

Abstract
Recombinant proteins are the mainstay of biopharmaceuticals. A key challenge in the manufacturing and formulation of protein biologic products is the tendency for the active pharmaceutical ingredients to aggregate, resulting in irreversible drug loss, and an increase in immunogenicity risk. While the molecular mechanisms of protein aggregation have been discussed extensively in the literature, knowledge gaps remain in connecting the phenomenon in the context of immunogenicity of biotherapeutics. In this review, we discussed factors that drive aggregation of pharmaceutical recombinant proteins, and highlighted methods of prediction and mitigation that can be deployed through the development stages, from formulation to bioproduction. The purpose is to stimulate new dialogs that would bridge the interface between physical characterizations of protein aggregates in biotherapeutics and the functional attributes of the immune system.
Keywords: monoclonal antibodies, high-concentration protein formulations, anti-drug antibodies, T cell epitopes, bioprocessing
I. Introduction
The rapid expansion of biologic drugs in recent years has led to a pressing need for early and precise control of product quality, in which protein aggregation remains a key challenge because of its implications in potency and safety [1]. Understanding the molecular mechanisms of protein aggregation is critical for developing mitigation strategies. Proteins aggregate through three main mechanisms, broadly defined by the seeding entity: native monomers, denatured proteins, and pre-existing aggregates [2, 3]. Monomers of native proteins can self-associate into oligomers through complementarity of charge-charge interactions, or through covalent linkages formed between hydrophilic and hydrophobic residues on the protein exterior. Low molecular weights, non-covalent oligomers can revert to their native states, but as the oligomers increase in size over time, the associations become irreversible. It is assumed that in any given protein product there exists a denatured fraction [4], which has a propensity to undergo irreversible aggregation. Because protein conformation is a dynamic phenomenon, partially unfolded proteins may refold under certain conditions. However, the free energy and kinetics generally favor aggregation rather than refolding [5]. Native protein monomers can also aggregate by adhering to pre-existing protein oligomers, contaminants, or vessel surface, rapidly expand via nucleation.
Aggregates can be classified as soluble and insoluble [6]. Soluble aggregates have low molecular mass and may be reversible. A small amount of soluble aggregates, between 5 to 10%, for example, may be acceptable in biologic products, because the belief is that it is generally impractical to eliminate aggregates below these levels [7]. When protein aggregation exceeds the solution solubility limit, aggregates become irreversible and precipitate out of solution. The tolerable quantity of insoluble aggregates appears to correlate with the size of particulates detected in the protein product upon reconstitution. Particles as low as 150 μm in diameter in injectable products may be detected visually [8]. Perhaps paradoxically, particles that below 150 μm are more likely to elicit immune reactions [9-11]. Particles with a hydrodynamic radius of ≤ 50-100μm are generally considered subvisible [6], with those that are ≥ 10 μm occlude blood flow [12]. Described in the U.S. Pharmacopeia (USP) chapter <788> is a subvisible particle counting method, which sets the acceptable limit of particulate matter in a container of ≤ 100 mL to be 6000 particles ≥10 μm and 600 particles ≥ 25 μm. Based on the standards, 10 μm is most often invoked as a limit in analytical regulatory guidance. Because many recently developed protein products are administered subcutaneously or intramuscularly, limiting aggregates based on the 10 μm threshold to avoid blood vessel occlusion has become less relevant. Furthermore, aggregates ≤10 μm possess a greater risk in product stability and immunogenicity [9-11, 13, 14]. Therefore, the U.S. Food and Drug Administration (FDA) has tighten the regulatory scrutiny of aggregates below 10 μm in biologic products [9].
Beyond the classifications based on the aggregation mechanisms, solubility, and size, protein aggregation can be delineated as intrinsic and extrinsic. Intrinsic protein aggregation arises within the protein formulation during the synthesis and purification steps. Extrinsic aggregation, in contrast, results from the contacts of protein with external sources during processing, such as glass surfaces inside containers, stainless steel of bioprocessing equipment, or silicon oil droplets inside pre-filled syringes. Because these two categories intertwine and collectively contribute to protein aggregates detected downstream, specific attribution to each is often not done. In this review, we will focus on intrinsic protein aggregation, in which prediction and mitigation approaches can be applied as early as in the drug development process. Summarized in Figure 1 are the main aspects and motivations of our discussion.
Figure 1.
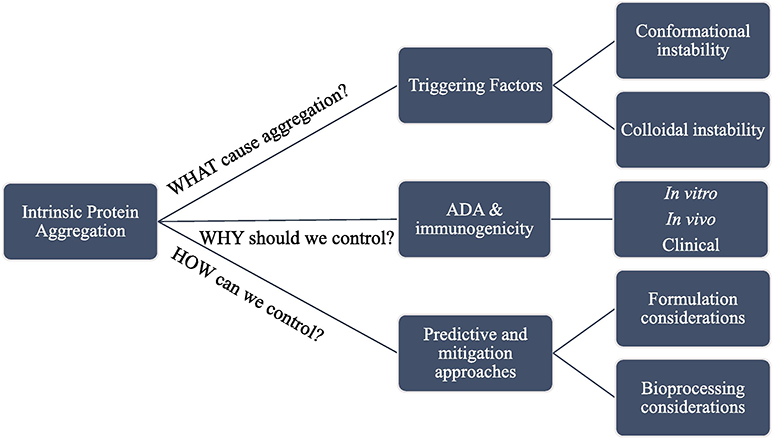
Intrinsic protein aggregation evaluation and mitigation from multiple aspects
In the following narrative, we discussed intrinsic protein aggregation and its driving factors from conformational and colloidal stability perspectives. The relationships between protein aggregation and immunogenicity are examined across in vitro, in vivo and clinical studies. We highlight the advantages and the uncertainties associated with aggregation prediction, and mitigating strategies at the formulation and bioprocessing stages. The issues are reflected in the context of monoclonal antibodies (mAbs), the mainstay of the current biotherapeutics.
II. Intrinsic Protein Aggregation
1. Conformational and Colloidal Stability
Protein aggregation is evaluated through the assessment of conformational and colloidal stability. Proteins are driven by thermodynamics to retain their native conformations at the lowest energy level to attain a high stability [15, 16]. In hydrated environments, hydrophobic amino acid residues prefer to position themselves inside of the core while hydrophilic residues stay in the outer shell. Not all apolar residues are completely buried in the interior; water molecules tend to organize into lose structures around the hydrophobic moieties [17]. The system entropy increases as structured water are released into the bulk solvent upon protein folding, surface adsorption or collapse of hydrophobic patches [18]. Together with a decrease in enthalpy due to bond formations among amino residues, the increase in entropic cost reduces the system’s Gibb’s free energy [19, 20]. The thermodynamic driving force facilitates conformational changes, which enable further colloidal instability and eventually would result in aggregation of the protein.
Colloidal stability is a function of the intermolecular interactions between protein molecules [21, 22]. The forces include steric repulsion, short-range attraction, and electrostatic interactions [17]. The stability is usually determined through the osmotic second viral coefficient (B22), which can be measured with static light scattering and ultracentrifugation as a function of protein solution concentrations [21-23]. Light scattering techniques are used to detect particulates in protein products based on the Brownian motion in diluted systems [24]. The velocity of the motion relates inversely to the size of particle insofar aggregates diffuse slower than protein monomers [25]. B22 is derived from the slope of linear regression of light scattering intensity and protein concentration. Negative B22 values indicate net attractive protein-protein interactions (PPI), which dominate in protein aggregates, while positive B22 values denote the opposite [26].
Although the use of the B22 coefficient to predict aggregation has been confirmed in various studies, the correlation does not hold in systems where conformation change is the main driving force of aggregation [27]. The deviation is due in part to the presumption that interactions among native proteins and partially unfolded proteins are equivalent [27]. Furthermore, negative B22 values poorly predict the aggregate propensity as exemplified in Saito et al.’ s study in which three antibodies with different sequences and the same B22 value exhibited different aggregation profiles [28]. Nevertheless, conformation and colloidal stability remains a useful perspective in modeling aggregation.
2. Factors triggering intrinsic protein aggregation
Protein aggregation can be caused by several factors, which have been analyzed in multiple publications. Summarized in Table 1 are the potential impacts of major protein and solution factors on the aggregation of mAbs and Fc-fusion protein reported in the last five years.
Table 1.
Recent reports on the impact of major factors on intrinsic aggregation of mAbs and Fc-fusion proteins.
| Factor | Cofactors | Type of Protein | Mode of instability |
Reported effects | References |
|---|---|---|---|---|---|
| pH | mAbs | conformational | Low pH (2.7) increases IgG hydrophobicity and induces aggregation. | [29] | |
| pH | mAbs | conformational | pH-shift from 2 to 7 induces irreversible aggregates. | [30] | |
| pH | mAbs | conformational | IgG subclasses respond differently to pH-induced aggregation. | [31] | |
| pH | mAbs | conformational | Nivolumab is susceptible to aggregation (30%) at pH 3.5. IgG4 aggregation is dependent on the Fc region. Propensity to acid-induced aggregation: IgG4 > IgG2 > aglyco-IgG1 > IgG1. |
[32] | |
| pH | Temp | Fc fusion protein | conformational | Reducing pH 7, 30°C to pH 6.7, 27°C significantly decreases aggregation. | [33] |
| Temp | pH | Fc fusion protein mAb |
conformational | Low temperature and/or low pH in a mammalian cell culture reduce misfolded and aggregated proteins. | [34] |
| pH | Excipient | mAbs | colloidal | Isoelectric precipitation of plasma proteins leads to dextrose-mediated mAbs aggregation. | [35] |
| Freeze-thaw | pH | mAbs Fc fusion protein |
conformational | Adjusting protein solution (frozen at pH 3) in a higher pH buffer (pH 7) during thawing increases aggregation. | [36] |
| Freeze-thaw | mAbs | conformational | Slow thawing induces highest aggregate and subvisible particle levels. Fast freeze and fast thaw are better for mAbs. |
[37] | |
| Chemical modification | pH | mAbs | colloidal | Deamination increases aggregation at low pH 3.8. | [38] |
| Chemical modification | pH/Temp | mAbs Fc fusion proteins |
conformational colloidal | Met oxidation induces aggregation at low pH and high temperature. Trp oxidation and Asn deamidation induces covalent crosslinked and attractive colloidal interactions. |
[39] |
| Chemical modification | Biotherapeutic protein | conformational | Asn deamidation causes less unfolding at the aggregation hotspots identified with NMR compared to methionine oxidation. | [40] | |
| Buffer | Ion | bispecific Ab | colloidal | His + Glu at low ionic strength stabilize antibody and reduce aggregation. | [41] |
| Ion | pH | mAbs | colloidal | Accumulation of citrate ions at the protein surface decreases protein-protein repulsion (compared to acetate buffer). | [42] |
| Ion | mAbs | conformational colloidal | Sodium citrate buffer reduces protein stability compared to sodium acetate buffer. | [43] |
The combined effects of these factors can be explained from the conformational and colloidal stability standpoints. In Table 1, pH is repeatedly reported to have diverse effects on intrinsic protein aggregation. At pH at the alkaline or acidic extremes or after significant pH shifts, aggregation propensity increase due to PPI changes from charge alterations that induce partial unfolding [29, 30, 32]. In certain cases, as solution pH approaches the protein isoelectric point (pI), protein solubility is reduced and leads to aggregation. In other cases, therapeutic proteins aggregate due to isoelectric precipitation of non-therapeutic components. Luo and Zhang revealed that 5% dextrose (a common diluent) induced the precipitation of plasma proteins (pI 5.5-6.7) when mixed with mAbs formulations (pH 6) [35]. Consequently, the therapeutic mAbs formed insoluble aggregates from binding to precursor precipitations. The observation of this phenomenon led the FDA to prohibit the use of 5% dextrose as diluent in intravenous formulations of Herceptin, Avastin, and Remicade [44]. Furthermore, the effect of pH on aggregation propensity varies among the antibody subclasses [31, 32]. In acidic pH, IgG1 remains monomeric whereas IgG2 and IgG4 tend to oligomerize. When the solutions are neutralized, IgG2 partially reverts to being monomeric whereas IgG4 forms aggregates.
With regard to the influence of ionic species on protein aggregation, it was recently reported that mAbs formulated in sodium acetate buffer exhibited higher conformational and colloidal stabilities than those in sodium citrate buffer [42, 43]. Compared to acetate anions, citrate anions have higher valency, which increases the shielding of positive charges and reduces intermolecular repulsive forces. The preferential binding of citrate ions over acetate ions also explains the smaller change in Gibbs free energy when mAbs were formulated in citrate buffer.
Chemical modification is another intrinsic factor that impacts colloidal and conformational stabilities of proteins. Alam et al. reported an increase in aggregation upon deamidation and oxidation modification on antibodies [38, 39]. Deamidation of asparagine (Asn) is a common modification in antibody drug design [38]. The deamidated side chain, isoaspartic acids, carries a negative charge at neutral pH, via the formation of hydrophobic cyclic succinimide intermediates [45, 46]. The changes in net charge and hydrophobicity of the antibody would affect the molecule colloidal stability. As a result, increased in turbidity corresponding to high-molecular-weight species were observed in deamidated antibodies. Notably, deamidation enhanced aggregation in a pH-dependent manner [38]. While the underlying mechanism remains to be fully understood, this finding agreed with most of the studies reported in Table 1 that the aggregation-triggering factors acted synergistically rather than independently.
Kent et al. reported another example of the collective effect of different factors on protein aggregation [36]. The study revealed that pH is a more significant factor relative to temperature in increasing aggregation upon in freeze-thaw mAbs [36]. When the mAbs were allowed to thaw completely before buffer adjustment, the shift in pH did not alter the aggregation profile. Together, pH and temperature changes affect the thermal unfolding transition in proteins and hence increase aggregation.
Overall, protein aggregation is a complicated phenomenon involving several mechanisms, classifications and contributing factors. In order to highlight the need for protein aggregation control, in the next section, we will discuss the relationship between aggregation and clinical immunogenicity as well as the gaps in the current correlation models.
III. The Relevance of Protein Aggregation in Clinical Immunogenicity
1. Interaction with the immune system
For the purpose of this review, immunogenicity is defined as the propensity of a protein biotherapeutic to elicit undesirable harmful immune responses towards itself. Due to how biologics are historically synthesized, species difference is often perceived as a primary cause for immunogenicity. Over the past two decades recombinant technology has evolved to the extent many recently approved monoclonal antibodies are “humanized” or “fully human”, with the latter developed using libraries of human immunoglobulin sequences. Nevertheless, studies have shown that even these human-like antibodies can induce of anti-drug antibodies (ADAs) [47]. Most likely, these biologics do not trigger the immune responses through recognition of xenogeneic epitopes by the human B and T cell repertories. As such, protein aggregation has been postulated as a drug-intrinsic factor in ADA formation. Because protein aggregates contain multiple monomers, the repetitive patterns of amino acids can mimic ligands of scavenger and other receptors, leading to enhanced uptake by antigen-presenting cells (APCs), which detect microbial antigens in a similar manner [48-50].
Upon processing by APCs, the generation of ADA against protein biologics is proposed to occur through T-cell dependent and independent pathways [51]. APCs process and present peptide fragments to T cell receptors (TCRs) on naïve T cells through major histocompatibility complex (MHC) molecules. Once the binding between the MHC molecules and TCRs is established, APCs will form secondary connections with T cells through costimulatory molecules such as B7, CD80, and CD86. Activated T cells then proliferate and release cytokines, which participate in the communication between activated T cells and B cells. Mature B cells will secrete memory B cells and long-lived plasma cells, which generate ADAs (mostly IgG1). In the T cell independent pathway, protein aggregates can directly activate B cells, which lead to short-lived plasma cells and eventually ADAs (mostly IgG2b, IgG3, or IgM). ADAs are classified into non-neutralizing and neutralizing subtypes. Non-neutralizing ADAs bind to epitopes that do not interfere with the interaction of the protein drug with its target. Neutralizing ADAs on other hand interfere with a drug at the active site, hence reducing its efficacy [52]. The development and validation of ADAs detection assays are beyond the scope of this discussion, but can be found in a final guidance for industry issued by the FDA in January 2019 [53].
2. Aggregation, ADAs and immunogenicity
The relationships between protein aggregation, ADAs and immunogenicity have been examined extensively in in vitro, in vivo and clinical studies. Most in vitro mechanistic studies follow a classical experimental design, in which protein aggregation is induced under accelerated and exaggerated stress conditions followed by exposure to selected immune cells. Rombach-Riegraf et al. induced subvisible aggregation in antibodies through shear stress, heat, shaking, and freeze-thaw cycles to study the effects of these aggregates on activating dendritic cells (DCs) in vitro [54]. Purported as the most potent APCs, DCs play a central role in CD4+ T cell activation and polarization, which can determine the immunogenicity of a given therapeutic antibody. The researchers found that subvisible aggregates upregulate co-stimulatory molecules on DCs, which led to induction of humoral immune responses in vitro [54]. The density of particles correlated with the degree of DCs maturation. Using the MHC-associated Peptide Proteomics (MAPPs) assay, which is used to locate peptide fragments exhibiting strong binding to MHC molecules on human DCs, it was concluded that the antigen presentation altered as a function of the number of particulates generated in the denatured antibody [54]. Therefore, aggregation appears to be an upstream predisposing factor in eliciting immune responses against biologics at the antigen presentation stage.
Ahmadi et al. highlighted the contribution of aggregation to the immunogenicity of two approved mAbs: rituximab and trastuzumab, again using CD4+ T cell proliferation and DCs activation as in vitro readouts [10]. Rituximab, a chimeric IgG1, is a highly immunogenic recombinant protein, and the presence of aggregates in the test formulations did not result in further immune activation in vitro [44]. Conversely, even a low level of subvisible aggregates trastuzumab, a humanized IgG1, elicited increased cytokines and costimulatory signals on DCs relative to un-adulterated drug formulations [44]. A caveat of these in vitro experiments is that protein aggregation is generated under exaggerated conditions, and the level of particulates may not be relevant in actual products within the shelf life specified. Additionally, there is an inherent limitation in extrapolating in vitro measurements to immunogenicity risk in vivo; the responses of the DC-T cell axis in cultures take place without perturbations by other mechanisms in an intact immune system.
Animal models are essential for evaluating immunogenicity of candidate protein drugs prior to human trials. Kijanka et al. examined the effect of the size of murine mAbs aggregates injected subcutaneously in BALB/c mice [14]. Low pH and elevated temperatures were used to induce aggregates at three levels: soluble oligomers, submicron size particles and micron size particles. The submicron size group was rated as most immunogenic, with 58% the mice developed measurable ADAs in serum, possibly due to an enhanced DCs uptake. The samples enriched in soluble oligomers and micron size particles did not trigger significant ADA in this mouse model. Somali et al. investigated the effect of aggregate levels on immunogenicity by administering several aggregate-containing formulations (5-500 μg aggregates) of mouse mAbs to C57BL/6J and BALB/c mice [55]. Compared to the control formulation in which induced aggregates were removed using ultracentrifugation, formulations with particles generated higher titers of ADAs in the mouse serum [55]. The formulations enriched with high levels of aggregates induced lower ADA responses, presumably because of development of immunological tolerance. In contrast, the formulations with a lower particulate content, which is harder to detect, exhibited a higher immunogenicity risk. As predicted, the formulations elicited higher ADA responses generated more rapid clearance. A rapid drug clearance directly reduces the drug efficacy, which explains the loss of treatment response over time.
Transgenic mouse models have been developed to improve the translational relevance of in vivo immunogenicity data [56-58]. Van Beers et al. found that aggregates of a recombinant human interferon beta formulation elicits neutralizing ADAs in wild-type mice but not in transgenic mice expressing a mini-repertoire of human IgG1 antibodies, thus highlighting the significance of humanized mouse models in immunogenicity prediction [58]. Bessa et al. revealed in their human transgenic mouse model with human IgH-γ1, Igκ and Igλ transgenes that only soluble human mAb covalent aggregates with a high degree of neoepitopes (generated from chemical modifications) resulted in ADAs [59]. Conversely, non-covalent aggregates did not elicit immunogenicity in the same model. While it appears that transgenic models are superior than wild-type animals in predicting immunogenicity in humans, the reliability is likely to dependent highly on the specific drugs and the nature of the aggregates tested. Regardless of wildtype or transgenic phenotypes, animal models cannot accurately mimic the human immune system by nature. Even nonhuman primates (NHP), which share the highest homology to humans, are inadequate in immunogenicity prediction. Van Meers et al. investigated all mAbs approved by the European Union by 2010 and noted ADA response variations between NHP and humans in 59% of the cases [60]. Hence, the immunogenicity of a drug is only definitely determined from examining treated patients.
There have been reports of incidences of post-approval adverse outcomes due to ADA responses [61-64]. For instance, ADA development against adalimumab (Humira) is correlated with a diminished drug serum level and a loss of treatment response in several hundreds patients with rheumatoid arthritis [61, 62]. In another study, shortened overall survival in patients with metastatic melanoma could be attributed to ADA against ipilimumab (Yervoy), warranting critical assessment of immunogenicity risk for checkpoint inhibitors, a rapidly growing class of therapeutics [63]. Although the effect of aggregation on ADAs and the relationship between ADAs and clinical immunogenicity are often investigated independently, the contribution of protein aggregation to immunogenicity is established [9]. Altogether, these reports call for better aggregation detection techniques and preventive methods. Methods used in detecting post-production aggregation, including dynamic light scattering (DLS), size exclusion chromatography (SEC), microscopic flow imaging, and advanced derivations of these, have been reviewed recently [65, 66]. Given the common goal of developing end-to-end, Quality-by-design, manufacturing philosophy, preemptive and mitigation will be the focus in the second half of this review.
IV. Prediction and Mitigation of Intrinsic Protein Aggregation
In considering protein aggregation in the context of formulation and production, it is worth noting that high concentration protein formulations (HCPFs) are gaining in the share of the overall mAb products [67]. Historically, mAbs formulations are administered at relatively low concentrations (<10mg/mL) in large volumes through intravenous infusion. In contrast, HCPFs (50-150mg/mL for mAbs) are developed as sustained release subcutaneous depots for the purpose of increasing compliance by extending the intervals of administration [28, 68]. Inherent in the design, HCPFs tend to be highly viscous, thereby are more likely to aggregate. It is therefore not unexpected that the majority of the recent literature on aggregation prediction, detection and/or prevention concerned HCPFs. The following narrative reflects recent updates on both low and high concentration protein formulations.
1. Formulation Development Considerations
It is generally assumed that protein aggregation in biopharmaceuticals can be predicted and prevented early in the development phase through careful design of formulation factors, including concentration of the active ingredient, buffers, and excipients. The concentration of the protein drug directly affects colloidal stability of the system and thus its aggregation tendency. As discussed above, colloidal stability is typically reported with the osmotic second viral coefficient, which is measured by static or dynamic light scattering. A conventional method is plotting light scattering intensity against a range of protein concentrations to establish a linear regression function (B22), which can be used to extrapolate aggregation behavior of protein at a given concentration. However, the validity of this form of linear regression has only been established in low concentration protein formulations (< 10mg/mL) [69]. In high concentration systems (>50 mg/ml), the changes in protein particle size and light scattering may behave in nonlinear fashions [69-71]. Protein-protein interactions, in terms of steric repulsions, van de Waal’s attractions, and long-range electrostatic interactions, are exaggerated in HCPFs. Kumar et al. revealed that in highly concentrated protein products, aggregation is predominantly governed by short-range hydrophobic interactions, while long-range electrostatic forces facilitate aggregation in diluted formulations [72]. Thus, it would not be appropriate to apply the same linear predictive method to analyze aggregation behaviors in highly concentrated products.
In an attempt to resolve this discrepancy, Hofmann et al. constructed a model that would allow identifying intrinsic protein aggregation over a range of protein concentrations [69]. The method entailed first establishing a linear fit in the low concentration range (<10 mg/mL), followed by identifying the deviations between measured scattering and extrapolated scattering intensities of a concentrated system (40mg/mL) outside the fitted range. When comparing the colloidal stability predicted with the optimized model and real-time stability data from DLS and SEC techniques (10 to 20-week readouts), the predictive method was found to be useful in qualitatively ranking aggregation tendencies among Fc-fusion protein, single domain antibody and monoclonal antibody formulations at different pH values. The study represents on-going efforts to optimize protein aggregation prediction at the formulation screening stage.
Because protein stability is generally dependent on pH and ionic strength, a critical step in the formulation stage is to screen for optimal buffering and salt compositions. A common approach is to examine protein thermal unfolding under various pH values and ionic conditions using differential scanning fluorimetry (DSF). Conventional DSF involves hydrophobic fluorescence dyes such as SYPRO Orange or 1-anilino-8-naphthalenesulfonate (ANS), which are quenched in a hydrophilic environment, but become fluorescent when exposed to hydrophobic patches in denatured proteins [73]. As the protein unfolds by inductive heating, protein hydrophobic residues are exposed, leading to an increase in fluorescence. The unfolding transition midpoint temperature (Tm) reflects the conformational stability. Using real-time DSF, Moggridge et al. showed that human IgG (10mg/mL) dissolved in different phosphate and sulfonic acid buffers remained thermally stable in pH 5.0 to 10 [74]; the human IgG maintained the highest thermal stability in 1,4-Piperazinediethanesulfonic acid (PIPES) buffer at pH 7.0. A possible explanation is that, according to the Hofmeister series, sulfonate ions exert greater protein stabilization effects (“salting in”) than phosphate ions in standard phosphate buffers. In the presence of a free-radical generator which oxidizes methionine, cysteine, tryptophan, tyrosine, and histidine residues of proteins, the Tm of the IgG decreased by 8°C, suggesting a reduction in protein stability [74]. Therefore, it is important to control the level of oxidizers in a protein formulation because upon amino acids oxidization, more interactive patches are revealed for aggregation.
While DSF can be a powerful tool for high-throughput screening of aggregation, a limitation is that additives in the formulation may exhibit autofluorescence or interact with the reporter dye. Kroeger et al. reported that the DSF shift of the SYPRO dye was induced by its interaction with ethylenediaminetetraacetic acid (EDTA), a common excipient, rather than with the unfolded protein [75]. To overcome such interference, the newest generation of DSF instrument, the Prometheus NT.48 (NanoTemper Technologies INC.), was developed to detect domain-specific unfolding transitions and aggregation temperatures in a label-free fashion [76]. Instead of using a fluorescent dye, Prometheus monitors the intrinsic fluorescence of hydrophobic amino acids, such as tryptophan and tyrosine, in the protein of interest [77, 78]. The instrument can complete the scanning of a wide range of protein concentrations (5 ug to 150 mg/mL) within minutes [76]. Because only a few microliters of the sample are required, bench methods can be scale-up for monitoring stability in the production stage.
In addition to optimizing concentration, buffer, and ionic strength, additives such as chaotropes and kosmotropes can be used to stability proteins in solutions. Chaotropes (“order disrupter”) such as magnesium chloride and denaturants such as urea improve protein solubility and reduce aggregate propensity by destabilizing a protein’s intramolecular hydrogen bonding and remove the entropic incentive of hydrophobic collapses [79-81]. Urea interacts with hydrogen atoms and displaces the bulk water, leading to a lower freedom gain from releasing the water structures to the bulk water, which drives hydrophobic collapses. As a result, the system is shifted away from an aggregation inclination. On the other hand, kosmotropes (“order maker”) such as magnesium sulfate stabilize the formation of water structure and foster hydrophobic aggregation [82, 83]. Determination of additive concentrations is critical because a high concentration of kosmotropes will salt out protein into solution, and a high concentration of chaotropes will denature native proteins [84]. Because the interaction between salt and protein is electrostatic-based, determining an appropriate level of salt depends heavily on pH, protein concentration and pI [85]. Moreover, dissolving aggregation induced by covalent linkages requires reducing agents such as dithiothreitol and β-mercaptoethanol [86]. Thiols on these agents will reduce non-native disulfide linkages among cysteine residues, thereby dissolve covalent aggregates. However, these strong reducing agents are rarely included in the final formulation because they also disrupt the disulfide bonds required for native protein conformation.
2. Bioprocessing Considerations
2.1. Upstream bioproduction
Post-production removal of aggregates can be costly. Therefore, the most common approach is to optimize process parameters to reduce aggregation during bioproduction. Paul et al. utilized Design-of-experiment (DoE) to optimize mAbs bioprocessing parameters through monitoring protein aggregation [87]. This was a unique approach insofar the previous efforts emphasized increasing yield by optimizing glycosylation rather than direct control of the aggregation process. The work involves systemically resolving the effects of temperature, pH, osmolarity, agitation, antifoam and valproic acid (VPA, a common culture additive) on mAbs aggregation in Chinese Hamster ovary (CHO) cell cultures [87]. The authors studied both soluble aggregates and aggregate particles. pH was identified to have a minimal impact compared to other parameters, possibly due to the narrow range (6.8, 7.2, and 7.6) compatible with CHO cells tested. VPA, added to enhance protein expression in mammalian cell cultures, was found to be the sole additive that induced soluble aggregates. Increased aggregates were found at high temperatures held for an extended duration, which led to the recommendation that a temperature-shift should be programmed after 72h. A temperature shift below 32°C reduced aggregate particles, while the shift occurring at 32-34°C and 33-35°C increased particle formation.
Kaisermayer et al. also used DoE to identify optimal temperatures and pH for cultivating erythropoietin fusion protein (Epo-Fc) in CHO cells [33]. Unlike Paul et al. study, Kaisermayer et al. focused on the interplay between pH and temperature on aggregation. From 28.5 °C to 38.5 °C, aggregation propensity was higher at high pH compared to low pH. At high pH level, increasing temperature slightly reduced protein aggregate percentages. In contrast, at low pH, increasing temperature increased protein aggregation. Adjusting the temperature and pH from 37°C, pH 7.05 to 30 °C, pH 6.75 synergistically reduced Epo-Fc protein aggregation from 75% to 0.7%. This result corresponded with a recent patent suggesting therapeutic proteins production in a mammalian cell culture cultivated at reduced temperature (27 °C - ≤ 30 °C) and reduced pH (6.8 - ≤ 7) significantly reduced protein misfolding and aggregation [34]. Taken together, these recent reports suggested that the standardized 37 °C, pH 7.0 might not be the optimal cell culture for therapeutic protein synthesis. Rather, a slight reduction of temperature and pH at the right timing can minimize protein aggregation and improve product quality. These studies also highlight the importance of utilizing a systematic DoE approach in optimizing bioprocessing because of the nonlinear and cross-reactive effects among parameters.
Although mammalian cell cultures (predominantly CHO cells) are the current standard hosts for therapeutic protein synthesis, many cell-free protein synthesis (CFPS) methods are being developed as high-throughput and enhanced controlled strategies. In CFPS methods, enzymatic components are extracted from cell lysate and then used to facilitate translation, transcription and protein folding with an addition of external essential substrates (such as nucleotides, DNA, mRNA, energy factors, etc.). Over the past few years, the development of CFPS has gradually moved from prokaryote-based to eukaryote-based processes, and from synthesizing small protein fragments to synthesizing intact monoclonal antibodies. The increase in size and complexity also increases the risk of aggregation.
In 2017, Martin et al. was the first to report a CHO-based cell-free platform that was used to synthesize mAbs [88]. A key finding of the study was that the heavy chains formed more aggregates when synthesized independent of the light chains, suggesting that the latter assist the folding and stability of the former [88, 89]. Thus, adding a light chain plasmid first and delaying the addition of a heavy chain plasmid by 6 hours from the time of protein synthesis detection helped minimize aggregation propensity in this CHO-based CFPS [88]. No significant impact of light chain to heavy chain plasmid ratio on protein aggregation was observed. A shortcoming of this study was that protein aggregation was evaluated using a single method (western blot). Orthogonal methods, such as circular dichroism spectroscopy or SEC, are necessary to identify the types of aggregates, and to relate the level of aggregation to process parameters. In comparison to cell-based bioreactors, cell-free platforms allow more flexibility for in situ manipulation and direct sampling, as protein synthesis occurs outside cells. However, CFPS entails a different scope of parameters, of which effects on aggregation need to be delineation prior to implementation at industrial scale.
2.2. Downstream bioprocessing
Aggregation can be a significant impediment during protein purification. The standard antibody purification entails sequentially capturing the API with a Staphylococcal protein A-resin chromatography, contamination removal, elution with an acidic treatment (pH 3.5), and pH neutralization, polishing and filtration [90, 91]. Protein A (or protein G) is widely used for its reversible and specific binding to the Fc domain on antibodies and Fc-containing proteins [92-94]. The protein adsorption and desorption process with protein A-resin column often change the hydration layer around the purified protein, which alters protein conformation and increases aggregation propensity [95, 96]. In addition, proteins can denature during the acidic elution step, thereby increasing the risk of aggregation.
It has been proposed that the chromatography binding and elution process can also be exploited to remove aggregation from the final yield [97, 98]. Yu et al. revealed that mAb aggregates from upstream cell cultures would bind to protein A resin more strongly than mAb monomers [97]. Furthermore, aggregates that involved both Fab-Fab and Fc-Fc interactions between antibodies were shown to be more compact and may bind to more protein A ligands than aggregates of Fab-Fab. As such, the difference in protein A affinities provides an opportunity to remove antibody aggregates at the initial step of purification. Dileo et al. found that an optimized pH and protein A-resin selection process decreased 25-30% of mAb aggregates and lowered the high molecular weight aggregates from 18% in clarified culture to 12% in protein A elution product [99]. It was also suggested adding low-pH washes (but above that of pH required for elution) prior to elution can improve the final product quality. While pH drifts can induce aggregation, a well-conceived pH transition regime can be advantageous.
Alternatives to protein A chromatography have been developed. Inomata et al. reported a resin column employing an alkaline-tolerant RNA aptamer which could specifically bind to IgG and dissociate upon chelation at neutral pH [100]. Scheffel et al. developed a protein A derived domain, ZCa, which bound and eluted antibody in a Calcium-dependent manner using EDTA at neutral pH [101, 102]. Successful commercialization of these new technologies would hinge on consistent demonstrations of improved yields compared to existing methods.
V. Summary
ADA are associated with negative therapeutic outcomes in patients, presumably due to accelerating clearance and/or neutralizing target binding of the API. Because of the diversity of therapeutic proteins and the complexities of the human immune system, it has been challenging to construct a set of consensus properties by which the risks of developing ADA can be defined. Advances in ADA risk assessment will depend on converging understanding of the immunological mechanisms and physical/chemical attributes of API driving aggregation. Attempts to resolve the latter have been made in the development of computational methods for identifying aggregation hotspots in protein sequence, which have been revised elsewhere [103-105]. The current review centered on factors that drive intrinsic protein aggregation, and development of predictive models that can be applied at the formulation and bioprocessing stages. Prediction methods should be evaluated based on transferability of key parameters across diluted and concentrated systems. Applying linear models trained using low concentration formulations to high protein concentration formulations would likely yield invalid results. To this end, high-throughput approaches, in which concentrations spanning several logs can be employed in model training, will lead to more robust models. Such predictive models will be a key component in effective systematic strategies aimed to reduce immunogenicity risk of biotherapeutics.
Acknowledgements
This work was supported in part by NIH grant R21 AI139828 and DOD grant KC170029.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Roberts CJ, Protein aggregation and its impact on product quality. Current Opinion in Biotechnology, 2014. 30: p. 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Philo JS and Arakawa T, Mechanisms of protein aggregation. Curr Pharm Biotechnol, 2009. 10(4): p. 348–51. [DOI] [PubMed] [Google Scholar]
- 3.Lumry R and Eyring H, Conformation Changes of Proteins. The Journal of Physical Chemistry, 1954. 58(2): p. 110–120. [Google Scholar]
- 4.Roberts CJ, Therapeutic protein aggregation: mechanisms, design, and control. Trends in Biotechnology, 2014. 32(7): p. 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg ME, Rudolph R, and Jaenicke R, A kinetic study of the competition between renaturation and aggregation during the refolding of denatured-reduced egg white lysozyme. Biochemistry, 1991. 30(11): p. 2790–2797. [DOI] [PubMed] [Google Scholar]
- 6.Narhi LO, et al. , Classification of protein aggregates. J Pharm Sci, 2012. 101(2): p. 493–8. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, et al. , Immunogenicity of protein aggregates—Concerns and realities. International Journal of Pharmaceutics, 2012. 431(1): p. 1–11. [DOI] [PubMed] [Google Scholar]
- 8.Shabushnig JG, et al. , A Proposed Working Standard for Validation of Particulate Inspection in Sterile Solutions. 1994. [Google Scholar]
- 9.FDA, Guidance for industry: immunogenicity assessment for therapeutic protein products. 2014.
- 10.Ahmadi M, et al. , Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm Res, 2015. 32(4): p. 1383–94. [DOI] [PubMed] [Google Scholar]
- 11.Singh SK, et al. , An industry perspective on the monitoring of subvisible particles as a quality attribute for protein therapeutics. J Pharm Sci, 2010. 99(8): p. 3302–21. [DOI] [PubMed] [Google Scholar]
- 12.USP< 788>. Particulate matter in Injections. 2017, USP. [Google Scholar]
- 13.Ratanji KD, et al. , Editor’s Highlight: Subvisible Aggregates of Immunogenic Proteins Promote a Th1-Type Response. Toxicological Sciences, 2016. 153(2): p. 258–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kijanka G, et al. , Submicron Size Particles of a Murine Monoclonal Antibody Are More Immunogenic Than Soluble Oligomers or Micron Size Particles Upon Subcutaneous Administration in Mice. J Pharm Sci, 2018. 107(11): p. 2847–2859. [DOI] [PubMed] [Google Scholar]
- 15.Anfinsen CB, Principles that Govern the Folding of Protein Chains. Science, 1973. 181(4096): p. 223–230. [DOI] [PubMed] [Google Scholar]
- 16.Chi EY, et al. , Physical Stability of Proteins in Aqueous Solution: Mechanism and Driving Forces in Nonnative Protein Aggregation. Pharm Res, 2003. 20(9): p. 1325–1336. [DOI] [PubMed] [Google Scholar]
- 17.Dill KA, Dominant forces in protein folding. Biochemistry, 1990. 29(31): p. 7133–7155. [DOI] [PubMed] [Google Scholar]
- 18.Ratner BD, Chapter I.1.6 - Role of Water in Biomaterials, in Biomaterials Science (Third Edition), Ratner BD, et al. , Editors. 2013, Academic Press, p. 55–59. [Google Scholar]
- 19.Sticke DF, et al. , Hydrogen bonding in globular proteins. Journal of Molecular Biology, 1992. 226(4): p. 1143–1159. [DOI] [PubMed] [Google Scholar]
- 20.Privalov PL and Makhatadze GI, Contribution of Hydration to Protein Folding Thermodynamics: II. The Entropy and Gibbs Energy of Hydration. Journal of Molecular Biology, 1993. 232(2): p. 660–679. [DOI] [PubMed] [Google Scholar]
- 21.Quigley A and Williams DR, The second virial coefficient as a predictor of protein aggregation propensity: A self-interaction chromatography study. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 2015. 96: p. 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts D, et al. , The role of electrostatics in protein-protein interactions of a monoclonal antibody. Molecular pharmaceutics, 2014. 11(7): p. 2475–2489. [DOI] [PubMed] [Google Scholar]
- 23.Neal BL, Asthagiri D, and Lenhoff AM, Molecular origins of osmotic second virial coefficients of proteins. Biophys J, 1998. 75(5): p. 2469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stetefeld J, McKenna SA, and Patel TR, Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophysical reviews, 2016. 8(4): p. 409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassan PA, Rana S, and Verma G, Making Sense of Brownian Motion: Colloid Characterization by Dynamic Light Scattering. Langmuir, 2015. 31(1): p. 3–12. [DOI] [PubMed] [Google Scholar]
- 26.Ferreira G, et al. , Identifying Key Residues That Drive Strong Electrostatic Attractions between Therapeutic Antibodies. The Journal of Physical Chemistry B, 2019. [DOI] [PubMed] [Google Scholar]
- 27.Sahin E, et al. , Comparative Effects of pH and Ionic Strength on Protein-Protein Interactions, Unfolding, and Aggregation for IgG1 Antibodies. J Pharm Sci, 2010. 99(12): p. 4830–4848. [DOI] [PubMed] [Google Scholar]
- 28.Saito S, et al. , Behavior of Monoclonal Antibodies: Relation Between the Second Virial Coefficient (B2) at Low Concentrations and Aggregation Propensity and Viscosity at High Concentrations. Pharm Res, 2012. 29(2): p. 397–410. [DOI] [PubMed] [Google Scholar]
- 29.Lopez E, et al. , Low pH Exposure During Immunoglobulin G Purification Methods Results in Aggregates That Avidly Bind Fcγ Receptors: Implications for Measuring Fc Dependent Antibody Functions. Frontiers in immunology, 2019. 10: p. 2415–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imamura H and Honda S, Kinetics of Antibody Aggregation at Neutral pH and Ambient Temperatures Triggered by Temporal Exposure to Acid. The Journal of Physical Chemistry B, 2016. 120(36): p. 9581–9589. [DOI] [PubMed] [Google Scholar]
- 31.Skamris T, et al. , Monoclonal Antibodies Follow Distinct Aggregation Pathways During Production-Relevant Acidic Incubation and Neutralization. Pharm Res, 2016. 33(3): p. 716–728. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, et al. , Acid-induced aggregation propensity of nivolumab is dependent on the Fc. mAbs, 2016. 8(6): p. 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaisermayer C, et al. , Biphasic cultivation strategy to avoid Epo-Fc aggregation and optimize protein expression. Journal of Biotechnology, 2016. 227: p. 3–9. [DOI] [PubMed] [Google Scholar]
- 34.Gomes JM and Hiller GW, Use of low temperature and/or low pH in cell culture. 2018, Google Patents. [Google Scholar]
- 35.Luo S and Zhang B, Dextrose-mediated aggregation of therapeutic monoclonal antibodies in human plasma: Implication of isoelectric precipitation of complement proteins. mAbs, 2015. 7(6): p. 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kent KP, Schroeder CE, and Sharma C, Solution pH jump during antibody and Fc-fusion protein thaw leads to increased aggregation. Journal of Pharmaceutical Analysis, 2018. 8(5): p. 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desai KG, et al. , Impact of manufacturing-scale freeze-thaw conditions on a mAb solution. BioPharm Int, 2017. 30(2): p. 30–36. [Google Scholar]
- 38.Alam ME, et al. , Deamidation Can Compromise Antibody Colloidal Stability and Enhance Aggregation in a pH-Dependent Manner. Molecular pharmaceutics, 2019. 16(5): p. 1939–1949. [DOI] [PubMed] [Google Scholar]
- 39.Alam ME, et al. , Unique impacts of methionine oxidation, tryptophan oxidation and asparagine deamidation on antibody stability and aggregation. J Pharm Sci, 2019. [DOI] [PubMed] [Google Scholar]
- 40.Bandi S, et al. , 2D NMR Analysis of the Effect of Asparagine Deamidation Versus Methionine Oxidation on the Structure, Stability, Aggregation, and Function of a Therapeutic Protein. Mol Pharm, 2019. 16(11): p. 4621–4635. [DOI] [PubMed] [Google Scholar]
- 41.Majumder S, Wang W, and Alphonse Ignatius A, Impact of Buffers on Colloidal Property and Aggregation Propensities of a Bispecific Antibody. J Pharm Sci, 2019. 108(3): p. 1139–1147. [DOI] [PubMed] [Google Scholar]
- 42.Barnett GV, et al. , Specific-Ion Effects on the Aggregation Mechanisms and Protein-Protein Interactions for Anti-streptavidin Immunoglobulin Gamma-1. The Journal of Physical Chemistry B, 2015. 119(18): p. 5793–5804. [DOI] [PubMed] [Google Scholar]
- 43.Oyama H, et al. , Relation of colloidal and conformational stabilities to aggregate formation in a monoclonal antibody. J Pharm Sci, 2019. [DOI] [PubMed] [Google Scholar]
- 44.Avastin®, Herceptin®, Rituxan® and Remicade® prescribing information. US Food and Drug Administration [Google Scholar]
- 45.Yan Q, et al. , Structure Based Prediction of Asparagine Deamidation Propensity in Monoclonal Antibodies. mAbs, 2018. 10(6): p. 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geiger T and Clarke S, Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem, 1987. 262(2): p. 785–94. [PubMed] [Google Scholar]
- 47.van Brummelen EMJ, et al. , Antidrug Antibody Formation in Oncology: Clinical Relevance and Challenges. The oncologist, 2016. 21(10): p. 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szomolanyi-Tsuda E and Welsh RM, T-cell-independent antiviral antibody responses. Current Opinion in Immunology, 1998. 10(4): p. 431–435. [DOI] [PubMed] [Google Scholar]
- 49.Rudra JS, et al. , Immune responses to coiled coil supramolecular biomaterials. Biomaterials, 2010. 31(32): p. 8475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrick BJ, et al. , Predicting Hemagglutinin MHC-II Ligand Analogues in Anti-TNFα Biologics: Implications for Immunogenicity of Pharmaceutical Proteins. PLOS ONE, 2015. 10(8): p. e0135451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moussa EM, et al. , Immunogenicity of Therapeutic Protein Aggregates. J Pharm Sci, 2016. 105(2): p. 417–430. [DOI] [PubMed] [Google Scholar]
- 52.Sorensen PS, et al. , Is the treatment effect of IFN-β restored after the disappearance of neutralizing antibodies? Multiple Sclerosis Journal, 2008. 14(6): p. 837–842. [DOI] [PubMed] [Google Scholar]
- 53.FDA, Guidance for industry: Immunogenicity Testing of Therapeutic Protein Products - Developing and Validating Assays for Anti-Drug Antibody Detection. 2019.
- 54.Rombach-Riegraf V, et al. , Aggregation of Human Recombinant Monoclonal Antibodies Influences the Capacity of Dendritic Cells to Stimulate Adaptive T-Cell Responses In Vitro. PLOS ONE, 2014. 9(1): p. e86322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shomali M, et al. , Dose Levels in Particulate-Containing Formulations Impact Anti-drug Antibody Responses to Murine Monoclonal Antibody in Mice. J Pharm Sci, 2015. 104(5): p. 1610–1621. [DOI] [PubMed] [Google Scholar]
- 56.Braun A, et al. , Protein Aggregates Seem to Play a Key Role Among the Parameters Influencing the Antigenicity of Interferon Alpha (IFN-α) in Normal and Transgenic Mice. Pharm Res, 1997. 14(10): p. 1472–1478. [DOI] [PubMed] [Google Scholar]
- 57.Hermeling S, et al. , Antibody response to aggregated human interferon alpha2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci, 2006. 95(5): p. 1084–1096. [DOI] [PubMed] [Google Scholar]
- 58.van Beers MMC, et al. , Aggregated Recombinant Human Interferon Beta Induces Antibodies but No Memory in Immune-Tolerant Transgenic Mice. Pharm Res, 2010. 27(9): p. 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bessa J, et al. , The Immunogenicity of Antibody Aggregates in a Novel Transgenic Mouse Model. Pharm Res, 2015. 32(7): p. 2344–2359. [DOI] [PubMed] [Google Scholar]
- 60.van Meer PJ, et al. , Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs, 2013. 5(5): p. 810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartelds GM, et al. , Development of Antidrug Antibodies Against Adalimumab and Association With Disease Activity and Treatment Failure During Long-term Follow-up. JAMA, 2011. 305(14): p. 1460–1468. [DOI] [PubMed] [Google Scholar]
- 62.Bartelds GM, et al. , Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Annals of the Rheumatic Diseases, 2007. 66(7): p. 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kverneland AH, et al. , Development of anti-drug antibodies is associated with shortened survival in patients with metastatic melanoma treated with ipilimumab. Oncoimmunology, 2018. 7(5): p. e1424674–e1424674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalluri SR, et al. , Interferon-beta specific T cells are associated with the development of neutralizing antibodies in interferon-beta treated multiple sclerosis patients. Journal of Autoimmunity, 2018. 88: p. 83–90. [DOI] [PubMed] [Google Scholar]
- 65.Doss HR, et al. , Streamlining the polishing step development process via physicochemical characterization of monoclonal antibody aggregates. Journal of Chromatography A, 2019. 1598: p. 101–112. [DOI] [PubMed] [Google Scholar]
- 66.Das TK, Protein particulate detection issues in biotherapeutics development--current status. AAPS PharmSciTech, 2012. 13(2): p. 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gervasi V, et al. , Parenteral protein formulations: An overview of approved products within the European Union. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 2018. 131: p. 8–24. [DOI] [PubMed] [Google Scholar]
- 68.Garidel P, et al. , High-concentration protein formulations: How high is high? European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 2017. 119: p. 353–360. [DOI] [PubMed] [Google Scholar]
- 69.Hofmann M, et al. , Prediction of Protein Aggregation in High Concentration Protein Solutions Utilizing Protein-Protein Interactions Determined by Low Volume Static Light Scattering. J Pharm Sci, 2016. 105(6): p. 1819–1828. [DOI] [PubMed] [Google Scholar]
- 70.Scherer TM, et al. , Intermolecular Interactions of IgG1 Monoclonal Antibodies at High Concentrations Characterized by Light Scattering. The Journal of Physical Chemistry B, 2010. 114(40): p. 12948–12957. [DOI] [PubMed] [Google Scholar]
- 71.Barnett GV, et al. , Identifying protein aggregation mechanisms and quantifying aggregation rates from combined monomer depletion and continuous scattering. Analytical Biochemistry, 2016. 511: p. 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumar V, et al. , Impact of short range hydrophobic interactions and long range electrostatic forces on the aggregation kinetics of a monoclonal antibody and a dual-variable domain immunoglobulin at low and high concentrations. International Journal of Pharmaceutics, 2011. 421(1): p. 82–93. [DOI] [PubMed] [Google Scholar]
- 73.Hellman LM, et al. , Differential scanning fluorimetry based assessments of the thermal and kinetic stability of peptide-MHC complexes. Journal of immunological methods, 2016. 432: p. 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moggridge J, et al. , Sensitive Detection of Immunoglobulin G Stability Using in Real-Time Isothermal Differential Scanning Fluorimetry: Determinants of Protein Stability for Antibody-Based Therapeutics. Technology in cancer research & treatment, 2017. 16(6): p. 1533034617714149–1533034617714149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kroeger T, et al. , EDTA aggregates induce SYPRO orange-based fluorescence in thermal shift assay. PLOS ONE, 2017. 12(5): p. e0177024–e0177024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strutz W, Exploring Protein Stability by NanoDSF. Biophys J, 2016. 110(3, Supplement 1): p. 393a. [Google Scholar]
- 77.Simon J, et al. , Protein denaturation caused by heat inactivation detrimentally affects biomolecular corona formation and cellular uptake. Nanoscale, 2018. 10(45): p. 21096–21105. [DOI] [PubMed] [Google Scholar]
- 78.Martin L, Maschberger M, and Breitsprecher D, Thermal Unfolding of Antibodies with the Prometheus NT.48. BioTechniques, 2015. 59(4): p. 251. [Google Scholar]
- 79.Wu S-L and Karger BL, [2] Hydrophobic interaction chromatography of proteins, in Methods in Enzymology. 1996, Academic Press; p. 27–47. [DOI] [PubMed] [Google Scholar]
- 80.Foster TM, et al. , Thermal stability of low molecular weight urokinase during heat treatment. III. Effect of salts, sugars and Tween 80. International Journal of Pharmaceutics, 1996. 134(1): p. 193–201. [Google Scholar]
- 81.Edwin F, Valkya Sharma Y, and Jagannadham MV, Stabilization of Molten Globule State of Papain by Urea. Biochemical and Biophysical Research Communications, 2002. 290(5): p. 1441–1446. [DOI] [PubMed] [Google Scholar]
- 82.Moelbert S, Normand B, and De Los Rios P, Kosmotropes and chaotropes: modelling preferential exclusion, binding and aggregate stability. Biophys Chem, 2004. 112(1): p. 45–57. [DOI] [PubMed] [Google Scholar]
- 83.Zhang Y and Cremer PS, Chemistry of Hofmeister Anions and Osmolytes. Annual Review of Physical Chemistry, 2010. 61(1): p. 63–83. [DOI] [PubMed] [Google Scholar]
- 84.Neagu A, Neagu M, and Dér A, Fluctuations and the Hofmeister effect. Biophys J, 2001. 81(3): p. 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schermeyer M-T, et al. , Characterization of highly concentrated antibody solution - A toolbox for the description of protein long-term solution stability. mAbs, 2017. 9(7): p. 1169–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bondos SE and Bicknell A, Detection and prevention of protein aggregation before, during, and after purification. Analytical Biochemistry, 2003. 316(2): p. 223–231. [DOI] [PubMed] [Google Scholar]
- 87.Paul AJ, et al. , Identification of process conditions influencing protein aggregation in Chinese hamster ovary cell culture. Biotechnol Bioeng, 2018. 115(5): p. 1173–1185. [DOI] [PubMed] [Google Scholar]
- 88.Martin RW, et al. , Development of a CHO-Based Cell-Free Platform for Synthesis of Active Monoclonal Antibodies. ACS Synthetic Biology, 2017. 6(7): p. 1370–1379. [DOI] [PubMed] [Google Scholar]
- 89.Ishii Y, et al. , Comparison of Antibody Molecules Produced from Two Cell Lines with Contrasting Productivities and Aggregate Contents. Biological and Pharmaceutical Bulletin, 2015. 38(2): p. 306–316. [DOI] [PubMed] [Google Scholar]
- 90.Hjelm H, Hjelm K, and Sjöquist J, Protein a from Staphylococcus aureus. Its isolation by affinity chromatography and its use as an immunosorbent for isolation of immunoglobulins. FEBS Letters, 1972. 28(1): p. 73–76. [DOI] [PubMed] [Google Scholar]
- 91.Ey PL, Prowse SJ, and Jenkin CR, Isolation of pure IgG1, IgG2a and IgG2b immunoglobulins from mouse serum using protein A-Sepharose. Immunochemistry, 1978. 15(7): p. 429–436. [DOI] [PubMed] [Google Scholar]
- 92.Sjodahl J, Structural studies on the four repetitive Fc-binding regions in protein A from Staphylococcus aureus. Eur J Biochem, 1977. 78(2): p. 471–90. [DOI] [PubMed] [Google Scholar]
- 93.Deisenhofer J, Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry, 1981. 20(9): p. 2361–70. [PubMed] [Google Scholar]
- 94.Lian L-Y, et al. , Mapping the interactions between streptococcal protein G and the Fab fragment of IgG in solution. Nature structural biology, 1994. 1(6): p. 355. [DOI] [PubMed] [Google Scholar]
- 95.Gagnon P, et al. , Transient conformational modification of immunoglobulin G during purification by protein A affinity chromatography. Journal of Chromatography A, 2015. 1395: p. 136–142. [DOI] [PubMed] [Google Scholar]
- 96.Mazzer AR, et al. , Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. Journal of Chromatography A, 2015. 1415: p. 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu D, et al. , Molecular perspective of antibody aggregates and their adsorption on Protein A resin. Journal of Chromatography A, 2016. 1457: p. 66–75. [DOI] [PubMed] [Google Scholar]
- 98.Yada T, et al. , Choosing the right protein A affinity chromatography media can remove aggregates efficiently. Biotechnology Journal, 2017. 12(1): p. 1600427. [DOI] [PubMed] [Google Scholar]
- 99.DiLeo M, et al. , Choices of capture chromatography technology in antibody manufacturing processes. Journal of Chromatography B, 2017. 1068-1069: p. 136–148. [DOI] [PubMed] [Google Scholar]
- 100.Inomata E, et al. , Alkaline-tolerant RNA aptamers useful to purify acid-sensitive antibodies in neutral conditions. Biochimie, 2018. 145: p. 113–124. [DOI] [PubMed] [Google Scholar]
- 101.Scheffel J, et al. , Optimization of a calcium-dependent Protein A-derived domain for mild antibody purification. mAbs, 2019. 11(8): p. 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanje S, et al. , Protein Engineering Allows for Mild Affinity-based Elution of Therapeutic Antibodies. J Mol Biol, 2018. 430(18 Pt B): p. 3427–3438. [DOI] [PubMed] [Google Scholar]
- 103.Gil-Garcia M, et al. , Combining Structural Aggregation Propensity and Stability Predictions To Redesign Protein Solubility. Molecular pharmaceutics, 2018. 15(9): p. 3846–3859. [DOI] [PubMed] [Google Scholar]
- 104.Walsh I, et al. , PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Research, 2014. 42(W1): p. W301–W307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sankar K, et al. , AggScore: Prediction of aggregation-prone regions in proteins based on the distribution of surface patches. Proteins: Structure, Function, and Bioinformatics, 2018. 86(11): p. 1147–1156. [DOI] [PubMed] [Google Scholar]