

CONSPECTUS
De novo protein design represents an attractive approach for testing and extending our understanding of metalloprotein structure and function. Here, we describe our work on the design of DF (Due Ferri or two-iron in Italian), a minimalist model for the active sites of much larger and more complex natural diiron and dimanganese proteins. In nature, diiron and dimanganese proteins protypically bind their ions in 4-Glu, 2-His environments, and they catalyze diverse reactions, ranging from hydrolysis, to O2-dependent chemistry, to decarbonylation of aldehydes. In the design of DF, the position of each atom—including the backbone, the first-shell ligands, the second-shell hydrogen-bonded groups, and the well-packed hydrophobic core—was bespoke using precise mathematical equations and chemical principles. The first member of the DF family was designed to be of minimal size and complexity and yet to display the quintessential elements required for binding the dimetal cofactor. After thoroughly characterizing its structural, dynamic, spectroscopic, and functional properties, we added additional complexity in a rational stepwise manner to achieve increasingly sophisticated catalytic functions, ultimately demonstrating substrate-gated four-electron reduction of O2 to water. We also briefly describe the extension of these studies to the design of proteins that bind nonbiological metal cofactors (a synthetic porphyrin and a tetranuclear cluster), and a Zn2+/proton antiporting membrane protein.
Together these studies demonstrate a successful and generally applicable strategy for de novo metalloprotein design, which might indeed mimic the process by which primordial metalloproteins evolved. We began the design process with a highly symmetrical backbone and binding site, by using point-group symmetry to assemble the secondary structures that position the amino acid side chains required for binding. The resulting models provided a rough starting point and initial parameters for the subsequent precise design of the final protein using modern methods of computational protein design. Unless the desired site is itself symmetrical, this process requires reduction of the symmetry or lifting it altogether. Nevertheless, the initial symmetrical structure can be helpful to restrain the search space during assembly of the backbone.
Finally, the methods described here should be generally applicable to the design of highly stable and robust catalysts and sensors. There is considerable potential in combining the efficiency and knowledge base associated with homogeneous metal catalysis with the programmability, biocompatibility, and versatility of proteins. While the work reported here focuses on testing and learning the principles of natural metalloproteins by designing and studying proteins one at a time, there is also considerable potential for using designed proteins that incorporate both biological and nonbiological metal ion cofactors for the evolution of novel catalysts.
Graphical Abstract
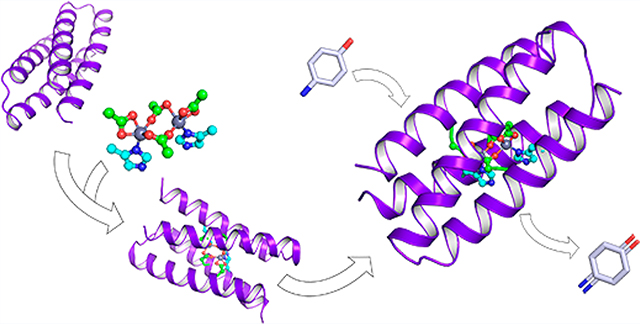
■ INTRODUCTION
For over a century, chemists have been able to design a wide range of small molecules with precisely predetermined structures and functions that have shaped our chemical, pharmaceutical, and agricultural industries. Molecular functions ranging from binding to catalysis depend critically on precise design. However, until recently proteins were considered far too complex to ever be designed by humans. Nevertheless, starting in the 1980s with the ability to create synthetic genes and the ever-growing understanding of protein structure, we felt that it should be possible to design proteins from scratch rather than by modification of existing natural proteins and named this approach de novo protein design.1 Initially, the goal of de novo design was to construct a protein that folded in solution and to critically characterize its properties to confirm the design principles.2,3
As de novo protein design became increasingly feasible, we turned our attention to function, including the design of metalloproteins.4 Metalloproteins are capable of catalyzing a remarkable array of reactions, and a given metal ion can be used in a number of oxidative, reductive, and hydrolytic transformations in different enzymes.5 Metalloprotein activity represents a partnership between the metal cofactor and the protein matrix: the metal brings a modicum of nondiscriminate chemical reactivity, while the protein stabilizes it in solution and directs its unique reactivity.
While our groups have designed a number of proteins that bind porphyrins6–11 and iron–sulfur centers,12,13 here we focus primarily on bridged dinuclear cofactors, such as carboxylate-bridged diiron centers, which catalyze a variety of O2-dependent reactions.14 These studies represented the crossing of a number of special milestones in de novo protein design including premier examples of a de novo-designed metalloprotein whose structure was confirmed by crystallography and NMR,15,16 the use of parametric approaches to design complex functional proteins,14 computational algorithms to facilitate simultaneous positive and negative protein design,17 the demonstration of robust, saturable catalytic activities,18–21 and the design and structural characterization of functionally rich metalloproteins22 (Figure 1). Since the principles to construct metalloproteins have significantly advanced, in perspective, it should be possible to extend our general approach to the design of proteins for a variety of practical applications. Moreover, our helical bundles are attractive targets for directed evolution of metalloenzymes that combine the efficiency of non-natural transitional metal catalysts with the high programmability, selectivity, and biocompatibility of proteins.
Figure 1.

Milestones in the development of DF proteins.
■ DIMETAL CENTERS IN O2-UTILIZING PROTEINS
Dimetal sites are widely used throughout biology to catalyze hydrolytic as well as redox-active processes.5,23–25 Although structurally diverse, they have some common features: they are rich in Glu/Asp and His ligands, and the metal ions are generally bridged by water (also OH− or O2−) or carboxylate-containing side chains. We were particularly drawn to the O2-utilizing proteins, which include hydroxylases, fatty acid desaturases, radical-generating ribonucleotide reductases, catalases, ferritins, and aldehyde decarbonylases. The di-Mn or di-Fe sites of all these proteins are housed within an antiparallel four-helix bundle that is generally embedded into a much larger and far more complex structure.14,15
The catalytic mechanisms of individual diiron proteins represent variations on a theme, in which binding of substrate to the reduced di-Fe2+ cofactor triggers O2 binding, which rapidly generates an end-on or bridging di-Fe3+ peroxo-intermediate. 24–27 This intermediate can react directly with substrates or be converted into other kinetically competent superoxo species or ferryl (Fe4+) species.24,26,27 The protein adjusts to the large change in cofactor charge by shifting carboxylate ligands and changing the protonation of ligating water. We wished to determine the extent to which these features that steer reactivity could be captured in simple model proteins. However, before we could address this question, we needed to develop methods for high-precision design of complex, functional proteins as our first objective.
■ PRECISE DESIGN OF DIMETAL PROTEINS
In our study,14,15 we endeavored to design a minimal four-helix diiron protein, not by modification of the sequence of a natural counterpart but rather by starting from first principles. We used a parametric approach, applying an analytic equation to define the backbone of the overall structural target. Prior to our work on DF, we had already combined the α-helix-describing equations with a D2 operator to construct antiparallel four-helix bundles.28 Also, Harbury and co-workers had used Crick’s equation to design both right- and left-handed coiled coils.29 We asked whether similar parametric equations might be used to design a more complex protein.
We began by examining the backbone parameters that define the geometry of the active site bundles of natural diiron proteins and then building a miniature, idealized diiron protein that embodies this bundle (Figure 2).15 First, we built a D2-symmetric four-helix bundle of identical, unconnected 21-residue helices. Only six adjustable parameters were used to create the initial backbone. To provide a Glu4His2 liganding environment, the D2 symmetry was relaxed to C2 leading to homodimeric helix–loop–helix motifs. Helices 1 and 2 contained a single Glu ligand and a Glu-Xxx-Xxx-His motif, respectively. The final bundle geometry was dictated by (1) coordination requirements of the Glu4His2-diiron site (Figure 2a–d), (2) suitable helical packing angles and distances, (3) precise 2-fold symmetry between the helical hairpins, and (4) introduction of Asp and Tyr residues to form second-shell H-bonds to the liganding His and Glu (Figure 2e,f). This extensive network of H-bonds stabilizes the primary ligands and modulates the reactivity of the cluster. In summary, the intended binding site dictated the overall tertiary structure of the protein. This approach differed fundamentally from other approaches, which repurpose existing protein scaffolds to bind a given cofactor.
Figure 2.

Crucial steps in DF1 de novo design. (a,b) Coordination environment of the diiron-binding site. (c,d) Diiron site into the four-helix bundle. (e,f) Second shell H-bond interactions. (g) Hydrophobic core and interhelical loop.
The next step was to choose well-packed hydrophobic side chains at the remaining core positions to stabilize the folded state of the protein (Figure 2g). This process was facilitated by using the side chain repacking algorithm of Dejarlais and Handel,30 which had recently been introduced to redesign the sequence of small natural proteins.30,31 Moreover, we had already applied these algorithms to the de novo design of proteins with novel backbones, accomplishing the first computational design of a protein that folded into a precisely predetermined structure,2,32 and a small protein that bound a natural peptide.33,34 In the final steps, an interhelical loop and the remaining surface-facing side chains were introduced (Figure 3a).
Figure 3.

(a) DF1 sequence and its intended secondary structure. Metal-binding residues in orange; hydrophobic core residues in green. (b,c) Dimetal site and H-bond second-shell interactions in the X-ray structure of di-Zn2+-DF1 (PDB-ID 1ec5).
Remarkably, the first designed sequence, DF1, folded into a very stable dimeric four-helix bundle: for the first time, a de novo metalloprotein showed an experimental crystal structure in perfect agreement with the intended design.15 The protein was crystallized as the di-Zn2+ derivative, as a redox-inert surrogate of the diferrous state. The DF1 crystal structure deviated 1.6 Å from the designed model. The deviations are greatest near the ends of helices (far from the binding site), and indeed they lower to 1.0 Å in proximity of the metal site. Importantly, the network of hydrogen-bonding second-shell ligands and the metal-binding site were realized precisely as in the intended design (Figure 3b,c).
Metalloproteins with functionally interesting sites tend to bind their metal cofactors in coordinately unsaturated geometry, leaving room for substrates. Similarly, the DF1 dimetal center (Figure 3b,c) presents two adjacent vacant sites for interaction with water or substrates. By contrast, “structural” metal-binding sites tend toward highly regular and coordinately saturated geometry that stabilizes a given protein fold. Thus, we determined the solution NMR structure of the apo-form of DF1 to establish whether the protein was imposing its structure onto the metal cofactor or vice versa. The apo-structure was nearly identical to the holo-structure, maintaining the same packing. The six coordinating and the four second-shell residues were largely preorganized with angstrom-level accuracy even in the absence of the metal cofactor.16 This finding demonstrated that (1) a sufficient set of H-bonds and salt bridges is required to achieve a preorganized binding site in the apolar core and (2) a proton-exchange mechanism should be engineered to facilitate metal binding.
As we next endeavored to engineer substrate-binding and catalytic activity into DF1 (Figure 1), we had to face the tradeoff between protein stability and function.16 The desired changes were highly destabilizing, as they involved burial of additional polar groups and mutations of buried Leu side chains to small helix-breaking Gly residues.16,35 To compensate, we needed to introduce stabilizing substitutions at positions distant from the active site. Ultimately, a highly stable protein, DF3, was designed,19,36 which had four glycines lining the access sites and an idealized αR–αL–β interhelical loop,37 featuring a network of hydrogen-bonded side chain/main chain interactions (Figure 4a).
Figure 4.

(a) Peptide sequences of the DF family. Coordinating residues are in orange, active site access in cyan, H-bond second-shell interactions in magenta (X stands for propargyl glycine and aha stands for 6-azidohexanoic acid). (b–e) Different DF protein constructs.
The thermodynamic stability of DF could also be improved by building fully asymmetric single-chain proteins. This was accomplished by cross-linking two helix-loop-helix monomers (Figure 4a,b) using a click reaction between alkyne- and azide-containing residues (DF-C1, Figure 4a,c).21,38 We also computationally designed and bacterially expressed a single-chain version (DFsc),39 a 114-residue scaffold with three loops connecting the four helices (Figure 4a,d).
In an alternate approach, we designed DFtet, which consisted of four disconnected helices that could be combinatorially assembled to facilitate evaluation of multiple sequence variants (Figure 4a,e).17,18,40 The active site was retained as in the other DF constructs, and DFtet was used to rapidly evaluate candidate sequence changes, which were subsequently introduced into DFsc or DF3. To increase stability, the helices of DFtet were extended to 33 residues. By engineering the electrostatic helix–helix interfaces, we developed two-component A2B2,17 and three-component AAABB2 heterotetramers,18,40 which assembled with very high specificity.18,40 To facilitate the design, we developed a Monte Carlo algorithm that explicitly evaluated the electrostatic interactions in the desired heterotetramer, as well as in all the other alternative topologies. To the best of our knowledge, this was the first use of a computational algorithm to design a sequence that not only stabilized the desired structure (positive design) but also destabilized undesired outcomes (negative design). Since then, highly sophisticated methods that incorporate machinelearning have been developed for positive and negative design of coiled coils.41
■ FROM STRUCTURE TO FUNCTION
Precisely as designed, the bespoke site consisted of two metals surrounded by four Glu ligands. Additionally, two His ligands were oriented trans to unoccupied ligand-binding sites for water, O2 and organic substrates. However, access to the binding site was blocked by Leu residues at positions 13 (and the symmetry-related 13′) and 9/9′, which we define as the “inner” and “outer” region of the substrate access site (Figure 5).15,42
Figure 5.

Substrate access sites in the X-ray structure of di-Mn2+-DF1 (PDB-ID 1ovr) and di-Mn2+-DF1-L13G (PDB-ID 1lt1).
We therefore replaced the inner Leu residues with Ala to create a small substrate-access cavity. The crystal structure of di-Mn2+-DF1-L13A indeed showed that a dimethyl sulfoxide molecule, bridging the two metal ions, occupies the cavity (DMSO rvdW = 2.54 Å).43 Further bulk-decreasing mutations to Gly resulted in a large increase in the hydration of the binding site (rvdW ≈ 5.9 Å). The dimetal site was bridged by either a single or a pair of water molecules in different dimers within the asymmetric unit.35 Small but apparently significant (vide infra) structural changes accompanied the variation in sequence and bound ligands. The two helices that defined the inner site moved slightly closer in di-Mn2+-DF1-L13A, while the increased hydration of di-Mn2+-DF1-L13G lead to an expansion of the interhelix distance. Furthermore, these two helices slide back and forth by up to 1.0 Å to modulate the metal–metal distance in response to these changes.
To probe further the extent to which the DF1 variants imposed their structure on the dimetal cofactor, we examined structures of di-Zn2+, di-Cd2+, di-Co2+, di-Mn2+, and di-Fe2+, each with different ligand-binding preferred geometries.42 Overall, there was little change in the protein ligands other than small shifts of carboxylates and the above-mentioned shifts of backbone atoms. We also examined the thermodynamic cost of carving a substrate-access channel into DF1; a Leu-to-Gly mutation destabilized the dimer by >10 kcal/mol! Although the destabilization caused by side chain substitutions extracted a large thermodynamic price, the structural and thermodynamic stability of DF1 proved sufficient to allow systematic evaluation of sequence–structure–reactivity relationships.
■ THE ENVIRONMENT OF THE DIIRON SITE DICTATES ITS REACTIVITY
The Ferroxidase Reaction
The simplest reaction catalyzed by diiron proteins is the two-electron ferroxidase reaction, in which the two Fe2+ ions reduce O2 to generate peroxide plus a diferric center (Scheme 1). The resulting di-Fe3+ center often recruits a stabilizing hydroxide or oxo (O2−) group that bridges the two metal ions.
Scheme 1.

Ferroxidase Reaction Catalyzed by DFsc
The DF proteins also catalyze the ferroxidase reaction, whose rate tends to increase as the bulk of the residues lining the inner and outer substrate-access channel decreases, reaching rates similar to natural diiron proteins when all four Leu residues (Figure 5) are converted to Ala. Solomon and coworkers have examined the electronic structure of the diferrous site and the detailed mechanism of its reaction with O2.20,44–47 In DFsc and DF2, O2 reacts with the diferrous center 1 to form a 1,2-peroxo-bridged diferric intermediate 2, which rapidly decays to form an oxo or hydroxo-bridged diferric species, 3.44,45,48 Surprisingly, however, as the bulk of the channel-lining residues are further reduced to Gly in G4DFsc, the rate of the reaction decreases from ∼2 s−1 to ∼0.02 s−1.46 Spectroscopic studies showed that substitution of four-Ala for four-Gly residues increases the coordination numbers of the diferrous site from 4/5 to 5/5. The extra ligand is likely a water, which changes the O2 reaction to favor end-on binding at a single Fe2+.46 This binding-mode slows the reaction, because the ferrous ions are only weakly antiferromagnetically coupled by μ−1,3 carboxylate bridges, thereby eliminating an efficient superexchange pathway for electron transfer. A second-shell Tyr, which forms a H-bond to a terminal Glu ligand (Figure 3b,c) also played a critical role in determining the rate of this reaction.
The Oxidase Reaction
As we decreased the bulk of the channel-lining residues, the site became sufficiently large to bind and react with organic substrates. Systematic exploration showed that the 4-Gly variant of DFtet was a good catalyst of the O2-dependent oxidation of 4-aminophenol, 4, to the corresponding quinonemonoimine 5 (Scheme 2) at over 1000-fold the rate of the corresponding background reaction.18
Scheme 2.

Proposed Catalytic Cycle for the 4-Aminophenol Oxidation by DFtet
The product of the oxidation of 4 is the highly reactive 5 (Scheme 3a). We therefore quenched 5 with 3-aminoaniline 8, which first reacts to form an initial adduct 9 that is then oxidized by a second mole of 5 to create the spectroscopically observed product 10 (Scheme 3b,c).
Scheme 3.

Four-Electron Oxidative Coupling Reactions Involving 4-Aminophenol
The presence of four Gly at the inner/outer access sites in DF3 resulted in a designed phenol oxidase whose structure was confirmed by solution NMR.19 We benchmarked DF3 using the well-studied oxidation of 3,5-di-tert-butylcatechol (6) to quinone 7 (Scheme 4).
Scheme 4.

Proposed Catalytic Cycle for Catechol Oxidation by DF3
Similarly to alternative oxidase and plastid terminal oxidase,49 DF3 catalyzed this reaction with saturation kinetics (KM = 2.1 × 10−3 M for 6, kcat = 0.22 s−1, ambient O2). Notably, the highly reactive p-phenylenediamine and o-phenylenediamine were very poor substrates for the enzyme, likely due to their low affinity to the diferric center.19
As we introduced asymmetry into the active site of the DF proteins, the oxidase mechanism changed significantly. The less symmetrical protein G4DFsc oxidizes 4 and displays substrate-dependent reductive activation of oxygen. The diferrous form of G4DFsc binds the phenol, priming the site for binding of O2 in an end-on manner and triggering the subsequent oxidase reaction within the active site.46,47
The effect of asymmetry is even more pronounced in DF-C121 (Figure 4a,c), an asymmetric version of DF3 with an active site modeled after toluene mono-oxygenase (TOMO). DF-C1’s second-shell ligands were changed to those in TOMO, and a single Thr and Phe were introduced into the inner and outer region of the substrate access site, respectively. DF-C1 also oxidizes 4, but the final products are entirely different from other DF proteins under our standard assay conditions. The intermediate 5 (Scheme 3a) was quenched with 8, thus preventing the formation of a heterogeneous mixture of products when it is released from the active site, as observed when DF3 reacted with 4 in the absence of 8.
However, in DF-C1 neither the initially formed quinonemonoimine 5 nor H2O2 are released from the active site. Instead, the protein catalyzes a remarkable cascade of reactions in which three molecules of 4 are oxidatively coupled to form a derivative of Brandowsky’s dye (11) via tandem two-electron oxidative coupling reactions (similar to the first step of Scheme 3b) and a two-electron oxidation of the central ring to the quinone-imine (Scheme 5).
Scheme 5.

Six-Electron Oxidation of 4-Aminophenol Catalyzed by DF-C1
A possible mechanism for the first step of the reaction involves four-electron reduction of O2 to water (taking two electrons each from 4 and di-Fe2+). After coupling additional molecules of 4, the resulting diferric center is reduced to the resting diferrous state to generate the central quinonemonoimine of 11. A remarkable feature of the reaction is the lack of side-products associated with nonspecific oxidative side-reactions and hydrogen peroxide release.
DF-C1 is therefore able to store the reactive oxidation intermediate in the purposely designed binding pocket. The docking model revealed that the proposed intermediate, the 4AP dimer, properly fits into DF-C1, thanks to the stabilizing role of Phe9 by π-stacking interactions (Figure 6a,b). Interestingly, when the 4AP dimer was docked into DF3, a broad range of accessible binding modes was observed in the wider access site (Figure 6c), making 4AP dimerization highly unlikely to take place within the protein.
Figure 6.

Best docking models of the 4AP dimeric intermediate in complex with (a,b) DF-C1 (front and side view, respectively) and (c) DF3 (front view). Reproduced with permission from ref 21. Copyright 2017 John Wiley and Sons.
Thus, DF-C1 mimics natural proteins that guide substrates through reactive species to create stable products without releasing any highly reactive or toxic species.
The Oxygenase Reaction
An even more intriguing result has been obtained by introducing an additional histidine ligand into G4DFsc to mimic the active site of the p-aminobenzoate N-oxygenase,20 which catalyzes hydroxylation of arylamines. The design of this variant was a challenging exercise in protein design, because a single His introduction into DFsc did not afford a stable protein. However, stability could be rescued by introducing a network of second-shell H-bonds to the primary His ligand. The resulting protein, 3His-G4DFsc, has very weak ferroxidase and oxidase activity but catalyzes multiple rounds of conversion of p-anisidine 12 to p-hydroxylamino-anisole 13 (Scheme 6). The diferrous site in 3His-G4DFsc is 5/6 coordinate, and the very weak antiferromagnetic coupling between the two iron ions explains its slow ferroxidase activity (the single open coordination site limits O2 to end-on ligation to a single iron and the low coupling impedes electron transfer from the other iron).46,47 The oxygenase mechanism involves binding of 12 in proximity of the diferrous site, likely through H-bond interactions between the amine group of p-anisidine and a protein residue close to the site. This binding slightly increases the antiferromagnetic coupling and allows formation of an end-on peroxo intermediate. Electrophilic attack on the amine generates the hydroxylamine intermediate 13, which is further oxidized to the nitroso 14 thereby regenerating the reduced diferrous center.46,47
Scheme 6.

Proposed Catalytic Cycle for the p-Anisidine Oxygenation by 3His-G4DFsc
Stabilization of Radical Species through Binding to di-Zn2+−3His-A2DFsc
Enzymes use binding energy to stabilize reactive intermediates that are otherwise inaccessible in aqueous native conditions. To test whether the DF proteins might be capable of stabilizing otherwise reactive semiquinone radical species, we chose the 3,5-di-tert-butyl-semiquinone radical anion (SQ●−, 15), a one-electron oxidized intermediate in the redox triad between catechol 6 and quinone 7 (H2Q/SQ●−/Q) (Scheme 7).
Scheme 7.

Stabilization of a Semiquinone Radical in 3His-A2DFsc
As discussed above, di-Fe-DF3 catalyzes the oxidation of 6 to 7 (Scheme 3) without a detectable SQ●− intermediate. We rationalized that the redox-inert di-Zn2+ derivative might instead stabilize SQ●−. Semiquinones are strong chelating ligands and hence would be specifically stabilized by binding to the Zn2+, relative to the weakly coordinating quinone or catechol. Moreover, the burial of the hydrophobic substituents would help stabilize the SQ●− in the active site.
The di-Zn2+−3His-A2DFsc protein indeed bound tightly to the radical semiquinone anion form of 6 and stabilized it.50 Solution NMR in conjunction with molecular dynamics and QM/MM calculations showed the substrate chelating a single Zn2+ in the active site (Figure 7). Spectroelectrochemical redox titrations showed that the protein stabilized the semiquinone by reducing the midpoint potential for its formation via the one-electron oxidation of the catechol by approximately 400 mV (9 kcal/mol−1). Furthermore, a set of SQ●− derivatives was examined to disentangle the factors directing the binding affinity and thus the specificity of the designed cleft. Any electronic role could be revealed for the electron-donating t-butyl groups, as binding did not take place for other more donating substituents. Only 4-t-butylcathecol semiquinone could bind the protein but with a much lower yield than its di-t-butyl analogue, leading to the conclusion that a single hydrophobic t-butyl group dramatically affected the binding energy of the finely tuned DF cleft. Hence, the stability of a radical species was drastically stabilized by harnessing its binding-energy to the metalloprotein.
Figure 7.

View of di-Zn2+−3His-A2DFsc stabilizing the t-butyl groups of SQ●− (shown as sticks in magenta) with the hydrophobic residues lining the substrate access site (shown as spheres). Reproduced with permission from ref 50. Copyright 2016 Springer Nature.
■ BEYOND DIIRON PROTEINS
Porphyrin-Binding Proteins
Complementary work from our laboratories on porphyrin-binding peptides and proteins further illustrates our modular inside-out approach to protein design, in which the target cofactor and its environment drive the design of both the tertiary structure and sequence of a protein. In early work with the Dutton lab, we designed the first de novo four-helix heme-binding helical bundle proteins using a sequence-based approach.6,7 However, these “maquettes” have evaded structural determination, largely due their dynamical properties. With the exception of shorter covalently linked peptide–heme complexes studied in Naples,51,52 and of one apo-protein that showed a hydrophobically collapsed binding site with no space for binding heme,53 structures were lacking for designed heme-binding proteins. We therefore recently extended the methods used in the design of DF1 (Figure 2) to porphyrin-binding proteins. We purposefully included a folded core remote from the ligand-binding site and optimized its sequence and structure in concert with the binding to ensure appropriate coupling between the free energy of folding and specific binding. While the principles were the same as followed for DF1, the process was facilitated by the availability of Rosetta.54 The resulting protein, PS1, bound the intended synthetic porphyrin target Zn2+[m-tetrakis(trifluoromethyl)-porphyrinate)], and its NMR structure was in sub-angstrom agreement with the design (Figure 8a).55 The structure of the apo-protein retained the remote core packing of the holo, predisposing a flexible binding region for the desired ligand-binding geometry. These results illustrate the unification of core packing and binding site definition as a central principle of ligand-binding protein design.
Figure 8.

(a) NMR structure of PS1 (PDB-ID 5tgy). (b) X-ray structure of 4DH2 (PDB-ID 5wlm) and close-view of the tetranuclear zinc cluster (histidines have been hidden for clarity). (c) Rocker tetrameric model (PDB-ID 2muz, 4p6j), depicted in a membrane (gray rectangle).
Symmetry and Frustrated Symmetry in Protein Design
Symmetry can greatly facilitate design of helical bundles that bind symmetrical cofactors. Extending the methods used for DF, we recently designed and structurally characterized a fourhelix bundle that binds a non-natural tetranuclear cluster, consisting of four Zn2+ and four carboxylate oxygens, situated at the vertices of a distorted cube-like structure.56,57 The site also includes four His ligands and 16 polar side chains in a fully connected hydrogen-bonded network (Figure 8b). Similar to DFtet, the designed proteins have apolar cores at the top and bottom of the bundle, which drive the assembly of the bundle.
Interesting properties can result when an assembly cannot stably pack in a fully symmetrical bundle, as seen in our design of a Zn2+/proton transporter (named Rocker), intended to oscillate between two conformations as in the alternating access mechanism.22 Key to our success was the observation that in the apo-form the Glu ligands in DF proteins were largely protonated and binding of Zn2+ displaced these protons, providing a means to achieve thermodynamic coupling. Indeed, our transmembrane channel is able to rock between two energetically equivalent conformations to facilitate coupled hopping of Zn2+ and protons through the transporter. X-ray crystallography and solid-state NMR showed that the overall helical bundle was formed from two pairs of tightly interacting helices that interact weakly along a more dynamic interface.22 Vesicle flux experiments show that as Zn2+ ions diffuse down their concentration gradients, protons are antiported (Figure 8c).
■ OUTLOOK
The development of DF proteins spans from the very earliest days of de novo protein design to the current, rapidly expanding design of metalloproteins bearing both natural and artificial cofactors.58–63 We learned many lessons of interest to contemporary protein design, from the use of parametric equations for designing functional proteins to the construction of functionally essential hydrogen-bonded networks. The DF proteins also illustrate stability–function compensation,16,64 which placed increasingly stringent demands on design as functional demands increase. We adopted the philosophy that if we understand proteins we should be able to design them from scratch, and we focused on purposeful design and rigorous evaluation of proteins one-at-a-time. Therefore, we have avoided the use of large libraries or directed evolution, which can be most helpful when the initially designed sequences are close to a solution but require optimization through a more stochastic search of sequence space.54,65 Indeed, in an impressive contribution, Hilvert, Kuhlman, and co-workers recently described the use of directed evolution to discover a highly efficient Zn2+-dependent esterase in a de novo designed protein scaffold.66
The methods used in construction of DF also provide a valuable link between coordination complexes assembled from small molecule ligands and the study of highly evolved modern-day proteins. Coordination complexes are prepared through self-assembly of metal ions with organic ligands, while our approach similarly starts with peptides as ligands, with loops optionally added at later stages of the design process (Figure 2, 4). In small transition metal complexes, the geometric positioning of the liganding groups is achieved through clever synthesis of multivalent ligands. By contrast, in de novo design, positioning of the ligands is achieved through protein folding. Thus, tight interdigitation of core side chains structurally restrains the first- and second-shell packing around the metal-binding site. In our de novo-designed proteins, outer-sphere ligands can be easily incorporated. The construction of deep substrate-binding pockets, a hallmark of proteins, has been accomplished in DF proteins. Structural characterization and docking experiments of substrates carried out on DF proteins revealed that it is possible to fine-tune the binding pocket width to obtain different specificity. Finally, while we focus here on helical bundles, we have used similar approaches to design small miniproteins rich in beta structure.12,13 In summary, the de novo construction of metalloproteins by intra- or intermolecular assembly of peptide units holds considerable promise for understanding the mechanisms of natural proteins as well as the design of proteins with complex functions, also able to bind metal ions not commonly used in nature.
ACKNOWLEDGMENTS
We are grateful to all members of the Lombardi and DeGrado Labs, who made the work reported in this Account possible. The research was mainly supported by NIH, MIUR and EU funding.
Biographies
Angela Lombardi (Master degree in Industrial Chemistry; Ph.D. in Chemical Science, University of Napoli Federico II) is a professor of inorganic and bioinorganic chemistry at the University Federico II of Napoli. Angela’s research interests mainly focus on the design of peptide-based metalloprotein models able to promote oxidative reactions and with potential applications in biosensing and diagnostics. Angela’s research interests also include bioactive peptides, such as antimicrobial peptides and hormones.
Fabio Pirro received his B.S. and M.S. degrees in Chemistry from the University of Napoli Federico II. In 2017, he started the graduate program in Chemical Sciences under the supervision of Professor Angela Lombardi in Napoli. In the same year, he spent six months in San Francisco in the DeGrado Lab as visiting graduate. His research focuses on the development of de novo designed metalloproteins bearing natural and unnatural metal cofactors.
Ornella Maglio is a researcher at the Institute of Biostructures and Bioimaging of the CNR in Napoli. She graduated in Industrial Chemistry in 1989 from the University of Napoli Federico II. In 1992, she received her Ph.D. in Chemical Sciences. In 1998, she was visiting fellow at the National Institute of Medical Research of London, in the group of Annalisa Pastore. Her research interests include design, synthesis, and structural characterization by NMR of bioactive peptides and metalloprotein models.
Marco Chino received his B.S. and M.S. degrees in Chemistry from University of Napoli Federico II. In 2014, he received his Ph.D. in Chemical Sciences under the supervision of Professor Angela Lombardi, after spending six months in San Francisco in the DeGrado Lab. He is now a fixed-term researcher in the artificial metalloenzymes group at the Department of Chemical Sciences in Napoli. His research interests include computational protein design, bioinorganic chemistry, biosensors, and protein–protein interactions.
William (Bill) DeGrado (B.A. Kalamazoo College, Ph.D. in organic chemistry University of Chicago, with Emil T. (Tom) Kaiser and Ferenc Kézdy) worked at DuPont Central Research and DuPont Merck Pharmaceutical Company from 1981 to 1996. He moved to the Department of Biochemistry and Biophysics of University of Pennsylvania in 1996, and in 2011 to University of California, San Francisco. He and Angela Lombardi began their collaboration in 1995 during her sabbatical year at DuPont Merck.
Footnotes
The authors declare no competing financial interest.
Published as part of the Accounts of Chemical Research special issue “Artificial Metalloenzymes and Abiological Catalysis of Metalloenzymes”.
■ REFERENCES
- (1).Regan L; Degrado WF Characterization of a Helical Protein Designed from 1st Principles. Science 1988, 241, 976–978. [DOI] [PubMed] [Google Scholar]
- (2).Walsh STR; Cheng H; Bryson JW; Roder H; DeGrado WF Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kuhlman B; Dantas G; Ireton GC; Varani G; Stoddard BL; Baker D Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science 2003, 302, 1364–1368. [DOI] [PubMed] [Google Scholar]
- (4).DeGrado WF; Summa CM; Pavone V; Nastri F; Lombardi A De novo design and structural characterization of proteins and metalloproteins. Annu. Rev. Biochem. 1999, 68, 779–819. [DOI] [PubMed] [Google Scholar]
- (5).Crichton RR Biological Inorganic Chemistry: A New Introduction to Molecular Structure and Function In Biological Inorganic Chemistry; 3rd ed.; Crichton R., Ed.; Academic Press, 2019. [Google Scholar]
- (6).Choma CT; Lear JD; Nelson MJ; Dutton PL; Robertson DE; DeGrado WF Design of a Heme-Binding 4-Helix Bundle. J. Am. Chem. Soc. 1994, 116, 856–865. [Google Scholar]
- (7).Robertson DE; Farid RS; Moser CC; Urbauer JL; Mulholland SE; Pidikiti R; Lear JD; Wand AJ; DeGrado WF; Dutton PL Design and Synthesis of Multi-Heme Proteins. Nature 1994, 368, 425–431. [DOI] [PubMed] [Google Scholar]
- (8).Korendovych IV; Senes A; Kim YH; Lear JD; Fry HC; Therien MJ; Blasie JK; Walker FA; DeGrado WF De Novo Design and Molecular Assembly of a Transmembrane Diporphyrin-Binding Protein Complex. J. Am. Chem. Soc. 2010, 132, 15516–5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Faiella M; Maglio O; Nastri F; Lombardi A; Lista L; Hagen WR; Pavone V De Novo Design, Synthesis and Characterisation of MP3, A New Catalytic Four-Helix Bundle Hemeprotein. Chem. - Eur. J 2012, 18, 15960–15971. [DOI] [PubMed] [Google Scholar]
- (10).Vitale R; Lista L; Cerrone C; Caserta G; Chino M; Maglio O; Nastri F; Pavone V; Lombardi A An artificial heme-enzyme with enhanced catalytic activity: evolution, functional screening and structural characterization. Org. Biomol. Chem. 2015, 13, 4859–4868. [DOI] [PubMed] [Google Scholar]
- (11).Caserta G; Chino M; Firpo V; Zambrano G; Leone L; D’Alonzo D; Nastri F; Maglio O; Pavone V; Lombardi A Enhancement of Peroxidase Activity in Artificial Mimochrome VI Catalysts through Rational Design. ChemBioChem 2018, 19, 1823–1826. [DOI] [PubMed] [Google Scholar]
- (12).Lombardi A; Marasco D; Maglio O; Di Costanzo L; Nastri F; Pavone V Miniaturized metalloproteins: Application to iron-sulfur proteins. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 11922–11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nanda V; Rosenblatt MM; Osyczka A; Kono H; Getahun Z; Dutton PL; Saven JG; DeGrado WF De novo design of a redox-active minimal rubredoxin mimic. J. Am. Chem. Soc. 2005, 127, 5804–5805. [DOI] [PubMed] [Google Scholar]
- (14).Summa CM; Lombardi A; Lewis M; DeGrado WF Tertiary templates for the design of diiron proteins. Curr. Opin. Struct. Biol. 1999, 9, 500–508. [DOI] [PubMed] [Google Scholar]
- (15).Lombardi A; Summa CM; Geremia S; Randaccio L; Pavone V; DeGrado WF Retrostructural analysis of metalloproteins: Application to the design of a minimal model for diiron proteins. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 6298–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Maglio O; Nastri F; Pavone V; Lombardi A; DeGrado WF Preorganization of molecular binding sites in designed diiron proteins. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3772–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Summa CM; Rosenblatt MM; Hong JK; Lear JD; DeGrado WF Computational de novo design, and characterization of an A(2)B(2) diiron protein. J. Mol. Biol. 2002, 321, 923–938. [DOI] [PubMed] [Google Scholar]
- (18).Kaplan J; DeGrado WF De novo design of catalytic proteins. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 11566–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Faiella M; Andreozzi C; de Rosales RTM; Pavone V; Maglio O; Nastri F; DeGrado WF; Lombardi A An artificial diiron oxo-protein with phenol oxidase activity. Nat. Chem. Biol. 2009, 5, 882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Reig AJ; Pires MM; Snyder RA; Wu YB; Jo H; Kulp DW; Butch SE; Calhoun JR; Szyperski TA; Solomon EI; DeGrado WF Alteration of the oxygen-dependent reactivity of de novo Due Ferri proteins. Nat. Chem. 2012, 4, 900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chino M; Leone L; Maglio O; D’Alonzo D; Pirro F; Pavone V; Nastri F; Lombardi A A De Novo Heterodimeric Due Ferri Protein Minimizes the Release of Reactive Intermediates in Dioxygen-Dependent Oxidation. Angew. Chem., Int. Ed. 2017, 56, 15580–15583. [DOI] [PubMed] [Google Scholar]
- (22).Joh NH; Wang T; Bhate MP; Acharya R; Wu YB; Grabe M; Hong M; Grigoryan G; DeGrado WF De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 2014, 346, 1520–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Marsh ENG; Waugh MW Aldehyde Decarbonylases: Enigmatic Enzymes of Hydrocarbon Biosynthesis. ACS Catal 2013, 3, 2515–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang W; Liang AD; Lippard SJ Coupling Oxygen Consumption with Hydrocarbon Oxidation in Bacterial Multi-component Monooxygenases. Acc. Chem. Res. 2015, 48, 2632–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jasniewski AJ; Que L Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Solomon EI; Park K Structure/function correlations over binuclear non-heme iron active sites. JBIC, J. Biol. Inorg. Chem. 2016, 21, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Acheson JF; Bailey LJ; Brunold TC; Fox BG In-crystal reaction cycle of a toluene-bound diiron hydroxylase. Nature 2017, 544, 191–195. [DOI] [PubMed] [Google Scholar]
- (28).Ho SP; Degrado WF Design of a 4-Helix Bundle Protein - Synthesis of Peptides Which Self-Associate into a Helical Protein. J. Am. Chem. Soc. 1987, 109, 6751–6758. [Google Scholar]
- (29).Harbury PB; Plecs JJ; Tidor B; Alber T; Kim PS High-resolution protein design with backbone freedom. Science 1998, 282, 1462–1467. [DOI] [PubMed] [Google Scholar]
- (30).Desjarlais JR; Handel TM De-Novo Design of the Hydrophobic Cores of Proteins. Protein Sci. 1995, 4, 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Su A; Mayo SL Coupling backbone flexibility and amino acid sequence selection in protein design. Protein Sci. 1997, 6, 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bryson JW; Desjarlais JR; Handel TM; DeGrado WF From coiled coils to small globular proteins: Design of a native-like three-helix bundle. Protein Sci. 1998, 7, 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lombardi A; Bryson JW; Ghirlanda G; DeGrado WF Design of a synthetic receptor for the calmodulin-binding domain of calcineurin. J. Am. Chem. Soc. 1997, 119, 12378–12379. [Google Scholar]
- (34).Ghirlanda G; Lear JD; Lombardi A; DeGrado WF From synthetic coiled coils to functional proteins: Automated design of a receptor for the calmodulin-binding domain of calcineurin. J. Mol. Biol. 1998, 281, 379–391. [DOI] [PubMed] [Google Scholar]
- (35).DeGrado WF; Di Costanzo L; Geremia S; Lombardi A; Pavone V; Randaccio L Sliding helix and change of coordination geometry in a model Di-Mn-II protein. Angew. Chem., Int. Ed. 2003, 42, 417–420. [DOI] [PubMed] [Google Scholar]
- (36).Torres Martin de Rosales R; Faiella M; Farquhar E; Que L; Andreozzi C; Pavone V; Maglio O; Nastri F; Lombardi A Spectroscopic and metal-binding properties of DF3: an artificial protein able to accommodate different metal ions. JBIC, J. Biol. Inorg. Chem. 2010, 15, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lahr SJ; Engel DE; Stayrook SE; Maglio O; North B; Geremia S; Lombardi A; DeGrado WF Analysis and design of turns in alpha-helical hairpins. J. Mol. Biol. 2005, 346, 1441–1454. [DOI] [PubMed] [Google Scholar]
- (38).Chino M; Leone L; Maglio O; Lombardi A Designing Covalently Linked Heterodimeric Four-Helix Bundles. Methods Enzymol. 2016, 580, 471–499. [DOI] [PubMed] [Google Scholar]
- (39).Calhoun JR; Kono H; Lahr SJ; Wang W; DeGrado WF; Saven JG Computational design and characterization of a monomeric helical dinuclear metalloprotein. J. Mol. Biol. 2003, 334, 1101–1115. [DOI] [PubMed] [Google Scholar]
- (40).Marsh ENG; DeGrado WF Noncovalent self-assembly of a heterotetrameric diiron protein. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5150–5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Grigoryan G; Reinke AW; Keating AE Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 2009, 458, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Geremia S; Di Costanzo L; Randaccio L; Engel DE; Lombardi A; Nastri F; DeGrado WF Response of a designed metalloprotein to changes in metal ion coordination, exogenous ligands, and active site volume determined by X-ray crystallography. J. Am. Chem. Soc. 2005, 127, 17266–17276. [DOI] [PubMed] [Google Scholar]
- (43).Di Costanzo L; Wade H; Geremia S; Randaccio L; Pavone V; DeGrado WF; Lombardi A Toward the de novo design of a catalytically active helix bundle: A substrate-accessible carboxylate-bridged dinuclear metal center. J. Am. Chem. Soc. 2001, 123, 12749–12757. [DOI] [PubMed] [Google Scholar]
- (44).Bell CB; Calhoun JR; Bobyr E; Wei PP; Hedman B; Hodgson KO; DeGrado WF; Solomon ET Spectroscopic Definition of the Biferrous and Biferric Sites in de Novo Designed Four-Helix Bundle DFsc Peptides: Implications for O2 Reactivity of Binuclear Non-Heme Iron Enzymes. Biochemistry 2009, 48, 59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wei PP; Skulan AJ; Wade H; DeGrado WF; Solomon EI Spectroscopic and computational studies of the de novo designed protein DF2t: Correlation to the biferrous active site of ribonucleotide reductase and factors that affect O2 reactivity. J. Am. Chem. Soc. 2005, 127, 16098–16106. [DOI] [PubMed] [Google Scholar]
- (46).Snyder RA; Betzu J; Butch SE; Reig AJ; DeGrado WF; Solomon EI Systematic Perturbations of Binuclear Non-heme Iron Sites: Structure and Dioxygen Reactivity of de Novo Due Ferri Proteins. Biochemistry 2015, 54, 4637–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Snyder RA; Butch SE; Reig AJ; DeGrado WF; Solomon EI Molecular-Level Insight into the Differential Oxidase and Oxygenase Reactivities of de Novo Due Ferri Proteins. J. Am. Chem. Soc. 2015, 137, 9302–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wade H; Stayrook SE; DeGrado WF The structure of a designed diiron(III) protein: Implications for cofactor stabilization and catalysis. Angew. Chem., Int. Ed. 2006, 45, 4951–4954. [DOI] [PubMed] [Google Scholar]
- (49).Berthold DA; Stenmark P Membrane-bound diiron carboxylate proteins. Annu. Rev. Plant Biol. 2003, 54, 497–517. [DOI] [PubMed] [Google Scholar]
- (50).Ulas G; Lemmin T; Wu YB; Gassner GT; DeGrado WF Designed metalloprotein stabilizes a semiquinone radical. Nat. Chem. 2016, 8, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).D’Auria G; Maglio O; Nastri F; Lombardi A; Mazzeo M; Morelli G; Paolillo L; Pedone C; Pavone V Hemoprotein models based on a covalent helix-heme-helix sandwich. 2. Structural characterization of CoIII mimochrome I Δ and Λ isomers. Chem. - Eur. J. 1997, 3, 350–362. [Google Scholar]
- (52).Di Costanzo L; Geremia S; Randaccio L; Nastri F; Maglio O; Lombardi A; Pavone V Miniaturized heme proteins: crystal structure of Co(III)-mimochrome IV. JBIC, J. Biol. Inorg. Chem. 2004, 9, 1017–1027. [DOI] [PubMed] [Google Scholar]
- (53).Huang SS; Gibney BR; Stayrook SE; Dutton PL; Lewis M X-ray structure of a Maquette scaffold. J. Mol. Biol. 2003, 326, 1219–1225. [DOI] [PubMed] [Google Scholar]
- (54).Huang PS; Boyken SE; Baker D The coming of age of de novo protein design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (55).Polizzi NF; Wu Y; Lemmin T; Maxwell AM; Zhang SQ; Rawson J; Beratan DN; Therien MJ; DeGrado WF De novo design of a hyperstable non-natural protein-ligand complex with sub-accuracy. Nat. Chem 2017, 9, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhang SQ; Chino M; Liu LJ; Tang YZ; Hu XZ; DeGrado WF; Lombardi A De Novo Design of Tetranuclear Transition Metal Clusters Stabilized by Hydrogen-Bonded Networks in Helical Bundles. J. Am. Chem. Soc. 2018, 140, 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chino M; Zhang SQ; Pirro F; Leone L; Maglio O; Lombardi A; DeGrado WF Spectroscopic and metal binding properties of a de novo metalloprotein binding a tetrazinc cluster. Biopolymers 2018, 109, e23339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Key HM; Dydio P; Clark DS; Hartwig JF Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Nastri F; Chino M; Maglio O; Bhagi-Damodaran A; Lu Y; Lombardi A Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Schwizer F; Okamoto Y; Heinisch T; Gu YF; Pellizzoni MM; Lebrun V; Reuter R; Kohler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (61).Zaytsev DV; Morozov VA; Fan JF; Zhu XC; Mukherjee M; Ni SS; Kennedy MA; Ogawa MY Metal-binding properties and structural characterization of a self-assembled coiled coil: Formation of a polynuclear Cd-thiolate cluster. J. Inorg. Biochem. 2013, 119, 1–9. [DOI] [PubMed] [Google Scholar]
- (62).Flores M; Olson TL; Wang D; Edwardraja S; Shinde S; Williams JC; Ghirlanda G; Allen JP Copper Environment in Artificial Metalloproteins Probed by Electron Paramagnetic Resonance Spectroscopy. J. Phys. Chem. B 2015, 119, 13825–13833. [DOI] [PubMed] [Google Scholar]
- (63).Teare P; Smith CF; Adams SJ; Anbu S; Ciani B; Jeuken LJC; Peacock AFA pH dependent binding in de novo hetero bimetallic coiled coils. Dalton T 2018, 47, 10784–10790. [DOI] [PubMed] [Google Scholar]
- (64).Shoichet BK; Baase WA; Kuroki R; Matthews BW A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Dou JY; Vorobieva AA; Sheffler W; Doyle LA; Park H; Bick MJ; Mao BC; Foight GW; Lee MY; Gagnon LA; Carter L; Sankaran B; Ovchinnikov S; Marcos E; Huang PS; Vaughan JC; Stoddard BL; Baker D De novo design of a fluorescence-activating beta-barrel. Nature 2018, 561, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Studer S; Hansen DA; Pianowski ZL; Mittl PRE; Debon A; Guffy SL; Der BS; Kuhlman B; Hilvert D Evolution of a highly active and enantiospecific metalloenzyme from short peptides. Science 2018, 362, 1285–1288. [DOI] [PubMed] [Google Scholar]