

There is a Blood Commentary on this article in this issue.
Abstract
The 2016 revision of the World Health Organization classification of tumors of hematopoietic and lymphoid tissues is characterized by a closer integration of morphology and molecular genetics. Notwithstanding, the myelodysplastic syndrome (MDS) with isolated del(5q) remains so far the only MDS subtype defined by a genetic abnormality. Approximately half of MDS patients carry somatic mutations in spliceosome genes, with SF3B1 being the most commonly mutated one. SF3B1 mutation identifies a condition characterized by ring sideroblasts (RS), ineffective erythropoiesis, and indolent clinical course. A large body of evidence supports recognition of SF3B1-mutant MDS as a distinct nosologic entity. To further validate this notion, we interrogated the data set of the International Working Group for the Prognosis of MDS (IWG-PM). Based on the findings of our analyses, we propose the following diagnostic criteria for SF3B1-mutant MDS: (1) cytopenia as defined by standard hematologic values, (2) somatic SF3B1 mutation, (3) morphologic dysplasia (with or without RS), and (4) bone marrow blasts <5% and peripheral blood blasts <1%. Selected concomitant genetic lesions represent exclusion criteria for the proposed entity. In patients with clonal cytopenia of undetermined significance, SF3B1 mutation is almost invariably associated with subsequent development of overt MDS with RS, suggesting that this genetic lesion might provide presumptive evidence of MDS in the setting of persistent unexplained cytopenia. Diagnosis of SF3B1-mutant MDS has considerable clinical implications in terms of risk stratification and therapeutic decision making. In fact, this condition has a relatively good prognosis and may respond to luspatercept with abolishment of the transfusion requirement.
Introduction
The World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues was revised in 2016.1,2 While several novel molecular findings with diagnostic and/or prognostic importance have been incorporated into this revision, a closer integration of morphology and molecular genetics is still needed for many hematologic malignancies.
According to the WHO classification of myeloid neoplasms, myelodysplastic syndromes (MDSs) are a group of clonal disorders characterized by morphologic dysplasia in hematopoietic cells, ineffective hematopoiesis, and peripheral cytopenias.3 In the last few years, the ascertainment of clonal nature has become feasible in clinical practice with the use of massive parallel sequencing for identification of somatic gene mutations.4 Mutated driver genes include those of RNA splicing, DNA methylation, histone modification, transcription regulation, DNA repair, signal transduction, and cohesin complex.5,6
Defining the genetic basis is clinically relevant not only in the diagnostic approach to MDS but also in the prognostication and therapeutic decision making.4 This paradigm is represented by the MDS with isolated del(5q), the only MDS subtype currently defined by a genetic abnormality.3 Del(5q) is a disease-defining genetic lesion, as haploinsufficiency of several genes mapping on the deleted chromosomal region, including CSNK1A1 and RPS14, explains the molecular pathophysiology of the disease.7,8 It also predicts response to lenalidomide, which induces the ubiquitination and degradation of CSNK1A1, abolishing the selective advantage of hematopoietic cells carrying del(5q).9,10
Approximately half of MDS patients carry somatic mutations in spliceosome genes, and of these, SF3B1 is the most commonly mutated one. The SF3B1 gene encodes the splicing factor 3b subunit 1 and is typically mutated in MDS with ring sideroblasts (RS).11,12 The revised WHO classification specifically accounts for this genetic lesion, and a diagnosis of MDS-RS can now be made if RS comprise as few as 5% of nucleated red cells and a somatic mutation of SF3B1 is present.3 Several lines of evidence support recognition of somatic SF3B1 mutation as a disease-defining genetic lesion. In fact, it (1) most often represents a founding genetic lesion, (2) is a major determinant of disease phenotype, (3) has an independent prognostic value on survival and risk of progression to acute myeloid leukemia (AML), and (4) may predict response to specific agents.13-17
In this report, we analyzed the available evidence supporting the recognition of SF3B1-mutant MDS as a distinct nosologic entity. To validate our proposal, we interrogated the data set of the International Working Group for the Prognosis of MDS (IWG-PM), including 3479 patients with known SF3B1 mutation status from 18 centers or networks.
Current principles of MDS classification
According to the WHO classification, MDSs are currently categorized according to the number of cytopenias at presentation, the number of lineages manifesting dysplasia, and the percentage of RS and blasts in the bone marrow and peripheral blood (Table 1).3 While only 1 genetic abnormality, del(5q), is used to define a specific MDS subtype [ie, MDS with isolated del(5q)], selected cytogenetic abnormalities are recognized as “MDS defining” in a cytopenic patient, as they provide presumptive evidence of MDS even in the absence of definitive morphologic features.
Table 1.
Diagnostic criteria for MDS entities
| Name | Dysplastic lineages | Cytopenias | RS (%)* | BM and PB blasts (%) | Cytogenetics† |
|---|---|---|---|---|---|
| MDS-SLD | 1 | 1 or 2 | <15/<5‡ | BM <5, PB <1, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS-MLD | 2 or 3 | 1-3 | <15/<5‡ | BM <5, PB <1, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS-RS | |||||
| MDS-RS-SLD | 1 | 1 or 2 | ≥15/≥5‡ | BM < 5, PB <1, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS-RS-MLD | 2 or 3 | 1-3 | ≥15/≥5‡ | BM <5, PB <1, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with isolated del(5q) | 1-3 | 1-2 | None or any | BM <5, PB <1, no Auer rods | Del(5q) alone or with 1 additional abnormality except −7 or del(7q) |
| MDS-EB | |||||
| MDS-EB-1 | 0-3 | 1-3 | None or any | BM 5-9 or PB 2-4, no Auer rods | Any |
| MDS-EB-2 | 0-3 | 1-3 | None or any | BM 10-19 or PB 5-19 or Auer rods | Any |
| MDS-U | |||||
| 1% blood blasts | 1-3 | 1-3 | None or any | BM <5, PB = 1,§ no Auer rods | Any |
| SLD and pancytopenia | 1 | 3 | None or any | BM <5, PB <1, no Auer rods | Any |
| Defining cytogenetic abnormality | 0 | 1-3 | <15|| | BM <5, PB <1, no Auer rods | MDS-defining abnormality |
| Refractory cytopenia of childhood | 1-3 | 1-3 | None | BM <5, PB <2 | Any |
BM, bone marrow; MDS-EB, MDS with excess blasts; MDS-U, MDS, unclassifiable; PB, peripheral blood.
RS as percentage of marrow erythroid elements.
Cytogenetics by conventional karyotype analysis.
If SF3B1 mutation is present.
One percent PB blasts must be recorded on ≥2 separate occasions.
Cases with ≥15% RS by definition have significant erythroid dysplasia and are classified as MDS-RS-SLD.
MDS-RS is subdivided into a condition with single (erythroid)–lineage dysplasia (MDS-RS-SLD) and a condition with multilineage dysplasia (MDS-RS-MLD).3,18
SF3B1 mutation is critical to the pathophysiology of myelodysplasia and RS
SF3B1 mutation is an initiating genetic lesion in MDS
Several lines of evidence are consistent with the notion that SF3B1 mutation may be an initiating genetic event and that primitive lympho-myeloid hematopoietic stem cells represent the propagating cells in SF3B1-mutant MDS.6,11-13,15,19,20
Previous reports showed that SF3B1 mutations are typically heterozygous and the overall median variant allele frequency (VAF) is ∼40%.6,11-13 These data have been confirmed by the analysis of VAF reported in the IWG data set, which showed median values for the observed variants ranging from 35% to 43%.
Computational prediction in MDS-RS patients with ≥1 recurrent driver mutations based on targeted sequencing data, coupled with mutational analysis of the SF3B1 gene in hematopoietic stem/progenitor cells, demonstrated that the SF3B1 mutation may occur alone or as the first event in most cases, whereas it appears to be secondary to other oncogenic mutations in a minority of cases,15,19,20 In these latter subjects, most frequently SF3B1 mutations are occurring on the background of TET2-, DNMT3A-, or ASXL1-mutated age-related hematopoietic clones (Figure 1).14,15
Figure 1.
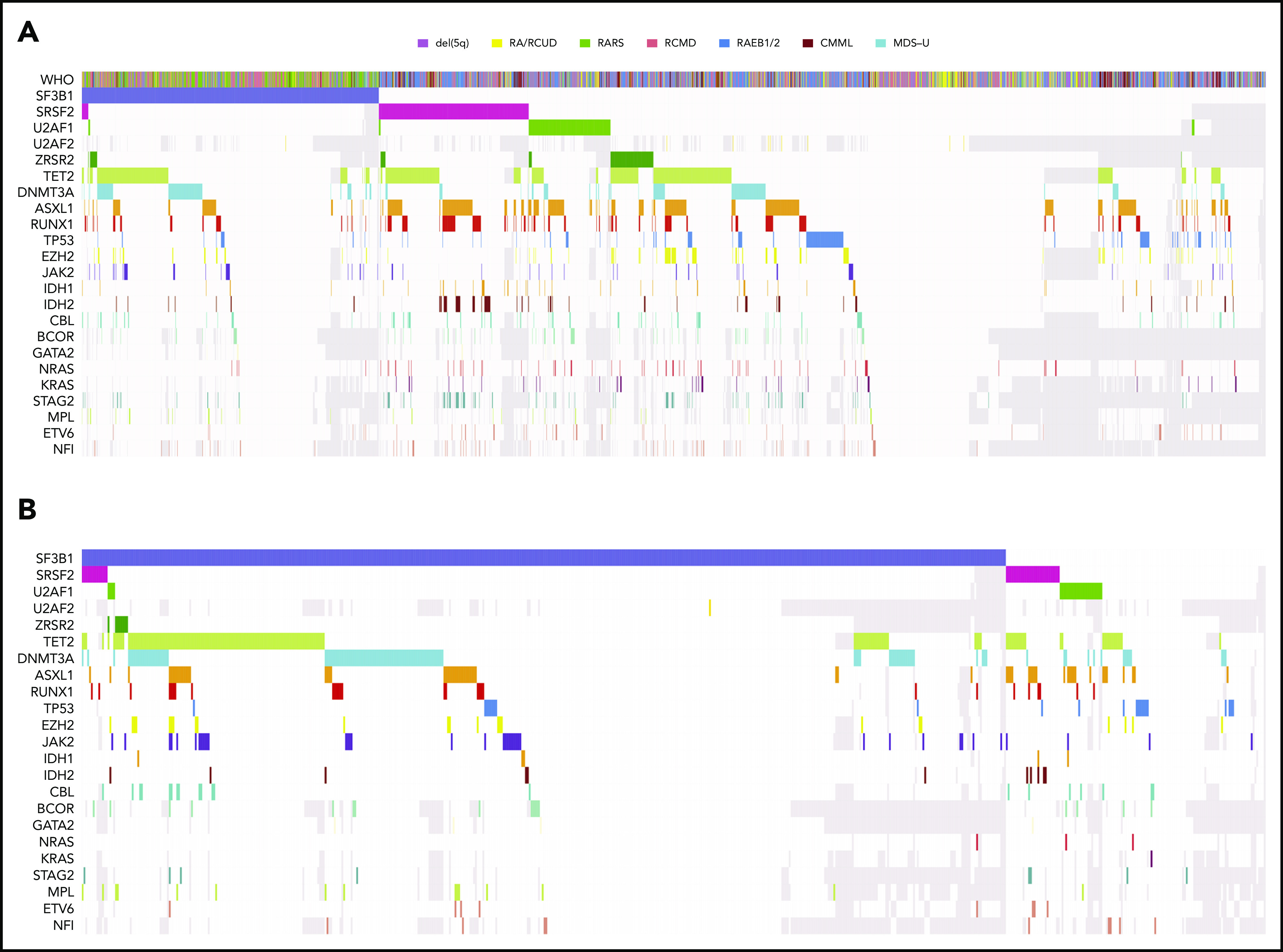
Patterns of the mutations observed in MDS patients reported to the data set of the International Working Group for MDS. (A) Distribution of somatic lesions in the analyzed genes according to the WHO category. Each column represents an individual patient sample. (B) Distribution of somatic lesions in the analyzed genes in patients with MDS-RS with or without SF3B1 mutation. CMML, chronic myelomonocytic leukemia; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RCUD, refractory cytopenia with unilineage dysplasia; unk, unknown.
Phenotypic and functional evidence also indicated that the most primitive lymphomyeloid hematopoietic stem cells (Lin−CD34+CD38−CD90+CD45RA−) represent the origin of the mutated SF3B1 clone in MDS-RS as well as the rare MDS propagating cells.15,20 Mutations identified in the hematopoietic stem cell compartment were also present in downstream myeloid and erythroid progenitor cells.15
Relationships among SF3B1 mutation, aberrant messenger RNA (mRNA) splicing, and RS
The strong association between SF3B1 mutation and myelodysplasia with RS was evident since the first reports.11,12 A subsequent study provided evidence that, when accounting for cases assigned to nonsideroblastic WHO categories, SF3B1 mutation had a positive predictive value of 98% for disease phenotype with RS.13 These data are consistent with a causal relationship between SF3B1 mutation and bone marrow RS.
Following these genotype-phenotype correlation analyses, investigations were then performed to explore the abnormal biologic pathways and networks downstream of the mutation. Studies on cell lines and primary human cells showed that the mutant SF3B1 protein retains altered function, resulting in deregulated expression and splicing of key genes and pathways in myelodysplastic hematopoietic stem and progenitor cells.21,22 Conditional knockin mouse models of the most common SF3B1 mutation, Sf3b1(K700E), confirmed that Sf3b1(K700E) mice develop macrocytic anemia, erythroid dysplasia, and long-term hematopoietic stem cell expansion.23,24
RNA-sequencing studies in SF3B1-mutated cells provided evidence that most of the aberrant splicing events selectively observed in SF3B1-mutated samples are caused by misrecognition of 3′ splice sites, resulting in a frameshift.16,25 These studies also indicated that ∼50% of the aberrant mRNAs induced by SF3B1 mutations undergo degradation by a nonsense-mediated mRNA decay (NMD) pathway, resulting in downregulation of canonical transcripts and protein expression.16,25 In addition, it is also possible that NMD-insensitive aberrant transcripts are translated into aberrant proteins with altered function.16,25,26
Two genes involved in mitochondrial iron metabolism synthesis, PPOX and ABCB7, were found to be significantly downregulated in SF3B1-mutated samples. As PPOX encodes protoporphyrinogen oxidase, which catalyzes the dehydrogenation of protoporphyrinogen IX to form protoporphyrin IX, it is likely that haploinsufficiency of this gene may induce defective heme synthesis and iron accumulation into the mitochondria. ABCB7, the causative gene of congenital sideroblastic anemia with cerebellar ataxia, uniformly showed reduced expression in SF3B1-mutated samples, consequent to abnormal splicing and NMD.16,27 Forced ABCB7 expression was found to restore erythroid colony growth and decreased mitochondrial ferritin expression level in CD34+ cells from MDS-RS, supporting the hypothesis that ABCB7 is implicated in the phenotype of this disorder.28,29
SF3B1 mutation is a major determinant of disease phenotype in MDS
SF3B1 mutation is associated with a highly homogeneous disease phenotype and distinctive demographic features
Patients with SF3B1-mutant MDS show a homogeneous disease phenotype characterized by erythroid dysplasia with RS and ineffective erythropoiesis.13,14 Furthermore, cases with multilineage dysplasia according to current WHO morphological criteria have only very mild dysplasia in granulocytic or megakaryocytic lineage without significant effects on peripheral cytopenia (Figure 2).14
Figure 2.

Tridimensional scatterplot of SF3B1-mutated and unmutated MDS with RS according to bone marrow dysplastic features. Red dots identify MDS associated with SF3B1 mutation, whereas blue dots identify MDS unmutated for SF3B1. The degree of dysmyelopoiesis and dysmegakaryopoiesis is measured as percentage of lineage dysplastic cells.14
These observations are fully confirmed by interrogating the IWG registry. Patients reported in this data set were originally classified according WHO criteria 2008. These analyses clearly show that SF3B1 mutations are enriched in the refractory anemia with RS (RARS) category, accounting for 82% of cases, as well as in the refractory cytopenia with multilineage dysplasia (RCMD)-RS category (75%) (Table 2). In addition, SF3B1 mutations are also reported in 9% of patients with refractory cytopenia with unilineage dysplasia (RCUD) or RCMD. It must be noted that most of these patients harboring an SF3B1 mutation and ≥5% RS are expectedly reclassified into the category of MDS-RS according to 2016 WHO criteria.3,18 In addition, we took advantage of the large IWG data set to explore the relationships among SF3B1 mutation type, VAF, and disease phenotype. No significant association was found between the most common SF3B1 mutations or VAF and WHO categories (P = .11 and P = .08, respectively).
Table 2.
Characteristics of 3479 patients with known SF3B1 mutation status within the IWG data set
| Variable | SF3B1 WT | SF3B1 mutated | P |
|---|---|---|---|
| Number of patients | 2684 | 795 | |
| Sex | |||
| Female | 978 (36) | 349 (44) | <.001 |
| Male | 1706 (64) | 446 (56) | |
| Age (y) at sample, median (range) | 69 (18-99) | 72 (34-94) | <.001 |
| <40 | 61 (2) | 3 (<1) | <.001 |
| 40-49 | 131 (5) | 22 (3) | |
| 50-59 | 326 (12) | 78 (10) | |
| 60-69 | 822 (31) | 205 (26) | |
| 70-79 | 959 (36) | 348 (44) | |
| 80-89 | 313 (12) | 125 (16) | |
| ≥90 | 15 (1) | 6 (1) | |
| Unknown | 57 (2) | 8 (1) | |
| WHO 2008 | |||
| Del(5q) | 91 (3) | 20 (3) | <.001 |
| RARS | 60 (2) | 273 (34) | |
| RA/RCUD | 238 (9) | 21 (3) | |
| RCMD | 520 (19) | 18 (2) | |
| RCMD-RS | 56 (2) | 171 (22) | |
| RAEB-1 | 412 (15) | 49 (6) | |
| RAEB-2 | 426 (16) | 28 (4) | |
| Unknown | 735 (27) | 206 (26) | |
| FAB | |||
| RA | 611 (23) | 61 (8) | <.001 |
| RARS | 103 (4) | 352 (44) | |
| RAEB | 763 (28) | 86 (11) | |
| RAEB-T | 48 (2) | 5 (1) | |
| CMML | 61 (2) | 4 (1) | |
| Unknown | 1098 (41) | 287 (36) | |
| Blast %, median (IQR) | 4.0 (1, 9.0) | 2.0 (1.0, 4.0) | <.001 |
| <5 | 1347 (50) | 635 (80) | <.001 |
| 5-10 | 649 (24) | 94 (12) | |
| 11-20 | 486 (18) | 33 (4) | |
| 21-30 | 23 (1) | 2 (<1) | |
| Unknown | 179 (7) | 31 (4) | |
| Hemoglobin (g/dL), median (IQR) | 9.9 (8.7, 11.3) | 9.5 (8.6, 10.5) | <.001 |
| <8.0 | 307 (11) | 102 (13) | 0.001 |
| 8.0-9.99 | 1000 (37) | 353 (44) | |
| 10.0-11.99 | 774 (29) | 249 (31) | |
| ≥12.0 | 447 (17) | 34 (4) | |
| Unknown | 156 (6) | 57 (7) | |
| ANC (×109/L), median (IQR) | 1.6 (0.8, 3.3) | 2.73 (1.7, 4.24) | <.001 |
| <0.5 | 262 (10) | 20 (3) | <.001 |
| 0-5 to 0.99 | 393 (15) | 43 (5) | |
| 1.0-1.8 | 415 (15) | 96 (12) | |
| ≥1.8 | 940 (35) | 410 (52) | |
| Unknown | 674 (25) | 226 (28) | |
| Platelets (×109/L), median (IQR) | 93 (48, 171) | 261 (150, 378) | <.001 |
| <50 | 639 (24) | 41 (5) | <.001 |
| 50-100 | 668 (25) | 60 (8) | |
| 100- 149 | 410 (15) | 76 (10) | |
| 150-449 | 662 (25) | 422 (53) | |
| ≥450 | 74 (3) | 118 (15) | |
| Unknown | 231 (9) | 78 (10) | |
| IPSS-R | |||
| Very low | 263 (10) | 152 (19) | <.001 |
| Low | 610 (23) | 352 (44) | |
| Intermediate | 531 (20) | 86 (11) | |
| High | 391 (14) | 45 (6) | |
| Very high | 320 (12) | 9 (1) | |
| Unknown | 569 (21) | 151 (19) | |
| IPSS-R cytogenetic risk | |||
| Very good | 79 (3) | 38 (5) | <.001 |
| Good | 1681 (63) | 608 (76) | |
| Intermediate | 322 (12) | 93 (12) | |
| Poor | 154 (6) | 14 (2) | |
| Very poor | 271 (10) | 7 (1) | |
| Unknown | 177 (7) | 35 (4) |
Values are reported as n (%) of patients unless otherwise indicated.
ANC, absolute neutrophil count; CMML, chronic myelomonocytic leukemia; IPSS-R, revised International Prognostic Scoring System; IQR, interquartile range; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RAEB-T, RAEB in transformation; WT, wild-type.
In agreement with previous findings, when compared with SF3B1-unmutated MDS, SF3B1-mutated MDS shows significantly lower hemoglobin values, consistent with a high degree of ineffective erythropoiesis, higher neutrophil and platelet counts, and lower bone marrow blasts (P < .001) (Table 2). It is worth noting that 89% and 86% of patients with SF3B1-mutant MDS have normal or nearly normal neutrophil and platelet counts (ie, ANC >1.0 × 109/L, and platelet count >100 × 109/L) at the time of the registration into the IWG data set.
Compared with the whole MDS population, SF3B1-mutated MDS displays a significantly higher prevalence of females, resulting in a male to female ratio close to 1:1 (Table 2). Notably, a similar profile is also typically observed in the only genetically defined MDS subtype, MDS with del(5q).18 In addition, individuals with SF3B1-mutated MDS have a disease onset at a significantly older age than those with SF3B1-unmutated MDS (P < .001) (Table 2).
WHO classification criteria fail to segregate distinct subsets within SF3B1-mutant MDS
Previous reports suggested that the current WHO classification criteria do not allow identification of distinct subsets within SF3B1-mutated MDS, supporting the notion that SF3B1 mutation is a major determinant of disease phenotype in MDS.14,30,31 In fact, the threshold of 15% for RS failed to stratify the prognosis of SF3B1-mutated patients.14,31 In addition, single- or multilineage dysplasia did not show effect on survival or risk of disease progression within SF3B1-mutated patients.14 This observation is fully confirmed by the analysis of IWG data set that clearly shows that the presence of a single or a multilineage dysplasia according to WHO morphological criteria does not have any impact on survival of patients with SF3B1-mutated MDS (P = .4) (Figure 3A). Conversely, in agreement with previous reports,14 the occurrence of an excess blasts significantly affects survival of patients with SF3B1-mutated MDS (P < .001) (Figure 3B), suggesting that clonal evolution may overcome the prognostic advantage of SF3B1 mutation.
Figure 3.

Effect of current WHO classification criteria on OS of patients with SF3B1-mutated MDS. (A) OS of patients with SF3B1-mutated MDS according to the presence of single-lineage (blue curve, n = 267) or multilineage (red curve, n = 171) dysplasia (P = .4). (B) OS of patients with SF3B1-mutated MDS according to bone marrow blasts <5% (blue curve, n = 341) or ≥5% (red curve, n = 85) (P < .001).
Taken together, these results suggest that SF3B1 mutation is the major determinant of disease phenotype, irrespective of current WHO classification criteria. In agreement with this conclusion, a previous study adopting unsupervised hierarchical clustering analyses showed that SF3B1 mutation is recognized as a hierarchically high classification criterion identifying a highly homogeneous group of patients and that within the group of MDS with RS 2 subsets were segregated according to SF3B1 mutation status.30
SF3B1 mutation is a favorable prognostic factor
When analyzing the whole MDS study population, several studies suggested that SF3B1 mutations had a positive prognostic value on overall survival (OS) and risk of disease progression. Some conflicting results were obtained when these analyses were adjusted for phenotypic covariates, mostly due to high collinearity of genotype- and phenotype-related variables.11,13,14,30
An analysis on the largest cohort of SF3B1-mutated MDS patients so far reported showed that the mutation retained an independent positive prognostic value in multivariable analyses including demographic and disease-related factors. The independent prognostic value of SF3B1 mutations was confirmed when the analyses were focused on sideroblastic categories. By contrast, within MDS with excess blasts, the mutation did not retain significant effect on survival and risk of disease progression.14
These findings are confirmed by the analysis of IWG data set that shows that SF3B1 mutation identifies a subgroup of MDS with favorable prognosis (P < .001) (Figure 4A). A stratified analysis within IPSS-R categories32 indicates that this positive prognostic value is significant within very low and low IPSS-R categories (P = .002), whereas it is not retained within intermediate (P = .66) and high- or very high-risk groups (P = .11). Notably, the positive prognostic value of SF3B1 mutation is also confirmed within the categories of RARS (P < .001) and RCMD-RS (P = .003) (Figure 4B-C). In addition, in order to estimate the prognostic effect of the mutation in 2016 MDS-RS categories, we generated 2 groups of patients: (1) RARS and SF3B1-mutated RCUD and (2) RCMD-RS and SF3B1-mutated RCMD. Compared with the respective 2016 categories, these groups comprised occasional patients with SF3B1-mutation and <5% RS. The positive prognostic value of SF3B1 mutation was fully confirmed within the categories of single-lineage (P < .001) and multi-lineage dysplasia (P = .003) (Figure 4D-E). No significant effect of SF3B1 mutation type and VAF was observed on survival. Taken together, these data suggest that within MDS-RS, SF3B1 mutation represents a classification criterion stronger than single- or multilineage dysplasia and concur to support the recognition of MDS with mutated SF3B1 as a distinct disease entity.
Figure 4.
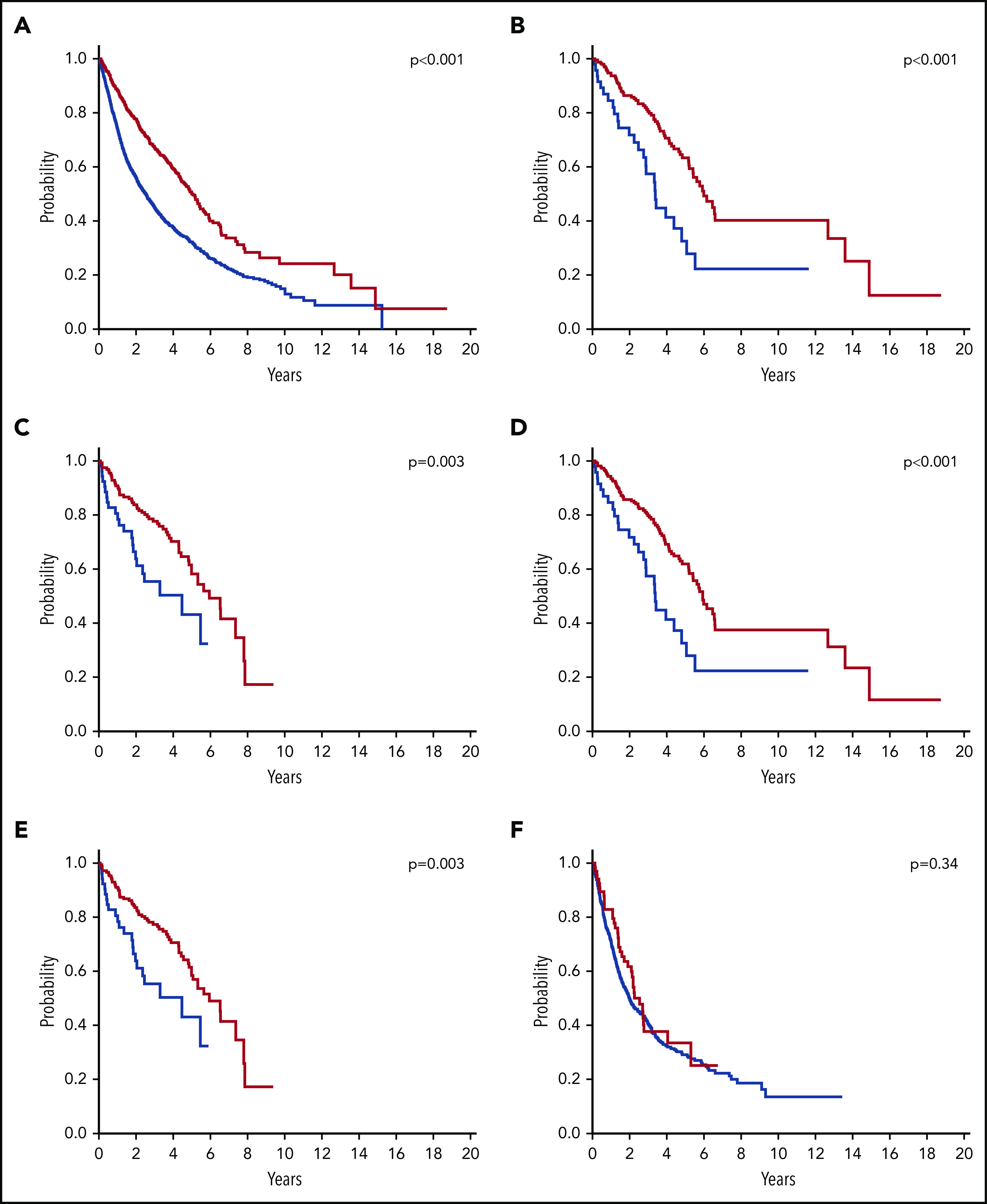
OS of patients with MDS classified according to SF3B1 mutation status. (A) OS of the whole MDS population according to SF3B1 mutation status. Patients with SF3B1-mutated MDS (red curve, n = 769) have a significantly longer survival compared with SF3B1-unmutated MDS patients (blue curve, n = 2555) (P < .001). (B) OS of SF3B1-mutated (red curve, n = 267) and unmutated (blue curve, n = 54) patients with RARS (P < .001). (C) OS of SF3B1-mutated (red curve, n = 171) and unmutated (blue curve, n = 56) patients with RCMD-RS (P = .003). (D) OS of patients with SF3B1-mutated RARS or RCUD (red curve, n = 287) compared to SF3B1-unmutated patients with RARS (blue curve, n = 54) (P < .001). This group overlaps the category of MDS-RS-SLD according to 2016 WHO criteria, except that it comprises occasional patients with SF3B1-mutation and <5% RS. (E) OS of patients with SF3B1-mutated RCMD-RS or RCMD (red curve, n = 189) compared to SF3B1-unmutated patients with RCMD-RS (blue curve, n = 56) (P = .003). This group overlaps the category of MDS-RS-MLD according to 2016 WHO criteria, except that it comprises occasional patients with SF3B1-mutation and <5% RS.(F) OS of SF3B1-mutated (red curve, n = 77) and unmutated patients (blue curve, n = 823) with MDS-EB (P = .34).
Analysis of the IWG data set confirmed that the mutation did not retain significant effect on survival and risk of disease progression within MDS with excess blasts (Figure 4F), suggesting that subclonal mutations driving clonal evolution may overcome the prognostic advantage of SF3B1 mutation.
SF3B1 mutation constrains the spectrum of genetic events driving clonal progression
The available evidence suggests that progression to higher-risk MDS or AML occurs with a relatively low frequency in SF3B1-mutated MDS and is driven by a restricted repertoire of cooperating genetic lesions.6,14
The IWG data set enabled us to validate and expand these observations by testing the prognostic value of cooccurring cytogenetic abnormalities and somatic mutations in the largest cohort of SF3B1-mutant MDS reported so far. Overall, only 3% of patients with MDS and SF3B1 mutation reported in the IWG data set had a poor- or very poor-risk karyotype according to IPSS-R stratification (Table 2). This figure decreased to 1% in patients without excess blasts. Within these latter, a significant effect of IPSS-R poor or very poor cytogenetic risk compared with very low, low, or intermediate risk groups was noticed on OS (P = .032, P = .007, and P = .049, respectively). Within IPSS-R poor or very poor cytogenetic risk, the negative prognostic value of monosomy 7 was fully confirmed (n = 7, P < .001).
A recent comprehensive transcriptomic analysis showed that a high proportion of SF3B1-mutated cases clustering in the category with high risk of leukemic transformation showed overexpression of EVI1, resulting from aberrant gene fusions, including NRIP1-EVI1 and RUNX1-EVI1, or 3q26 abnormality.33 Accordingly, in a recent study on genomic classification of AML, a clustering of SF3B1-mutated cases has been also reported in AML with inv(3) or t(3;3).34 Thirteen SF3B1-mutated patients in the IWG data set harbored an inv(3) or t(3;3). These subjects showed markedly lower OS (median, 27 vs 60 months) and higher risk of AML evolution (5-year cumulative incidence, 75% vs 40%) compared with SF3B1-mutated patients without chromosome 3q26 abnormalities, though these differences did not reach statistical significance (P = .13 and P = .11, respectively).
Overall, SF3B1 mutation is associated with a restricted spectrum of subclonal mutations driving clonal progression (Figure 1). According to the available evidence, mutations in epigenetic regulators, including TET2, DNMT3A, and ASXL1, did not affect survival of MDS with SF3B1 mutation.14 Conversely, RUNX1 mutations have been reported to be significantly associated with increased risk of disease evolution.6,14
We tested the prognostic value of the number of mutations and the most frequent cooccurring or biologically relevant mutated genes in SF3B1-mutant MDS within the IWG data set. When focusing the analysis on SF3B1-mutant MDS without excess blasts, the number of cooccurring mutations (ie, isolated SF3B1 mutation vs 1, 2, or 3 additional mutations) did not significantly affect OS (P values ranging from .90 to .07) (Figure 5A). The prognostic value of RUNX1 mutations was confirmed highly significant on both OS and cumulative incidence of AML evolution (P < .001) (Figure 5B-C). In addition, significant effects on OS were noticed for mutations in EZH2 (P = .003) (Figure 5D), previously reported associated with increased risk of developing transfusion dependency in SF3B1-mutated MDS,14 and in NF1 (P = .003), a functional target of mutant SF3B1-associated splicing.16 The effect of RUNX1 and EZH2 mutations was confirmed in a multivariable analysis adjusted for IPSS-R risk categories (hazard ratio [HR] = 2.66, P < .001 and HR = 2.25, P = .001, respectively), whereas NF1 mutations did not retain statistical significance (HR = 1.43, P = .50).
Figure 5.
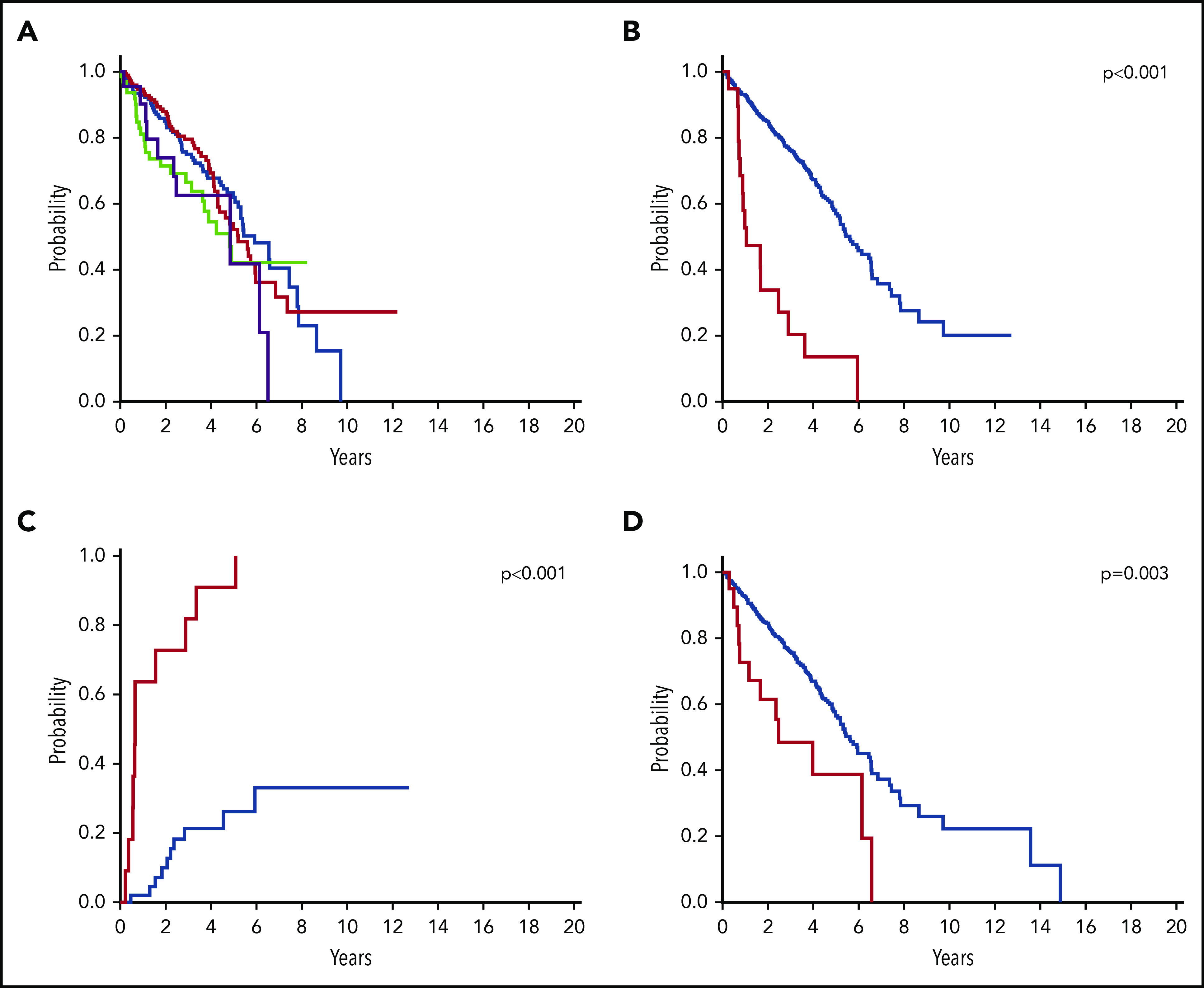
OS of patients with SF3B1-mutant MDS according to additional somatic mutations. (A) OS by isolated SF3B1 mutation (n = 201, blue curve) vs SF3B1 mutation associated with additional somatic mutations within SF3B1-mutated MDS without excess blasts (SF3B1 plus 1 additional mutation, n = 192, red curve; 2 additional mutations, n = 66, green curve; ≥3 additional mutations, n = 23, purple curve) (including patients sequenced for all of the following 15 genes: SF3B1, TET2, DNMT3A, SRSF2, ASXL1, RUNX1, TP53, EZH2, JAK2, U2AF1, IDH1, IDH2, CBL, NRAS, and ETV6). (B-C) OS and cumulative incidence of AML evolution of SF3B1-mutated MDS without excess blasts according to RUNX1 mutation status (mutated, n = 21, red curve; unmutated, n = 505, blue curve) (P < .001). Cumulative incidence of AML evolution was estimated with a competing risk approach, considering death for any cause as a competing event. (D) OS of SF3B1-mutated MDS without excess blasts according to EZH2 mutation status (mutated, n = 20, red curve; unmutated, n = 499, blue curve) (P = .003).
In addition, a significant cooccurrence has been reported between SF3B1 mutations and JAK-STAT pathway–activating mutations, including the classical JAK2 (V617F) and less frequently CALR or MPL mutations.13,14,35-38 This mutation pattern is typically associated with the myelodysplastic/myeloproliferative (MDS/MPN) with RS and thrombocytosis (MDS/MPN-RS-T), currently recognized by the WHO classification as a distinct disease entity.39 The available evidence suggests that SF3B1 mutations act as initiating lesions, responsible for myelodysplastic features (ie, ineffective erythropoiesis and RS), whereas JAK2, MPL, or CALR mutations drive the emergence of subclones conferring the myeloproliferative phenotype.14,36 Within the IWG data set, a significantly higher prevalence of JAK2 and MPL mutations was observed in SF3B1-mutated compared with SF3B1-unmutated MDS (Figure 6A). Although these patients did not fulfill WHO criteria for a diagnosis of MDS/MPN-RS-T, a significantly higher platelet count was found in SF3B1-mutated patients carrying either JAK2 or MPL mutation compared with those wild type for these comutations (P < .001).
Figure 6.

Frequency of cooccurring or mutually exclusive mutated genes in SF3B1-mutated or unmutated MDS in the IWG data set. (A) Most frequent cooccurring or mutually exclusive mutated genes in SF3B1-mutant MDS in the IWG data set. Red and blue bars represent relative frequencies (percentage) of mutated genes in SF3B1-mutated and SF3B1-wild-type MDS, respectively. Red or blue gene labels indicate significantly higher frequencies of the comutated gene in SF3B1-mutated or SF3B1-wild-type MDS, respectively (P values ranging from .019 to <.001). (B) Most frequent cooccurring or mutually exclusive mutated genes in SF3B1-wild-type vs SF3B1-mutant MDS-RS in the IWG data set. Blue and red bars represent relative frequencies (percentage) of mutated genes in SF3B1-wild-type and SF3B1-mutant MDS-RS, respectively. Blue or red gene labels indicate significantly higher frequencies of the comutated gene in SF3B1-wild-type or SF3B1-mutant MDS-RS, respectively (P values ranging from .047 to .002).
Clinical features and outcomes of patients with MDS with RS without SF3B1 mutation
According to current WHO criteria, ∼20% of MDS-RS cases do not harbor the SF3B1 mutation.5,6,11-14 The available evidence suggests that SF3B1-unmutated MDS-RS has clinical features and outcome significantly different from SF3B1-mutated MDS, with a significantly higher prevalence of myeloid and megakaryocyte dysplasia (Figure 2) and reduced survival.14 These findings are fully confirmed by the interrogation of the IWG data set that showed that the SF3B1-negative MDS-RS group had a significantly shorter survival compared with the SF3B1-mutated group (Figure 4B-C). While no specific mutation profile was identified in this subset, a significantly higher prevalence of mutations in TP53 was reported.14 Mutation patterns of SF3B1-unmutated MDS-RS within the IWG data set are reported in Figures 1B and 6B.
Although the molecular basis of this subset remains to be clarified, at present, it seems rational to confirm SF3B1-unmutated cases with RS within the distinct category of MDS-RS according to current WHO classification criteria.18
Relationship between SF3B1 mutation and del(5q)
SF3B1 mutations have been reported in ∼20% of patients classified with the category of MDS with isolated del(5q), associated with a variable proportion of RS.5,6,13,14 These cases are classified within the category of MDS with isolated del(5q) according to current WHO criteria (Table 1).18
The reported cooccurrence of SF3B1 and del(5q) is consistent with the prevalence of this genotype within the IWG data set (Table 2). We analyze the clinical outcome of patients with MDS with isolated del(5q) according to SF3B1 mutation status within the IWG-PM data set, and no significant difference in OS was noticed (P = .57). In addition, no significant effect of the presence or absence of del(5q) on survival of SF3B1-mutated MDS without excess of blasts was found (P = .40).
A study combining single hematopoietic stem and progenitor cell and DNA mutational analysis by targeted sequencing and exome sequencing provided evidence that del(5q) usually precedes recurrent driver mutations in isolated del(5q) MDS, whereas in cases of ring sideroblastic anemia, del(5q) may be either preceded or be followed by SF3B1 mutation.19 Although genetic ontogeny of these myelodysplastic clones might inform the classification process and determine whether a case with concomitant del(5q) and SF3B1 mutation should be more appropriately classified as MDS with isolated del(5q) or MDS with mutated SF3B1, in many cases, clonal hierarchy cannot be easily and unequivocally solved in the everyday clinical practice. Therefore, at present, it seems sensible that these cases should be classified according to current WHO criteria with the category of MDS with isolated del(5q).18 Additional information useful to the classification of these cases might derive from studies investigating the effect of this genotype and clonal hierarchy on response to lenalidomide and luspatercept.
Proposed diagnostic criteria for MDS with mutated SF3B1
According to the available evidence and the results of the IWG data set analysis, the following classification criteria are proposed for the MDS with mutated SF3B1: (1) cytopenia defined by standard hematologic values40; (2) somatic SF3B1 mutation; (3) isolated erythroid or multilineage dysplasia (RS are not required for the diagnosis); (4) bone marrow blasts <5% and peripheral blood blasts <1%; and (5) WHO criteria for MDS with isolated del(5q), MDS/MPN-RS-T, or other MDS/MPN and primary myelofibrosis or other MPN are not met. Due to their significant negative prognostic value and distinctive interaction with SF3B1 mutations, the following genetic lesions represent robust exclusion criteria for the proposed entity (Table 3): (1) poor-risk genetic features, including monosomy 7, inv(3) or abnormalities of chromosome 3q26, resulting in aberrant gene fusions and overexpression of EVI1, and complex karyotype (≥3 chromosomal abnormalities); and (2) cooccurring mutations in RUNX1 and/or EZH2.
Table 3.
Proposed diagnostic criteria for the MDS with mutated SF3B1
| Cytopenia defined by standard hematologic values |
| Somatic SF3B1 mutation |
| Isolated erythroid or multilineage dysplasia* |
| Bone marrow blasts <5% and peripheral blood blasts <1% |
| WHO criteria for MDS with isolated del(5q), MDS/MPN-RS-T or other MDS/MPNs, and primary myelofibrosis or other MPNs are not met |
| Normal karyotype or any cytogenetic abnormality other than del(5q); monosomy 7; inv(3) or abnormal 3q26, complex (≥3) |
| Any additional somatically mutated gene other than RUNX1 and/or EZH2† |
RS are not required for the diagnosis.
Additional JAK2V617F, CALR, or MPL mutations strongly support the diagnosis of MDS/MPN-RS-T.
Clinical and hematological features and survival of patients classified according to the proposed criteria are reported in Table 4 and Figure 7.
Table 4.
Clinical and hematological features of 495 patients within the IWG cohort classified according to the proposed entity of MDS with mutated SF3B1
| Patients, n (%) | |
|---|---|
| Sex | |
| Female | 212 (43) |
| Male | 283 (57) |
| Age (y) at sample, median (range) | 70 (11, 99) |
| <40 | 0 (0) |
| 40-49 | 10 (2) |
| 50-59 | 50 (10) |
| 60-69 | 115 (23) |
| 70-79 | 236 (48) |
| 80-89 | 79 (16) |
| ≥90 | 2 (<1) |
| Unknown | 3 (1) |
| WHO 2008 | |
| RARS | 238 (48) |
| RA/RCUD | 15 (3) |
| RCMD-RS | 156 (32) |
| RCMD | 17 (3) |
| Unknown | 68 (14) |
| IPSS-R | |
| Very low | 130 (26) |
| Low | 269 (54) |
| Intermediate | 21 (4) |
| High | 3 (1) |
| Very high | 0 (0) |
| Unknown | 72 (15) |
| IPSS-R cytogenetic risk group | |
| Very good | 26 (5) |
| Good | 415 (84) |
| Intermediate | 54 (11) |
| Poor | 0 (0) |
| Very poor | 0 (0) |
| Hemoglobin (g/dL), median (IQR) | 9.8 (8.7, 11.1) |
| <8.0 | 51 (10) |
| 8.0-9.99 | 216 (44) |
| 10.0-11.99 | 174 (35) |
| ≥12.0 | 19 (4) |
| Unknown | 35 (7) |
| ANC (×109/L), median (IQR) | 1.89 (0.9-3.6) |
| <0.5 | 4 (1) |
| 0.5-0.99 | 14 (3) |
| 1.0-1.8 | 49 (10) |
| ≥1.8 | 271 (55) |
| Unknown | 157 (32) |
| Platelets (×109/L), median (IQR) | 115 (56, 238) |
| <50 | 12 (2) |
| 50-100 | 12 (2) |
| 100-149 | 49 (10) |
| 150-449 | 290 (59) |
| ≥450 | 89 (18) |
| Unknown | 43 (9) |
Figure 7.
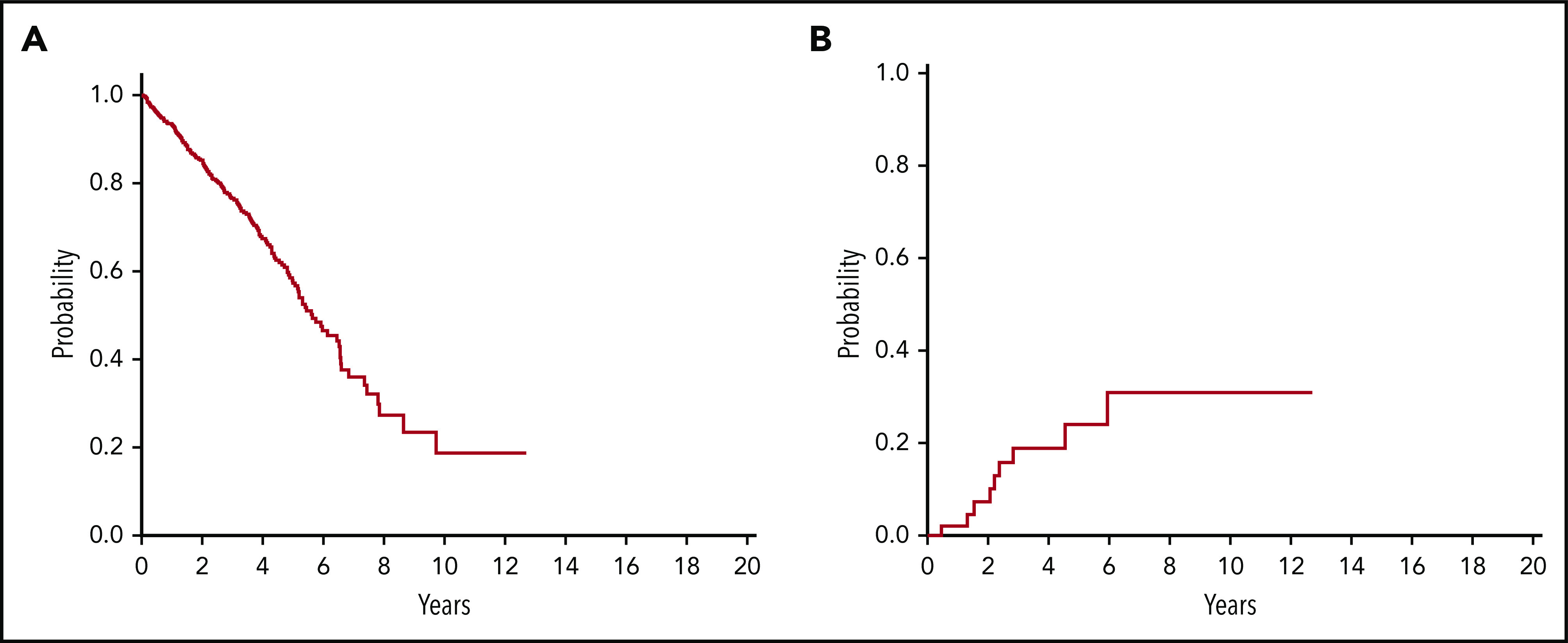
Survival and risk of leukemic evolution of patients classified within the proposed entity of MDS with mutated SF3B1. (A) OS of patients classified within the proposed entity of MDS with mutated SF3B1 (n = 486). (B) Cumulative incidence of AML evolution of evaluable patients (n = 52) classified within the proposed entity of MDS with mutated SF3B1. Cumulative incidence of AML evolution was estimated with a competing risk approach, considering death for any cause as a competing event.
Significance of SF3B1 mutation in clonal hematopoiesis of indeterminate potential and clonal cytopenia of undetermined significance (CCUS)
SF3B1 have been reported as driver mutated genes in a fraction of individuals with clonal hematopoiesis of indeterminate potential.41,42 In these subjects without any hematologic phenotype, median VAF of driver mutations was typically significantly lower than that observed in patients receiving a diagnosis of MDS.43 Whether these studies intercepted a very early phase of the evolutionary trajectory of SF3B1-mutated clones preceding clinical expressivity, or whether additional genetic events are required to promote their expansion, remains to be clarified.
In addition, SF3B1 mutations were detected in a fraction of patients with idiopathic cytopenia of undetermined significance not fulfilling diagnostic criteria for MDS (CCUS).44-46 Preliminary observations suggested that in these patients, SF3B1 mutations were highly predictive of developing MDS with RS,46 suggesting that this genetic lesion in cytopenic patients might provide presumptive evidence of MDS even in the absence of definitive morphological features, as previously acknowledged for selected cytogenetic abnormalities, including del(5q).3,47,48 However, prospective studies are warranted to validate these observations and establish the value of SF3B1-mutated clones in the context of cytopenia of undetermined significance, and patients with these features should be carefully monitored and repeated tests, including bone marrow examination, should be performed to reach a conclusive diagnosis.
Functional consequences of SF3B1 mutation are candidate therapeutic targets
Emerging experimental and clinical evidence suggests that SF3B1 mutation and its functional consequences on erythropoiesis are candidate targets for therapeutic intervention.
SF3B1-mutant patients have a high degree of ineffective hematopoiesis that results in elevated erythroferrone levels and inappropriately low serum hepcidin, as typically observed in congenital iron-loading anemias due to ineffective erythropoiesis.26,49 Transforming growth factor-β superfamily ligand traps have been found to reduce aberrant Smad2/3 signaling and enhance late-stage erythropoiesis in animal models of ineffective erythropoiesis.50-52
Luspatercept is a recombinant fusion protein that binds transforming growth factor-β superfamily ligands to reduce Smad2/3 signaling. In a phase 2 study, luspatercept was found to be effective for the treatment of anemia in lower-risk MDS.53 In a subsequent phase 3, placebo-controlled study on transfusion-dependent patients with MDS-RS, luspatercept treatment abolished the transfusion requirement in ∼40% of cases.17 The fact that >90% of these patients carried a somatic mutation of SF3B1 indicates that this drug can be particularly effective in SF3B1-mutant MDS-RS.
Several compounds can modulate RNA splicing by a direct interaction with the SF3b complex.54,55 Emerging experimental evidence suggests that cancer cells bearing point mutations in the RNA splicing factor–encoding genes are dependent on wild-type spliceosome function, thus resulting in the preferential killing of spliceosome-mutant cells.55 These data demonstrate the therapeutic potential of splicing modulation in spliceosome-mutant cancers and clinical studies are ongoing.
Conclusions and open questions
The available evidence and the findings of our analyses indicate that SF3B1-mutant MDS represents a distinct entity, mainly characterized by ineffective erythropoiesis, relatively good prognosis, and potential response of anemia to luspatercept treatment.
A limited number of concomitant genetic abnormalities are associated with poor outcome and represent exclusion criteria for the proposed nosologic entity. Cooccurrence of JAK-STAT pathway–activating mutations is typically associated with thrombocytosis, indicating the diagnosis of MDS/MPN-RS-T. A fraction of patients with SF3B1 mutation have relative or absolute monocytosis, indicating chronic myelomonocytic leukemia, but the concurrent genetic lesions driving this phenotype remain to be clarified.
In patients with CCUS, SF3B1 mutation is almost invariably associated with subsequent development of overt MDS with RS, suggesting that this mutation might be included among the genetic lesions that provide presumptive evidence of MDS even in the absence of definitive morphological features.
Finally, SF3B1-unmutated MDS-RS appears to be a more heterogeneous group with less favorable prognosis and a largely obscure molecular basis, and additional efforts are warrant to fully elucidate the pathophysiology of these disorders.
Acknowledgments
The authors thank the MDS Foundation for its support of the IWG-PM.
This study was additionally supported by the Associazione Italiana per la Ricerca sul Cancro, Milan, Italy (investigator grant 20125 [L.M.]; 5x1000 project 21267 and the International Accelerator Program project 22796 [M.C.]), Bloodwise (grant 13042 [J.B., A.P.]), and NIH/NCI SPORE in Myeloid Malignancies grant 1P50CA206963.
Authorship
Contribution: L.M. and M.C. conceived this special report; K.S. and D.N. performed statistical analyses of the IWG data set; E.P., P.L.G., S.O., and E.H.-L. contributed to interpretation of the data and report design; R.B., J.B., D.T.B., P.J.C., B.L.E., P.F., T.H., M.H., J.H.J., R.S.K., J.P.M., M.J.W., M.F., G.G.-M., T.A.G., A.K., M.M., A.P, D.A.S., M.R.S., M.A.S., D.P.S., S.T., F.T., and P.V. collected clinical and molecular data; A.A.V.d.L., D.H., and H.T. were responsible for curation of the IWG data set; and all authors contributed to manuscript preparation and approved its content.
Conflict-of-interest disclosure: R.B. received consulting fees from AbbVie, Astex, Celgene, Daiichi-Sankyo, Forty Seven, and NeoGenomics; honoraria for serving on steering and data safety monitoring committees for Celgene; and research funding from Celgene and Takeda. B.L.E. received research funding from Celgene and Deerfield; plays a consulting role from GRAIL; plays an advisory role and holds equity in Skyhawk Therapeutics and Exo Therapeutics. M.H. received honoraria from Novartis, Pfizer, and PriME Oncology; plays a consulting or advisory role for AbbVie, Bayer Pharma AG, Daiichi Sankyo, Novartis, and Pfizer; and received research funding (to institution) from Astellas, Bayer Pharma AG, BergenBio, Daiichi Sankyo, Karyopharm, Novartis, Pfizer, and Roche. M.R.S received research funding from Astex, Takeda, TG Therapeutics; Equity–Karyopharm; consulting fees for AbbVie, BMS, Celgene, Incyte, Karyopharm, Ryvu, Sierra Oncology, Takeda, TG Therapeutics. D.P.S. received institutional research funding from H3 Biosciences and consulting fees for Celgene. The remaining authors declare no competing financial interests.
The International Working Group for the Prognosis of MDS (IWG-PM) operates under the aegis of the MDS Foundation (https://www.mds-foundation.org/) and includes all the investigators who are willing to collaborate for improving our understanding of the pathophysiology of MDS and the treatment of these disorders.
Correspondence: Luca Malcovati, Department of Molecular Medicine, University of Pavia, Pavia 27100, Italy; e-mail: luca.malcovati@unipv.it; and Mario Cazzola, Fondazione IRCCS Policlinico San Matteo, 27100 Pavia, Italy; e-mail: mario.cazzola@unipv.it.
REFERENCES
- 1.Swerdlow SH, Campo E, Harris NL, et al. , eds.. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon, France: IARC; 2017. [Google Scholar]
- 2.Cazzola M. Introduction to a review series: the 2016 revision of the WHO classification of tumors of hematopoietic and lymphoid tissues. Blood. 2016;127(20):2361-2364. [DOI] [PubMed] [Google Scholar]
- 3.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 4.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122(25):4021-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haferlach T, Nagata Y, Grossmann V, et al. . Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Gerstung M, Malcovati L, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium . Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616-3627, quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebert BL, Pretz J, Bosco J, et al. . Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451(7176):335-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider RK, Ademà V, Heckl D, et al. . Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell. 2014;26(4):509-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.List A, Dewald G, Bennett J, et al. ; Myelodysplastic Syndrome-003 Study Investigators . Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456-1465. [DOI] [PubMed] [Google Scholar]
- 10.Krönke J, Fink EC, Hollenbach PW, et al. . Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. 2015;523(7559):183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papaemmanuil E, Cazzola M, Boultwood J, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium . Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida K, Sanada M, Shiraishi Y, et al. . Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64-69. [DOI] [PubMed] [Google Scholar]
- 13.Malcovati L, Papaemmanuil E, Bowen DT, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium and of the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative . Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118(24):6239-6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malcovati L, Karimi M, Papaemmanuil E, et al. . SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126(2):233-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mortera-Blanco T, Dimitriou M, Woll PS, et al. . SF3B1-initiating mutations in MDS-RSs target lymphomyeloid hematopoietic stem cells. Blood. 2017;130(7):881-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiozawa Y, Malcovati L, Gallì A, et al. . Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun. 2018;9(1):3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenaux P, Platzbecker U, Mufti GJ, et al. . Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140-151. [DOI] [PubMed] [Google Scholar]
- 18.Hasserjian RP, Orazi A, Brunning R, et al. . Myelodysplastic syndromes: overview In: Swerdlow SH, Campo E, Harris NL, et al., eds.. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon, France: IARC; 2017:98-106. [Google Scholar]
- 19.Woll PS, Kjällquist U, Chowdhury O, et al. . Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo [published corrections appear in Cancer Cell. 2014;25(6):861 and Cancer Cell. 2015;27(4):603-605]. Cancer Cell. 2014;25(6):794-808. [DOI] [PubMed] [Google Scholar]
- 20.Mian SA, Rouault-Pierre K, Smith AE, et al. . SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun. 2015;6(1):10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolatshad H, Pellagatti A, Fernandez-Mercado M, et al. . Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells [published correction appears in Leukemia. 2015;29(8):1798]. Leukemia. 2015;29(5):1092-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellagatti A, Armstrong RN, Steeples V, et al. . Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood. 2018;132(12):1225-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obeng EA, Chappell RJ, Seiler M, et al. . Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell. 2016;30(3):404-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mupo A, Seiler M, Sathiaseelan V, et al. . Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia. 2017;31(3):720-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darman RB, Seiler M, Agrawal AA, et al. . Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 2015;13(5):1033-1045. [DOI] [PubMed] [Google Scholar]
- 26.Bondu S, Alary AS, Lefèvre C, et al. . A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome. Sci Transl Med. 2019;11(500):eaav5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolatshad H, Pellagatti A, Liberante FG, et al. . Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia. 2016;30(12):2322-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boultwood J, Pellagatti A, Nikpour M, et al. . The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS One. 2008;3(4):e1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikpour M, Scharenberg C, Liu A, et al. . The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia. 2013;27(4):889-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malcovati L, Papaemmanuil E, Ambaglio I, et al. . Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124(9):1513-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patnaik MM, Hanson CA, Sulai NH, et al. . Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119(24):5674-5677. [DOI] [PubMed] [Google Scholar]
- 32.Greenberg PL, Tuechler H, Schanz J, et al. . Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiozawa Y, Malcovati L, Gallì A, et al. . Gene expression and risk of leukemic transformation in myelodysplasia [published correction appears in Blood. 2018;132(8):869-875]. Blood. 2017;130(24):2642-2653. [DOI] [PubMed] [Google Scholar]
- 34.Papaemmanuil E, Gerstung M, Bullinger L, et al. . Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malcovati L, Della Porta MG, Pietra D, et al. . Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114(17):3538-3545. [DOI] [PubMed] [Google Scholar]
- 36.Broséus J, Alpermann T, Wulfert M, et al. ; MPN and MPNr-EuroNet (COST Action BM0902) . Age, JAK2(V617F) and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia. 2013;27(9):1826-1831. [DOI] [PubMed] [Google Scholar]
- 37.Broseus J, Florensa L, Zipperer E, et al. . Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica. 2012;97(7):1036-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klampfl T, Gisslinger H, Harutyunyan AS, et al. . Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390. [DOI] [PubMed] [Google Scholar]
- 39.Orazi A, Hasserjian RP, Cazzola M, Thiele J, Malcovati L Myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon, France: IARC; 2017:93-94. [Google Scholar]
- 40.Greenberg PL, Tuechler H, Schanz J, et al. . Cytopenia levels for aiding establishment of the diagnosis of myelodysplastic syndromes. Blood. 2016;128(16):2096-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal S, Fontanillas P, Flannick J, et al. . Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genovese G, Kähler AK, Handsaker RE, et al. . Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malcovati L, Cazzola M. The shadowlands of MDS: idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematology Am Soc Hematol Educ Program. 2015;2015:299-307. [DOI] [PubMed] [Google Scholar]
- 44.Kwok B, Hall JM, Witte JS, et al. . MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126(21):2355-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cargo CA, Rowbotham N, Evans PA, et al. . Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126(21):2362-2365. [DOI] [PubMed] [Google Scholar]
- 46.Malcovati L, Gallì A, Travaglino E, et al. . Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292-2302. [DOI] [PubMed] [Google Scholar]
- 48.Vardiman JW, Thiele J, Arber DA, et al. . The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951. [DOI] [PubMed] [Google Scholar]
- 49.Ambaglio I, Malcovati L, Papaemmanuil E, et al. . Inappropriately low hepcidin levels in patients with myelodysplastic syndrome carrying a somatic mutation of SF3B1. Haematologica. 2013;98(3):420-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suragani RN, Cadena SM, Cawley SM, et al. . Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408-414. [DOI] [PubMed] [Google Scholar]
- 51.Dussiot M, Maciel TT, Fricot A, et al. . An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat Med. 2014;20(4):398-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fenaux P, Kiladjian JJ, Platzbecker U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood. 2019;133(8):790-794. [DOI] [PubMed] [Google Scholar]
- 53.Platzbecker U, Germing U, Götze KS, et al. . Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338-1347. [DOI] [PubMed] [Google Scholar]
- 54.Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seiler M, Yoshimi A, Darman R, et al. . H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24(4):497-504. [DOI] [PMC free article] [PubMed] [Google Scholar]