

Abstract
The recent clinical success of cancer immunotherapy with checkpoint blockade has led to renewed interest into the development of immune modulatory agents with the capacity to activate anti-tumor T cell responses. Standardization of optimized in vitro assays for efficient assessment of immune function of such new drugs is thus needed to facilitate clinical development of the optimal drug candidates.
Here, we describe an optimized version of T cell suppression assay designed to test the effect of immunomodulatory agents on T cell function and activation. We apply this assay to investigate the agonist activity of the T cell co-stimulatory molecule glucocorticoid-induced TNFR-related protein (GITR). We detail a protocol for concurrent assessment of multiple levels of T cell functional modulation upon GITR engagement, including T cell priming, activation and effector function, in a single assay. As human GITR agonist antibodies are currently under development, availability of standardized cell-based functional assays of GITR agonism is instrumental to translate anti-GITR therapy into the clinical setting.
1. Introduction
The unprecedented clinical success of immune checkpoint blockade for the treatment of cancer has triggered a substantial interest in developing drugs that can modulate T cell responses to cancer (Khalil et al., 2015). Since the FDA approval of CTLA-4 blockade with ipilimumab in 2011 and PD-1 blockade with pembrolizumab and nivolumab in 2014, immunotherapy is now at the cutting edge of cancer care (Chen & Han, 2015; Wolchok et al., 2013). Despite the success of these drugs leading the durable responses in the clinic, the majority of cancer patients do not benefit from current immunotherapies. With the exception of few particularly sensitive disease types, such as Hodgkin lymphoma, the clinical success rate of approved checkpoint blockade therapies remains relatively low, with durable clinical responses seen in 20–40% of patients treated with single-agent immunotherapy and in up to 60% of patients treated with combination regimens (Zappasodi, Merghoub, & Wolchok, 2018; Zappasodi, Wolchok, & Merghoub, 2018). Thus, there is a need for novel and more effective immunotherapies. Currently, there are several immune modulatory agents at various stages of clinical development that either block alternative inhibitory T cell checkpoints (e.g., LAG-3, TIM-3) or activate T cell co-stimulatory receptors (e.g., GITR, OX40, 4–1BB) (Khalil et al., 2015). Other forms of immunotherapies are also under development, such as adoptive cell transfer, with tumor-infiltrating lymphocytes (TILs), chimeric antigen receptor (CAR) T cells, or T cell receptor (TCR)-transgenic T cells, cytokines and vaccines. While some therapies target the tumor cells directly (e.g., CAR T cells), others act indirectly by enhancing pre-existing tumor immunity (cytokines, immune-modulating antibodies) or inducing de novo T cell responses (vaccines). There has been some success of these agents as monotherapies; however, it is becoming apparent that the combination of these agents together or with conventional therapies may provide greater clinical outcomes (Zappasodi, Merghoub, & Wolchok, 2018). Further improvement of immunotherapies is possible through rational design of pre-clinical and clinical trials with a focus on the optimal means to combine two or more therapies. In addition, optimization of molecular design, regimens and combinations of approved immunotherapies, guided by our current understanding of their mechanisms of action, has the potential to increase the response rates in patients in the clinic.
Prior to moving a new drug into pre-clinical or clinical evaluation, the design, screen and ultimate choice of the compound must undergo rigorous testing in order to maximize the likelihood of anti-tumor efficacy in in vivo animal models, and achievement of clinical benefit in patients. As part of this process, proper in vitro assays are needed to efficiently screen the drug candidates with the desired biologic activity. In order to optimize an assay specifically for a target molecule expressed by T cells, several considerations must be taken into account. First, the target molecule must be expressed by T cells in the culture conditions chosen as part of the selected assay(s). It is also important to determine which cell subsets (e.g., CD4+, CD8+ T cells, regulatory T cells (Tregs)) express the target molecule. Second, knowledge of the expression pattern of the target molecule over time during T cell activation may be needed to develop the proper assay conditions, including the definition of the appropriate length of in vitro incubation with the immunomodulatory agent for maximal effects and readout detectability. Lastly, it is important to consider that in vitro assays, while useful to determine the functional activity of immunomodulatory agents on immune cells, may not always anticipate the level of in vivo efficacy, as observed for example with PD-1 blocking antibodies (Wang et al., 2014).
In this chapter, we describe an optimized version of T cell suppression assay designed to test the capacity of immune co-stimulatory agents to enhance priming and activation of T cells in the presence of Tregs. More specifically, we describe a T cell functional assay optimized to test the activity of agents stimulating the T cell co-stimulatory molecule glucocorticoid-induced TNFR-related protein (GITR). GITR is expressed at high baseline levels on Tregs and upregulated on activated CD4+ and CD8+ effector T cells (Teff) (Nocentini, Ronchetti, Petrillo, & Riccardi, 2012; Schaer, Murphy, & Wolchok, 2012). Thus, engagement of GITR affects both Teff and Tregs. The assays described here were optimized to test GITR stimulation using a recombinant human GITR ligand (rhGITRL) to overcome the suppressive effects of Tregs on CD4+ and CD8+ Teff activation. Concurrently, these assays allow to test direct agonistic effects of GITR simulation on activation and effector function of T cells, in the absence of Tregs. It is important to note that a good commend and knowledge of flow cytometry is necessary in order to perform these assays since the readout for this procedure is analyzed by flow cytometry.
2. Functional assays for T cell co-stimulatory molecules: Principles
T cell priming (de novo activation of T cell responses) requires concurrent activation of two signals in T cells: recognition of peptides bound to MHC molecules through the TCR, and CD28 co-stimulation via CD80/CD86 on antigen presenting cells (APCs). The fate of activated T cells is further modulated by additional interactions between co-signaling (co-stimulatory/co-inhibitory) receptors on T cells and their ligands generally expressed on APCs (Chen & Flies, 2013). It is now clear that co-signaling molecules regulate T cell activation, effector function, survival and memory development. The availability of in vitro assays for the characterization of the effects of T cell co-signaling molecules is thus critical to understand their biology and develop therapeutics mimicking their function.
T cell activation can be measured by assessing (1) proliferation, (2) up-regulation of activation markers (e.g., IL2RA/CD25) and (3) production of effector cytokines (IFN-γ, TNF-α). To test the contribution of co-stimulatory receptor engagement to these effects, T cells are typically activated with agonist anti-CD3 antibodies, for the activation of signal 1, and sub-optimal concentrations of agonist anti-CD28 antibodies, for a signal 2 that can be improved by engaging the co-stimulatory molecules (e.g., GITR) under investigation. Conventional immunosuppressive Tregs can be added for a more stringent version of the assay in which to test the capacity of the co-stimulatory agents in exam to bypass Treg suppression and activate Teff even in their presence. If this is true, it may be important to further dissect the mechanism of action of the co-stimulatory agent under investigation in the Teff:Treg co-culture assay and test whether it acts on Teff, Tregs or both. This can be assessed by pre-incubating Tregs or Teff with the co-stimulatory agent in exam before the use of these cell subsets in the co-culture assay. If selective engagement of the co-stimulatory receptor on one cell type can reproduce the effects obtained in the co-culture system, it means that the co-stimulatory pathway has a unique or predominant cellular target.
Technically, T cell proliferation can be assessed by staining T cells with a fluorescent tracking dye, such as carboxyfluorescein succinimidyl ester (CFSE), before starting the culture and by monitoring dilution of the dye in daughter cells as cells get activated and divide over time. These are typically very bright and stable dyes that stain lymphocytes with minimal variation. With cell division, the amount of dye in each cell is halved and this produces discrete shifts or peaks in fluorescence intensity associated with each cell generation. These discrete peaks in fluorescence intensity are highly amenable to mathematical modeling for quantifying cell proliferation. Importantly, dilution of the fluorescent tracking dyes in proliferating T cells nicely correlates with up-regulation of activation markers and production of effector cytokines. Human T cells generally need to be incubated with anti-CD3 and anti-CD28 antibodies for 3–5 days to detect discrete proliferation peaks. Treg suppression can be measured in these assays by co-culturing CFSE-labeled Teff (target cells) in the presence of Tregs (suppressor cells) and monitoring inhibition of proliferation and activation of target cells (Fig. 1A). In these suppression assays, Tregs can also be labeled with a fluorescent tracking dye—different from the one used to label Teff—such as CellTrace Violet dye (CTV), to confidently exclude these cells from the target Teff gate and monitor concurrent changes in Tregs from the same co-cultures (Fig. 1A). As T cell activation is influenced by cell concentration, it is important that Treg suppression assay cultures are properly controlled for cell number. Therefore, in addition to testing CFSE-labeled target Teff alone to measure baseline proliferation in the absence of Tregs, a condition where Teff are co-cultured with an equal amount of CTV-labeled Teff should be included to control for metabolic/nutrient competition (Fig. 1A). This allows to reliably assess and distinguish actual T-cell suppression from metabolic/nutrient competition, which can also result in inhibition of target Teff proliferation, although through different mechanisms. Teff capacity to undergo proliferation upon anti-CD3 and anti-CD28 stimulation may vary from donor to donor. Therefore, it is advisable to plate extra wells with Teff to check CFSE dilution starting on day 3 after stimulation to understand when to stop the incubation and acquire the samples. Following such incubation, cells are stained with a cocktail of fluorochrome-labeled antibodies specific for the T cell subsets in culture (CD4 and/or CD8) and co-stimulatory molecules (e.g., CD25) and a viability dye thus allowing for concurrent flow cytometry-based analysis of T cell proliferation and activation. Fluorescent cell tracking dyes are compatible with fixation/permeabilization buffers for detection of intracellular proteins, including cytokines and transcription factors. For a more comprehensive analysis of cytokine production, culture supernatants can be subjected to multiplex ELISA covering cytokine specificities across multiple T cell differentiation lineages (Fig. 1B).
Fig. 1.
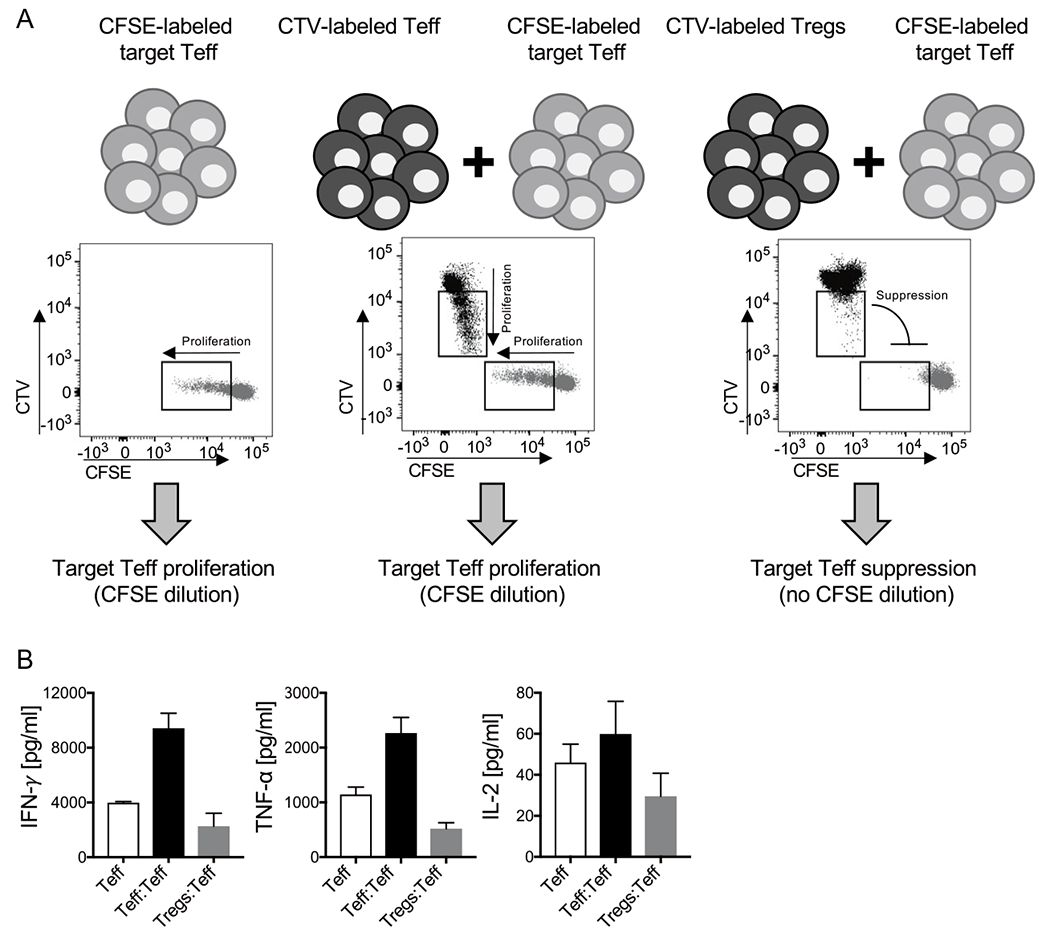
Principle of Treg suppression assay. CFSE-labeled Teff are cultured alone, or with an equal amount of CTV-labeled Teff or Tregs and stimulated for 4 days with 1 μg/mL plate-bound anti-CD3 (clone SP34) and 0.1 μg/mL soluble anti-CD28 (clone CD28.2). Cultures are then analyzed for proliferation by FACS (A) and the indicated cytokines are quantified in culture supernatants by Luminex-based bead multiplex immunoassay (B). (A) Representative bivariate FACS plots (CFSE (FITC) vs CTV (Pacific Blue)) of live single CD4+ T cells in each culture condition are reported.
Here we detail a basic protocol for assessing the effects of GITR co-stimulation on proliferation and activation of human Teff cultured alone or in the presence of human Tregs isolated from healthy donor peripheral blood. CD4+ Teff and Tregs are isolated by magnetic bead purification from peripheral blood mononuclear cells (PBMC) obtained by gradient centrifugation of whole peripheral blood. Alternatively, Teff and Treg subsets can be FACS-sorted from PBMC using proper combinations of fluorochrome-conjugated anti-CD4 and anti-CD25 antibodies. If Teff and Tregs are FACS-sorted, it is important that anti-CD4 and anti-CD25 antibodies conjugated with the same fluorochromes are included in the FACS panel for the acquisition of the assay. We have optimized these assays to provide suboptimal co-stimulation through the CD28 signaling pathway, which can be further improved by the addition of GITR stimulation. By promoting Teff function and reducing Treg suppression, GITR agonist antibodies have shown potent preclinical activity in multiple mouse tumor models (Cohen et al., 2010; Ko et al., 2005; Turk et al., 2004). These promising preclinical results have led to the development of human GITR agonist antibodies, which are currently under clinical investigation. Availability of standardized cell-based functional assays of GITR agonism is thus instrumental to properly translate anti-GITR therapy into the clinical setting.
3. Functional assays for T-cell co-stimulatory molecules: Protocol
3.1. Reagents and instruments
Agonist anti-human CD3 antibody (clone SP34, BD Biosciences)
Agonist anti-human CD28 antibody (clone CD8.2, BD Biosciences) rhGITRL (Genscript)
Lymphocyte Separation Medium (Corning)
CD4+CD25+ Human Regulatory T Cell Isolation Kit (Miltenyi)
MACS separation columns (LD and LS, Miltenyi)
MidiMACS separator (Miltenyi)
CFSE and CTV labeling dyes (Invitrogen)
RPMI 1640 supplemented with 10% FBS, 4mM glutamine, 1mM sodium pyruvate and 50 μM β-mercaptoethanol
Ca2+/Mg2+ free PBS
U bottomed 96-well plates
15 and 50mL falcon tubes
FcR blocking reagent (Miltenyi)
Refrigerated centrifuge
3.2. Day 1: Coating plates with stimulatory antibodies
The day before the assay, add 50 μL of 2 μg/mL anti-CD3 in PBS to the required number of wells of a U-bottomed 96-well plate. Add 50 μL of 20 μg/mL rhGITRL in PBS or 50 μL PBS for GITR stimulation or control, respectively, for a final concentration of 1 μg/mL anti-CD3 and 10 μg/mL rhGITRL. Mix well, seal the plate with parafilm and incubate at 4 °C for at least 18 h.
3.3. Day 0: Isolation of mononuclear cells from human peripheral blood by density gradient centrifugation
Dilute one part of peripheral blood with three parts of Ca2+/Mg2+ free PBS.
Carefully layer 30 mL diluted blood on 15 mL Lymphocyte Separation Medium (Corning) in a 50 mL conical tube; be sure to maintain a clear interphase between the blood and the lymphocyte separation medium. Mixing of the gradient with the blood prior to centrifugation may result in poor recovery.
Centrifuge at 400 × g for 30 min at room temperature without brake.
Aspirate the upper layer as much as possible leaving the mononuclear cell layer undisturbed at the interphase.
Transfer the mononuclear cell layer into a new 50 mL tube, fill with Ca2+/Mg2+ free PBS to the 50mL mark, mix, and centrifuge at 300 × g for 5 min.
Discard the supernatant and resuspend in an appropriate volume of media for counting the mononuclear cells by hemocytometer.
3.4. Day 0: Teff and Treg immunomagnetic purification
Teff and Tregs are purified from total PBMC using the Miltenyi CD4+ CD25+ Human Regulatory T Cell Isolation Kit following the manufacturer’s instructions. This procedure includes a first step for CD4+ T cell enrichment by negative selection followed by separation of CD25+ CD4+ Tregs and CD25− CD4+ Teff using immunomagnetic beads conjugated to an anti-CD25 antibody (Fig. 2A). This method allows to enrich in Foxp3-expressing cells within the CD25+ Treg fraction (Fig. 2B), which displays inhibitory function (Fig. 1).
Fig. 2.
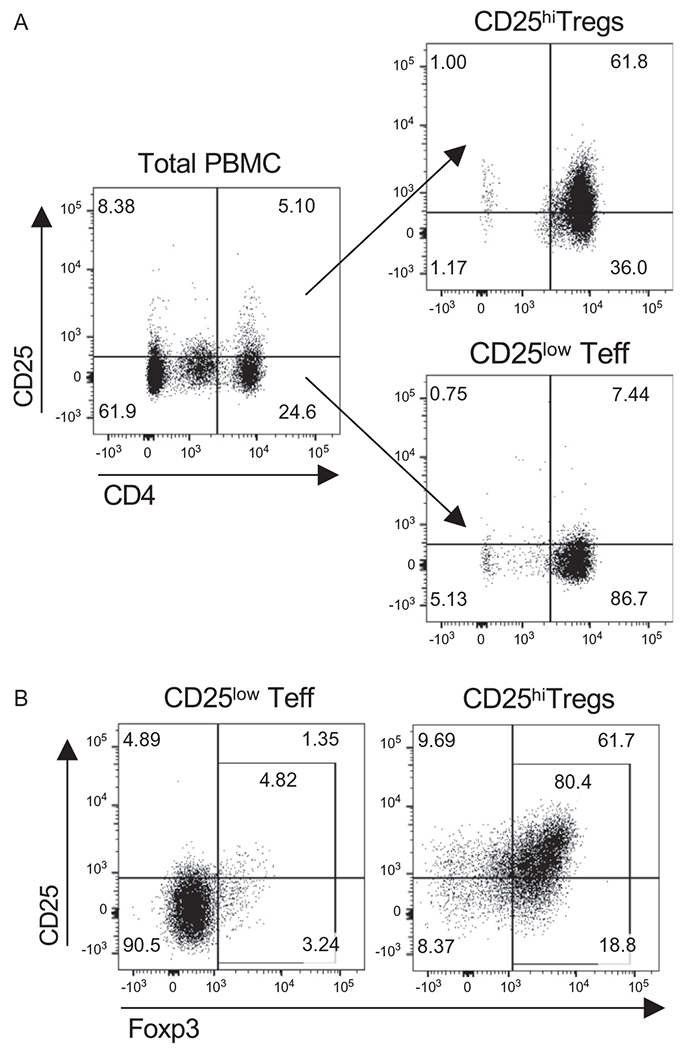
Immunomagnetic purification of human Tregs. Representative example of Treg and Teff purification from healthy donor derived PBMC using CD4+CD25+ Human Regulatory T Cell Isolation Kit (Miltenyi). (A) FACS plots gated on live single cells showing expression of CD25 and CD4 before (left) and after Tregs (right, top) and Teff (right, bottom) purification. (B) Intracellular staining of Foxp3 in purified Teff and Tregs. FACS plots showing Foxp3 and CD25 expression in CD4+ T cells from Teff and Treg purified fractions.
3.5. Day 0: Assay set up
Determine Teff and Treg cell number.
Label target Teff with CFSE: adjust the volume of Teff to 107/mL in serum-free RPMI 1640 medium and add CFSE to a final concentration of 5–10 μM. After 10 min incubation at 37 °C, quench free CFSE with a double amount of FBS, top up with fresh cold complete RPMI 1640, supplemented with 10% FBS, 4mM glutamine, 1 mM sodium pyruvate and 50μM β-mercaptoethanol, and pellet the cells (300 × g for 5min). Resuspend CFSE-labeled cells at the desired concentration in complete RPMI 1640.
Label Tregs and another set of Teff with CTV: adjust the cell volume to 5 × 106/mL in serum-free RPMI 1460 medium and add CTV to a final concentration of 5–10 μM. After 20 min incubation at 37 °C, quench free CTV with a double amount of FBS, top up with fresh cold complete RPMI 1640 and pellet the cells (300 × g for 5min). Resuspend CTV-labeled cells at the desired concentration in complete RPMI 1640.
Plate CTV-labeled cells in 100 μL complete RPMI 1640 per well in the antibody-coated U bottomed 96-well plate. Add 100 μL complete RPMI 1640 per well to the wells where CFSE-labeled target Teff have to be cultured alone. In general, plating Teff and Treg at 1:1 ratio is sufficient to detect suppression. Depending on the recovered quantity of Tregs, determine the number of Tregs to use per well. A number of Teff ranging from 30,000 to 100,000 can be efficiently stimulated in U-bottomed 96-well plate. Therefore, it is recommended to plate a number of Tregs per well within that range. If multiple Treg:Teff ratios need to be tested, perform serial dilution of CTV-labeled Tregs (and CTV-labeled Teff as control) directly in the culture plate.
Plate CFSE-labeled target Teff in 50 μL complete RPMI 1640 medium in each well (same number as Tregs for the 1:1 ratio). In case of multiple Treg:Teff ratios to be tested, the number of target Teff has to be fixed.
Add 50 μL of 0.4 μg/mL anti-CD28 to all the wells that need to be stimulated for a final concentration of 0.1 μg/mL.
Additional control for FACS compensation and set up: plate extra wells with CFSE-labeled or CTV-labeled Teff alone stimulated with anti-CD3 and anti-CD28 for CFSE and CTV single stain controls and to check proliferation between day 3–5 incubation. Plate extra wells with CFSE-labeled and CTV-labeled Teff (1:1 ratio) co-incubated with anti-CD3 and anti-CD28 for isotype control staining. Plate extra wells with CFSE-labeled and CTV-labeled Teff (1:1 ratio) and leave them unstimulated to gate the top CFSE and CTV peak of non-proliferative cells.
Incubated plates at 37 °C in a humidified incubator for 3–5 days.
3.6. Day 3-5: FACS analysis
Before proceeding with the following steps, prepare FACS buffer (2mM EDTA and 1% BSA in Ca2+/Mg2+ free PBS), a 2 × FcR blocking reagent solution (Miltenyi) and a 2 × surface antibody cocktail as described below.
Collect 100 μL culture supernatant from each well and store at −80 °C for cytokine analysis by multiplex ELISA.
Add 100 μL FACS buffer/well and spin down the plate (2000 × rpm for 3 min at 4 °C).
Resuspend cell pellets in 50 μL of 2 × FcR blocking reagent (Miltenyi) in FACS buffer and incubate for 10 min on ice.
Add 50 μL of 2 × surface antibody cocktail diluted in FACS buffer, mix and incubate for 30min on ice in the dark. FACS-based suppression assays have the advantage to allow for concurrent assessment of T-cell proliferation and expression of maturation/activation, as a further measure of functional changes in target cells (Fig. 3). Besides staining for CD4 and with a viability dye, it is thus desirable to include antibodies for such markers, such as CD25. Results shown in Fig. 3B were obtained by using a nine-color FACS panel (Table 1), which can be acquired with a flow cytometry equipped with three lasers (488, 633, and 405 nm).
Add 100 μL FACS buffer per well and spin the plate (2000 × rpm for 3 min at 4 °C).
Discard the supernatant and resuspend with 200 μL FACS buffer per well for another wash (2000 × rpm for 3 min at 4 °C).
Resuspend in 50 μL FACS buffer and transfer to collection tubes for FACS acquisition.
Fig. 3.
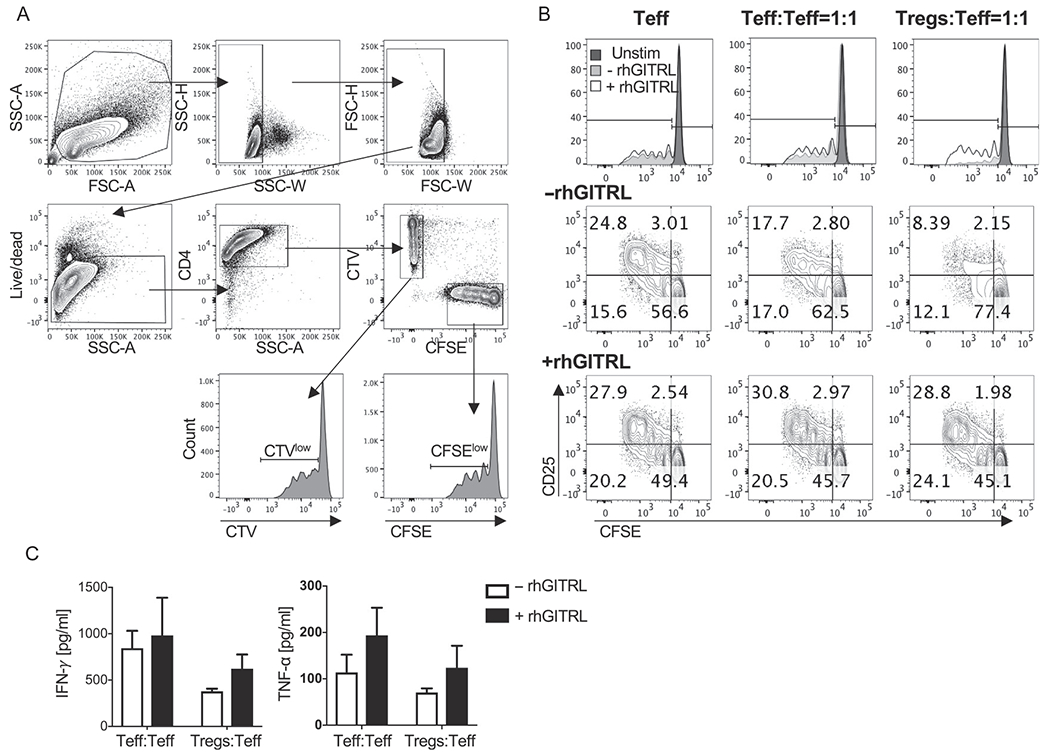
Effects of GITR agonism in Treg suppression assay. Donor-derived CFSE-labeled Teff were cultured alone, or with an equal amount of CTV-labeled Teff or Tregs and stimulated for 4 days with anti-CD3 and anti-CD28 in the presence or the absence of rhGITRL. (A) Representative gating strategy of proliferating CFSE-labeled and CTV-labeled CD4+Teff. CFSE-labeled and CTV-labeled cells are gated among live single CD4+ T cells. Proliferating CFSElowand CTVlow cells are then gated among the CFSE+ and CTV+ CD4+ cell subsets in the respective histogram plot. (B) Representative FACS analysis of proliferation (CFSE dilution) and activation (CD25 up-regulation) in target Teff cultured alone or with an equal amount of CTV-labeled Teff or Tregs and stimulated with (+rhGITRL) or without (−rhGITRL) rhGITRL. (C) Quantification of the indicated cytokines in the supernatants from Teff:Teff and Tregs:Teff co-cultures as shown in (B).
Table 1.
Example of FACS antibody panel for assay acquisition.
| Channel | Target | Dilution |
|---|---|---|
| FITC | CFSE | – |
| PE | GITR | 1:50 |
| PE-TexasRed | CD45RA | 1:50 |
| PerCP-Cy5.5 | PD-1 | 1:50 |
| PE-Cy7 | CD62L | 1:50 |
| Pacific Blue | CTV | – |
| AmCyan | Viability | 1:500 |
| AlexaFluor700 | CD4 | 1:50 |
| APC-Cy7 | CD25 | 1:50 |
Highlighted in gray are the non-lineage/non-viability markers that need to be substituted with fluorochrome matched isotype controls in the isotype control samples described in Section 4.
3.7. Multiplex ELISA for cytokine quantification in culture supernatant
As a further readout of T cell co-stimulation, relevant cytokines can be quantified in the culture supernatants collected at the end of the culture incubation. This can be assessed by standard or multiplex ELISA. Fig. 3C shows representative cytokines quantified using the Luminex-based bead multiplex immunoassay Th1/Th2/Th9/Th17 Cytokine 18-Plex Human ProcartaPlex™ Panel (Invitrogen), which is particularly suitable to investigate changes in T cell function (Zappasodi et al., 2018). This type of immunoassays uses the xMAP® technology (multi-analyte profiling beads) for the simultaneous detection and quantification of multiple cytokines in the same sample. All Luminex-based instruments are compatible with this analysis.
4. FACS compensation, set up and data analysis
Flow cytometry is an essential part of an immunologist toolbox. Understanding the principles of flow cytometry is essential for monitoring immune responses in preclinical and clinical studies. This section will not cover the principles or basic operations of flow cytometry but instead is tailored to users of intermediate and advance levels. A proficient understanding of multicolor flow cytometry including compensation, knowledge of the FACS Diva and FlowJo softwares is assumed.
4.1. Single stain controls
In general, compensation beads are suitable to properly compensate multicolor flow cytometry antibody panels. However, given the nature of this assay, we recommend using fresh human PBMC for single stain controls and pre-activated T cells to control for CFSE and CTV single staining.
Add 100,000–200,000 cells in 100 μL of FACS buffer to each well in a U-bottomed 96-well plate. Plate a well for each fluorochrome included in the FACS panel plus one unstained control. For CTV and CFSE single stain controls, use pre-labeled cells.
Add 1 × (use manufacturer recommended amount) of fluorochrome-conjugated antibody to the appropriate well.
Mix thoroughly by pipetting several times.
Incubate plates for 30min on ice in the dark.
Add 100 μL FACS buffer to each well and spin the plate (2000 × rpm for 3 min at 4 °C).
Discard supernatant and resuspend cells in 200 μL FACS buffer and spin the plate again (2000 × rpm for 3 min at 4 °C).
Discard supernatant and resuspend cells in 200 μL FACS buffer and transfer to collection tubes for FACS acquisition.
Keep tubes in the dark until ready for use.
4.2. Isotype controls
To ensure proper gating of the appropriate T cell populations and their activation markers, it is recommended to prepare additional isotype control samples for each antibody that is not a lineage marker. For example, in the experiment outlined in Section 3 (Fig. 3B), we recommend staining one control sample using pre-activated CFSE:CTV T-cell co-cultures with fluorochrome-matched isotype controls in place of the non-lineage/non-viability markers (Table 1, bold) in addition to the lineage and viability staining included in the acquisition panel.
4.3. Gating strategy and data analysis
Fig. 3A shows the representative gating strategy of live single CFSE-labeled and CTV-labeled CD4+ T cells in co-culture, acquired on an LSRII flow cytometer (BD Biosciences) using BD FACSDiva software (BD Biosciences) and analyzed with the Flow software version 10 (FlowJo, LLC). To gate on the appropriate T cell populations, first gate on the lymphocyte population according to the physical parameters FSC and SSC, then gate on single cells using SSC-H vs SSC-W and FSC-H vs FSC-W (Fig. 3A). Next, gate the live cells using eFluor506 vs SSC-A. The live cells can then be sub-gated into CD4+ T cells (CD4 vs SSC-A), where a clear separation of CFSE-labeled and CTV-labeled T cells can be detected on a bivariate plot (CFSE (FITC) vs CTV (Pacific Blue)). Each T cell population can then be gated and analyzed individually for cell proliferation and activation (Fig. 3). The unstimulated control is used to gate on the CFSElow and CTVlow populations, which represent the cells that have undergone one or more rounds of division (Fig. 3B). In addition to proliferation, T cell activation can also be assessed by examining relative expression of the activation markers. Activation markers can be analyzed by gating on the percentage of positive cells using the isotype control or by median fluorescence intensity (MFI).
5. Concluding remarks
The methods describe here are useful to assess Teff and Treg functions. The experiments were designed in a suboptimal setting to examine how the addition of immunomodulatory agents to the assay modifies Teff and Treg behavior. In this protocol, we used GITRL as the immunomodulatory agent. Since GITR stimulation is known to affect both Teff and Tregs, this assay can be used to test two variables: (1) the effect of GITR stimulation on modulating the suppressive function of Tregs and (2) the effect of GITR stimulation on modulating the activation state of effector T cells. In the data shown in Fig. 3B, suppression of Teff proliferation and CD25 expression can be observed when Teff were co-cultured with Tregs at a 1:1 ratio (−rhGITRL). The addition of rhGITRL to this co-culture was able to reverse this suppression. Moreover, the experiment setup described here can be modified to a simpler version where only CFSE or CTV labeled Teff (CD4+ or CD8+) are stimulated. Fig. 3B shows that addition of rhGITRL to CFSE-labeled Teff cultured either alone or in the presence of an equal number of Teff (Teff:Teff) enhances Teff proliferation and activation. The co-stimulatory effects of GITR agonism on Teff proliferation, activation and inhibition of Treg suppression are paralleled by increases in pro-inflammatory cytokine production (Fig. 3C). Thus, the assay described here provides a useful tool in deciphering several levels of T cell and Treg function that can be nicely complemented with the assessment of cytokine production in the supernatants from the same cultures.
In case T-cell antigen specificity is known, this assay can be further modified to use cognate antigen stimulation in place of anti-CD3 polyclonal activation. For example, common viral epitopes (e.g., from Cytomegalovirus, Epstein-Barr and Influenza viruses) can be used to re-stimulate donor-derived PBMC. In this setting, peptide-pulsed irradiated APCs may be used as scaffold for crosslinking the immunomodulatory antibodies in exam, providing even more physiological co-stimulatory signals compared to plastic-bound antibodies.
Upon in vitro selection with these assays, candidate agents with the desired immunomodulatory function can be further studied in proper in vivo model systems to assess therapeutic activity (e.g., in suitable humanized mouse tumor models) and safety (e.g., in primates, in case of cross-reactivity of the drug with the target molecule in these species), before initiating the clinical development.
Acknowledgments
We thank the Ludwig Institute for Cancer Research, Leap Therapeutics, the Swim Across America, the Parker Institute for Cancer Immunotherapy, the Breast Cancer Research Foundation and Hazen Polsky Foundation for supporting this research. This study was supported in part by NIH grants R01CA056821, R01CA215136-02, P01CA33049, and P01CA59350, and MSKCC Cancer Center Core Grant P30CA008748.
Competing interests
R.Z. is inventor on patent applications related to work on GITR, PD-1 and CTLA-4. R.Z. is consultant for Leap Therapeutics. T.M. is consultant for Leap Therapeutics, Immunos Therapeutics and Pfizer, and co-founder of Imvaq therapeutics. T.M. has equity in Imvaq therapeutics. T.M. reports grants from Bristol-Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics, Aprea. T.M. is inventor on patent applications related to work on oncolytic viral therapy, alphavirus-based vaccines, neo-antigen modeling, CD40, GITR, OX40, PD-1 and CTLA-4.
References
- Chen L, & Flies DB (2013). Molecular mechanisms of T cell co-stimulation and co-inhibition. Nature Reviews. Immunology, 13, 227–242. 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, & Han X (2015). Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. The Journal of Clinical Investigation, 125, 3384–3391. 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, et al. (2010). Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One, 5, e10436 10.1371/journal.pone.0010436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil DN, et al. (2015). The new era of cancer immunotherapy: Manipulating T-cell activity to overcome malignancy. Advances in Cancer Research, 128, 1–68. 10.1016/bs.acr.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Ko K, et al. (2005). Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. Journal of Experimental Medicine, 202, 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocentini G, Ronchetti S, Petrillo MG, & Riccardi C (2012). Pharmacological modulation of GITRL/GITR system: Therapeutic perspectives. British Journal of Pharmacology, 165, 2089–2099. 10.1111/j.1476-5381.2011.01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DA, Murphy JT, & Wolchok JD (2012). Modulation of GITR for cancer immunotherapy. Current Opinion in Immunology, 24, 217–224. 10.1016/j.coi.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk MJ, et al. (2004). Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. The Journal of Experimental Medicine, 200, 771–782. 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, et al. (2014). In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunology Research, 2, 846–856. 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, et al. (2013). Development of ipilimumab: A novel immunotherapeutic approach for the treatment of advanced melanoma. Annals of the New York Academy of Sciences, 1291, 1–13. 10.1111/nyas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappasodi R, Merghoub T, & Wolchok JD (2018). Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell, 33, 581–598. 10.1016/j.ccell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappasodi R, Wolchok JD, &Merghoub T (2018). Strategies for predicting response to checkpoint inhibitors. Current Hematologic Malignancy Reports, 13, 383–395. 10.1007/s11899-018-0471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappasodi R, et al. (2018). Non-conventional inhibitory CD4(+)Foxp3(−)PD-1(hi) T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell, 33, 10.1016/j.ccell.2018.05.009 1017–1032.e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]