

Abstract
Aims
Hepatitis C virus (HCV) infection is monitored by the host innate immunity that includes the endogenous interferon (IFN), which up‐regulates IFN‐stimulated genes (ISGs). HCV is both hepatotropic and lymphotropic, but HCV replication in lymphoid cells is a controversial issue. Here, we analyzed the mRNA levels of the ISGs in B cells of HCV‐infected patients during antiviral therapy and investigated the effects of viral eradication.
Methods
One hundred and eighty‐one patients with chronic hepatitis C and 26 healthy volunteers were enrolled in this study. Levels of HCV RNA and mRNA of ISGs in B cells isolated from the patients were monitored before, during, and after antiviral therapy.
Results
HCV RNA was detected in B cells of 133/175 (76.0%) patients who achieved sustained virologic response (SVR) before therapy was started. The positive ratio of HCV RNA in B cells was higher in patients with genotype 1 and the non‐major genotype of interleukin 28B. HCV RNA in B cells of most patients disappeared 1 week after antiviral therapy was started. The baseline expression of ISG mRNA was significantly higher in the patients than in the healthy volunteers. Levels of ISG mRNA were increased and remained high throughout the IFN‐based therapy. In contrast, levels of ISG mRNA in patients who achieved SVR were significantly decreased 1 week after the IFN‐free therapy was started and remained low during the therapy.
Conclusions
These results suggested that IFN‐free therapy potentially eradicated HCV in the B cells, leading to the down‐regulation of endogenous ISGs. The level of ISG mRNA could be used as a marker for viral eradication in B cells.
Keywords: anti‐hepatitis C virus DAA (directly acting antivirals), B cell, hepatitis C virus, interferons, tissue tropism
List of Abbreviations
- ASV
asunaprevir
- CH‐C
chronic hepatitis C
- DAAs
directly acting antivirals
- DCV
daclatasvir
- G1
genotype 1
- G2
genotype 2
- HCV
Hepatitis C virus
- IPS‐1
IFN‐β promotor stimulator 1.
- ISGs
interferon‐stimulated genes
- LDV
ledipasvir
- MxA
myxovirus A
- PEG‐IFN
pegylated interferon
- PKR
protein kinase
- RBV
ribavirin
- RIG‐I
retinoic acid‐inducible gene I
- SE
standard error
- SOF
sofosbuvir
- TVR
telaprevir
1. INTRODUCTION
Hepatitis C virus (HCV) is not only a hepatotropic but also a lymphotropic virus. 1 Since HCV infection persists in 70% of all cases, it provokes chronic hepatitis C (CH‐C) and further liver cirrhosis or hepatocellular carcinoma. 2 Even if the CH‐C patients infected with HCV genotype 1 (G1) were treated with pegylated interferon (PEG‐IFN) and ribavirin (RBV), the efficacy of this therapy was only about 50% until several years ago. 3 The therapy for CH‐C has been greatly improved by using direct acting antivirals (DAAs). It was initiated with telaprevir (TVR) with ordinary PEG‐IFN/RBV (DAA‐IFN therapy) in 2011. Currently, the combination therapy of DAA without IFN, including asunaprevir (ASV)/daclatasvir (DCV), ledipasvir (LDV)/sofosbuvir (SOF), and glecaprevir/pibrentasvir, is used as a standard therapy worldwide. 4 , 5 , 6 , 7 Furthermore, effective treatment of HCV‐infected patients with decompensated cirrhosis with SOF/velpatasvir, with or without RBV, has been approved. 8
Recent clinical studies revealed that some factors, such as resistance associated viruses (RAVs) especially in HCV‐NS3, NS5A, and NS5B, were correlated with the effectiveness of the therapy. 9 Host factors, including interleukin 28B (IL28B) single nucleotide polymorphisms (SNPs), age, degree of hepatic fibrosis, sexuality, hepatic steatosis, and innate immunity, were also identified to be correlated with the efficacy of the treatment. 10
Our previous report clarified that HCV replication and/or association with B cells and presence of HCV in B cells were correlated with immunologic disorders and resistance to IFN‐based therapy. 11 Once the virus is recognized, endogenous interferon‐stimulated genes (ISGs) are produced in the uninfected cells. 12 ISGs, such as myxovirus A (MxA) and OAS2, of the humoral immunity system were reported to suppress viral replication and reproduction in HCV infection. 13 ISG levels increased after administering IFN, and hence, it could be concluded that antiviral therapy with IFN causes exogenous ISG production. In contrast, IFN‐free therapy does not directly stimulate the endogenous pathway related to ISGs. Thus, studies on the dynamics of ISGs after viral eradication should be demonstrated in HCV‐infected patients who receive IFN‐free therapy. It is difficult to monitor HCV replication in B cells due to lower viral replication in B cells than that in hepatocytes. 14 Monitoring the dynamics of ISGs in B cells of HCV‐infected patients would enable us to analyze viral eradication in B cells during DAA therapy.
In this study, levels of ISG mRNA in B cells of CH‐C patients were monitored during antiviral therapy to investigate the effects of viral eradication on the innate immune system. This study evaluated the antiviral response in B cells of CH‐C patients who achieved sustained virologic response (SVR) during antiviral therapy and the correlation between viral eradication and ISG mRNA expression in B cells.
2. METHODS
2.1. Study subjects
One hundred and thirty‐eight Japanese CH‐C patients with G1 and high viral load (once ≥5.0 log copies/mL during the observation) and 43 CH‐C patients with G2 were enrolled in this study. This study was performed in the Showa University Hospital from 2005 to 2019. The patients with CH‐C and healthy subjects in the Showa University Hospital from 2005 to 2019 and the patients with CH‐C in the Showa University Koto Toyosu Hospital from 2014 to 2019 were enrolled for this study. The subjects were analyzed before, during, and 8 weeks after the therapy. Eligibility criteria indicated that all the patients who received antiviral therapy and agreed to participate in this study with their signatures could be enrolled. After DAA therapy, as described below, SVR, achieved by the patient, was indicated by negative HCV RNA 24 weeks after the end of the therapy. Samples from only those patients who achieved SVR were further analyzed. Data of the patients who did not achieve SVR were excluded from this study.
The CH‐C patients with G1 were treated with TVR + PEG‐IFN + RBV (DAA‐IFN therapy; n = 33) in the Showa University Hospital, ASV + DCV [n = 44 (n = 25 in the Showa University hospital and n = 19 in the Showa University Koto Toyosu Hospital)], and SOF/LDV [n = 61 (n = 34 in the Showa University Hospital and n = 27 in the Showa University Koto Toyosu Hospital)], while the CH‐C patients with G2 were treated with SOF + RBV [n = 43 (n = 17 in the Showa University Hospital and n = 26 in the Showa University Koto Toyosu Hospital)]. Twenty‐six healthy volunteers without history of chronic hepatitis were also enrolled in this study as a control group. Diagnosis of HCV infection was defined as the detection of anti‐HCV antibody and HCV RNA in the serum of the patient. Genotyping of IL28B (rs8099917) was performed using the TaqMan 5′ allelic discrimination assay (Applied Biosystems, Foster City, CA), in accordance with the TaqManGTXpress Master Mix Protocol (Applied Biosystems, Foster City, CA). Written informed consent was obtained from each participant. This research project was approved by a suitably constituted Ethics Committee of our institution, and it confirmed that the project was in accordance to the provisions of the Declaration of Helsinki in 1975. This research was approved by the ethical committees of the Showa University Hospital (the approval number is 660) and Showa University Koto Toyosu Hospital (the approval number is 14H007).
2.2. Isolation of B cells
B cells were isolated using an auto‐MACS Pro Separator ver.2.0.0 (Miltenyi Biotec K.K., Bergisch Gladbach, Germany), as previously described. 11 Briefly, peripheral blood mononuclear cells (PBMCs) were obtained from whole blood samples (30 mL) by centrifugation. Non‐B cells were labeled using a cocktail of specific antibodies that were conjugated to biotin, and the mixture was adsorbed on MACS microbeads to capture the biotin‐labelled cells. The cell‐microbead mixture was then passed through the auto‐MACS magnetic column, and B cells (the flow‐through) were collected. The blood samples were taken at four time‐points before therapy; 1, 8, or 16 weeks after therapy was started; and 8 weeks after the end of the therapy.
2.3. Quantitation of HCV RNA in B cells
Total RNA from each cellular compartment was extracted using the AllPrep DNA/RNA/Protein Mini kit (Qiagen, Duesseldorf, Germany). HCV‐RNA levels were determined in each RNA sample (50 ng) by real‐time reverse transcriptase‐polymerase chain reaction (RT‐PCR) using the primers, as previously described 15 ; this assay had a detection range over 1.0‐8.0 log copies. Samples were scored positive for HCV when titers exceeded 1.0 log copies/100 ng; this threshold was excluded for lymphoid cells contaminated with serum HCV RNA.
2.4. Evaluation of levels of the ISG mRNA
Using the cDNA obtained from each sample, the mRNA levels of ISGs (MxA, ISGF3, IFITM, and OAS2) were analyzed by real‐time PCR, and those of β‐actin were determined as internal controls. The ISG primer kit was purchased (Takara Bio, Shiga, Japan). At each time‐point, ISG mRNA level was adjusted to the level of the internal control and quantified, as previously described. 16
2.5. Statistical analysis
The mean of continuous variables, with and without normal distributions, was compared using analysis of variance and the non‐parametric Wilcoxon test. Comparison of discontinuous variables was performed by the chi‐squared test or Fisher's exact test. A P value of <0.05 was considered statistically significant. Values with normal distributions were expressed as mean ± standard error (SE). For variables that were not distributed normally, data were transformed into log values as required. All statistical analyses were performed using the JMP Pro ver. 14 software (SAS Institute, Cary, NC, USA).
3. RESULTS
3.1. Positive rates of HCV RNA in B cells were correlated with HCV genotypes and IL28B SNPs
Among the 138 CH‐C patients with G1, the rate of SVR in patients who received DAA‐IFN therapy was 90.9% (30/33), and that in patients who received IFN‐free therapy was 93.2% (41/44) for the ASV + DCV therapy and 100% (61/61) for the LDV/SOF therapy. Among the 41 CH‐C patients with G2, the rate of SVR in patients who received IFN‐free therapy was 95.3% (41/43). The positive rate of HCV RNA in B cells of the patients who achieved SVR was 133/175 (76.0%). Baseline characteristics of all patients with CH‐C and those HCV G1 patients who achieved SVR are shown in Table 1. There was no significant difference in the male/female ratio, age, log HCV RNA in the serum (log/mL), platelet count, and levels of alanine aminotransferase (ALT) and γ‐GTP between the patients with HCV genotypes 1 and 2 (Table 1). The positive rates of HCV RNA in B cells were significantly higher in CH‐C patients with G1 than in those with G2 (P < 0.001) [G1, 110/132 (83.3%); G2, 20/40 (50.0%)].
Table 1.
Clinical characteristics of HCV‐infected SVR patients
| DAA‐IFN (N = 30) | SOF/LDV (N = 61) | ASV + DCV (N = 41) | Genotype 1 (N = 132) | Genotype 2 (N = 41) | P value | |
|---|---|---|---|---|---|---|
| Male/total population | 17/30 (56.7%) | 27/61 (44.3%) | 18/41 (43.9%) | 62/132 (47.0%) | 18/41 (43.9%) | n.s |
| Age, years (median) | 59(29‐75) | 66(46‐83) | 67(24‐83) | 63(24‐83) | 65(41‐81) | n.s |
| Log HCV RNA in serum before therapy (log/ml) [mean ± SE] | 6.28 ± 0.14 | 5.92 ± 0.12 | 5.97 ± 0.12 | 6.02 ± 0.07 | 5.98 ± 0.21 | n.s |
| Positivity of HCV RNA in B cells | 29/30 (96.7%) | 46/61 (75.4%) | 35/41 (96.7%) | 110/132 (83.3%) | 20/40 (50.0%)a | <0.001 |
| Platelets (× 104/mm3) | 17.5 ± 1.2 | 17.0 ± 0.9 | 16.5 ± 1.0 | 16.9 + 0.6 | 17.6 ± 1.0 | n.s |
| ALT | 47.0 ± 6.9 | 53.0 ± 5.6 | 54.7 ± 5.9 | 51.8 ± 3.5 | 64.1 ± 7.5 | n.s |
| γ‐GTP | 32.3 ± 10.0 | 44.5 ± 8.0 | 62.2 ± 8.4 | 47.5 ± 5.0 | 49.4 ± 8.6 | n.s |
Data not analyzed in one patient.
Since the genotype of IL28B SNPs (rs8099917) was reported as an essential genetic factor to predict the treatment efficacy of IFN‐based therapy in CH‐C patients 17 , 18 , 19 and the natural clearance of the virus after acute HCV infection, 19 it might be correlated with the viral load of HCV in B cells. We then analyzed HCV RNA levels in the serum and B cells of CH‐C patients with G1 and major (n = 85) and non‐major (n = 47) IL28B genotypes. The backgrounds, biochemical parameters, and serum levels of HCV RNA in both groups did not show significant differences, while the positive rate of HCV RNA in B cells was significantly higher in the patients with the non‐major genotype than in those with the major genotype (P = 0.01) (Table 2). These results indirectly indicated that HCV G1 propagated in B cells more than HCV G2 and HCV could persist in B cells of patients with non‐major IL28B genotype. The positive rate of HCV RNA in B cells was not correlated with the pretreatment levels of various biochemical parameters, such as ALT and γ‐GTP, in the serum (data not shown).
Table 2.
Clinical characteristics of HCV G1‐infected SVR patients
| IL28B major (N = 85) | IL28B non‐major (N = 47) | P value | |
|---|---|---|---|
| Male/total population | 40/85 (47.1%) | 22/47 (46.8%) | n.s |
| Age, years [mean ± SE] | 61.7 ± 1.6 | 61.4 ± 2.2 | n.s |
| Log HCV RNA in serum before therapy (log/ml) [mean ± SE] | 5.95 ± 0.09 | 6.16 ± 0.12 | m.s |
| Positivity of HCV RNA in B cells | 66/85 (77.6%) | 44/47(93.6%) | 0.01 |
| Platelets (×104/mm3) | 16.3 ± 0.7 | 18.1 ± 1.0 | n.s |
| ALT | 48.5 ± 4.3 | 57.9 ± 5.6 | n.s |
| γ‐GTP | 41.6 ± 6.2 | 58.5 + 8.5 | n.s |
3.2. Baseline expression of ISG mRNA was up‐regulated in B cells of patients with CH‐C
The expression levels of ISG mRNA in B cells of patients with CH‐C were first analyzed and compared with those in B cells of healthy volunteers. Figure 1 shows that the expression levels of MxA, ISGF3, IFITM, and OAS‐2 mRNA in B cells of CH‐C patients with G1 were significantly higher than those in B cells of healthy volunteers, while the mRNA expression levels of ISGF3 and IFITM in B cells of CH‐C patients with G2 were higher than those in B cells of the control subjects. The levels of ISG mRNA in B cells of CH‐C patients with G1 were higher than those in B cells of CH‐C patients with G2 (Figure 1).
Figure 1.
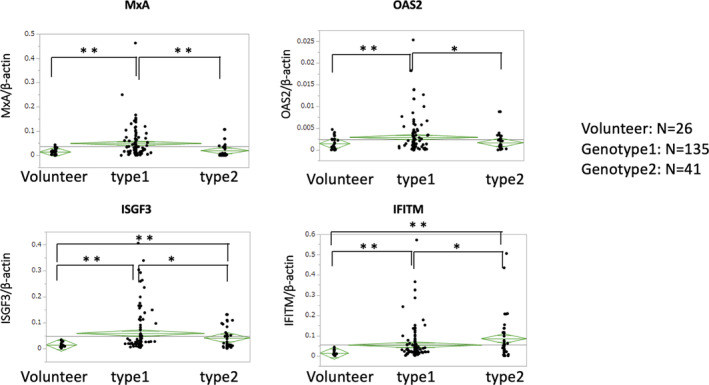
Comparison of the mRNA expression levels of ISGs in B cells of CH‐C patients and healthy volunteers. Each green diamond indicates the average and 95% confidence interval of each group. Statistical significance is determined by analysis of variance. A P value of <0.05 is considered statistically significant (*P < 0.05, **P < 0.01)
On evaluating the effects of sex and age on the levels of ISG mRNA, we found that neither was correlated with ISG mRNA in both CH‐C patients and healthy volunteers. To evaluate the host factors that affected pretreatment ISG levels, the factors were compared based on the pretreatment levels of various biochemical parameters, such as ALT and γ‐GTP, in the serum. As a result, it was found that the mRNA levels of all ISGs in B cells were not correlated with these scores (data not shown).
3.3. Dynamics of viral titer in the sera and B cells and the detection rate of HCV RNA in B cells of CH‐C patients who achieved SVR during antiviral therapy
We then analyzed the dynamics of viral titer in both the sera and B cells of CH‐C patients who achieved SVR after antiviral therapy. Although HCV RNA was detected in the sera of all patients after 1 week of either DAA‐IFN or IFN‐free therapy without significant differences, levels of HCV RNA in sera of patients treated with the SOF/LDV tended to be higher than those of patients treated with other therapies at week 2. HCV RNA levels were reached undetectable in almost all the patients at the time of 8 weeks after the treatment (Figure 2A). On the other hand, HCV RNA in B cells before therapy was detected more in CH‐C patients with G1 (110/132:83.3%) than in CH‐C patients with G2 (20/40:50.0%). HCV RNA in B cells was detectable in 4/30 (13.3%) patients 1 week after receiving DAA‐IFN therapy, while HCV RNA in B cells was undetectable in all patients who received IFN‐free therapy (Figure 2B).
Figure 2.
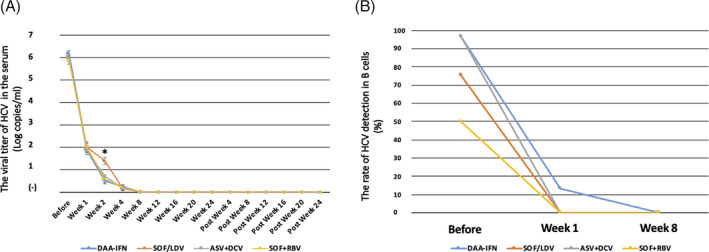
The time course of average viral titers in the sera and the detection rate of HCV RNA in B cells of patients who achieved SVR after receiving the DAA‐IFN and IFN‐free therapies. A, Changes in the viral titer in the sera of HCV G1 and G2 patients who achieved SVR during antiviral therapy, including the DAA‐IFN (n = 30), SOF/LDV (n = 41), ASV + DCV (n = 61), and SOF + RBV (n = 41) therapies, are presented. Data are shown as means ± SE, and statistical significance was determined using a two‐tailed non‐paired Student's t test. A P value of <0.05 was considered to be statistically significant (* P < 0.05). B, The rate of detection for HCV RNA in B cells of the HCV G1 and G2 patients who achieved SVR before and after antiviral therapy, including the DAA‐IFN, SOF/LDV, ASV + DCV, and SOF + RBV therapies, is presented
3.4. The time course of mRNA levels of all ISGs during antiviral therapy
During DAA‐IFN therapy, which was received by the HCV G1 patients who achieved SVR, the levels of all ISGs were significantly increased 1 week after the therapy was started and were decreased at the end of the therapy to levels less than those before the therapy was started (Figure 3). During IFN‐free therapy, which was received by the HCV G1 patients who achieved SVR, the expression levels of mRNA of most ISGs, except ISGF3, in B cells were significantly decreased 1 week after the treatment was started. This level was maintained 8 weeks after the end of the therapy (Figure 3A‐C). This pattern of expression of the ISG mRNA showed the same trend in B cells of CH‐C patients with G2 during IFN‐free therapy of SOF + RBV (Figure 3D). However, the pretreatment viral titer of HCV RNA in B cells did not significantly correlate with the expression levels of ISG mRNA (data not shown).
Figure 3.
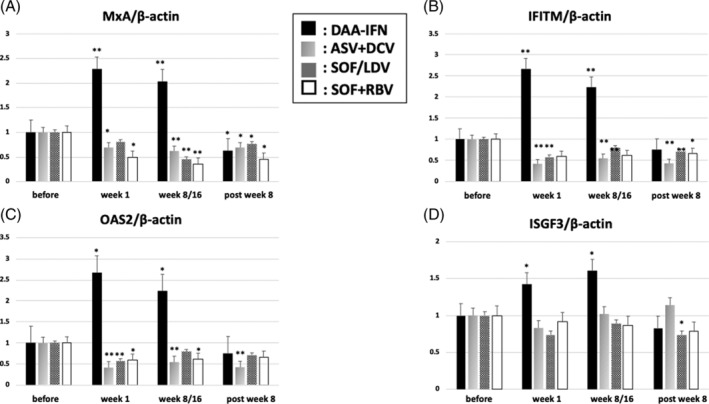
The course of mRNA expression levels of ISGs in patients who achieved SVR during the DAA‐IFN and IFN‐free therapies. The mRNA levels of ISGs, including, A, MxA, B, IFITM, C, OAS2, and, D, ISGF3, are presented during DAA‐IFN, ASV + DCV, SOF/LDV therapies for G1 patients, and SOF + RBV therapy for G2 patients. All the mRNA levels are normalized to levels of β actin mRNA. The initial ratio of the mRNA expressions between each ISG and β actin is set arbitrarily at 1.0. Data are shown as means ± SE, and statistical significance was determined using a two‐tailed non‐paired Student's t test. A P value of <0.05 is considered statistically significant (*P < 0.05, **P < 0.01)
4. DISCUSSION
HCV is not only a hepatotropic but also a lymphotropic virus, and this finding has been supported by the results of several reports that demonstrated the association of HCV with B cells both in vitro and in vivo. 11 , 20 , 21 Furthermore, our previous studies indicated that HCV RNA in B cells was an independent factor that correlated with at least one of the markers for lymphoproliferative disorders (LPDs) in multivariate analysis. 11 Therefore, infection and/or association of B cells with HCV may lead to LPDs. However, whether HCV infects B cells is still not universally accepted. 22 In this study, we confirmed that HCV RNA was detected in B cells of 83.3% and 50% of CH‐C patients with G1 and G2, respectively, as previously described (Table 1). 11 , 14 This result suggested that HCV G1 was more lymphotropic than HCV G2; however, the detailed mechanism remains unknown. Recently, Chen et al proved that the viral envelope and 5'‐UTR sequences of the lymphotropic HCV strain were responsible for its lymphotropic nature. 20 Difference in the sequences in these regions between HCV G1 and G2 may play an important role in the determination of lymphotropism. The limitations of this study were that the sample size of patients with HCV G2 (n = 42) was smaller than that of patients with HCV G1 (n = 132) and no patients with other genotypes were enrolled. We could not conclude that lymphotropism was correlated with the HCV genotypes. Further international studies are required to derive this conclusion and determine a generalization of the lymphotropic nature of HCV. We also showed that the positive ratio of HCV RNA in B cells was higher in patients with the non‐major IL28B genotype (Table 2). This result allowed us to speculate that the host innate immunity was correlated with HCV infection of B cells of CH‐C patients.
After HCV infection of hepatocytes, HCV RNA is rapidly recognized by retinoic acid‐inducible gene I (RIG‐I), which is an intracellular viral sensor. Its signal is transmitted to the nuclei of hepatocytes through an adaptor molecule, IFN‐β promotor stimulator 1 (IPS‐1). Subsequently, IFN‐α, IFN‐β, and IFN‐λ are produced; the ISGs are consequently induced via the JAK‐STAT pathway to prevent viral proliferation. 23 , 24 We observed that mRNA levels of almost all the ISGs, which were investigated in B cells of CH‐C patients (especially MxA), were significantly higher in CH‐C patients than in healthy volunteers. These results suggested that HCV potentially infected B cells, leading to the stimulation of IFN signaling.
We previously reported that HCV RNA was detected more frequently in B cells than in CD4+ and CD8+ T cells and other cells in 75 patients (63%, 16%, 14%, and 17%, respectively). 14 Moreover, the HCV RNA titer was significantly higher in B cells, and HCV infection of B cells acted as a predictive marker for worse outcomes to PEG‐IFN and RBV therapy. 11
In another study, it was reported that HCV infection of B cells induced genetic mutations of immunoglobulins or decreased the antibody producing ability on culturing B cell lines. 25 , 26 , 27
Among approximately 300 ISGs, MxA is known as an important and specific ISG induced only by type I IFN without any correlation with cytokines. 28 This characteristic of MxA makes it different from other ISGs. Exogenously administered IFN evokes a higher level of ISGs in effector cells to establish an antiviral environment. 3 The mRNA levels of all ISGs were significantly increased 1 week after DAA‐IFN therapy was started, compared with those before the therapy was started. Then, the levels decreased to less than the pretreatment levels after the end of the therapy. The administration of PEG‐IFN induced exogenous ISG elevation via the JAK‐STAT pathway (Figure 3). Expression levels of the ISG mRNA were expected to be decreased after eradication of HCV from B cells.
To evaluate the time‐course of endogenous ISGs in B cells after the eradication of HCV, the subjects who received the IFN‐free therapy were further analyzed. During and after the IFN‐free therapy, which was received by CH‐C patients with both G1 and G2, the mRNA expression levels of most ISGs in B cells were significantly decreased (Figure 3). These results suggested that persistent infection of HCV caused the host cells to up‐regulate the endogenous ISGs, and antiviral therapy caused eradication of HCV in B cells and hepatocytes, resulting in down‐regulation of the endogenous ISGs in host cells.
Notably, the previous case‐control study and other meta‐analyses revealed that chronic HCV replication contributed to lymphomagenesis and established a specific role of HCV infection in the pathogenesis of diffuse large B‐cell lymphoma. 29 , 30 The limitation of our study was the small sample size, including no patients who evoked B‐cell lymphoma, but further investigation is required to analyze the correlation between the frequency of HCV positivity in B cells and development of B‐cell lymphoma. Meanwhile, the precise reason for the latter remains unclear. The present study showed that HCV RNA in B cells became undetectable in almost all patients treated with DAA 1 week after the therapy was started, whereas HCV RNA in the sera was still detectable in most patients at the same time (Figure 2). This result suggested that HCV eradication could be achieved more quickly in B cells than in hepatocytes.
Recently, one study analyzed the dynamics of mRNA expression levels of ISGs in patients who were treated with DAA again because they had previously failed to achieve SVR after treatment with PEG‐IFN/RBV. 31 According to that report, down‐regulation of ISGs in liver samples was rapidly observed, and it correlated with the decrease in HCV RNA in the sera after re‐treatment with ASV + DCV for 24 weeks. 31 Our study showed that HCV RNA followed the same trend in the PBMCs. Since the number of relapsed patients was low, statistical analysis to investigate the risk factors of HCV RNA in B cells of relapsed patients could not be performed.
In conclusion, DAA‐IFN therapy produced a strong antiviral environment in B cells by increasing ISG levels. IFN‐free therapy directly eradicated HCV in the cells, leading to down‐regulation of endogenous ISGs. New evidence here suggested that the level of ISG mRNA in B cells could be a marker for viral replication in B cells, and monitoring these levels might help in eradication of the virus.
CONFLICT OF INTERESTS
All the authors declare no conflicts of interests relating to the manuscript.
AUTHOR CONTRIBUTIONS
Conceptualization: Takayoshi Ito
Data curation: Jun Arai, Takayoshi Ito, Yuu Shimozuma, Manabu Uchikoshi, Yoko Nakajima, Masashi Sakaki, Shojiro Uozumi, Atsushi Kajiwara, Ikuya Sugiura, Yumi Otoyama, Toshikazu Kurihara, Junichi Eguchi, Norihiro Nomura, Dai Sakuma, Masashi Sato, Yoshio Deguchi, Hitoshi Yoshida
Formal Analysis: Takayoshi Ito
Funding Acquisition: Jun Arai
Investigation: Jun Arai, Takayoshi Ito, Hisako Nozawa
Methodology: Takayoshi Ito, Hisako Nozawa
Project Administration: Takayoshi Ito, Hitoshi Yoshida
Writing – Original Draft Preparation: Jun Arai
Writing – Review & Editing: Takayoshi Ito, Hitoshi Yoshida
All authors have read and approved the final version of the manuscript. Ito, T had full access to all of the data and takes complete responsibility for the integrity of the data and the accuracy of the data analysis.
TRANSPARENCY STATEMENT
The corresponding author, Ito, T, affirms that this manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; that any discrepancies from the study as planned have been explained.
ACKNOWLEDGMENTS
This research project was supported by the Showa University Research Grant for Young Researches to J.A.
STROBE Statement—checklist of items that should be included in reports of observational studies
| Item No | Recommendation | |
|---|---|---|
| Title and abstract | 1✓ | (a) Indicate the study's design with a commonly used term in the title or the abstract |
| (b) Provide in the abstract an informative and balanced summary of what was done and what was found | ||
| Introduction | ||
| Background/rationale | 2✓ | Explain the scientific background and rationale for the investigation being reported |
| Objectives | 3✓ | State specific objectives, including any prespecified hypotheses |
| Methods | ||
| Study design | 4✓ | Present key elements of study design early in the paper |
| Setting | 5✓ | Describe the setting, locations, and relevant dates, including periods of recruitment, exposure, follow‐up, and data collection |
| Participants | 6✓ | (a) Cohort study—Give the eligibility criteria, and the sources and methods of selection of participants. Describe methods of follow‐up |
| Case‐control study—Give the eligibility criteria, and the sources and methods of case ascertainment and control selection. Give the rationale for the choice of cases and controls | ||
| Cross‐sectional study—Give the eligibility criteria, and the sources and methods of selection of participants | ||
| (b) Cohort study—For matched studies, give matching criteria and number of exposed and unexposed | ||
| Case‐control study—For matched studies, give matching criteria and the number of controls per case | ||
| Variables | 7✓ | Clearly define all outcomes, exposures, predictors, potential confounders, and effect modifiers. Give diagnostic criteria, if applicable |
| Data sources/measurement | 8a✓ | For each variable of interest, give sources of data and details of methods of assessment (measurement). Describe comparability of assessment methods if there is more than one group |
| Bias | 9✓ | Describe any efforts to address potential sources of bias |
| Study size | 10✓ | Explain how the study size was arrived at |
| Quantitative variables | 11✓ | Explain how quantitative variables were handled in the analyses. If applicable, describe which groupings were chosen and why |
| Statistical methods | 12✓ | (a) Describe all statistical methods, including those used to control for confounding |
| (b) Describe any methods used to examine subgroups and interactions | ||
| (c) Explain how missing data were addressed | ||
| (d) Cohort study—If applicable, explain how loss to follow‐up was addressed | ||
| Case‐control study—If applicable, explain how matching of cases and controls was addressed | ||
| Cross‐sectional study—If applicable, describe analytical methods taking account of sampling strategy | ||
| (e) Describe any sensitivity analyses | ||
| Results | ||
| Participants | 13a✓ | (a) Report numbers of individuals at each stage of study—eg numbers potentially eligible, examined for eligibility, confirmed eligible, included in the study, completing follow‐up, and analyzed |
| (b) Give reasons for non‐participation at each stage | ||
| (c) Consider use of a flow diagram | ||
| Descriptive data | 14a✓ | (a) Give characteristics of study participants (eg demographic, clinical, social) and information on exposures and potential confounders |
| (b) Indicate number of participants with missing data for each variable of interest | ||
| (c) Cohort study—Summarize follow‐up time (eg, average and total amount) | ||
| Outcome data | 15a✓ | Cohort study—Report numbers of outcome events or summary measures over time |
| Case–control study—Report numbers in each exposure category, or summary measures of exposure | ||
| Cross‐sectional study—Report numbers of outcome events or summary measures | ||
| Main results | 16✓ | (a) Give unadjusted estimates and, if applicable, confounder‐adjusted estimates and their precision (eg, 95% confidence interval). Make clear which confounders were adjusted for and why they were included |
| (b) Report category boundaries when continuous variables were categorized | ||
| (c) If relevant, consider translating estimates of relative risk into absolute risk for a meaningful time period | ||
| Other analyses | 17✓ | Report other analyses done—eg, analyses of subgroups and interactions, and sensitivity analyses |
| Discussion | ||
| Key results | 18✓ | Summarize key results with reference to study objectives |
| Limitations | 19✓ | Discuss limitations of the study, taking into account sources of potential bias or imprecision. Discuss both direction and magnitude of any potential bias |
| Interpretation | 20✓ | Give a cautious overall interpretation of results considering objectives, limitations, multiplicity of analyses, results from similar studies, and other relevant evidence |
| Generalizability | 21✓ | Discuss the generalizability (external validity) of the study results |
| Other information | ||
| Funding | 22✓ | Give the source of funding and the role of the funders for the present study and, if applicable, for the original study on which the present article is based |
Give information separately for cases and controls in case‐control studies and, if applicable, for exposed and unexposed groups in cohort and cross‐sectional studies.
Note: An Explanation and Elaboration article discusses each checklist item and gives methodological background and published examples of transparent reporting. The STROBE checklist is best used in conjunction with this article (freely available on the Web sites of PLoS Medicine at http://www.plosmedicine.org/, Annals of Internal Medicine at http://www.annals.org/, and Epidemiology at http://www.epidem.com/). Information on the STROBE Initiative is available at www.strobe‐statement.org.
Arai J, Ito T, Shimozuma Y, et al. Decreased expression of interferon‐stimulated genes in B cells of patients with chronic hepatitis C during interferon‐free therapy potentially suggests the eradication of hepatitis C virus in the B cells: A cohort study. Health Sci Rep. 2020;9999:e176 10.1002/hsr2.176
Funding information the Showa University Research Grant for Young Researches to J.A.
REFERENCES
- 1. Sung VM, Shimodaira S, Doughty AL, et al. Establishment of B‐cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. J Virol. 2003;77:2134‐2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaneko S, Ikeda K, Matsuzaki Y, et al. Safety and effectiveness of sorafenib in Japanese patients with hepatocellular carcinoma in daily medical practice: interim analysis of a prospective postmarketing all‐patient surveillance study. J Gastroenterol. 2016;51:1011‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gale M Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939‐945. [DOI] [PubMed] [Google Scholar]
- 4. Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2013;368:1867‐1877. [DOI] [PubMed] [Google Scholar]
- 5. Kumada H, Suzuki Y, Ikeda K, et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology. 2014;59:2083‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizokami M, Yokosuka O, Takehara T, et al. Ledipasvir and sofosbuvir fixed‐dose combination with and without ribavirin for 12 weeks in treatment‐naive and previously treated Japanese patients with genotype 1 hepatitis C: an open‐label, randomised, phase 3 trial. Lancet Infect Dis. 2015;15:645‐653. [DOI] [PubMed] [Google Scholar]
- 7. Chayama K, Suzuki F, Karino Y, et al. Efficacy and safety of glecaprevir/pibrentasvir in Japanese patients with chronic genotype 1 hepatitis C virus infection with and without cirrhosis. J Gastroenterol. 2018;53:557‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takehara T, Sakamoto N, Nishiguchi S, et al. Efficacy and safety of sofosbuvir‐velpatasvir with or without ribavirin in HCV‐infected Japanese patients with decompensated cirrhosis: an open‐label phase 3 trial. J Gastroenterol. 2019;54:87‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fridell RA, Qiu D, Wang C, Valera L, Gao M. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS‐790052 in an in vitro replicon system. Antimicrob Agents Chemother. 2010;54:3641‐3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayashi N, Izumi N, Kumada H, et al. Simeprevir with peginterferon/ribavirin for treatment‐naïve hepatitis C genotype 1 patients in Japan: CONCERTO‐1, a phase III trial. J Hepatol. 2014;61:219‐227. [DOI] [PubMed] [Google Scholar]
- 11. Inokuchi M, Ito T, Nozawa H, et al. Lymphotropic hepatitis C virus has an interferon‐resistant phenotype. J Viral Hepat. 2012;19:254‐262. [DOI] [PubMed] [Google Scholar]
- 12. Saito T, Gale M Jr. Regulation of innate immunity against hepatitis C virus infection. Hepatol Res. 2008;38:115‐122. [DOI] [PubMed] [Google Scholar]
- 13. Itsui Y, Sakamoto N, Kurosaki M, et al. Expressional screening of interferon‐stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat. 2006;13:690‐700. [DOI] [PubMed] [Google Scholar]
- 14. Inokuchi M, Ito T, Uchikoshi M, et al. Infection of B cells with hepatitis C virus for the development of lymphoproliferative disorders in patients with chronic hepatitis C. J Med Virol. 2009;81:619‐627. [DOI] [PubMed] [Google Scholar]
- 15. Ito T, Yasui K, Mukaigawa J, Katsume A, Kohara M, Mitamura K. Acquisition of susceptibility to hepatitis C virus replication in HepG2 cells by fusion with primary human hepatocytes: establishment of a quantitative assay for hepatitis C virus infectivity in a cell culture system. Hepatology. 2001;34:566‐572. [DOI] [PubMed] [Google Scholar]
- 16. Arai J, Goto K, Tanoue Y, et al. Enzymatic inhibition of MICA sheddase ADAM17 by lomofungin in hepatocellular carcinoma cells. Int J Cancer. 2018;10:2575‐2583. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka Y, Nishida N, Sugiyama M, et al. Genome‐wide association of IL28B with response to pegylated interferon‐alpha and ribavirin therapy for chronic hepatitis C. Nature Genet. 2009;41:1105‐1109. [DOI] [PubMed] [Google Scholar]
- 18. Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment‐induced viral clearance. Nature. 2009;461:399‐401. [DOI] [PubMed] [Google Scholar]
- 19. Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon‐α and ribavirin therapy. Nat Genet. 2009;10:1100‐1104. [DOI] [PubMed] [Google Scholar]
- 20. Chen C‐L, Huang JY, Wang C‐H, et al. Hepatitis C virus has a genetically determined lymphotropism through co‐receptor B7.2. Nat Commun. 2017;8:13882 10.1038/ncomms13882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosa D, Saletti G, De Gregorio E, et al. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus‐associated B lymphocyte disorders. Proc Natl Acad Sci. 2005;102:18544‐18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stamataki Z, Shannon‐Lowe C, Shaw J, et al. Hepatitis C virus association with peripheral blood B lymphocytes potentiates viral infection of liver‐derived hepatoma cells. Blood. 2009;113:585‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dai B, Chen AY, Corkum CP, et al. Hepatitis C virus upregulates B‐cell receptor signaling: a novel mechanism for HCV‐associated B‐cell lymphoproliferative disorders. Oncogene. 2016;35:2979‐2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saito T, Owen DM, Jiang F, et al. Innate immunity induced by composition‐dependent RIG‐I recognition of hepatitis C virus RNA. Nature. 2008;24:523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sumpter R Jr, Loo YM, Foy E, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG‐I. J Virol. 2005;79:2689‐2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Machida K, Cheng KT, Sung VM, et al. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc Natl Acad Sci. 2004;101:4262‐4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Machida K, Cheng KT, Pavio N, et al. Hepatitis C virus E2‐CD81 interaction induces hypermutation of the immunoglobulin gene in B cells. J Virol. 2005;79:8079‐8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Angela S, Jurg F, Otto H, Peter S. Interferon‐regulated mx genes are not responsive to interleukin‐1, tumor necrosis factor, and other cytokines. J Virol. 1991;65:968‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nieters A, Kallinowski B, Brennan P, et al. Hepatitis C and risk of lymphoma: results of the European multicenter case‐control study EPILYMPH. Gastroenterology. 2006;131:1879‐1886. [DOI] [PubMed] [Google Scholar]
- 30. de Sanjose S, Benavente Y, Vajdic CM, et al. Hepatitis C and non‐Hodgkin lymphoma among 4784 cases and 6269 controls from the International Lymphoma Epidemiology Consortium. Clin Gastroenterol Hepatol. 2008;6:451‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alao H, Cam M, Keembiyehetty C, et al. Baseline intrahepatic and peripheral innate immunity are associated with hepatitis C virus clearance during DAA therapy. Hepatology. 2018. Epub ahead of print;68:2078‐2088. [DOI] [PMC free article] [PubMed] [Google Scholar]