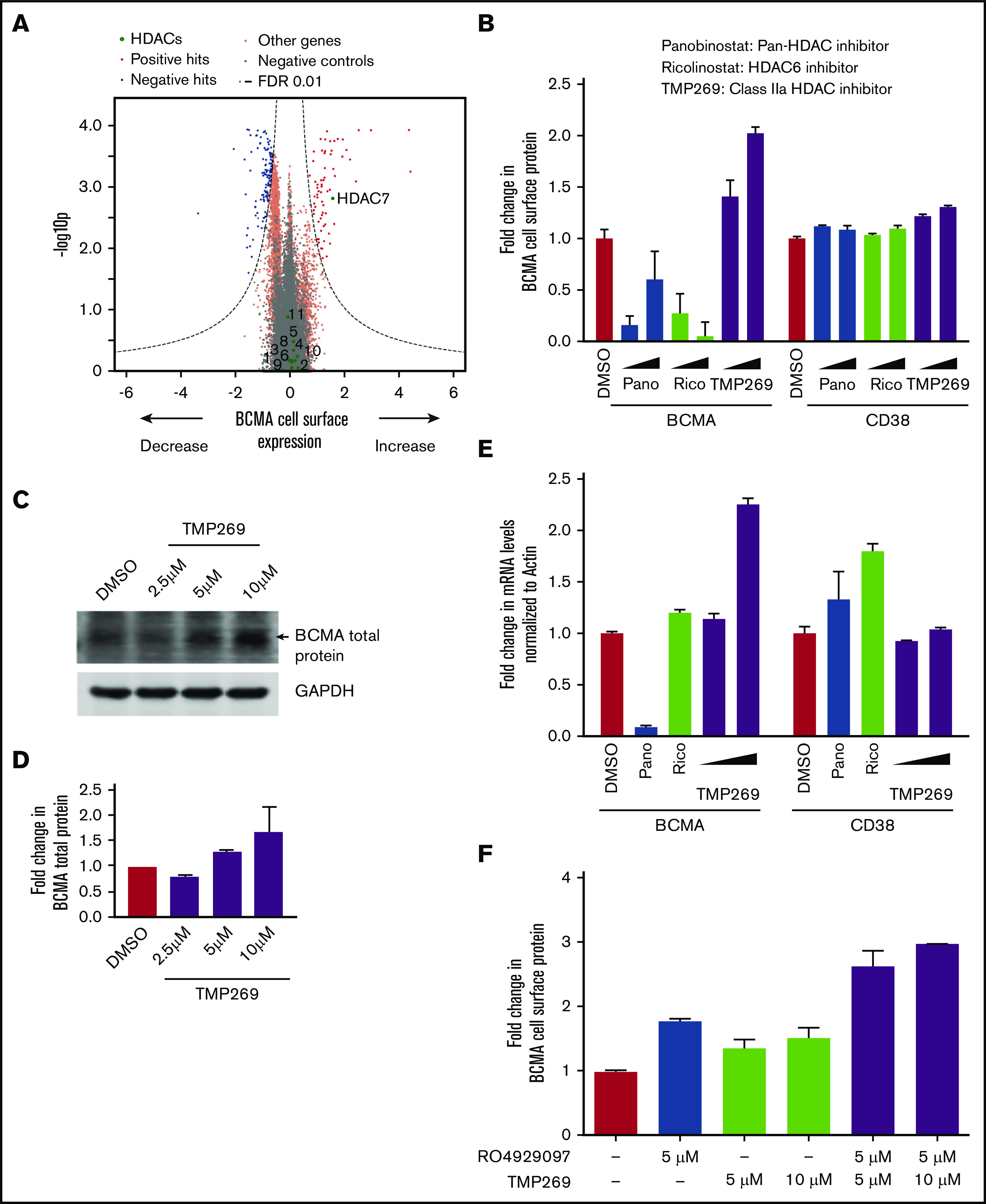
Class IIa HDAC inhibition increases transcription of BCMA. (A) Volcano plot indicating the BCMA expression phenotype and statistical significance for knockdown (CRISPRi) of human genes (orange dots) and quasi-genes generated from negative control sgRNA (gray dots). Genes in the HDAC family are shown as green dots and labeled with the HDAC number. (B) RPMI8226 cells were treated with increasing concentrations of the pan-HDAC inhibitor panobinostat (10 nM, 25 nM), the HDAC6-specific inhibitor ricolinostat (0.5 μM, 1 μM), the class II HDAC inhibitor TMP269 (5 μM, 10 μM), or DMSO for 48 hours and analyzed by using flow cytometry for cell surface expression of BCMA and CD38. Fold changes in protein levels were determined by normalizing to the DMSO-treated cells. Data points are means of 3 biological replicates, and error bars denote standard deviations. (C-D) Total protein extracts from RPMI8226 cells treated with 2.5 μM, 5 μM, and 10 μM of TMP269 for 48 hours were analyzed by using immunoblotting for expression levels of BCMA. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used to normalize differences in loading amounts. Data are represented as fold change relative to the total protein expression level after normalization with GAPDH. Data points are means of 2 technical replicates, and error bars denote standard deviations. (E) RPMI8226 cells treated with 10 nM of panobinostat, 0.5 μM of ricolinostat, and 5 μM and 10 μM of TMP269 for 48 hours were processed for quantitative polymerase chain reaction to determine transcript levels of BCMA and CD38. Fold changes in transcript levels with different drug treatments were determined after normalizing to the β-actin gene. Data are means of 2 biological replicates, and error bars denote standard deviations. (F) RPMI8226 cells were treated with DMSO, 5 μM and 10 μM of TMP269, and 5 μM of the γ-secretase inhibitor RO4929097 as single agents or in combination for 48 hours and analyzed by using flow cytometry for cell surface expression of BCMA. Fold changes in protein levels were determined by normalizing to the DMSO-treated cells. Data points are means of 3 biological replicates, and error bars denote standard deviations.
To investigate whether this change in BCMA levels is specific to HDAC7 inhibition, cells were treated with a pan-HDAC inhibitor, panobinostat,29 and an HDAC6-specific inhibitor, ricolinostat.30 The results showed no increase in BCMA transcript levels with these agents (Figure 3B,E). In fact, these compounds resulted in a decrease in BCMA cell surface levels, which can be rationalized by three facts. First, although panobinostat is a pan-HDAC inhibitor, it is not very potent for HDAC7, and it was used at nanomolar concentrations well below the 50% inhibitory concentration for HDAC7 (1.378 µM31) because we are limited by toxicity. Second, panobinostat inhibits several different HDACs with a range of varying cellular functions and will therefore cause pleiotropic effects. Second, both panobinostat and ricolinostat are very toxic to MM cells, likely resulting in a range of cellular changes due to toxicity, whereas no toxicity in MM cells was found with TMP269 treatment (supplemental Figure 2A).
Furthermore, treatment with TMP269 in K562 cells, which do not express BCMA, did not lead to an increase in BCMA expression (supplemental Figure 2B-C). These results indicate the potential for using class IIa HDAC inhibition to increase expression of BCMA in plasma cells in the context of BCMA-targeted immunotherapy.
Sec61 translocon regulates BCMA protein levels
Most integral plasma membrane proteins are inserted into membranes via the Sec61 translocon, located in the endoplasmic reticulum. It has been shown that inhibition of Sec61 affects correct localization of a subset of membrane proteins.32,33 Surprisingly, our CRISPRi screen identified that knockdown of genes in the SEC61 pathway, such as SEC61A1, SEC61G, SSR1, and OSTC, resulted in an increase (rather than a decrease) in BCMA cell surface levels (Figure 1C). This unexpected finding was confirmed in our validation screens in the panel of MM cell lines (Figure 2A). Conversely, SEC61A1 knockdown resulted in a decrease in CD38 cell surface levels, as would be expected for most membrane proteins.
To test whether pharmacologic inhibition of the Sec61 translocon complex phenocopies the genetic knockdown, cells were treated with SEC61 inhibitors, CT832 and PS3061.34 We observed that treatment of MM cell lines with increasing concentrations of these compounds results in an up to fivefold dose-dependent increase in cell surface BCMA levels and a decrease in CD38 levels with minimal cytotoxicity, as evidenced by flow cytometry (Figures 4A,D; supplemental Figure 3). Moreover, immunoblotting also showed a dose-dependent increase in total BCMA protein levels (Figure 4B,E; supplemental Figure 3) and up to a twofold increase in BCMA transcript levels (Figure 4C,F). Furthermore, treatment of cells with SEC61 inhibitors resulted in a decrease in TACI, which, like BCMA, belongs to the TNFRSF family of proteins, indicating that the SEC61 inhibition selectively upregulates BCMA levels. Combinatorial treatment with both γ-secretase and SEC61 inhibitors had cell line–specific outcomes, highlighting the heterogeneity of MM cell lines. An increase was observed in BCMA expression when both γ-secretase and SEC61 inhibitors were combined in AMO1 cells (Figure 4G) but not in KMS11 cells (Figure 6D).
Figure 4.
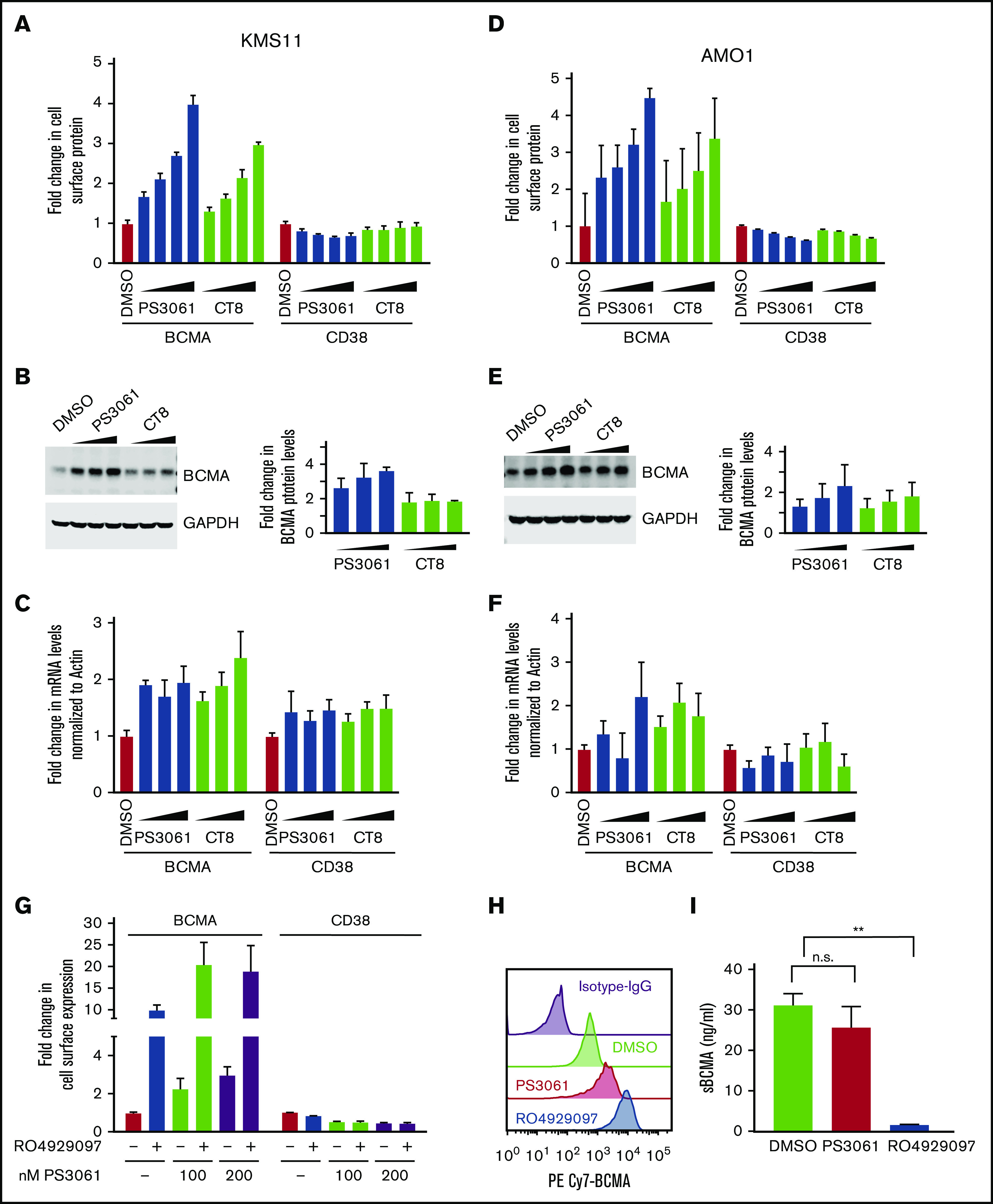
Sec61 inhibitors increase cell surface expression of BCMA in multiple myeloma cells. (A-C) KMS11 cells, (D-F) AMO1 cells. (A,D) KMS11 and AMO1 cells were treated with increasing concentrations of the Sec61 inhibitors CT8 and PS3061 (100, 200, 400, and 800 nM) or DMSO as a control for 24 hours. Cells were stained for cell surface expression of BCMA and CD38 and analyzed by using flow cytometry. Data are means of 3 biological replicates, and error bars denote standard deviations. (B,E) Total protein lysates from cells treated with increasing concentration of CT8 and PS3061 (200, 400, and 800 nM) for 24 hours were processed for western blotting. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used to normalize for differences in loading amounts. Data are represented as fold change relative to the normal protein expression level after normalization with GAPDH. Data points are means of 2 technical replicates, and error bars denote standard deviations. (C,F) Cells treated with increasing concentrations of PS3061 and CT8 (200, 400, and 800 nM) for 24 hours were processed for quantitative polymerase chain reaction to determine transcript levels of BCMA and CD38. Fold change in transcript levels were determined after normalizing to β-actin. Data are means of 2 biological replicates, and error bars denote standard deviations. (G) AMO1 cells were treated for 24 hours with the Sec61 inhibitor PS3061 (100 and 200 nM) and the γ-secretase inhibitor RO4929097 (5 μM) as single agents or in combination using DMSO as a control. Cells were stained for cell surface expression of BCMA and CD38 and analyzed by using flow cytometry. Data are means of 3 biological replicates, and error bars denote standard deviations. (H-I) KMS11 cells were treated with 800 nM of PS3061, 5 µM of RO4929097, and DMSO for 24 hours. (H) Drug-treated cells were analyzed by flow cytometry for cell surface expression of BCMA. Histograms indicate distribution of PE/CY7 BCMA in the drug-treated cells. Data are a representation of 2 biological replicates. (I) Concentration of sBCMA in the cell culture supernatant after drug treatment was measured by using enzyme-linked immunosorbent assay. Data are means of 2 biological replicates, and error bars denote standard deviations. **P < .005. n.s., not significant, 2-tailed, unpaired Student t test.
Figure 6.
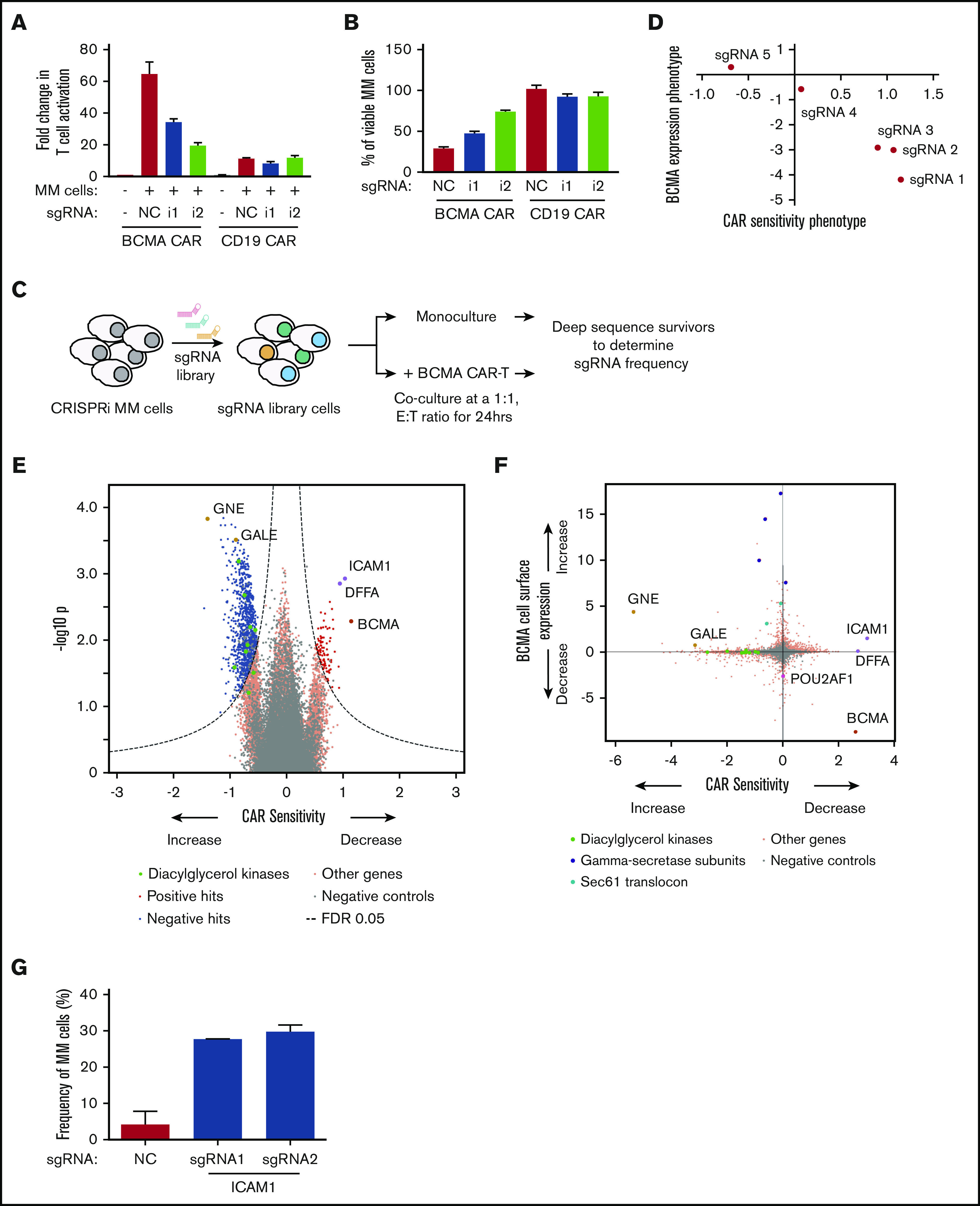
CRISPRi/CAR-T screen identifies genes modulating response to BCMA-targeted CAR-T cells. (A-B) CRISPRi AMO1 cells expressing BFP-sgRNA targeted toward BCMA or nontargeting control sgRNA were cocultured at a 1:1 ratio with BCMA– or CD19–GFP CAR-T cells. Fold changes in T-cell activation was determined by analyzing for cell surface expression of CD69 on T cells normalized to CD69 expression on resting T cells. Myeloma cell viability was determined by propidium iodide staining of BFP-positive myeloma cells. (C) Schematic representation of the CRISPRi/CAR-T cell coculture screen. (D) Comparison of BCMA expression phenotype to sensitivity toward BCMA-targeted CAR-T cells indicating different sgRNAs targeted toward BCMA. (E) Volcano plot indicating CAR-T cell sensitivity phenotypes and statistical significance of knockdown of human genes (orange dots) and quasi-genes from negative control sgRNAs (gray dots) in MM cells. Hit genes corresponding to functional categories are color-coded as labeled in the panel. (F) Comparison of BCMA expression phenotype from the CRISPRi primary screen (Figure 1) to sensitivity toward BCMA-targeted CAR T cells. Hit genes corresponding to functional categories are color-coded as labeled in the panel. (G) CRISPRi AMO1 cells expressing 2 independent sgRNAs targeting ICAM1 or nontargeting control sgRNA were cocultured overnight at a 1:1 ratio with BCMA-targeted CAR-T cells. Frequency of viable myeloma cells was determined by using flow cytometry.
To uncover the mechanism by which SEC61 inhibition increases BCMA levels, we first tested the possibility that a reduction in γ-secretase levels at the plasma membrane would reduce shedding of BCMA from the cell surface. Pharmacologic inhibition of either the γ-secretase or Sec61 resulted in increased membrane-bound BCMA as evidenced by flow cytometry (Figure 4H). To determine levels of sBCMA generated by γ-secretase processing, we performed sandwich enzyme-linked immunosorbent assay on cells treated with PS3061 or RO4929097. The results show that inhibition of γ-secretase activity significantly reduced sBCMA levels, whereas no significant changes were observed in sBCMA levels with SEC61 inhibition (Figure 4I). This scenario indicates that the increase in cell surface BCMA is not driven by reduced shedding of BCMA.
To validate our findings in primary patient cells, bone marrow mononuclear cells derived from various patients with MM (supplemental Table 5) were treated with increasing concentrations of TMP269 and RO4929097 for 24 hours. We then analyzed the expression of BCMA on plasma cells by using flow cytometry. An up to 2.5-fold increase was observed in cell surface BCMA with class IIa HDAC inhibition and an up to eightfold increase with γ-secretase inhibition (Figure 5). Similarly, treatment with the Sec61 inhibitor PS3061 increased cell surface levels of BCMA in primary patient cells approximately twofold, while not affecting cell surface levels of CD38. Furthermore, moderate effects on BCMA expression were observed on nonplasma cells in the patient samples (supplemental Figure 4).
Figure 5.
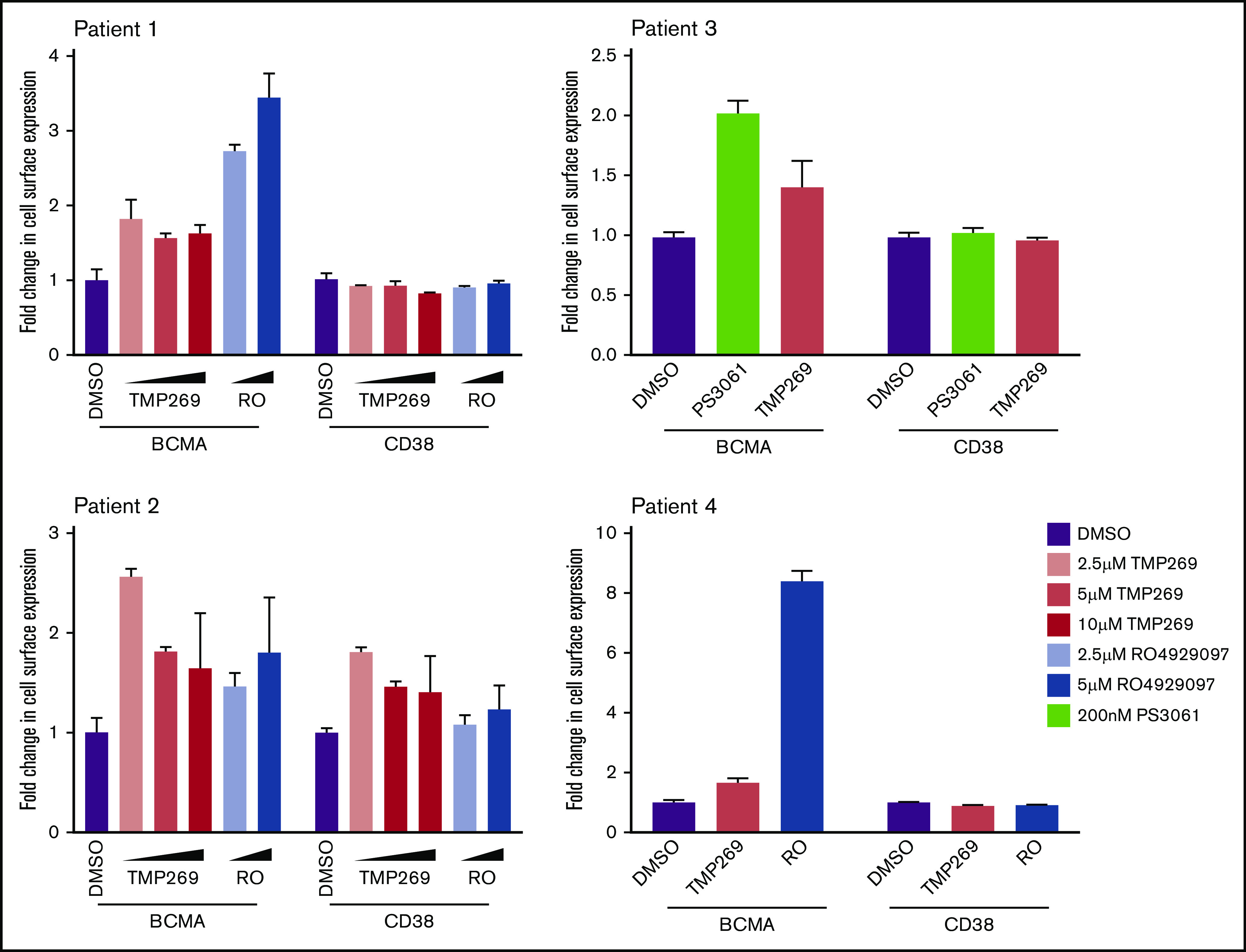
Pharmacologic inhibition of the validated hits in MM patient samples upregulate cell surface BCMA levels. Bone marrow–mononuclear cells isolated from bone marrow aspirates from various patients with MM were treated with indicated concentration of a class II HDAC inhibitor (TMP269), a γ-secretase inhibitor (RO4929097), and a Sec61 inhibitor (PS3061) for 24 hours. Cells were stained for cell surface CD138, BCMA, and CD38 and analyzed by using flow cytometry. Fold change in BCMA and CD38 levels were determined in CD138+ live cell populations. Data are means of 3 technical replicates, and error bars denote standard deviations.
Immunotherapy agents such as ADC are sensitive to changes in expression of target antigen.35 We found that BCMA knockdown reduced sensitivity of MM cell lines to a BCMA-targeting ADC (HDP101)21,36 (supplemental Figure 5A-C). Moreover, when cells were treated with either the γ secretase inhibitor RO4929097 or the Sec61 inhibitor PS3061 at concentrations increasing cell surface levels of BCMA (supplemental Figure 5D) in combination with HDP101, increased efficacy of the BCMA-targeted ADC on the MM cell lines was observed (supplemental Figure 5E).
Taken together, these findings show that our CRISPR screen was able to identify druggable targets that enhance the efficacy of immunotherapy agents.
BCMA CAR-T cell coculture screen reveals myeloma cell–intrinsic proteins that affect CAR-T efficacy
A systematic understanding of mechanisms causing resistance to CAR-T cell therapy will enable us to design potential combination therapies preempting resistance. Here, we used our CRISPR screening platform to identify genes or pathways that control sensitivity and resistance of MM cells to BCMA-targeted CAR-T cells.
First, BCMA CAR-T cells were generated by transducing CD8+ T cells with a lentiviral vector encoding a second-generation CAR incorporating an anti-BCMA single-chain variable fragment, 4-1BB costimulatory domain, and CD3-ζ signaling domain (see supplemental Methods). We validated the activation of BCMA CAR-T cells in the presence of AMO1 cells (Figure 6A) and their cytotoxicity against AMO1 cells (Figure 6B). Knockdown of BCMA in AMO1 cells using 2 independent sgRNAs (supplemental Figure 5) reduced both activation and cytotoxicity of BCMA-targeted CAR-T cells (Figure 6A-B). This finding indicates that the efficacy of the CAR-T cells depends on the cell surface levels of BCMA in MM cells.
Although the expression level of the target antigen is a major determinant of response to CAR-T cells, antigen-independent determinants are also likely. To systematically identify such determinants, a CRISPRi screen was conducted to identify genes or pathways in MM cells determining response to BCMA CAR-T cells (Figure 6C). AMO1 cells expressing the CRISPRi machinery and an sgRNA library targeting 12 838 genes (including kinases, phosphatases, cancer drug targets, apoptosis genes, mitochondrial genes, and transcription factors) were grown as monoculture or cocultured with BCMA CAR-T cells. Twenty-four hours later, the surviving cells were harvested and processed for next-generation sequencing.
Comparing BCMA cell surface levels with CAR-T sensitivity for different sgRNAs targeting BCMA, we observed that surface levels correlated with sensitivity to BCMA CAR-T cells (Figure 6D). This CRISPRi screen identified a substantial number of genes regulating sensitivity to CAR-T cells (Figure 6E; supplemental Table 6). For example, knockdown of subunits of the γ secretase complex and genes involved in the sialic acid biosynthesis pathways GALE and GNE, which change BCMA cell surface expression, also concordantly affected sensitivity to BCMA CAR-T cells (Figure 6E-F).
Furthermore, the screen identified a different category of genes, knockdown of which affected sensitivity of AMO1 cells to BCMA CAR-T cells without affecting cell surface levels of BCMA. These genes included ICAM1, which functions in T-cell activation,37 and DNA fragmentation factor subunit α (DFFA), which is required for caspase activation to trigger apoptosis.38 Knockdown of ICAM1 or DFFA resulted in decreased CAR-T cell sensitivity. Conversely, knockdown of genes belonging to the family of diacylglycerol kinases (DGK) caused increased sensitivity to BCMA CAR-T cells (Figure 6F). Using 2 individual sgRNAs targeting ICAM1, we were able to validate that knockdown of ICAM1 results in reduced sensitivity to BCMA CAR-induced cytotoxicity (Figure 6G).
This screen thus identified potential pathways that can regulate sensitivity to CAR-T cells either through changing the cell surface expression of the antigen or through an antigen-independent mechanism.
Discussion
The current study established that CRISPR-based screens enable a systematic and scalable approach to identifying mechanisms of antigen expression. Uncovering pathways that regulate BCMA transcription, translation and trafficking to the cell surface enabled us to propose effective treatment strategies in patients with low basal expression of BCMA or after relapse due to antigen loss. Clinical trials are ongoing to investigate the combination of γ-secretase inhibitor with BCMA CAR-T cell therapy (#NCT03502577). However, it is likely that not all patients would respond similarly to γ secretase inhibition, or resistance may also develop to this combination strategy. Therefore, identifying alternative ways to modulate BCMA, as we discover here, retains high clinical relevance. Our results also support the concept that strategies to upregulate antigen expression on cancer cells can enhance the efficacy of antigen-targeted immunotherapy agents.
Our findings also raise several mechanistic questions, which will be the subject of future studies. In particular, it is not clear how SEC61 inhibition results in increased cell surface levels of BCMA. SEC61 knockdown was previously shown to upregulate a subset of proteins.39 This effect on BCMA could be due to direct effects on the interaction of BCMA with the SEC61 complex, or due to indirect effects mediated by changes in other SEC61 clients.
Furthermore, our results indicate HDAC7 as a potential combination target for BCMA-targeted immunotherapy. This highlights the importance of inhibiting specific HDACs and provides a potential therapeutic use case for an HDAC7-specific inhibitor to enhance BCMA-targeted immunotherapy.
Using a complementary strategy, our CRISPRi/CAR-T coculture screen uncovered both antigen-dependent and antigen-independent mechanisms in MM cells that control the response to BCMA-targeted CAR-T cells. Recent screens for pathways controlling cytotoxicity of CAR-T cells in leukemia and lymphoma cells showed that death receptor signaling plays an important role,12 whereas our screens did not find the same pathway. This discrepancy could be due to differences in methodology, CAR target antigens (BCMA vs CD19), or the biology of the different cancer types. Our screen showed that knockdown of the DGK family of genes resulted in increased sensitivity to CAR-T cells. Previous groups have shown that inhibition of DGK-α activity in T cells using pharmacologic inhibitors induced T-cell activation, thus improving its cytotoxic activity on cancer cells.40,41 Our study indicates that knockdown of DGK kinases in MM cells increases sensitivity to T cells. This finding indicates that inhibitors against this family of kinases may be beneficial in CAR-T cell therapy of MM through a dual mechanism, acting by both increasing T-cell activity but also sensitizing MM cells to CAR-T cells.
Our study also identified genes in the sialic acid biosynthesis pathways GALE and GNE, knockdown of which sensitized MM cells to CAR-T cells. Although our BCMA expression screen identified these genes to upregulate BCMA, the effect size of sensitizing the cells to CAR-T cells was much higher than the change in BCMA expression, indicating that there could be additional mechanisms by which this pathway modulates response to CAR-T cells. Sialic acid blockade in cancer cells has been shown to create an immune-permissive tumor microenvironment increasing CD8+ T-cell activity.42 Therefore, inhibition of GALE and/or GNE could modulate the tumor microenvironment and increase sensitivity to CAR-T cells in an antigen-independent mechanism.
In conclusion, we present two complementary screening approaches to identify potential combinatorial treatments that can enhance the efficacy of immunotherapy through antigen-dependent and antigen-independent mechanisms. These strategies should be readily adaptable to a broad range of cancer cell types, antigens, and immunotherapy modalities.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank James Nunez, Marco Jost, Christina Liem, and Jonathan Weissman for sharing their unpublished CRISPRa construct; Diego Acosta-Alvear for sharing the mCherry-Luciferase RPMI8226 cell line; Axel Hyrenius-Wittsten and Joe Hiatt for input on T-cell culturing; Eric Chow and Derek Bogdanoff (UCSF Center for Advanced Technology) for support with next-generation sequencing; Sarah Elmes and Jane Gordon (UCSF Laboratory for Cell Analysis) for support with FACS; and Stratton Georgoulis, Molly Bassette, and Logan Hille for contributing to preliminary studies. For primary patient samples, the authors thank the Grand Multiple Myeloma Translational Initiative. They also thank members of M.K.'s laboratory for discussion and feedback on the manuscript.
This work was supported by a postdoctoral fellowship from the UCSF Program for Breakthrough Biomedical Research (P.R.), the National Institutes of Health, National Cancer Institute (K99/R00 CA181494 [M.K.] and R01 CA226851 [A.P.W.]), a Stand Up To Cancer Innovative Research Grant (M.K.), the UCSF Stephen and Nancy Multiple Myeloma Translational Initiative (M.K., K.T.R., and A.P.W.), and UCSF Breast Cancer Research Funds (J.T.).
Footnotes
Requests for original data not included in the manuscript and supplemental tables can be submitted to the corresponding author (Martin Kampmann; e-mail: martin.kampmann@ucsf.edu).
Authorship
Contribution: P.R., A.P.W., and M.K. designed the study; P.R., M.S., J.T.L., M.C., and K.T.R. developed the methodology; P.R., A.B.A., M.S., J.T.L., and M.C. performed the experiments; P.R., A.B.A., R.T., J.T., A.P.W., and M.K. analyzed the data; J.T.L., P.C., T.H., N.S., S.W.W., T.G.M., J.L.W., K.T.R., A.P., and J.T. provided material and technical support; P.R. and M.K. wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: M.K. has filed a patent application related to CRISPRi and CRISPRa screening (PCT/US15/40449); and serves on the Scientific Advisory Board of Engine Biosciences. A.P. and T.H. are employed at Heidelberg Pharma (A.P.) and Heidelberg Pharma Research GmbH (T.H.) and are working on the development of amatoxin-based ADCs (including HDP-101); Heidelberg Pharma holds patents concerning the conjugation of amatoxins to antibodies. K.T.R. is a cofounder and stockholder in Arsenal Biosciences; was a founding scientist/consultant and stockholder in Cell Design Labs (now a Gilead Company); and holds stock in Gilead. J.T. holds equity interests in Global Blood Therapeutics, Principia Biopharma, Kezar Life Sciences, and Cedilla Therapeutics; serves as a consultant for Cedilla Therapeutics; and is an inventor on a patent application that includes PS3061 (PCT/US2019/024731). A.P.W. is a member of the scientific advisory board and equity holder in Protocol Intelligence, LLC, and Indapta Therapeutics, LLC. N.S. has received research funding from Celgene/BMS, Janssen, Bluebird Bio, Sutro Biopharma, and TeneoBio; and is an advisor to Genentech, GSK, Amgen, Indapta Therapeutics, Sanofi, BMS, CareDx, and Kite. The remaining authors declare no competing financial interests.
Correspondence: Martin Kampmann, Institute for Neurodegenerative Diseases, University of California, San Francisco, 675 Nelson Rising Ln, San Francisco, CA 94158; e-mail: martin.kampmann@ucsf.edu.
References
- 1.Cho SF, Anderson KC, Tai YT, Targeting B. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. 2018;9:1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Susanibar Adaniya SP, Cohen AD, Garfall AL. Chimeric antigen receptor T cell immunotherapy for multiple myeloma: a review of current data and potential clinical applications. Am J Hematol. 2019;94(suppl 1):S28-S33. [DOI] [PubMed] [Google Scholar]
- 4.Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen AD, Garfall AL, Stadtmauer EA, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219-1226. [DOI] [PubMed] [Google Scholar]
- 7.Iorgulescu JB, Braun D, Oliveira G, Keskin DB, Wu CJ. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 2018;10(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nijhof IS, Casneuf T, van Velzen J, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016;128(7):959-970. [DOI] [PubMed] [Google Scholar]
- 9.Pont MJ, Hill T, Cole GO, et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019;134(19):1585-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han P, Dai Q, Fan L, et al. Genome-wide CRISPR screening identifies JAK1 deficiency as a mechanism of T-cell resistance. Front Immunol. 2019;10:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsui CK, Barfield RM, Fischer CR, et al. CRISPR-Cas9 screens identify regulators of antibody-drug conjugate toxicity. Nat Chem Biol. 2019;15(10):949-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dufva O, Koski J, Maliniemi P, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. 2020;135(9):597-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burr ML, Sparbier CE, Chan YC, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549(7670):101-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong MB, Wang G, Chow RD, et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell. 2019;178(5):1189-1204.e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert LA, Horlbeck MA, Adamson B, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159(3):647-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horlbeck MA, Gilbert LA, Villalta JE, et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife. 2016;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kampmann M, Bassik MC, Weissman JS. Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps [published correction appears in Nat Protoc. 2015;10(4):643]. Nat Protoc. 2014;9(8):1825-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian R, Gachechiladze MA, Ludwig CH, et al. CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron. 2019;104(2):239-255.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246-3248. [DOI] [PubMed] [Google Scholar]
- 21.Pahl A, Ko J, Breunig C, et al. HDP-101: preclinical evaluation of a novel anti-BCMA antibody drug conjugates in multiple myeloma. J Clin Oncol. 2018;36(suppl 15):e14527. [Google Scholar]
- 22.Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676-684. [DOI] [PubMed] [Google Scholar]
- 23.Jost M, Chen Y, Gilbert LA, et al. Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol Cell. 2017;68(1):210-223.e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao C, Inoue J, Imoto I, et al. POU2AF1, an amplification target at 11q23, promotes growth of multiple myeloma cells by directly regulating expression of a B-cell maturation factor, TNFRSF17. Oncogene. 2008;27(1):63-75. [DOI] [PubMed] [Google Scholar]
- 25.Laurent SA, Hoffmann FS, Kuhn PH, et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6(1):7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luistro L, He W, Smith M, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 2009;69(19):7672-7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding ZM, Liu YG. Effects of phosphoramidothiolate pesticides on rat erythrocyte membrane acetylcholinesterase [in Chinese]. Zhonghua Yu Fang Yi Xue Za Zhi. 1988;22(2):82-84. [PubMed] [Google Scholar]
- 28.Lobera M, Madauss KP, Pohlhaus DT, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9(5):319-325. [DOI] [PubMed] [Google Scholar]
- 29.Scuto A, Kirschbaum M, Kowolik C, et al. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008;111(10):5093-5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi SY, Kee HJ, Jin L, et al. Inhibition of class IIa histone deacetylase activity by gallic acid, sulforaphane, TMP269, and panobinostat. Biomed Pharmacother. 2018;101:145-154. [DOI] [PubMed] [Google Scholar]
- 32.Mackinnon AL, Paavilainen VO, Sharma A, Hegde RS, Taunton J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. eLife. 2014;3:e01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garrison JL, Kunkel EJ, Hegde RS, Taunton J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature. 2005;436(7048):285-289. [DOI] [PubMed] [Google Scholar]
- 34.Shah PS, Link N, Jang GM, et al. Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell. 2018;175(7):1931-1945.e1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins DM, Bossenmaier B, Kollmorgen G, Niederfellner G. Acquired resistance to antibody-drug conjugates. Cancers (Basel). 2019;11(3):E394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pahl A, Lutz C, Hechler T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov Today Technol. 2018;30:85-89. [DOI] [PubMed] [Google Scholar]
- 37.Wingren AG, Parra E, Varga M, et al. T cell activation pathways: B7, LFA-3, and ICAM-1 shape unique T cell profiles. Crit Rev Immunol. 1995;15(3-4):235-253. [DOI] [PubMed] [Google Scholar]
- 38.Gu J, Dong RP, Zhang C, McLaughlin DF, Wu MX, Schlossman SF. Functional interaction of DFF35 and DFF45 with caspase-activated DNA fragmentation nuclease DFF40. J Biol Chem. 1999;274(30):20759-20762. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen D, Stutz R, Schorr S, et al. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat Commun. 2018;9(1):3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noessner E. DGK-α: a checkpoint in cancer-mediated immuno-inhibition and target for immunotherapy. Front Cell Dev Biol. 2017;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riese MJ, Moon EK, Johnson BD, Albelda SM. Diacylglycerol kinases (DGKs): novel targets for improving T cell activity in cancer. Front Cell Dev Biol. 2016;4:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Büll C, Boltje TJ, Balneger N, et al. Sialic acid blockade suppresses tumor growth by enhancing T-cell-mediated tumor immunity. Cancer Res. 2018;78(13):3574-3588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.