

Abstract
The oral K+-sparing diuretic amiloride shows anti-cancer side-activities in multiple rodent models. These effects appear to arise, at least in part, through moderate inhibition of the urokinase-type plasminogen activator (uPA, Ki = 2.4 μM), a pro-metastatic trypsin-like serine protease that is upregulated in many aggressive solid malignancies. In applying the selective optimization of side-activity (SOSA) approach, a focused library of twenty two 6-substituted amiloride derivatives were prepared, with multiple examples displaying uPA inhibitory potencies in the nM range. X-ray co-crystal structures revealed that the potency increases relative to amiloride arise from increased occupancy of uPA’s S1β subsite by the appended 6-substituents. Leading compounds were shown to have high selectivity over related trypsin-like serine proteases and no diuretic or anti-kaliuretic effects in rats. Compound 15 showed anti-metastatic effects in a xenografted mouse model of late-stage lung metastasis.
Keywords: Urokinase plasminogen activator, uPA, Amiloride, Cancer, Metastasis, Anti-metastatic, Trypsin-like serine protease, Selective optimisation of side-activity
Amiloride 1 (Fig. 1, Scheme 1) is an oral K+-sparing diuretic with a history of safe clinical use spanning five decades.1,2 While it is well known that the drug exerts diuretic and anti-kaliuretic effects through inhibition of renal epithelial sodium channels (ENaC),3,4 amiloride has also repeatedly been shown to exhibit anti-cancer effects at high doses in a range of spontaneous and xenografted rodent tumor models (reviewed in Ref 5). The anti-cancer activity of amiloride has been attributed, at least in part, to moderate inhibition of the urokinase-type plasminogen activator (uPA, Ki = 7 μM).6
Fig. 1.
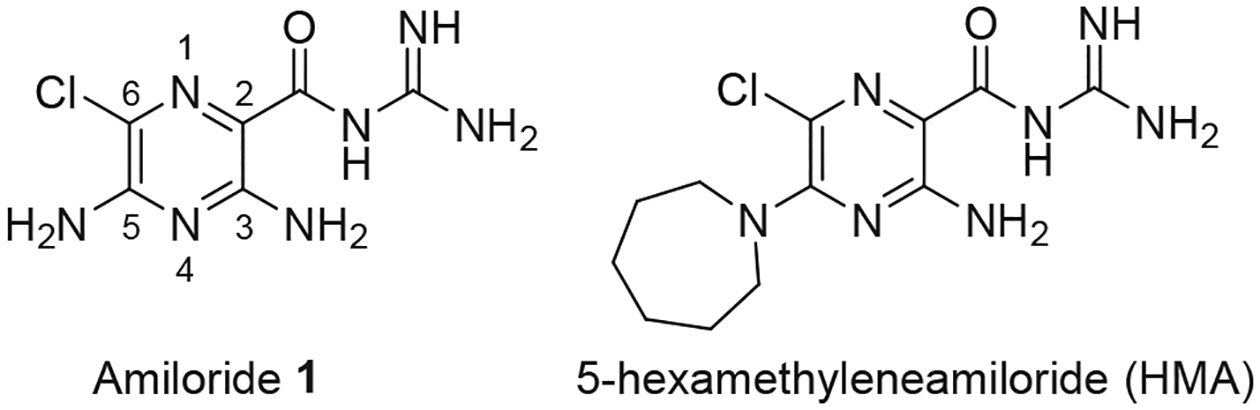
Chemical structures of amiloride 1 and 5-hexamethyleneamiloride (HMA).
Scheme 1.
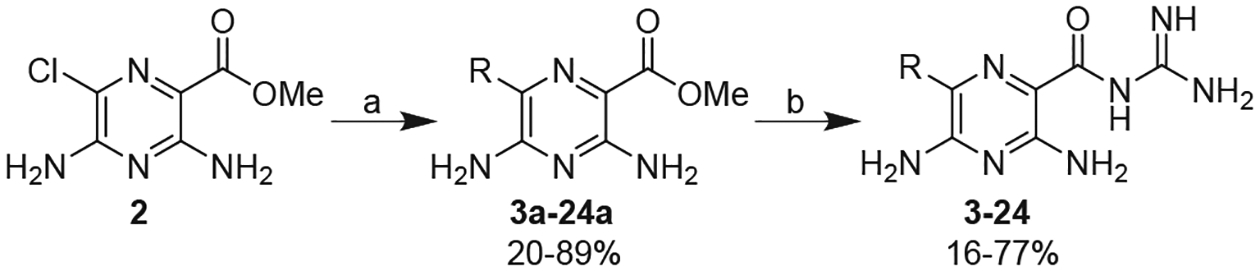
Synthesis of 6-substituted amiloride derivatives. Reagents and conditions: (a) Ar-B(OH)2, Pd(PPh3)4, K2CO3, Toluene/MeOH, reflux 0.5–48 h, (b) guanidine (2 M in MeOH), DMF, rt, 18 h or guanidine.HCl, Na(s), iPrOH, 1–4 h.
uPA is a trypsin-like serine protease (TLSP) that controls activation of the broad-spectrum TLSP plasmin. uPA and its cognate cell surface receptor uPAR are overexpressed in many aggressive malignancies, including breast, pancreatic, ovarian, head and neck, gastric and prostate cancers.7,8 In tumors that overexpress uPA/uPAR, uPA-mediated plasminogen activation turns on multiple downstream proteases (e.g. matrix metalloproteinases, cathepsins), leading to directed pericellular proteolysis and extracellular matrix remodeling.9,10 uPA upregulation is therefore a key determinant of cell invasiveness and metastasis in these tumors.11 The clinical significance of upregulated uPA is particularly evident in node-negative breast cancer, where increased tumor uPA is one of the strongest prognostic markers of progression-free and overall survival.12,13
Given its long history of clinical use, demonstrable in vivo anti-cancer effects and moderate activity against uPA, we reasoned that amiloride presents an excellent starting scaffold for a selective optimization of side activity (SOSA)14 campaign aimed at identifying derivatives with improved uPA potency and no diuretic/anti-kaliuretic effects, which could potentially be developed into an anti-metastasis drug for uPA-driven cancers. Analysis of the published X-ray co-crystal structure of amiloride bound to the uPA active site (PDB: 1F5L)15 suggested that uPA’s S1β subsite might be better filled by replacing the 6-chloro group of amiloride with bulkier (het)aryl substituents, leading to increased uPA inhibitory potency. Occupancy of S1β was also expected to confer target selectivity as this subsite is absent or reduced in size in closely related TLSPs.16
We recently explored these ideas with a series of 6-substituted derivatives of 5-hexamethyleneamiloride (HMA, Fig. 1), since like amiloride, HMA has been shown to exhibit anti-cancer effects in vivo.17 We found that appending 6-susbtituents to the HMA scaffold gave compounds with up to 110-fold higher uPA potency, excellent selectivity against serine protease off-targets, no diuretic/anti-kaliuretic effects and in vivo anti-metastatic activity.18 In the current work, we report on the activity of the parent 6-substituted amiloride series containing the 5-NH2 group.
Many previous reports have described structure-activity relationships of amiloride derivatives against a variety of biological targets,19–23 however, prior to our recent HMA work18 there have been few studies exploring derivatives carrying substituents at the 6-position.24 This is surprising given that 2-halopyrazines are well-suited to metal-catalysed cross-coupling chemistry.25 Here, we employed standard Suzuki-Miyaura reactions to couple (het)aryl-boronic acids to the commercially available 6-chloro-3,5-diaminopyrazine methyl ester 2, and converted the intermediate 6-substituted pyrazine methyl esters to acylguanidines (Scheme 1). A total of twenty two 6-(het)aryl amiloride derivatives were synthesized and screened for uPA inhibitory activity using a fluorogenic enzyme inhibition assay (Table 1).
Table 1.
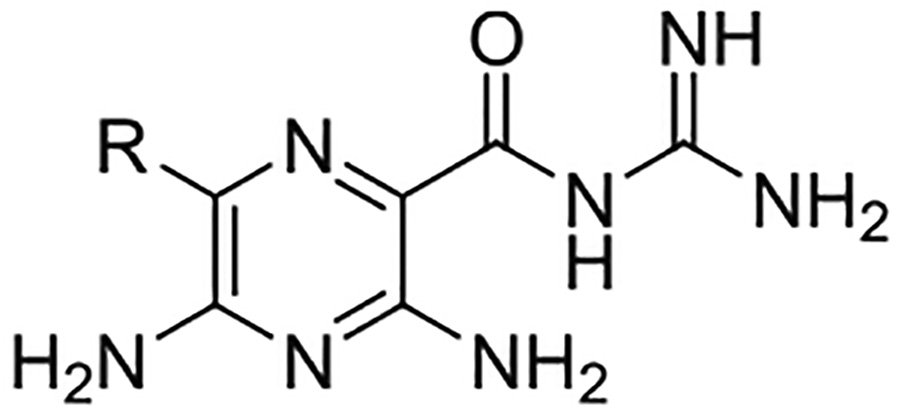
uPA inhibitory activity of amiloride 1 and 6-substituted amiloride derivatives 3-24, as determined by fluorogenic solution-phase enzyme inhibition assays.18 Values represent the mean ± SEM (n = 2 individual experiments).*Value taken from reference 18. Ki values were derived from IC50 values using the Cheng-Prusoff method.26
 | |||||
|---|---|---|---|---|---|
| Compound | R | uPA Ki (nM) | Compound | R | uPA Ki (nM) |
| 1 |  |
2,433 ± 192* | 15 | 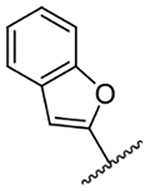 |
302 ± 4 |
| 3 | 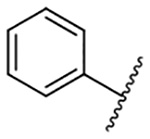 |
2,498 ± 58 | |||
| 4 | 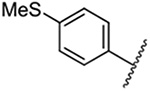 |
2,409 ± 61 | 16 | 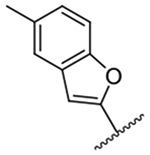 |
642 ± 88 |
| 5 | 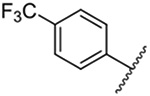 |
3,136 ± 30 | 17 | 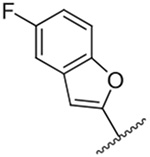 |
587 ± 44 |
| 6 | 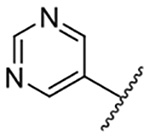 |
771 ± 22 | 18 | 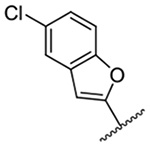 |
578 ± 36 |
| 7 | 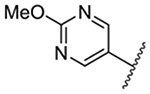 |
204 ± 14 | |||
| 8 | 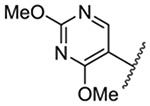 |
1,626 ± 12 | 19 | 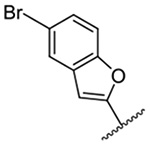 |
3,645 ± 254 |
| 9 | 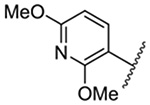 |
1,208 ± 20 | 20 | 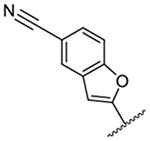 |
904 ± 93 |
| 10 | 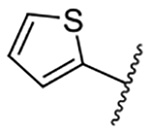 |
1,259 ± 14 | 21 | 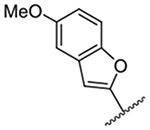 |
287 ± 14 |
| 11 | 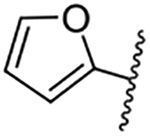 |
776 ± 101 | |||
| 12 | 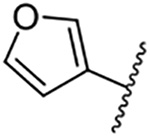 |
499 ± 19 | 22 | 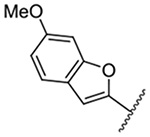 |
439 ± 14 |
| 13 | 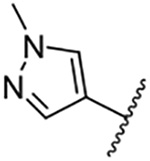 |
482 ± 4 | 23 | 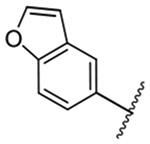 |
350 ± 61 |
| 14 | 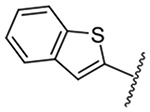 |
1,414 ± 79 | 24 | 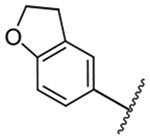 |
1,477 ± 38 |
Replacement of the 6-chloro group with phenyl 3 or 4-substituted phenyl substituents (4 and 5) did not improve potency relative to amiloride. Substitution with a 5-pyrimidine group 6 provided a ~3-fold increase in activity. Additional substitution of the pyrimidine at the 2-position with a methoxy group 7 produced a further ~4-fold increase in activity, affording the most potent derivative of the series (Ki = 204 nM). Addition of a second methoxy group 8 gave only a modest increase in activity relative to amiloride. Removal of a ring N to give the 2,3-dimethoxypyridyl derivative 9 provided a 2-fold increase. 5-Membered heterocycles were generally well tolerated with 2-thiophenyl 10 and 2-furanyl 11 showing 2-fold and 3.3-fold increases, respectively. Sub-500 nM potency was achieved with the 3-furanyl and N-methyl-1H-pyrazolyl derivatives 12 and 13. 2-Thiophenyl derivative 14 gave a modest 1.7-fold increase, whereas 2-benzofuranyl 15 showed 8-fold higher activity (15 Ki = 302 nM). The potency increases achieved with this series generally tracked with the trends seen with the corresponding 5-hexamethyleneamiloride derivatives we recently reported.18 An exception was seen with the 2,4-dimethoxy analog 8, which failed to recapitulate the > 55-fold potency increase seen with its corresponding HMA analog.18
The improved potency of 15 led to exploration of a small series of 5 and 6-substituted 2-benzofuranyl derivatives 16-22 to understand the effects of substituents around the benzofuran. However, none of the substituted benzofurans showed improved potency relative to 15. 5-Br substitution 19 was particularly unfavorable, causing a 1.5-fold decrease in activity relative to amiloride (~12-fold loss relative to 15). Altering the connectivity of the benzofuran to the pyrazine (i.e. 5-benzofuranyl 23) caused only a small decrease in potency relative to 15 and a slight increase was observed with the 2,3-dihydro-5-benzofuranyl 24 relative to amiloride.
The binding pose of the pyrazine acylguanidine unit in the S1 pocket of uPA was not significantly altered from that of amiloride in most cases by addition of the 6-substituents, as evidenced by X-ray co-crystal structures of 7, 13 and 15 bound to uPA (Figs. 2,3 and Fig. S1). An exception was observed with the dimethoxypyrimidine 8, where subtle shifting of the pyrazine core out of the S1 pocket lengthened the key salt bridge interaction between the guanidine and Asp189 relative to other derivatives (and 1), thus contributing to its reduced potency. Another common feature observed with 13 and 15 (and to a lesser extent 7) was an almost coplanar positioning of the pyrazine core with the 6-(het)aryl substituents, which appeared to limit interactions with the S1β subsite. This orientation differed from the corresponding HMA analogs, where larger dihedral angles about the pyrazine bond axis were seen, which appeared to confer better occupancy of S1β by the respective 6-(het)aryl groups.18 An exception to this was observed with compound 8, whose dimethoxypyrimidine had rotated 72° to project the 2-methoxy group away from S1β and towards bulk solvent. This was in contrast to its matched HMA analog, where a similar degree of rotation in the opposite direction allowed partial occupancy of S1β by the corresponding 2-methoxy group, leading to a considerable boost in potency (6-(2,4-dimethoxypyrimidin-5-yl)-5-(N,N-hexamethylene) amiloride Ki = 42 nM).18
Fig. 2.
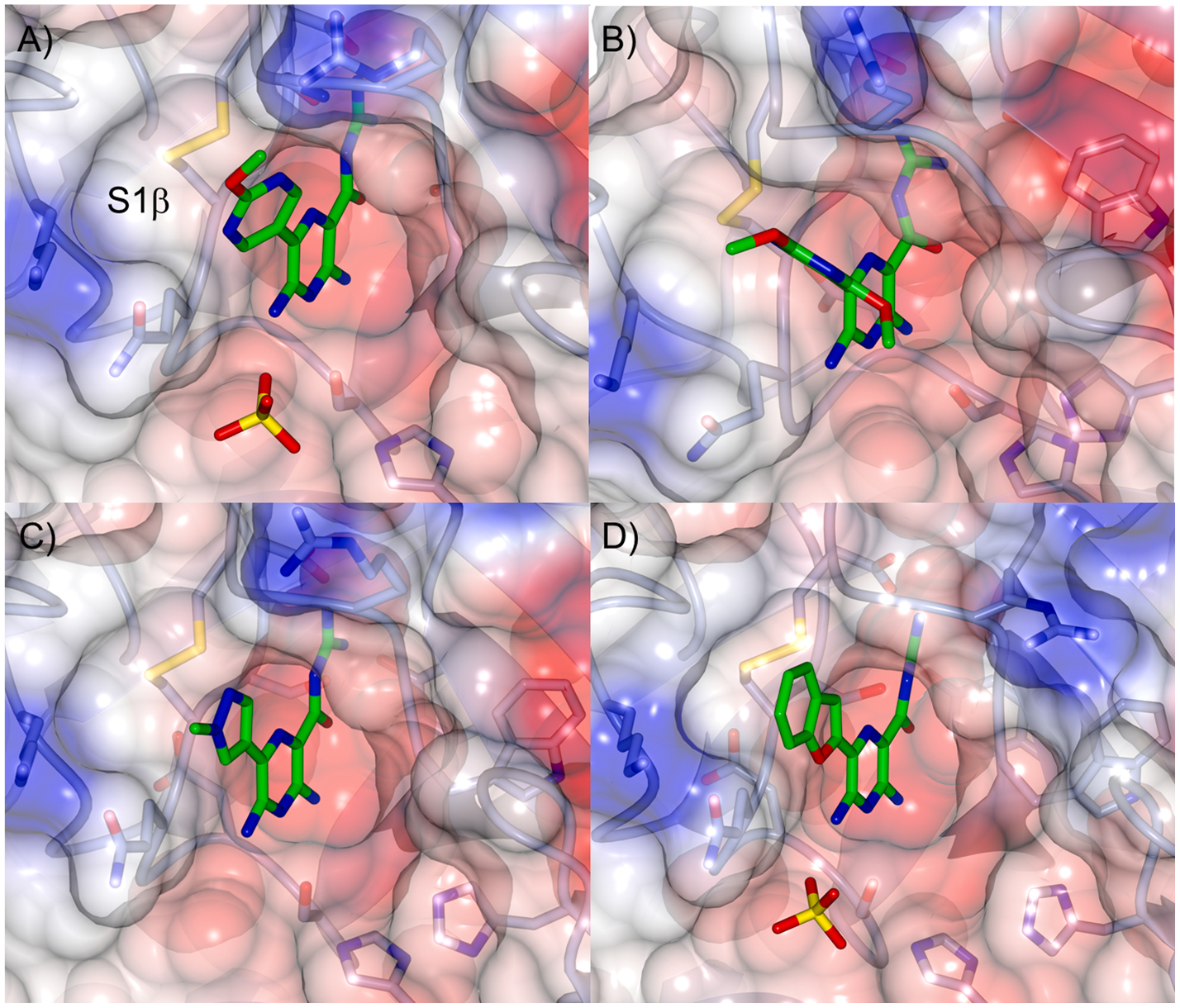
X-ray co-crystal structures of A) 7 (PDB 6AG2; 1.8 Å), B) 8 (PDB 6AG3; 2.5 Å), C) 13 (PDB 6AG7; 1.9 Å) and D) 15 (PDB 6AG9; 1.6 Å) bound to the active site of uPA. A sulfate of crystallization is shown, where detected. X-ray statistics are provided in the Supplementary Material.
Fig. 3.
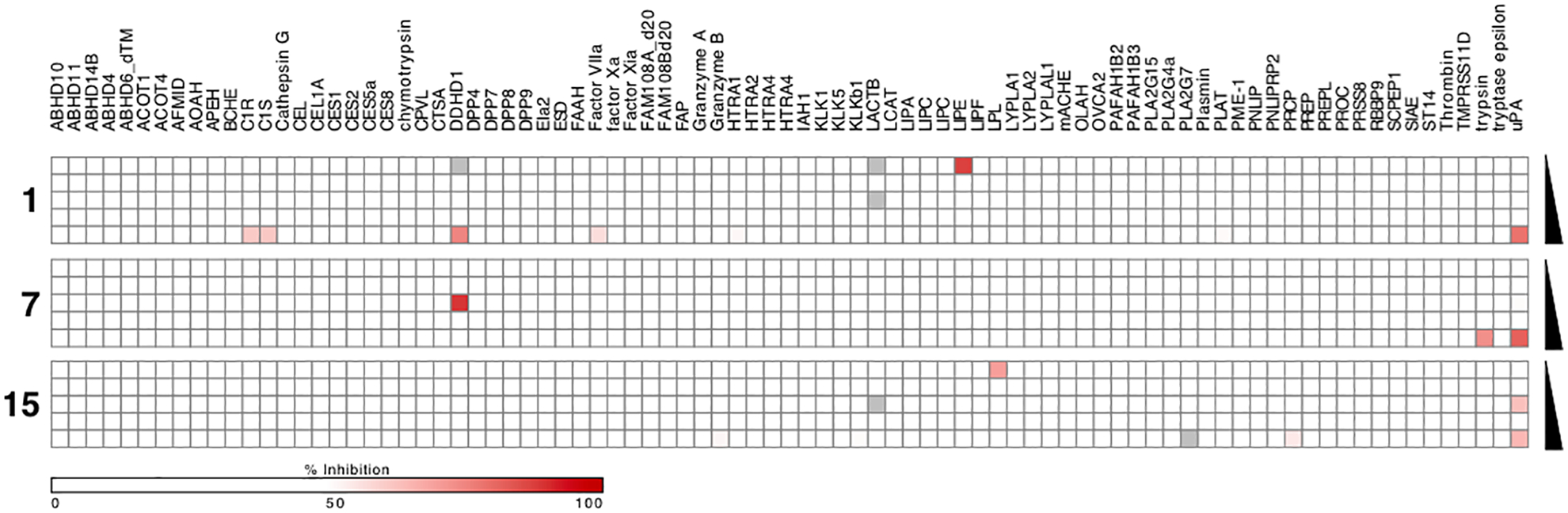
EnPlex assay showing comparative inhibition of 85 serine hydrolases by amiloride 1 and 6-substituted derivatives 7 and 15.27 Grey boxes indicate wells in which no fluorescence was detected. Wells where signal was detected at lower compound concentration without signal at higher concentrations are experimental artifacts. Black triangles indicate increasing compound concentrations in the range 5 nM-33 μM.
The gains in uPA potency (relative to amiloride) achieved with the 6-substituted amiloride series herein were relatively modest compared to the corresponding 6-substituted-5-azepane (HMA) series, where increases exceeding 110-fold were observed.18 The best inhibitor in the current series, 4-methoxypyrimidine 7, showed only a 12-fold improvement relative to amiloride. This trend can be explained in part by the decreased interactions seen between the 6-substitutents and the S1β subsite arising from their coplanarity with the pyrazine core. Additional van Der Waals interactions contributed by the azepane ring interacting with the S4 pocket of uPA presumably also play a role.
To explore uPA selectivity within the series, compound 15 was screened against a panel of closely related TLSPs using chromogenic enzyme inhibition assays (Table 2). A high degree of uPA selectivity was observed (> 40–100-fold), with IC50 not reached for any of the TLSPs tested. A similar degree of selectivity was observed with amiloride, as previously noted.18
Table 2.
Comparative uPA selectivity of amiloride 1 and 15 versus a panel of related trypsin-like serine proteases. IC50 values were determined using chromogenic solution-phase enzyme inhibition assays.18
| Enzyme | 1IC50 (μM) | 15IC50 (μM) |
|---|---|---|
| uPA | 2.4 | 0.43 |
| tPA | > 20 | > 20 |
| Plasmin | > 20 | > 20 |
| Thrombin | > 20 | > 20 |
| Trypsin | > 50 | > 50 |
| Plasma Kallikrein | > 50 | > 50 |
| FXa | > 50 | > 50 |
| FXla | > 50 | > 50 |
| Activated Protein C | > 50 | > 50 |
To further profile target selectivity, amiloride 1 and derivatives 7 and 15 were screened against 85 serine hydrolases using a high-throughput EnPlex assay (Fig. 3).27 Amiloride 1 showed slight inhibition of just a few enzymes (i.e. C1R, C1S, DDHD1, Factor VIIa and LACTB), consistent with previous reports6 and solution phase enzyme assay data (Table 2). High uPA selectivity was similarly observed for both 7 and 15, with trypsin and prolylcarboxypeptidase (PRCP) being the only enzymes showing slight inhibition across the serine hydrolase superfamily.
Species selectivity between human and mouse uPA (MuPA) is a known issue for the development of reversible, active-site inhibitors of human uPA (HuPA). This issue is particularly apparent for inhibitors that target the S1β subsite, where the improved potencies typically seen against the human enzyme are not recapitulated with MuPA.28 This trend was observed for the 2-benzofuranyl derivative 15, which showed 33-fold lower activity against the murine variant (MuPA IC50 = 14.2 μM vs HuPA IC50 = 0.43 μM). In contrast, amiloride with its smaller 6-chloro substituent showed equivalent potency against MuPA (IC50 = 2.3 μM) and HuPA (IC50 = 2.4 μM). Decreased potency against MuPA poses a potential problem for interpretation of results from human-mouse xenograft models of cancer, where the contribution of host MuPA to tumor growth and metastasis is unclear.29,30 Results from these models may therefore underestimate the anti-metastatic effects of candidate inhibitors in relation to their therapeutic potential in humans.31
Compounds 7 and 15 were screened for cytotoxicity alongside amiloride 1 against a panel of mammalian cell lines (Table 3). Amiloride did not inhibit the growth of any of the cells at concentrations up to 100 μM. The 2-benzofuranyl derivative 15 showed significant cytotoxicity across all cell lines but 7 was much less toxic. These effects align with the increased hydrophobicity of 15 (gLogD7.4 = 2.8) relative to 7 (gLogD7.4 = 0.8) and amiloride 1 (gLogD7.4 = 0.3) and mirror those seen with the related 6-HMA analogs.18
Table 3.
Cytotoxicity of 1, 7and 15 against a panel of human cell lines. IC50 values were determined using the MTS assay.18 Inhibitors were tested in quadruplicate at each concentration. Values represent the mean from 2 or more independent experiments.
| Compound | 1IC50 (μM) | |||
|---|---|---|---|---|
| MDA-MB-231 | HepG2 | HEK-293 | HT-1080 | |
| 1 | > 100 | > 100 | > 100 | > 100 |
| 7 | > 100 | > 100 | 58.5 | > 100 |
| 15 | 2.8 | 30.8 | 3.6 | 3.0 |
In our recent work,18 we demonstrated that 6-substituted HMA derivatives are poor inhibitors of epithelial sodium channels (ENaCs) in vitro and show no diuretic/anti-kaliuretic effects in a rat model, supporting the known trends for 5-alkyl substituted amilorides.19 Analogous experiments were performed here to assess whether mono-substitution at the 6-position also reduces amiloride-like activity.
Complete loss of ENaC activity was seen with compounds 7 and 15 at 10 μM (Fig. 4), revealing the high sensitivity of ENaCs towards substitution at amiloride’s 6-position. This trend aligns with a previous finding, where introduction of the larger 6-iodo group also reduced ENaC activity.32 To confirm that diminished ENaC activity corresponds to an absence of diuretic and anti-kaliuretic effects in vivo, derivatives 7 and 15 were examined alongside amiloride in an acetazolamide-induced rat model of diuresis/kaliuresis (Fig. 5).
Fig. 4.
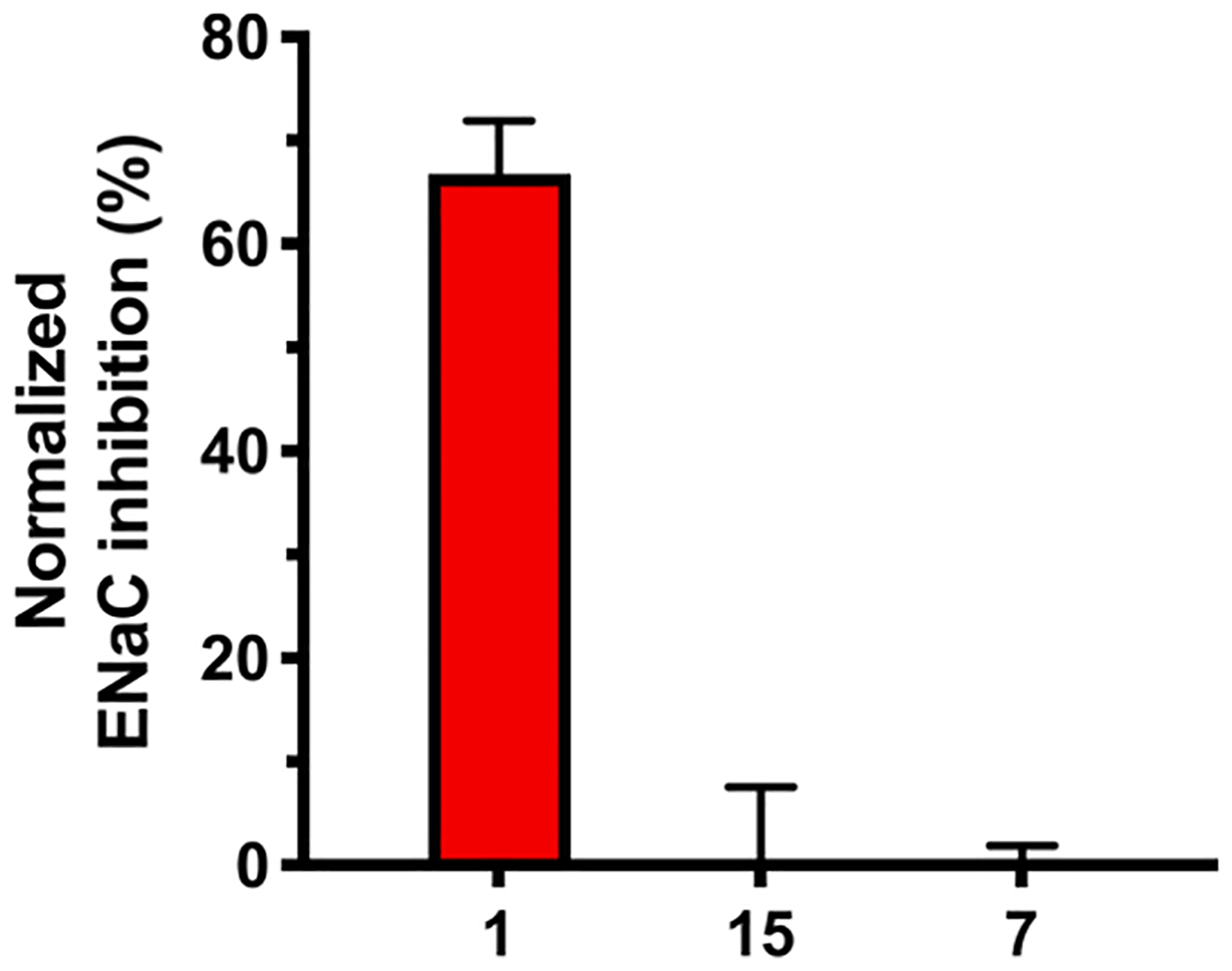
Inhibition of ENaCs by amiloride 1 and derivatives 7 and 15. All compounds were present at 10 μM. Data represent the mean ± SD (n = 4). Methods were as described in reference 18.
Fig. 5.
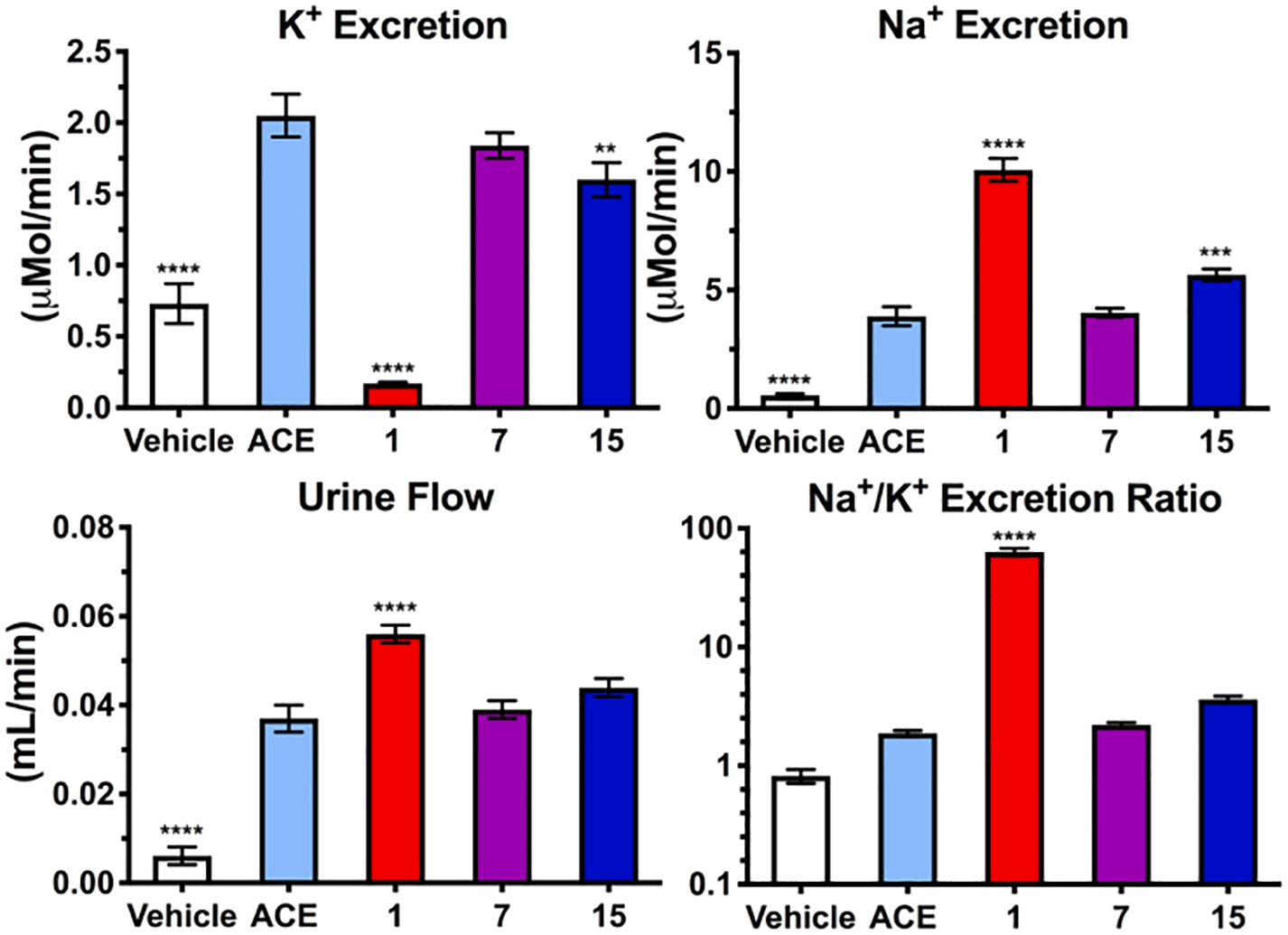
Effect of amiloride 1 and derivatives 7 and 15 on diuresis, naturesis and kaliuresis in Sprague Dawley rats. Rats were dosed with 25 mg/kg acetazolamide IV immediately prior to administration of 1.5 mg/kg of 1, 7 or 15 IP. Data represent the mean ± SEM (n = 8). **p = 0.001, ***p = 0.0005, ****p = 0.0001 relative to the acetazolamide control group (ACE). Methods were as described in reference 18.
As expected, amiloride 1 produced significant increases in urine volume and sodium excretion (34% and 159%, respectively) and decreased potassium excretion by 92% relative to acetazolamide-treated controls. In contrast, 2-benzofuranyl derivative 15 did not significantly increase diuresis and showed minimal effects on solute concentrations. Compound 7 showed no significant effect on diuresis or Na+/K+ excretion.
To explore the anti-metastatic effects of 6-substituted amilorides, compound 15 was evaluated alongside amiloride in an experimental late-stage lung metastasis model in mice (Fig. 6). Luciferase-tagged HT-1080 human fibrosarcoma cells were chosen for the model due to their high endogenous expression of uPA and uPAR.18 Inhibition of lung metastasis by amiloride 1 was not statistically significant, although a trend towards reduction appeared to be present. Compound 15 produced a statistically significant effect, decreasing metastatic lung burden by 32% (p = 0.0036).
Fig. 6.
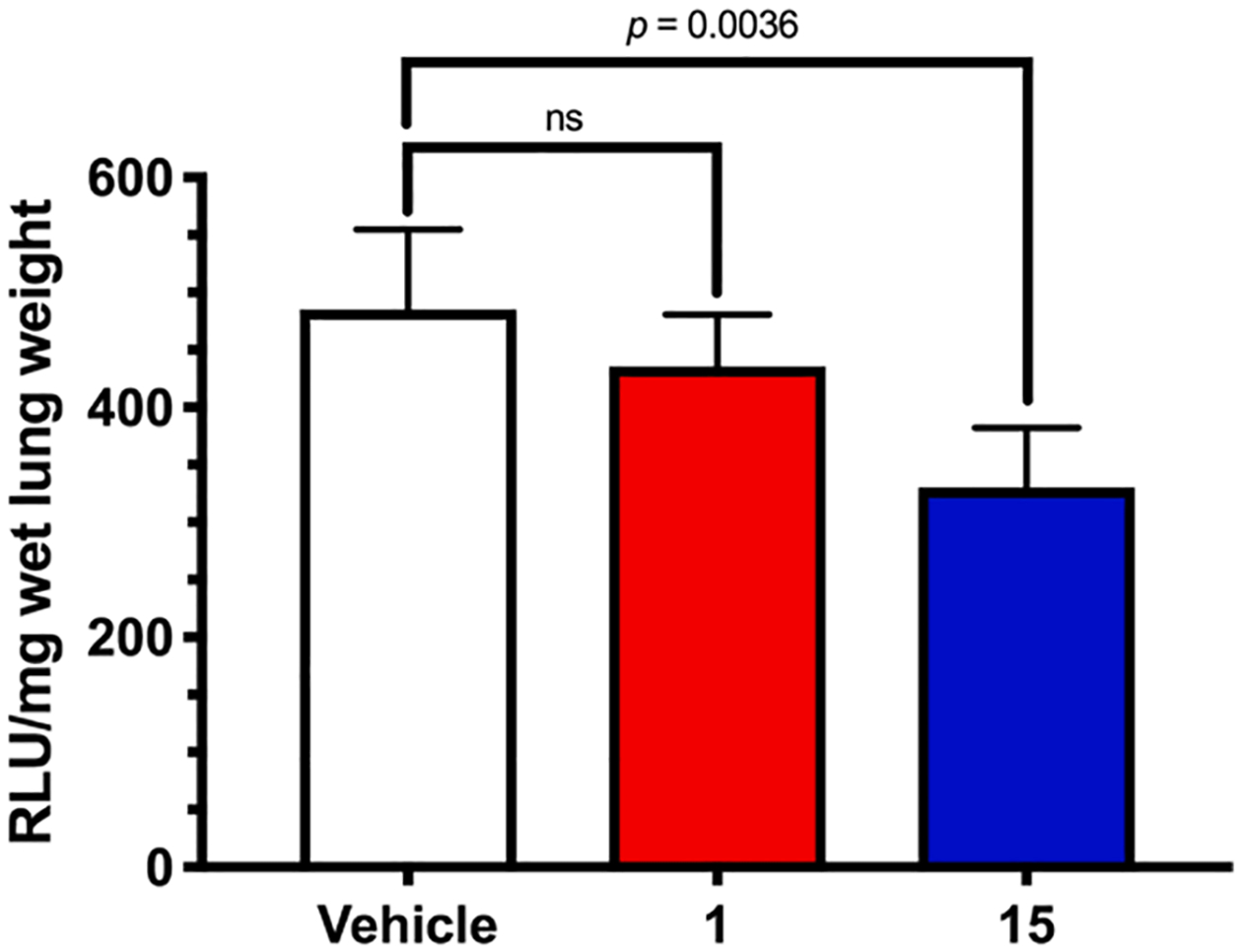
Effects of 1 and 15 on experimental lung metastasis formed in mice following tail vein injection of HT-1080RedFluc cells. Compounds were administered at 7.5 mg/kg/day IP for 21 days. Lung metastases were quantified using an endpoint luciferase activity assay of whole lung homogenates. RLU = relative luminescence units. Data represent the mean ± SEM (vehicle and 1, n = 4; 15, n = 6), ns = not significant. Methods as described in reference 18.
In summary, substitution of amiloride at the 6-position with (het) aryl groups was achieved using standard Suzuki-Miyaura cross-coupling chemistry followed by guanidinylation of the intermediate methyl esters. Screening of the focused library identified derivatives with improved activity (relative to amiloride) against uPA and excellent selectivity over related TLSP off-targets. Lead compounds showed an absence of diuretic and anti-kaliuretic effects and the 2-benzofuranyl derivative 15 significantly decreased lung metastasis in a commonly used mouse metastasis model. This work validates the SOSA approach for transforming amiloride into more potent uPA inhibitors and supports further efforts to develop the class into a novel anti-metastatic drug for non-cytotoxic therapy of uPA-driven cancer progression.
Supplementary Material
Acknowledgment
The authors thank Dr. Kara Vine-Perrow for assistance with the lung metastasis model.
Funding sources
This work was funded by a National Health and Medical Research Council (NHMRC, Australia) Project Grant (APP1100432) awarded to M. J. K., M.R. and M.H. and Health Research Council (HRC, New Zealand) Grant (16/361) to G.M.C. and M.J.K. National Key R&D Program of China (2017YFE0103200) and National Natural Science Foundation of Fujian province (2018J01729, China) funding to M.H. and L.J. is also acknowledged. D.C.J. acknowledges funding from NIH training grant T32 GM115327-Tan. D.A.B. and D.C.J. acknowledge funding from a MSKCC Core Grant P30 (CA008748, USA).
Abbreviations:
- C1r
complement C1r subcomponent
- C1s
Complement component 1s
- DDHD1
Phosphatidic acid-preferring phospholipase A1
- ENaC
Epithelial sodium channel
- HMA
5-hexamethyleneamiloride
- HuPA
Human urokinase plasminogen activator
- LACTB
Serine beta-lactamase-like protein
- MuPA
Mouse urokinase plasminogen activator
- PRCP
prolylcarboxypeptidase
- SOSA
Selective optimization of side-activity
- SAR
Structure-activity relationship
- TLSP
Trypsin-like serine protease
- uPA
Urokinase plasminogen activator
- uPAR
Urokinase plasminogen activator receptor
Footnotes
Appendix A. Supplementary data
X-ray data and refinement statistics, synthetic methods and compound characterization data are provided in the Supporting Information. Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2019.126753.
References
- 1.Lant AF, Smith AJ, Wilson GM. Clinical evaluation of amiloride, a potassium-sparing diuretic. Clin Pharmacol Ther. 1969;10(1):50–63. [DOI] [PubMed] [Google Scholar]
- 2.Cragoe EJJ, Woltersdorf OWJ, Bicking JB, Kwong SF, Jones JH. Pyrazine diuretics. II. N-amidino-3-amino-5-substituted 6-halopyrazinecarboxamides. J Med Chem. 1967;10(1):66–75. [DOI] [PubMed] [Google Scholar]
- 3.Kleyman TR, Cragoe EJJ. The mechanism of action of amiloride. Sem Nephrol. 1988;8(3):242–248. [PubMed] [Google Scholar]
- 4.Palmer BF. Regulation of Potassium Homeostasis. Clin J Am Soc Nephrol. 2015;10(6):1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthews H, Ranson M, Kelso MJ. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: an orally active anti-cancer drug waiting for its call-of-duty? Int J Cancer. 2011;129(9):2051–2061. [DOI] [PubMed] [Google Scholar]
- 6.Vassalli JD, Belin D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett. 1987;214(1):187–191. [DOI] [PubMed] [Google Scholar]
- 7.Ulisse S, Baldini E, Sorrenti S, D’Armiento M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets. 2009;9(1):32–71. [DOI] [PubMed] [Google Scholar]
- 8.Brungs D, Chen J, Aghmesheh M, et al. The urokinase plasminogen activation system in gastroesophageal cancer: a systematic review and meta-analysis. Oncotarget. 2017;8(14):23099–23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Can. 1997;72(1):1–22. [DOI] [PubMed] [Google Scholar]
- 10.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6(10):764–775. [DOI] [PubMed] [Google Scholar]
- 11.Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57(1):25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foekens JA, Peters HA, Look MP, et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res. 2000;60(3):636–643. [PubMed] [Google Scholar]
- 13.Look MP, van Putten WL, Duffy MJ, et al. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst. 2002;94(2):116–128. [DOI] [PubMed] [Google Scholar]
- 14.Wermuth CG. Selective optimization of side activities: another way for drug discovery. J Med Chem. 2004;47(6):1303–1314. [DOI] [PubMed] [Google Scholar]
- 15.Zeslawska E, Schweinitz A, Karcher A, et al. Crystals of the urokinase type plasminogen activator variant beta(c)-uPAin complex with small molecule inhibitors open the way towards structure-based drug design. J Mol Biol. 2000;301(2):465–475. [DOI] [PubMed] [Google Scholar]
- 16.Nienaber VL, Davidson D, Edalji R, et al. Structure-directed discovery of potent non-peptidic inhibitors of human urokinase that access a novel binding subsite. Structure. 2000;8(5):553–563. [DOI] [PubMed] [Google Scholar]
- 17.Luo J, Tannock IF. Inhibition of the regulation of intracellular pH: potential of 5-(N, N-hexamethylene) amiloride in tumour-selective therapy. Br J Cancer. 1994;70(4):617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckley BJ, Aboelela A, Minaei E, et al. 6-Substituted hexamethylene amiloride (HMA) derivatives as potent and selective inhibitors of the human urokinase plasminogen activator for use in cancer. J Med Chem. 2018;61:8299–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleyman TR, Cragoe EJ Jr. Amiloride and its analogs as tools in the study of ion transport. J Membr Biol. 1988;105(1):1–21. [DOI] [PubMed] [Google Scholar]
- 20.Kuduk SD, Di Marco CN, Chang RK, et al. Amiloride derived inhibitors of acid-sensing ion channel-3 (ASIC3). Bioorg Med Chem Lett. 2009;19(9):2514–2518. [DOI] [PubMed] [Google Scholar]
- 21.Ewart GD, Mills K, Cox GB, Gage PW. Amiloride derivatives block ion channel activity and enhancement of virus-like particle budding caused by HIV-1 protein Vpu. Eur Biophys J. 2002;31(1):26–35. [DOI] [PubMed] [Google Scholar]
- 22.Massink A, Louvel J, Adlere I, et al. 5’-Substituted amiloride derivatives as allosteric modulators binding in the sodium ion pocket of the adenosine A2A Receptor. J Med Chem. 2016;59(10):4769–4777. [DOI] [PubMed] [Google Scholar]
- 23.Snell HD, Gonzales EB. 5-(N, N-Hexamethylene) amiloride is a GABA-A ρ1 receptor positive allosteric modulator. Channels. 2016;10(6):498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patwardhan NN, Ganser LR, Kapral GJ, et al. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. MedChemComm. 2017;8(5):1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikishkin NI, Huskens J, Verboom W. Transition metal-catalyzed functionalization of pyrazines. Org Biomol Chem. 2013;11(22):3583–3602. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. [DOI] [PubMed] [Google Scholar]
- 27.Bachovchin DA, Koblan LW, Wu W, et al. A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nat Chem Biol. 2014;10(8):656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klinghofer V, Stewart K, McGonigal T, et al. Species specificity of amidine-based urokinase inhibitors. Biochemistry. 2001;40(31):9125–9131. [DOI] [PubMed] [Google Scholar]
- 29.Ploug M, Ostergaard S, Gardsvoll H, et al. Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry. 2001;40(40):12157–12168. [DOI] [PubMed] [Google Scholar]
- 30.Schweinitz A, Steinmetzer T, Banke IJ, et al. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J Biol Chem. 2004;279(32):33613–33622. [DOI] [PubMed] [Google Scholar]
- 31.Wang D, Yang Y, Jiang L, et al. Suppression of tumor growth and metastases by targeted intervention in urokinase activity with cyclic peptides. J Med Chem. 2019;62(4):2172–2183. [DOI] [PubMed] [Google Scholar]
- 32.Cuthbert AW, Fanelli GM. Effects of some pyrazinecarboxamides on sodium transport in frog skin. Br J Pharmacol. 1978;63(1):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.