

SUMMARY
Disruption of cyclophilin A (CypA)-capsid interactions affects HIV-1 replication in human lymphocytes. To understand this mechanism, we utilize human Jurkat cells, peripheral blood mononuclear cells (PBMCs), and CD4+ T cells. Our results show that inhibition of HIV-1 infection caused by disrupting CypA-capsid interactions is dependent on human tripartite motif 5a (TRIM5αhu), showing that TRIM5αhu restricts HIV-1 in CD4+ T cells. Accordingly, depletion of TRIM5αhu in CD4+ T cells rescues HIV-1 that fail to interact with CypA, such as HIV-1-P90A. We found that TRIM5αhu binds to the HIV-1 core. Disruption of CypA-capsid interactions fail to affect HIV-1-A92E/G94D infection, correlating with the loss of TRIM5αhu binding to HIV-1-A92E/G94D cores. Disruption of CypA-capsid interactions in primary cells has a greater inhibitory effect on HIV-1 when compared to Jurkat cells. Consistent with TRIM5α restriction, disruption of CypA-capsid interactions in CD4+ T cells inhibits reverse transcription. Overall, our results reveal that CypA binding to the core protects HIV-1 from TRIM5αhu restriction.
Graphical Abstract
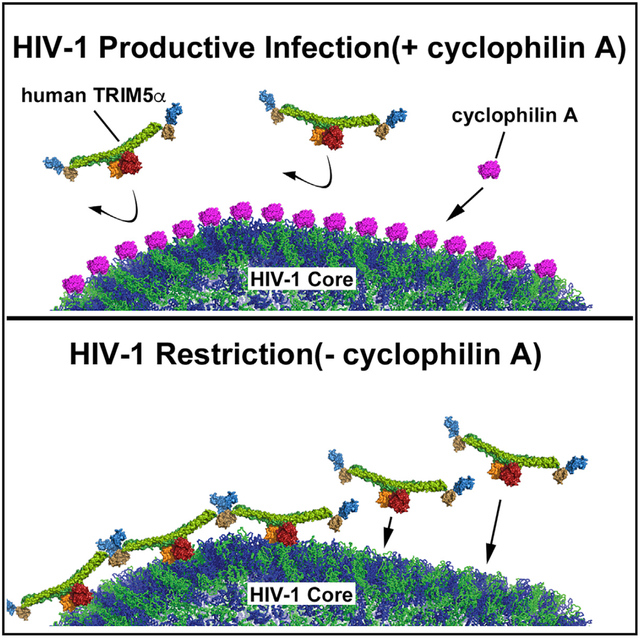
In Brief
Selyutina et al. show that, during HIV infection, the drug cyclosporine removes cyclophilin A bound to the HIV-1 core, facilitating the interaction between human TRIM5α and the core. This inhibits infection and explains the mechanism behind the long-standing observation that cyclosporine counteracts HIV.
INTRODUCTION
The early steps of human immunodeficiency virus (HIV-1) replication involve delivery of the viral core into the host cytoplasm. The viral core is a conical structure composed of ~ 1,500 monomers of the HIV-1 capsid protein, which protects the viral RNA genome (Pornillos et al., 2009; Sundquist and Hill, 2007). The capsid protein plays a role in uncoating, reverse transcription, and nuclear import. To perform these functions, the capsid protein has been shown to functionally interact with several host dependency factors, including cyclophilin A (CypA) (Luban et al., 1993), Nup153 (Brass et al., 2008; Buffone et al., 2018; Di Nunzio et al., 2013; König et al., 2008; Lee et al., 2010; Matreyek and Engelman, 2011; Matreyek et al., 2013; Zhou et al., 2008), RANBP2 (Di Nunzio et al., 2012; Meehan et al., 2014; Ocwieja et al., 2011), TNPO3 (Brass et al., 2008; Christ et al., 2008; König et al., 2008; Krishnan et al., 2010; Levin et al., 2010; Ocwieja et al., 2011; Thys et al., 2011; Valle-Casuso et al., 2012; Zhou et al., 2008), TNPO1 (Fernandez et al., 2019; Idica et al., 2017), and CPSF6 (Lee et al., 2010). Effective HIV-1 infection may occur only when these factors interact with the HIV-1 core at the appropriate time and in the appropriate order, thus raising the possibility that the initial binding of particular factors may modulate the subsequent binding of other factors.
The interaction of CypA with monomeric HIV-1 capsid was originally reported about 30 years ago (Luban et al., 1993), and it has been well established that this interaction modulates HIV-1 infection (Braaten et al., 1996; Braaten and Luban, 2001; Li et al., 2009; Schaller et al., 2011; Sherry et al., 1998; Sokolskaja et al., 2004; Thali et al., 1994; Towers et al., 2003). Although the ability of CypA to modulate HIV-1 infection is cell-type dependent, it is clear that the disruption of CypA-capsid interactions negatively affects HIV-1 infection of lymphocytes (Braaten and Luban, 2001; Li et al., 2009; Ptak et al., 2008; Sherry et al., 1998; Sokolskaja et al., 2004; Towers et al., 2003; Wainberg et al., 1988; Yin et al., 1998); however, the underlying mechanism remains elusive. This work will focus on the mechanism by which the disruption of CypA-capsid interactions negatively affects HIV-1 infection of lymphocytes.
To investigate the molecular events that result in a decrease in HIV-1 infection upon disruption of CypA-capsid interactions in lymphocytes, we used Jurkat cells, primary human peripheral blood mononuclear cells (PBMCs), and primary human CD4+ T cells as experimental models. Our experiments revealed that inhibition of HIV-1 infection caused by disrupting CypA-capsid interactions is dependent on the expression of human tripartite motif 5a (TRIM5αhu). Depletion of TRIM5αhu expression in Jurkat and CD4+ T cells rescued HIV-1 infection of viruses that cannot interact with CypA, such as HIV-1-P90A. These results suggest that TRIM5αhu restricts HIV-1 infection upon disruption of CypA-capsid interactions. In agreement, we found that TRIM5αhu binds to the HIV-1 core. Interestingly, TRIM5αhu does not bind to HIV-1 cores bearing the capsid mutant A92E (HIV-1-A92E) or G94D (HIV-1-G94D). Infection of HIV-1-A92E/G94D viruses was not affected by disruption of CypA-capsid interactions, which correlates with the loss of TRIM5αhu binding to HIV-1 cores bearing the A92E/G94D mutation. These experiments suggest that CypA protects the HIV-1 core from TRIM5αhu. Although the infectivity defect observed in Jurkat cells is small, disruption of CypA-interactions in PBMCs and CD4+ T cells causes a major defect in infectivity. In agreement with our hypothesis that CypA protects the core from TRIM5αhu, infection of PBMCs and CD4+ T cells by HIV-1-A92E viruses was insensitive to the disruption of CypA-capsid interactions. Consistent with a block imposed by TRIM5α proteins, disruption of CypA-capsid interactions in CD4+ T cells dramatically decreased the efficiency of reverse transcription. Overall, our investigations suggest a model in which CypA binding to the HIV-1 core protects the core from the restriction factor TRIM5αhu.
RESULTS
HIV-1 Infection of Jurkat Cells Is Sensitive to Cyclosporin A Treatment
To understand the role of CypA in HIV-1 infection of CD4+ T cells, we used the human T cell line Jurkat as an experimental model. Cyclosporin A (CsA) is a molecule that binds to CypA and thus blocks cellular CypA activity. To measure the effect of CsA treatment on HIV-1 infectivity, we infected Jurkat cells with HIV-1 tagged with green fluorescent protein pseudotyped with the vesicular stomatitis virus G (VSV-G) envelope (HIV-1-GFP) in the presence of 10 μM CsA. Consistent with results from previous studies (Braaten and Luban, 2001; Hatziioannou et al., 2005; Li et al., 2009; Ptak et al., 2008; Sokolskaja et al., 2004; Towers et al., 2003; Wainberg et al., 1988; Yin et al., 1998), HIV-1-GFP infection of human Jurkat T cells was sensitive to CsA treatment (Figure S1A, top panel). Experiments were repeated multiple times, and the fraction of GFP-positive cells normalized to the dimethyl sulfoxide (DMSO)-treated control is shown in Figure S1A (bottom panel). To further confirm the importance of CypA-capsid interactions in HIV-1 infection, we challenged Jurkat T cells with p24-normalized amounts of HIV-1-P90A-GFP and HIV-1-G89V-GFP, which are mutant versions of HIV-1 that disrupt CypA-capsid interactions (Gamble et al., 1996; Thali et al., 1994). Infection was determined by measuring the percentage of GFP-positive cells 48 h post-infection. We found that the mutant HIV-1 strains bearing capsid changes that disrupted CypA-capsid interactions were less infectious than wild-type HIV-1 (Figure S1B, top panel). Experiments were repeated multiple times, and the fraction of GFP-positive cells normalized to control is shown in Figure S1B (bottom panel). Although Jurkat cells failed to show a dramatic decrease in HIV-1 infectivity as a result of disrupted CypA-capsid interactions, we still used this cell line as a model to mechanistically understand the contribution of CypA to HIV-1 infection.
TRIM5αhu Binds to the HIV-1 Capsid
Previous studies have shown that CypA modulates the ability of rhesus TRIM protein, TRIM5αrh, to restrict HIV-1 infection (Berthoux et al., 2005; Burse et al., 2017; Keckesova et al., 2006; Lin and Emerman, 2008; Stremlau et al., 2006b), which is not surprising because both proteins interact with the HIV-1 core. One model is that TRIM5αrh cages the HIV-1 core using cooperative binding during infection (Diaz-Griffero et al., 2009; Ganser-Pornillos et al., 2011; Stremlau et al., 2006a). Because both CypA and TRIM5αrh bind to the HIV-1 core, it is possible that CypA modulates the binding of TRIM5αrh to the HIV-1 core or vice versa.
More recent studies have suggested that TRIM5αhu blocks HIV-1 infection (Jimenez-Guardeño et al., 2019; OhAinle et al., 2018), possibly by binding to the HIV-1 core. Therefore, we tested the ability of TRIM5αhu to bind to the HIV-1 core using our new capsid-binding assay that incorporates washing steps (Selyutina et al., 2018). This new capsid-binding assay has been extensively validated by using different capsid binding proteins (Selyutina et al., 2018). This capsid-binding assay utilizes capsid tubes that are stabilized by disulfide bonds, providing the necessary stability to sustain washing steps, which were not possible when using the capsid-binding assay that utilized in vitro assembled capsid-nucleocapsid (CA-NC) tubes. Using this new assay, we found that TRIM5αhu binds to the HIV-1 core (Figure 1A). TRIM5αhu showed a stronger interaction with the HIV-1 core when compared with TRIM5αrh (Figure 1A). To further test these results, we compared TRIM5αhu and TRIM5αrh binding to in vitro assembled HIV-1 CA-NC complexes (Figure 1B). Our results showed that TRIM5αhu interacts with the HIV-1 CA assembled tubes, suggesting that TRIM5αhu interacts with the HIV-1 core. One possibility is that CypA modulates the ability of TRIM5αhu to interact with the HIV-1 core in lymphocytes.
Figure 1. TRIM5αhu Binds to the HIV-1 Core.

Human 293T cells were transfected with plasmids expressing the wild-type human TRIM5α tagged with an HA epitope (TRIM5αhu-HA).
(A) 24 h post-transfection, cells were lysed in capsid binding buffer (CBB), as described in STAR Methods. Inputs containing different amounts of TRIM5αhu were prepared (INPUT). Cell extracts containing different amounts of TRIM5αhu-HA were mixed with 20 μL of stabilized HIV-1 capsid tubes (5 mg/mL) and incubated for 1 h at room temperature. Stabilized HIV-1 capsid tubes were collected by centrifugation and washed twice using CBB.
(B) Alternatively, cells were lysed in CBB with DTT, and a fraction of the cell lysate was stored (INPUT). Cell extracts were mixed with 5 μL of capsid-nucleocapsid (CA-NC) particles assembled in vitro and incubated for 1 h at room temperature. Subsequently, the mixture was spun through a 70% sucrose cushion. Pellets were resuspended in 1 3 Laemmli buffer (BOUND). INPUT and BOUND fractions were analyzed by western blotting using anti-HA and anti-p24 antibodies. As control, similar experiments were performed using the capsid binder, rhesus TRIM5α tagged with an HA epitope (TRIM5αrh-HA).
Experiments were repeated six times and a representative experiment is shown. The bound fraction relative to input for six independent experiments with standard deviations is shown. ***p < 0.0005 as determined by the unpaired t test.
TRIM5αhu Expression Is Necessary for the Inhibition of HIV-1 Infection Mediated by the Disruption of CypA-Capsid Interactions
Because of the ability of TRIM5αhu to bind to the HIV-1 core, we tested whether TRIM5αhu is involved in the blocking of HIV-1 infection mediated by the disruption of CypA-capsid interactions. For this purpose, we achieved knockdown (KD) of TRIM5αhu expression in Jurkat cells using a specific short hairpin RNA (shRNA) against TRIM5αhu (Figure 2A), as described in STAR Methods. To test for TRIM5αhu function, we challenged different sets of TRIM5αhu KD cells with increasing amounts of N-tropic murine leukemia virus GFP (N-MLV-GFP), which is restricted by TRIM5αhu. We found that Jurkat TRIM5αhu-KD cells were susceptible to N-MLV infection (Figure S2), whereas, in shRNA control cells, N-MLV-GFP infection was blocked (Figure S2). As a control, we also showed that TRIM5αhu KD cells were susceptible to B-tropic murine leukemia virus GFP (BMLV-GFP), which is not restricted by TRIM5αhu (Figure S2). These results confirmed that KD of TRIM5αhu expression was successfully achieved.
Figure 2. Expression of TRIM5αhu Is Necessary for the Inhibition of HIV-1 Infection Caused by the Disruption of CypA-Capsid Interactions.

(A) Human Jurkat cells stably expressing a specific shRNA against TRIM5αhu (Jurkat-TRIM5αhu-KD #1–#4) or the empty vector (Jurkat-Control #4 and #5) were analyzed for endogenous expression of TRIM5αhu by western blotting using antibodies against TRIM5αhu. Expression of GAPDH was used as a loading control.
(B) Jurkat-TRIM5αhu-KD and Jurkat-control cells were challenged with increasing amounts of HIV-1 expressing GFP as a reporter of infection (HIV-1-GFP) in the presence of 5 μM cyclosporin A (CsA). As a control, similar experiments were performed using DMSO.
(C) Jurkat-TRIM5αhu-KD and Jurkat-control cells were challenged with increasing amounts of p24-normalized HIV-1-GFP or HIV-1-P90A-GFP. Infection was determined at 48 h post-challenge by measuring the percentage of GFP-positive cells using a flow cytometer.
Each experiment was repeated at least three times and a representative experiment is shown. Statistical analysis of these experiments is shown in Figure S2B.
Next, we assessed the infection phenotype observed in Jurkat TRIM5αhu KD cells when CypA-capsid interactions are disrupted (Figures 2B and S2B). TRIM5αhu KD cells treated with CsA did not show marked inhibition of HIV-1-GFP infection compared with control cells (Figures 2B and S2B), suggesting that TRIM5αhu is involved in the inhibition of viral infection observed in cells that CypA-capsid interactions are disrupted.
HIV-1-P90A-GFP is a mutant form of HIV-1 in which CypA-capsid interactions are disrupted during infection. We tested whether HIV-1-P90A-GFP infection was inhibited in TRIM5αhu KD Jurkat cells. As shown in Figures 2C and S2B, we found that HIV-1-P90A-GFP infection was not inhibited in TRIM5αhu KD cells compared with control cells. This result supports our hypothesis that expression of TRIM5αhu is important for the infection phenotype observed when CypA-capsid interactions are disrupted in lymphocytes. It is possible that CypA prevents the binding of TRIM5αhu to the HIV-1 core via steric hindrance, thus protecting the core from the restriction factor during the early steps of infection.
CypA Prevents the Interaction of TRIM5αhu with the HIV-1 Core
To test whether CypA modulates the binding of TRIM5αhu to the HIV-1 core, we analyzed the binding of TRIM5αhu to the HIV-1 core in the presence of CsA, which binds to endogenously expressed CypA in cellular extracts and prevents CypA binding to the HIV-1 core. As shown in Figure 3A, TRIM5αhu binding to the HIV-1 core increased 3- to 4-fold in the presence of CsA. We also found that the binding of TRIM5αhu to mutant P90A HIV-1 cores was 5- to 6-fold higher than that of wild-type HIV-1 cores (Figure 3A) in the absence of CsA. These results suggest that CypA prevents the binding of TRIM5αhu to the HIV-1 core during infection. It is possible that CypA and TRIM5αhu compete with each other to interact with the incoming core. To test this hypothesis, we investigated whether the addition of exogenous CypA decreased TRIM5αhu
Figure 3. CypA Prevents the Binding of TRIM5αhu to the HIV-1 Core.

(A–C) Human 293T cells were transfected with plasmids expressing TRIM5αhu-HA. 24 h post-transfection, cells were lysed in capsid binding buffer (CBB) (INPUT). Cell extracts containing TRIM5αhu-HA were mixed with 20 μL of stabilized wild-type or P90A HIV-1 capsid tubes (5 mg/mL) in the presence of 10 mM CsA (A) or 10 μg recombinant CypA (B). (C) Cell extracts containing TRIM5αhu-HA were mixed with 20 μL of stabilized wild-type, A92E, or G94D HIV-1 capsid tubes (5 mg/mL) in the presence of 10 mM CsA.
(D and E) HT1080 cells (control or TRIM5αhu CRISPR/Cas9 knockout) were lysed in capsid binding buffer (CBB) (INPUT). Cell extracts were mixed with either 20 μL of stabilized wild-typeor P90A HIV-1 capsid tubes (5 mg/mL) in the presence of 10 μM CsA (D) or 20 μL of stabilized wild-type or P90A HIV-1 capsid tubes (5 mg/mL) (E). Mixtures were incubated for 1 h at room temperature. Stabilized HIV-1 capsid tubes were collected by centrifugation and washed twice using CBB. Pellets were resuspended in 1× Laemmli buffer (BOUND). INPUT and BOUND fractions were analyzed by western blotting using anti-HA, anti-TRIM5αhu, anti-CypA, and anti-p24 antibodies. Experiments were repeated three times and a representative experiment is shown. The bound fraction relative to input for three independent experiments with standard deviation is shown. *p < 0.005, **p < 0.001, ns indicates not significant as determined by the unpaired t test.
binding to the HIV-1 core. For this purpose, we assessed the ability of TRIM5αhu from cellular extracts to bind to the HIV-1 core in the presence of 10 mg of recombinant CypA protein. We found that the addition of recombinant CypA significantly decreased the binding of TRIM5αhu to the HIV-1 core (Figure 3B). As expected, recombinant CypA did not affect the binding of TRIM5αhu to HIV-1 cores bearing the P90A mutation. Overall, these results suggest that CypA protects the core from the restriction factor TRIM5αhu.
Mutant HIV-1 Strains that Are Impaired for Capsid-TRIM5αhu Interactions Are Insensitive to CsA Treatment
Previous studies have shown that infection of Jurkat cells by the HIV-1 strain bearing the capsid mutation A92E or G94D was less sensitive to CsA treatment when compared with wild-type HIV-1 (Hatziioannou et al., 2005; Sokolskaja et al., 2004). Consistent with these findings, we observed that HIV-1-A92EGFP infection of Jurkat cells was insensitive to CsA treatment (Figure S3). This insensitivity may occur due to HIV-1-A92EGFP inability to interact with TRIM5αhu. To explore this possibility, we tested whether TRIM5αhu interacts with HIV-1 cores containing the A92E mutation. We found that TRIM5αhu did not interact with mutant HIV-1 cores bearing the capsid change A92E (Figure 3C). We observed similar results using the capsid mutant G94D (Figure 3C), which showed a similar pattern of infectivity when compared to HIV-1-A92E-GFP viruses. As a control, we showed that the binding of TRIM5αhu to wild-type HIV-1 cores increased when CsA was added (Figure 3C). Overall, these results indicate that TRIM5αhu does not bind to mutant HIV-1 capsids bearing the change A92E or G94D, which may explain the reason that CsA treatment does not affect the infection of Jurkat cells by HIV-1-A92E-GFP or HIV-1-G94DGFP viruses.
Endogenously Expressed TRIM5αhu Binds to the HIV-1 Capsid
Next, we tested whether the endogenously expressed TRIM5αhu binds to the HIV-1 capsid. As shown in Figure 3D, endogenously expressed TRIM5αhu from cellular extracts derived from HT1080 cells binds to stabilized HIV-1 capsid tubes. This showed the ability of endogenously expressed TRIM5αhu to bind to HIV-1 capsid. As a control, we utilized HT1080 cells that were knocked out for expression of TRIM5αhu (HT1080-TRIM5αhu-KO). In agreement with our previous results, the use of CsA increased the binding of endogenously expressed TRIM5αhu to stabilized HIV-1 capsid tubes (Figure 3D). Furthermore, endogenously expressed TRIM5αhu also bound to stabilized HIV-1 capsid tubes bearing the P90A mutation (Figure 3E).
CypA-Capsid Interactions Are Required for HIV-1 Infection of Human PBMCs and CD4+ T Cells
To understand the role of CypA in HIV-1 infection of human primary cells, we challenged PBMCs and CD4+ T cells from different donors with HIV-1-GFP in the presence of increasing concentrations of CsA, which prevents the binding of CypA to HIV-1 capsid (Diaz-Griffero et al., 2006; Luban et al., 1993). As shown in Figure S4A, CsA treatment inhibited HIV-1 infection in PBMCs. Similarly, CsA inhibited HIV-1-GFP infection of human primary CD4+ T cells (Figure S4B). This result suggests that CypA-capsid interactions are required for HIV-1-GFP infection of human PBMCs and CD4+ T cells. To further corroborate these findings, we challenged human PBMCs and CD4+ T cells with increasing amounts of HIV-1-GFP particles bearing the capsid mutations P90A and G89V, both of which prevent CypA-capsid interactions (Gamble et al., 1996; Thali et al., 1994). Infection was determined by measuring the percentage of GFP-positive cells at 72 h post-infection. Wild-type and mutant HIV-1-GFP viruses were normalized by quantifying the levels of p24. We found that HIV-1-P90A-GFP and HIV-1-G89V-GFP were unable to infect human PBMCs or CD4+ T cells (Figures 4A and S5). Although these experiments were performed in PBMCs and CD4+ T cells obtained from six different donors, all of which showed similar results, we have only shown sample results obtained from three donors. Our results suggest that CypA-capsid interactions are required for HIV-1 infection in human primary PBMCs and CD4+ T cells. Importantly, the lack of CypA binding appeared to have a stronger effect on HIV-1 infection in primary human lymphocytes when compared with that of Jurkat cells (Figure 2), thus indicating that CypA is crucial for HIV-1 infection of human primary cells. Overall, these results suggest that the disruption of CypA-capsid interaction is detrimental for HIV-1 infection of PBMCs and CD4+ T cells.
Figure 4. CypA-Capsid Interactions Are Required for HIV-1 Infection of Human CD4+ T Cells.

(A–C) Purified CD4+ T cells were challenged with increasing amounts of p24-normalized HIV-1-GFP or HIV-1-P90A-GFP (A) and either HIV-1-A92E-GFP (B) or HIV-1-G94D-GFP (C). Alternatively, purified CD4+ T cells were challenged with increasing amounts of p24-normalized HIV-1-GFP or HIV-1-A92E-GFP in the presence of 5 μM cyclosporin A (CsA).
(D) DMSO was used as control. Infection was determined at 72 h post-challenge by measuring the percentage of GFP-positive cells.
Experiments were repeated three times per donor and a representative experiment is shown. The results of three independent experiments with standard deviation are shown; statistical analysis was performed using an intermediate value taken from the infection curves. **p < 0.001, ***p < 0.0005, ****p < 0.0001, ns indicates not significant as determined by the unpaired t test.
Disruption of HIV-1 Binding to TRIM5αhu Does Not Affect Viral Infection in Human PBMCs and CD4+ T Cells
Mutant HIV-1 bearing the capsid change A92E or G94D lacks the ability to interact with TRIM5αhu (Figure 3C). In order to study the role of TRIM5αhu during viral infection, we examined the ability of HIV-1-A92E-GFP and HIV-1-G94D-GFP to infect CD4+ T cells (Figures 4B and 4C). Consistent with results observed in Jurkat cells, infection levels by the mutant viruses were slightly affected when compared to that of wild-type HIV-1-GFP in primary human CD4+ T cells (Figures 4B and 4C). As a control, we also challenged CD4+ T cells with increasing concentrations of HIV-1-P90A-GFP, an HIV-1 mutant that is completely inhibited in CD4+ T cells (Figures 4B and 4C). Consistent with our previous binding experiments, these experiments suggest that infection of CD4+ T cells by HIV-1-A92E-GFP or HIV-1-G94D-GFP was not affected by TRIM5αhu, which correlated with the inability of TRIM5αhu to bind to HIV-1 cores bearing the mutation A92E or G94D. We also demonstrate that CsA does not affect the ability of HIV-1-A92E-GFP viruses to infect human primary CD4+ T cells (Figure 4D), suggesting that HIV-1-A92E-GFP viruses are insensitive to TRIM5αhu.
Depletion of CypA Expression in Human CD4+ T Cells Inhibits HIV-1 Infection
We have previously shown indirectly that CypA-capsid interactions are important for HIV-1 infection. To directly test the role of CypA in viral infection, we challenged CypA knockout CD4+ T cells with HIV-1-GFP (VSV-G pseudotyped virus) or HIV-1NL4–3-GFP (replication competent virus). To create CypA knockouts, CD4+ T cells were separately electroporated with CRISPR/Cas9 complexes containing five different guides against CypA (CypA-KO#1–5). Expression of CypA was analyzed by western blotting using anti-CypA antibodies at 3 days post-electroporation. As shown in Figure 5A, all five guides were able to eliminate the expression of CypA. The CypA knockout cells were then challenged with HIV-1-GFP (Figure 5B) or replication competent HIV-1NL4–3-GFP (Figure 5C). Consistent with our previous results, human CD4+ T cells depleted for CypA expression were resistant to HIV-1 infection. As a control, we used a CRISPR/Cas9 complex containing a non-targeting RNA guide (Figures 5B and 5C). As an additional control in experiments using HIV-1NL4–3-GFP virus, we used a CRISPR/Cas9 complex containing an RNA guide for CXCR4, which completely inhibits infection of the replication-competent virus HIV-1NL4–3-GFP (Figure 5C). These results demonstrate that CypA expression is essential for HIV-1 infection of human primary CD4+ T cells.
Figure 5. Depletion of CypA Expression in Human CD4+ T Cells Inhibits HIV-1 Infection.
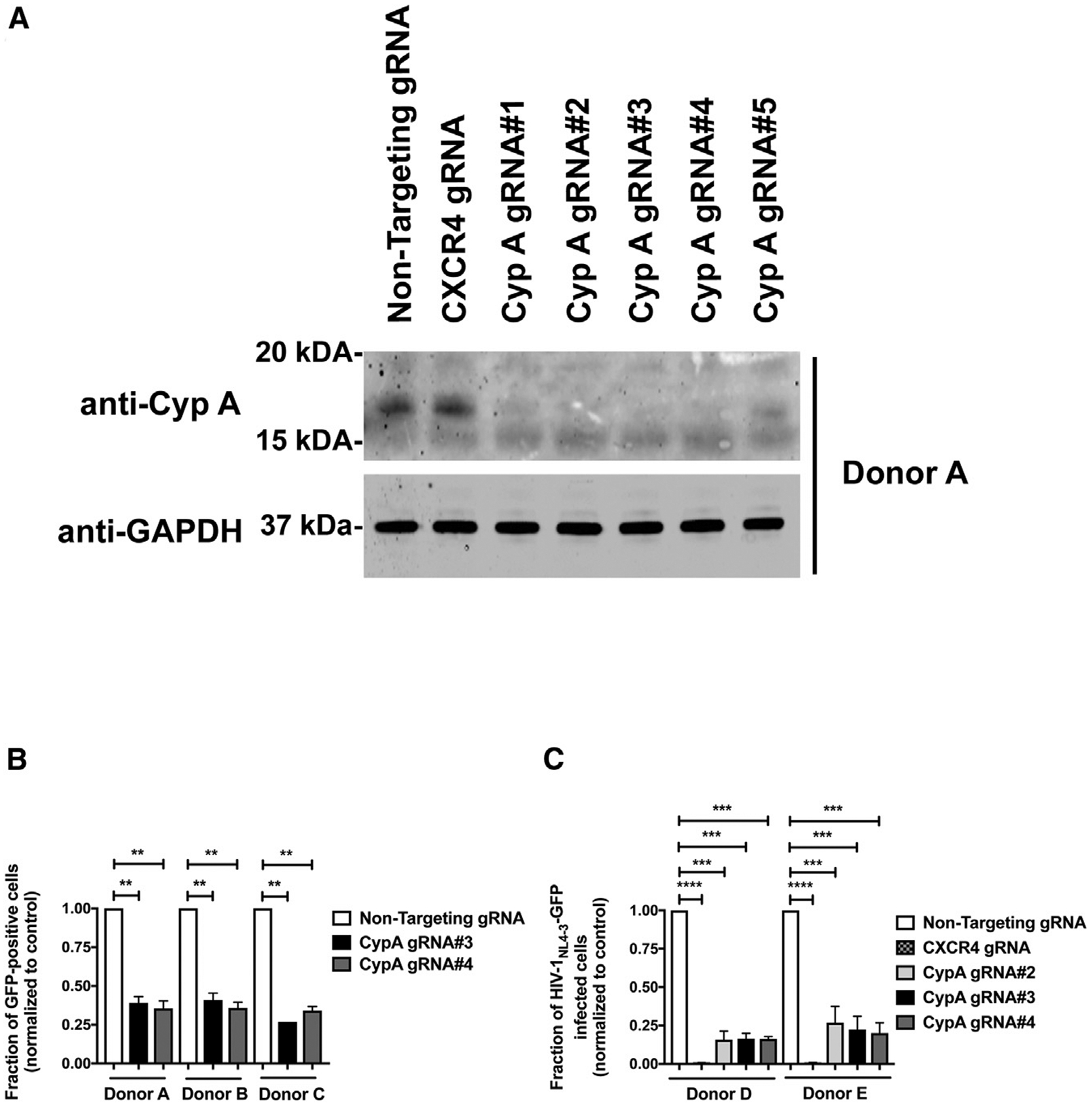
(A) Human primary CD4+ T cells from two different donors were knocked out for CypA expression using the CRISPR/Cas9 system, as described in STAR Methods. Briefly, CD4+ T cells were electroporated using five different guide RNAs (gRNAs) against CypA (gRNA#1–#5) together with the Cas9 protein. At 72 h post-electroporation, endogenous expression of CypA in CD4+ T was analyzed by western blotting using antibodies against CypA. For controls, we electroporated a gRNA against CXCR4 and a non-targeting gRNA. Expression of GAPDH was used as a loading control. Similar results were obtained using three different donors and a representative blot is shown.
(B and C) Human primary CD4+ T cells depleted for CypA expression were challenged with HIV-1-GFP pseudotyped with the VSV-G envelope (B) or with a replication-competent HIV-1 expressing GFP as a reporter of infection (HIV-1NL4–3-GFP) (C). Infection was determined at 72 h post-infection by measuring the percentage of GFP-positive cells. Experiments were performed in triplicates and results for three (B) or two (C) different donors with standard deviation are shown. **p < 0.001, ***p < 0.0005, ****p < 0.0001 determined by the unpaired t test.
Depletion of TRIM5αhu Expression in Human CD4+ T Cells Rescues Infection by HIV-1-P90A Viruses
Our results show that TRIM5αhu does not interact with A92E cores. In addition, we show that HIV-1-A92E viruses do not exhibit a replication defect in primary CD4+ T cells in the absence or presence of CsA. Altogether, these results suggest that TRIM5αhu blocks HIV-1 replication in primary CD4+ T cells where CypA-capsid interactions have been disrupted. To test whether TRIM5αhu blocks the infection of viruses that do not interact with CypA, we challenged TRIM5αhu knockout CD4+ T cells with increasing amounts of HIV-1-P90A viruses. As shown in Figure 6A, gRNA#6 and gRNA#7 efficiently depleted TRIM5αhu expression in CD4+ T cells. Consistent with our hypothesis, depletion of TRIM5αhu expression completely rescued infection of HIV-1-P90A viruses (Figure 6B). Considering that HIV-1-P90A-GFP does not bind CypA protein but does bind TRIM5αhu, these results demonstrate that TRIM5αhu is the restriction factor blocking infection of primary CD4+ T cells by HIV-1-P90A viruses. Furthermore, these experiments suggest that CypA protects the HIV-1 core from TRIM5αhu restriction.
Figure 6. Depletion of TRIM5αhu Expression in Human CD4+ T Cells Rescues Infection of HIV-1-P90A Viruses.
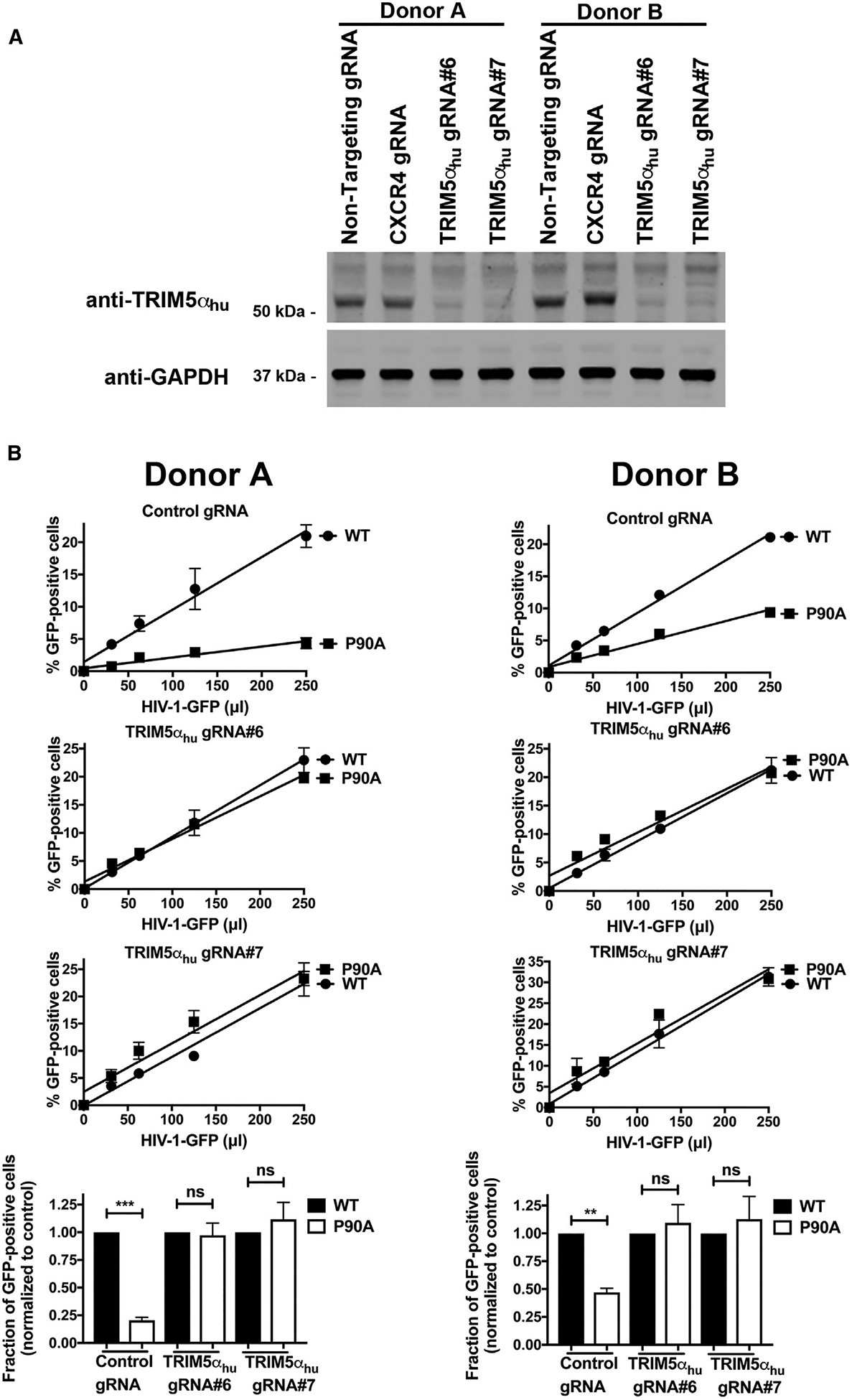
(A) Human primary CD4+ T cells from two different donors were knocked out for TRIM5αhu expression using the CRISPR/Cas9 system, as described in STAR Methods. Briefly, CD4+ T cells were electroporated using two different guide RNAs (gRNAs) against TRIM5αhu (gRNA#6 and #7) together with the Cas9 protein. 72 h post-electroporation, endogenous expression of TRIM5αhu in CD4+ T was analyzed by western blotting using antibodies against TRIM5αhu. For controls, we electroporated a gRNA against CXCR4 and a non-targeting gRNA. Expression of GAPDH was used as a loading control.
(B) Human primary CD4+ T cells depleted for TRIM5αhu expression were challenged with increasing amounts of HIV-1-GFP or HIV-1-P90A-GFP viruses. Infection was measured at 72 h post-infection by determining the percentage of GFP-positive cells.
Experiments were performed in duplicates and a representative experiment is shown. The results of two independent experiments normalized to wild-type HIV-1-GFP infection with standard deviation are shown. **p < 0.001, ***p < 0.0005, ns indicates not significant as determined by the unpaired t test.
CypA-Capsid Interactions Are Required for Efficient HIV-1 Reverse Transcription in Human CD4+ T Cells
Our analysis shows that HIV-1 strains bearing capsid mutations that disrupt CypA-capsid interactions are unable to infect human CD4+ T cells. To identify the step in the HIV-1 life cycle at which infection is disrupted, we challenged human CD4+ T cells with p24-normalized levels of wild-type and mutant HIV-1-GFP viruses and measured reverse transcription, nuclear import, and integration using real-time quantitative polymerase chain reaction (qPCR) (Figure S6). Infection was measured by determining the percentage of GFP-positive cells at 72 h post-infection (Figure S6A). Consistent with our previous results, HIV-1-P90AGFP and HIV-1-G89V-GFP strains showed weak infection of CD4+ T cells (Figure S6A). Reverse transcription was measured by determining the levels of early reverse transcripts (ERTs) and late reverse transcripts (LRTs) at 12 h post-infection (Figure S6B). Interestingly, reverse transcription was reduced for HIV-1-P90A-GFP and HIV-1-G89V-GFP, suggesting that CypA-capsid interactions were important for HIV-1 reverse transcription in CD4+ T cells. As a control, we used Nevirapine (Nev), which inhibits reverse transcription (Figure S6B). For nuclear import, we measured the levels of 2-long terminal repeat (2-LTR) circles at 36 h post-infection (Figure S6C). Consistent with a reverse transcription block, our results show that both viruses were blocked for nuclear import. Viral integration was determined using Alu-PCR at 66 h post-infection (Figure S6D). As a control, the integration inhibitor Raltegravir (Ral) was also used. As expected, the integration of HIV-1-P90A and HIV-1-G89V was blocked. Overall, these results suggest that HIV-1 requires interaction with CypA to undergo efficient reverse transcription in human CD4+ T cells. These results also suggest that, in the absence of CypA, HIV-1 particles are exposed to TRIM5αhu, which inhibits reverse transcription.
DISCUSSION
A few years after the discovery of HIV-1 capsid interaction with CypA (Luban et al., 1993), it was established that CsA treatment disrupted this interaction, which then affected HIV-1 infectivity. It was soon realized that CsA exerted varying effects on HIV-1 infection, depending upon the human cell lines used. Although CypA-capsid interactions have been studied for over 25 years, the mechanism by which CsA affects HIV-1 infection remains elusive. Here, we explore the role of CypA-capsid interactions in human lymphocytes.
Several recent studies have suggested that TRIM5αhu may function as an HIV-1 restriction factor (Jimenez-Guardeño et al., 2019; OhAinle et al., 2018). This led us to hypothesize that TRIM5αhu binds to the HIV-1 core, which was proven by our results. We also show that endogenously expressed TRIM5αhu binds to the HIV-1 core. Remarkably, we observed that TRIM5αhu showed stronger binding to the HIV-1 core than TRIM5αrh. Because TRIM5αhu and CypA both bind to the HIV-1 core, it is possible that these two factors compete for the HIV-1 core during infection—CypA may modulate the binding of TRIM5αhu to the HIV-1 core. We show that depletion of TRIM5αhu expression in Jurkat and primary CD4+ T cells rescue the HIV-1 infectivity phenotype caused by the disruption of CypA-capsid interactions. In cellular extracts, we also observed that CsA treatment increased binding of TRIM5αhu to the HIV-1 core. These results were consistent with our hypothesis that CypA modulates the binding of TRIM5αhu to the HIV-1 core. The results of the binding experiments suggest that CypA sterically hinders the binding of TRIM5αhu to the HIV-1 core, which could explain the observed infectivity phenotype when CypA-capsid interactions were disrupted in lymphocytes. Conversely, we also demonstrate that increasing the amounts of CypA prevents the binding of TRIM5αhu to the HIV-1 core. Based on these results, we propose that prevention of CypA binding allows TRIM5αhu to bind to the HIV-1 core, consequently inhibiting infection. While this manuscript was under revision, Kim et al. (2019) showed similar results using immunofluorescence microscopy.
Our results indicate that the interaction of TRIM5αhu with the HIV-1 core leads to restriction of infection. However, several previous studies have shown that overexpression of TRIM5αhu has little or no effect on HIV-1 infection (Li et al., 2006; Pham et al., 2010; Yap et al., 2005). One possibility is that restriction of HIV-1 infection by TRIM5αhu requires specific cellular cofactors expressed only in human lymphocytes. This cofactor may have been absent or present only in insufficient amounts in other human cell lines that were used for TRIM5αhu overexpression experiments. The search for these cofactors in the future is warranted. Alternatively, TRIM5αhu might be recruited to the HIV-1 core by other TRIM proteins through the formation of higher-order complexes (Diaz-Griffero et al., 2009): TRIM5α dimers are known to associate with other dimers and form higher-order self-association complexes. For example, TRIM5αrh forms higher-order complexes with human TRIM34 and TRIM6 (Li et al., 2011). A third possibility is that TRIM5αhu might be recruiting other human TRIM orthologs to the HIV-1 core, which are causing restriction. Future studies will explore the existence of other human TRIM orthologs that may form higher-order complexes with TRIM5αhu.
Mutations in the CypA-binding loop of the capsid, such as P90A and G89V, cause an infectivity defect similar to that caused by CsA treatment during wild-type HIV-1 infection of Jurkat and CD4+ T cells; however, infection of HIV-1 viruses bearing the capsid change A92E or G94D results in an infection that is insensitive to CsA treatment in Jurkat cells (Hatziioannou et al., 2005; Sokolskaja et al., 2004). Here, we show that this insensitivity is due to the lack of TRIM5αhu binding to HIV-1-A92E or HIV-1-G94D mutant cores. Normal viral infectivity correlated with the inability of TRIM5αhu to bind to HIV-1-A92E and HIV-1-G94D cores. Based on these results, it is possible that TRIM5αhu binds to the CypA-binding loop or in its proximity.
The disruption of CypA-capsid interactions had a stronger effect on viral infectivity in human primary cells compared with cell lines. Mutant HIV-1 (P90A or G89V) infection of PBMCs or CD4+ T cells was 10–20-fold weaker than that of wild-type HIV-1. Similarly, KD of CypA expression in human primary CD4+ T cells potently blocked HIV-1 infection. In contrast, infection of primary lymphocytes by HIV-1-A92E or HIV-1-G94D was not affected when compared to that of wild-type HIV-1. In addition, depletion of TRIM5αhu expression in CD4+ T cells rescued infectivity of HIV-1-P90A viruses. Taken together, these results show that TRIM5αhu blocks HIV-1 infection of human primary lymphocytes when CypA-capsid interactions are disrupted. Consistent with this idea, we observe that the inhibition of infection caused by the disruption of CypA-capsid interactions in CD4+ T cells occurred prior to HIV-1 reverse transcription.
Our work has shown that CypA protects the core from TRIM5αhu. Intriguingly, new- and old-world monkeys express a protein in which the TRIM domain of TRIM5α and CypA form a fusion protein (Brennan et al., 2008; Newman et al., 2008; Sayah et al., 2004). It is possible that evolution may have taken advantage of a positive factor, in this case CypA, and fused to the TRIM domain to develop a restriction factor (i.e., TRIMCyp) in response to CypA protecting the virus against TRIM5α.
Although this work explains the infection phenotype observed when disrupting CypA-capsid interactions during infection, it does not explain the role of CypA in HIV-1 infection. It has been suggested that CypA is involved in nuclear import and integration (Schaller et al., 2011). CypA has also been observed to enter the nucleus with capsid (Burdick et al., 2017). Future experiments will provide mechanistic insights on the works of CypA in HIV-1 infection.
Overall, this work shows the mechanism by which disruption of CypA-capsid interactions affect HIV-1 infection of human primary lymphocytes. Future experiments will explore whether a similar mechanism operates in macrophages or dendritic cells.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 | |
| Mouse monoclonal anti-hemagglutinin (HA) | Sigma-Aldrich | Cat# H3663; RRID:AB_262051 | |
| Mouse monoclonal anti-Cyclophilin A (human) | Abcam | Cat# ab58144; RRID:AB_941220 | |
| Rabbit monoclonal anti-TRIM5a (human, monkey) | Cell Signaling Technology | Cat# 14326S; RRID:AB_2798451 | |
| Mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Invitrogen (Thermo Fisher Scientific) | Cat# AM4300; RRID:AB_437392 | |
| Mouse monoclonal anti-HIV-1 p24 (183-H12-5C) | NIH AIDS Reagent Program | Cat# 3637; RRID:CVCL_9N48 | |
| Goat anti-Mouse IRDye 680LT | LI-COR | Cat# 925–68020; RRID:AB_2687826 | |
| Goat anti-Rabbit IRDye 680LT | LI-COR | Cat# 926–68021; RRID:AB_10706309 | |
| Goat anti-Mouse IRDye 800CW | LI-COR | Cat# 926–32210; RRID:AB_621842 | |
| Goat anti-Rabbit IRDye 800CW | LI-COR | Cat# 925–32211; RRID:AB_2651127 | |
| Rabbit monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Cell Signaling Technology | Cat #2118S; RRID:AB_437392 | |
| Bacterial and Virus Strains | |||
| E. Coli one-shoot BL21star (DE3) | Invitrogen | Cat#C601003 | |
| E. Coli DH5α competent cells | Zymo Research | Cat# T3007 | |
| N-MLV | Diaz-Griffero et al., 2008 | N/A | |
| B-MLV | Diaz-Griffero et al., 2008 | N/A | |
| HIV-1 NL4-3 | This paper | N/A | |
| HIV-1-G89V | This paper | N/A | |
| HIV-1-P90A | This paper | N/A | |
| HIV-1-A92E | This paper | N/A | |
| HIV-1-G94D | This paper | N/A | |
| HIV-1 NL4-3 Nef:IRES:GFP | Hultquist et al., 2016 | N/A | |
| Biological Samples | |||
| Healthy adult Peripheral Blood Mononuclear Cells (PBMCs) | LifeSource/Vitalant | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| Cyclosporin A | Millipore Sigma | Cat#: PHR1092; CAS: 59865-13-3 | |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#: D2438; CAS: 67-68-5 | |
| Difco LB Broth, Miller (Luria-Bertani) | Fisher Scientific | Cat#: BD244610 | |
| Isopropyl β-D-1-thiogalactopyranoside | Sigma-Aldrich | Cat#: I5502; CAS: 367-93-1 | |
| cOmplete EDTA-free protease inhibitor cocktail | Millipore Sigma | Cat#: 11873580001, | |
| Tris (TRIS(HYDROXYMETHYL)AMINOMETHANE) | Crystalgen | Cat#300-844-5000; CAS: 77-86-1 | |
| Sodium Chloride (NaCl) | Crystalgen | Cat# 300-747-5000; CAS: 767-14-5 | |
| β-Mercaptoethanol (BME) | Acros organics | Cat# 125470010; CAS: 60-24-2 | |
| 2-(N-morpholino) ethanesulfonic acid (MES) | Calbiochem | Cat# 475893; CAS: 4432-31-9 | |
| Magnesium Chloride (MgCl2) | Sigma-Aldrich | Cat# M2670; CAS: 7791-18-6 | |
| Potassium Chloride (KCl) | Fisher Scientific | Cat# BP366-1; CAS: 7447-40-7 | |
| Dithiothreitol (DTT) | VWR | Cat# 97061-340; CAS: 3483-12-3 | |
| D-(+)-Sucrose | VWR | Cat# 97061-432; CAS: 57-50-1 | |
| Human IL-2 | Cell Signaling Technology | Cat# 8907SF | |
| Phytohemagglutinin-L (PHA-L) | Millipore Sigma | Cat# 11249738001 | |
| Human IL-2 | MACS Miltenyi Biotec | Cat# 130-097-742 | |
| Anti-Human CD3 (UCHT1) | Tonbo Biosciences | Cat# 70-0038-U100; RRID:AB_2621475 | |
| Anti-Human CD28 (CD28.2) | Tonbo Biosciences | Cat# 70-0289-U100; RRID:AB_2621493 | |
| Cas9-NLS Recombinant Protein, purified, 40uM | MacroLab Berkeley | N/A | |
| Dulbecco’s Phosphate-Buffered Salt (PBS) Solution 1X | Corning | 21031CV | |
| EDTA, pH 8.0, 0.5M | Corning | 46034CI | |
| Critical Commercial Assays | |||
| QuikChange II site-directed mutagenesis kit | Agilent | Cat#: 200523 | |
| CD4+ T Cell Isolation Kit, human | MACS Miltenyi Biotec | Cat# 130-096-533 | |
| EasySep Human CD4+ T Cell Isolation Kit | STEMCELL | Cat# 17952 | |
| P3 Primary Cell 96-well Nucleofector Kit | Lonza | Cat# V4SP-3096 | |
| T Cell Activation/Expansion Kit, human | MACS Miltenyi Biotec | Cat# 130-091-441 | |
| QIAamp DNA Micro Kit | QIAGEN | Cat# 56304 | |
| Experimental Models: Cell Lines | |||
| Human: Jurkat E6.1 | ATCC | TIB-152 | |
| Human: 293T/17 | ATCC | CRL-11268 | |
| Human: HT1080 | ATCC | CCL-121 | |
| Oligonucleotides | |||
| All standard cloning primers for site-directed mutagenesis | Integrated DNA Technologies | N/A | |
| Early reverse transcripts (ERT) hRU5-F, 5′-GCCTCAATAAAGCTTGCCTTGA-3′ | Mbisa et al., 2009 | N/A | |
| Early reverse transcripts (ERT) hRU5-R, 5′-TGACTAAAAGGGTCTGAGGGATCT-3′ | Mbisa et al., 2009 | N/A | |
| ERT probe, 5′-(FAM)-AGAGTCACACAAC AGACGGGCACACACTA-(TAMRA)-3′ | Mbisa et al., 2009 | N/A | |
| Late Reverse Transcripts (LTR) MH531, 5′-TGTGTGCCCGTCTGTTGTGT-3′, | Butler et al., 2001 | N/A | |
| Late Reverse Transcripts (LTR) MH532, 5′-GAGTCCTGCGTCGAGAGAGC-3′ | Butler et al., 2001 | N/A | |
| LRT-Probe, 5′-(FAM)-CAGTGGCGCCCGAACAGGGA-(TAMRA)-3′ | Butler et al., 2001 | N/A | |
| 2-LTR circles MH535, 5′-AACTAGGGAACCCACTGCTTAAG-3′ | Butler et al., 2001 | N/A | |
| 2-LTR circles MH536, 5′-TCCACAGATCAAGGATATCTTGTC-3’ | Butler et al., 2001 | N/A | |
| 2-LTR probe MH603, 5′-(FAM)-ACACTAC TTGAAGCACTCAAG-GCAAGCTTT-(TAMRA)-3′ | Butler et al., 2001 | N/A | |
| Table S1 Primers and RNA guides | N/A | N/A | |
| Recombinant DNA | |||
| pET-11a | EMD Mollipore | Cat # 69436 | |
| Plasmid: CA A14C/E45C in pET-11a | Selyutina et al., 2018 | N/A | |
| Plasmid: CA A14C/E45C/P90A in pET-11a | Selyutina et al., 2018 | N/A | |
| Plasmid: CA A14C/E45C/A92E in pET-11a | This paper | N/A | |
| Plasmid: CA A14C/E45C/G94D in pET-11a | This paper | N/A | |
| pLKO1 | Sigma | Cat# 10878 | |
| pLKO1-shRNA-TRIM5ahu | Sigma | Cat# 13478 | |
| Plasmid: human TRIM5a-HA in pLPCX | Perron et al., 2007 | N/A | |
| Plasmid: rhesus TRIM5a-HA in pLPCX | Stremlau et al., 2006a | N/A | |
| Plasmid: Tat | Stremlau et al., 2006a | N/A | |
| Plasmid: Rev | Stremlau et al., 2006a | N/A | |
| Plasmid: VSVg | Stremlau et al., 2006 | N/A | |
| Other | |||
| HiTrap Q HP 5mL | GE Healthcare | Cat# 17115401 | |
| HiTrap SP HP 5mL | GE Healthcare | Cat# 17115201 | |
| Centriprep Centrifugal Filter Unit 30 kDa | Millipore Sigma | Cat# 4306 | |
| SnakeSkin Dialysis Tubing, 10KMWCO | Fisher Scientific | Cat# 68100 | |
| GE Healthcare Ficoll-Paque PLUS Media | Fisher Scientific | Cat# 45001-749 | |
| Hanks’ Balanced Salt solution | Sigma-Aldrich | Cat# H6648 | |
| LS Columns | MACS Miltenyi Biotec | Cat# 130-042-401 | |
| MS Columns | MACS Miltenyi Biotec | Cat# 130-042-201 | |
| SepMate PBMC Isolation Tubes | STEMCELL | Cat# 85450 | |
| Maxima Probe/ROX qPCR Master Mix (2X) | ThermoScientific | Cat#K0231 | |
| TURBO DNase (2 U/μL) | Invitrogen | Cat#AM2238 | |
| Platinum SuperFi DNA Polymerase | Invitrogen | Cat#12351050 | |
| iTaq Universal SYBR® Green Supermix | Biorad | Cat#1725124 | |
| 0.2 ml PCR 8-tube with attached clear flat caps, natural | USA Scientific | 1402–3900 | |
| Fetal Bovine Serum (FBS, heat-inactivated) | GIBCO | 16140–071 | |
| RPMI-1640 (high glucose) | Corning | MT10-017-CV | |
| Penicillin-streptomycin (5 mg/mL) | Corning | MT10-040-CV | |
| Sodium pyruvate (100×) | Corning | MT25-000-CI | |
| HEPES (100×) | Fisher / HyClone | SH3023701 | |
| DMEM (high glucose) | Corning | MT10-017-CV | |
LEAD CONTACT AND MATERIALS AVAILABILITY
This study generated the following new plasmids: pET11A-A14C/E45C/A92E, pET11A-A14C/E45C/G94D, HIV-1-P90A, HIV-1-G89V, HIV-1-A92E and HIV-1-G94D. Plasmids generated and used in this study are available upon request. Further information and requests for plasmids, resources, and reagents should be directed to and will be fulfilled by the lead contact Felipe Diaz-Griffero (Felipe.Diaz-Griffero@einsteinmed.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
All molecular cloning was carried out in E. coli DH5α competent cells. All recombinant proteins were expressed and purified from E. coli one-shot BL21star (DE3) strain. Both bacterial stains were routinely grown in Luria-Bertani (LB) medium at 37°C (or at the indicated temperatures) while shaking at 200 RPM.
Human cell lines
Human Jurkat lymphocytes cells obtained from the American type culture collection (ATCC) were grown at 37°C in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Human 293T/17 and HT1080 (ATCC) cells were grown at 37°C in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% FBS and 1% penicillin-streptomycin.
Human PBMCs
Adult Peripheral Blood Mononuclear Cells (PBMCs) from anonymous, healthy donors were isolated from donated buffy coat products, a post-centrifugation product from a single whole blood donation that contains white blood cells and platelets (Lifesource/Vitalant, Chicago, and New York Blood Center, New York).
METHOD DETAILS
Infection using HIV-1-GFP reporter viruses
Recombinant human immunodeficiency virus-1 (HIV-1) strains such as HIV-1-G89V, HIV-1-P90A, HIV-1-A92E and HIV-1-G94D expressing GFP and pseudotyped with VSV-G were prepared as previously described (Diaz-Griffero et al., 2008). To produce replication competent HIV-1NL4–3-GFP virus, we transfected human 293T/17 cells with a full molecular clone containing the sequence of HIV-1NL4–3 that expresses Nef-IRES-GFP as a reporter of infection (Hultquist et al., 2016). All viruses were titered by p24 levels and infectivity. Viral challenges were performed in 24-well plates by infecting 50,000 cells (Jurkat, PBMCs, or CD4+ T cells) per well. Infection was determined by measuring the percentage of GFP-positive cells using flow cytometry (BD FACSCelesta).
Capsid expression and purification
pET-11a vectors were used to express the HIV-1 capsid protein containing the A14C and E45C mutations. Point mutations P90A, A92E and G94D were introduced using the QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. All proteins were expressed in Escherichia coli one-shot BL21star (DE3) cells (Invitrogen), as previously described (Selyutina et al., 2018). Briefly, cells were inoculated in Luria-Bertani (LB) medium and cultured at 30°C until mid-log phase (A600,0.6–0.8). Protein expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) overnight at 18°C. Cells were harvested by centrifugation at 5,000 × g for 10 min at 4°C, and pellets were stored at −80°C until purification. Purification of capsids was carried out as follows. Pellets from 2 L cultures were lysed by sonication (Qsonica microtip: 4420; A = 45; 2 min; 2 s on; 2 s off for 12 cycles), in 40 mL of lysis buffer (50 mM Tris pH = 8, 50 mM NaCl, 100 mM β-mercaptoethanol, and Complete EDTA-free protease inhibitor tablets). Cell debris was removed by centrifugation at 40,000 × g for 20 min at 4°C. Proteins from the supernatant were precipitated by incubation with one-third the volume of saturated ammonium sulfate containing 100 mM β-mercaptoethanol for 20 min at 4°C, and centrifugation at 8,000 × g for 20 min at 4°C. Precipitated proteins were resuspended in 30 mL of buffer A (25 mM 2-(N-morpholino) ethanesulfonic acid (MES) pH 6.5 and 100 mM β-mercaptoethanol) and sonicated 2–3 times (Qsonica microtip: 4420; A = 45; 2 min; 1 s on; 2 s off). The protein sample was dialyzed three times in buffer A (2 h, overnight, 2 h), sonicated, diluted in 500 mL of buffer A, and was chromatographed sequentially on a 5 mL HiTrap Q HP column and on a 5 mL HiTrap SP FF column (GE Healthcare), which were both pre-equilibrated with buffer A. The capsid protein was eluted from the HiTrap SP FF column using a linear gradient of concentrations ranging from 0–2 M NaCl. The eluted fraction that had highest protein levels was selected based on absorbance at 280 nm. Pooled fractions were dialyzed three times (2 h, overnight, 2 h) in storage buffer (25 mM MES, 2 M NaCl, 20 mM β-mercaptoethanol). The sample was reduced to a concentration of 20 mg/ml using Centriprep Centrifugal Filter Unit and stored at −80°C.
Assembly of stabilized HIV-1 capsid tubes
One milliliter of monomeric capsid (5 mg/ml) was dialyzed in SnakeSkin dialysis tubing 10K MWCO (Thermo Scientific) using a buffer that is high in salt and contains a reducing agent (buffer 1: 50 mM Tris, pH 8, 1 M NaCl, 100 mM β-mercaptoethanol) at 4°C for 8 h. Subsequently the protein was dialyzed using buffer 1 without the reducing agent β-mercaptoethanol (buffer 2: 50 mM Tris, pH 8, 1 M NaCl) at 4°C for 8 h. The absence of β-mercaptoethanol in the second dialysis allows the formation of disulfide bonds between Cysteine 14 and 45 inter-capsid monomers in the hexamer. Finally, the protein was dialyzed using buffer 3 (20 mM Tris, pH 8,0, 40 mM NaCl) at 4°C for 8 h. Assembled complexes were kept at 4°C for up to 1 month.
Capsid binding assay protocol
Human HEK293T cells were transfected for 24 h with a plasmid expressing the protein of interest (TRIM5αhu or TRIM5αrh). The cell medium was completely removed and cells were scraped off the plate and lysed in 300 μL of capsid binding buffer (CBB: 10 mM Tris, pH 8,0, 1,5 mM MgCl2, 10 mM KCl). Cells were rotated at 4°C for 15 min and then centrifuged to remove cellular debris (21,000 3 g, 15 min, 4°C). Cell lysates were incubated with stabilized HIV-1 capsid tubes for 1 h at 25°C. Subsequently, the stabilized HIV-1 capsid tubes were centrifuged at 21,000 × g for 2 min. Pellets were washed 2–3 times by resuspension and centrifugation in CBB or phosphate-buffered saline (PBS). Pellets were resuspended in Laemmli buffer 1 × and analyzed by western blot using anti-p24 and the indicated antibodies.
Capsid binding assay using in vitro assembled HIV-1 CA-NC complexes
Capsid binding assays were performed as described earlier (Lienlaf et al., 2011). Briefly, HIV-1 CA-NC particles were assembled in vitro by incubating in buffer A (50 mM Tris (pH 8,0), 0.5 mM NaCl, 2 mg/ml DNA oligo-(TG)50) at 4°C overnight. Human HEK293T cells were transfected for 24 h with a plasmid expressing the protein of interest (TRIM5αhu or TRIM5αrh,). The cell medium was completely removed and cells were scraped off the plate and lysed in 300 μL of capsid binding buffer with DTT (CBB+: 10 mM Tris, pH 8,0, 1,5 mM MgCl2, 10 mM KCl, 0,5 mM DTT). Cells were rotated at 4°C for 15 min and then centrifuged to remove cellular debris (21,000 × g, 15 min, 4°C). Cell lysates were incubated with HIV-1 CA-NC complexes capsid tubes for 1 h at 25°C. The mixture was spun through a 70% sucrose cushion (70% sucrose, 1 × PBS) at 100,000 × g in an SW55 rotor (Beckman), 1 h, 4°C. After centrifugation pellets were resuspended in Laemmli buffer 1 × and analyzed by western blot using anti-p24 and anti-HA antibodies.
Preparation of PBMCs and CD4+ T cells
PBMCs from healthy donor whole blood were isolated by density gradient centrifugation using Ficoll-Paque Plus (GE Health Care). 40 mL of whole blood was centrifuged at 300 × g for 10 min, and the plasma layer was removed and replaced with Hank’s Balanced Salt solution (HBSS; Sigma Aldrich). The whole blood was then diluted to a ratio of 1:2 with HBSS with 20 mL of the diluted blood layered on top of 20 mL Ficoll-Paque Plus and centrifuged at 300 × g for 30 min. The resulting buffy coat layer was collected, washed twice with HBSS, and resuspended in RPMI medium containing 10% (vol/vol) FBS and 1% (vol/vol) penicillin-streptomycin, and activated with IL-2 (100 U/ml) (Human IL-2; Cell Signaling Technology, #8907SF) and phytohemagglutinin (1 μg/ml) for 3 days. CD4+ T cells were obtained via negative selection from PBMCs using a human CD4+ T cell isolation kit (MACS Miltenyi Biotec, #130-096-533). Per 1 × 107 total cells, PBMCs were resuspended in 40 μl CD4+ T cell isolation buffer (PBS, pH 7.2, 0.5% bovine serum albumin, and 2 mM EDTA). Ten microliters of CD4+ T cell biotin-antibody cocktail was then added and incubated at 4°C for 5 min. Thirty microliters of CD4+ T cell isolation buffer and 20 μl of CD4+ T cell microbead cocktail were then added and further incubated for 10 min at 4°C. Depending on the amount of PBMCs isolated, LS or MS Column attached to a MACS Separator was pre-washed with 3 mL or 6 mL of ice-cold CD4+ T cell isolation buffer, respectively. PBMC suspension was added to the column and the flow-through was collected in a 15 mL tube. The LS or MS Columns were then washed with 3 mL or 6 mL of ice-cold CD4+ T cell isolation buffer, respectively, and the flow-through was collected. The newly isolated CD4+ T cells were then centrifuged at 800 × g for 5 min and resuspended in RPMI medium supplemented with IL-2 (100 U/ml).
Knockdown of TRIM5α expression in Jurkat cells
Lentiviral particles were produced in human 293T/17 cells by co-transfection of 1 μg TAT, 1 μg REV, 1.5 μg VSVG, 1 mg HDM (codon optimized gag-pol), and 7.5 μg of the lentiviral vector pLKO1 or 7.5 μg the pLKO1 vector containing an shRNA that targets TRIM5α (pLKO1-shRNA-TRIM5α). Viruses were harvested at 48 h post-transfection and used to transduce Jurkat cells for 5 days at a 1:1 virus to complete media ratio. Transduced cells were selected in 0.15 μg/ml of puromycin. Cells that stably contained pLKO1 or pLKO1-shRNA-TRIM5α were maintained in puromycin. Expression of TRIM5α was evaluated by western blotting and by challenging the cells with N-tropic murine leukemia virus (N-MLV) and B-tropic murine leukemia virus (B-MLV).
Generation of TRIM5α knockout HT1080 cell line
Human TRIM5α knockout (KO) HT1080 cell line was generated by using the clustered regulatory interspaced short palindromic repeat (CRISPR)-Cas9 gene system (Edit-R CRISPR-Cas9 gene engineering with SMARTCas9 nucleases; GE Healthcare). The CRISPR genomic guide RNA sequence for human TRIM5α was designed to target exon 1 near the start codon. The guide RNA construct, human cytomegalovirus (hCMV)-Cas9, and CMV-GFP were co-transfected into HT-1080 cells. After 48 h of transfection, GFP-positive cells were single-cell sorted by using the FACSAria system (BD Biosciences, Franklin Lakes, NJ, USA). Clonal HT-1080 colonies were expanded and characterized for the loss of TRIM5α protein expression by western blot analysis using anti-TRIM5α antibodies and by challenging the cells with N-tropic murine leukemia virus (N-MLV) and B-tropic murine leukemia virus (B-MLV).
CRISPR-Cas9 Knock-outs in Primary CD4+ T Cells
Detailed protocols for the production of CRISPR-Cas9 ribonucleoprotein complexes (crRNPs) and primary CD4+ T cell editing have been previously published (Hultquist et al., 2016; Hultquist et al., 2019). Briefly, lyophilized crRNA and tracrRNA (Dharmacon) were suspended at a concentration of 160 μM in 10 mM Tris-HCl (7.4 pH), 150 mM KCl. 5μL of 160μM crRNA was mixed with 5μL of 16μM tracrRNA and incubated for 30 min at 37°C. The gRNA:tracrRNA complexes were then mixed gently with 10μL of 40μM purified Cas9-NLS protein (UC-Berkeley Macrolab) to form CRISPR-Cas9 ribonucleoprotein complexes (crRNPs). Five 3.5μL aliquots were frozen in 0.2 mL PCR tubes (USA Scientific) at −80°C until use. crRNA guide sequences used in this study were a combination of sequences derived from the Dharmacon pre-designed Edit-R library for gene knock-out and custom ordered sequences as indicated.
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque Plus (GE Health Care, #17-1440-02). PBMCs were washed with PBS 1× three times to remove platelets and suspended at a final concentration of 5*10^8 cells/mL in PBS 1×, 0.5% BSA, 2mM EDTA. Bulk CD4+ T cells were subsequently isolated from PBMCs by magnetic negative selection using an EasySep Human CD4+ T Cell Isolation Kit (STEMCELL, per manufacturer’s instructions). Isolated CD4+ T cells were suspended in complete Roswell Park Memorial Institute (RPMI), consisting of RPMI-1640 (Sigma) supplemented with 5mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Corning), 50 μg/mL penicillin/streptomycin (P/S, Corning), 5mM sodium pyruvate (Corning), and 10% fetal bovine serum (FBS, GIBCO). Media was supplemented with 20 IU/mL IL-2 (Miltenyi) immediately before use. For activation, bulk CD4+ T cells were immediately plated on anti-CD3 coated plates [coated for 12 hours at 4°C with 20μg/mL anti-CD3 (UCHT1, Tonbo Biosciences)] in the presence of 5μg/mL soluble anti-CD28 (CD28.2, Tonbo Biosciences). Cells were stimulated for 72 hours at 37°C / 5% CO2 prior to electroporation. After stimulation, cell purity and activation was verified by CD4/CD25 immunostaining and flow cytometry as previously described (Hultquist et al., 2016; Hultquist et al., 2019).
After three days of stimulation, cells were suspended and counted. Each electroporation reaction consisted of between 5×10^5 and 1×10^6 T cells, 3.5 μL RNP, and 20 μL electroporation buffer. crRNPs were thawed and allowed to come to room-temperature. Immediately prior to electroporation, cells were centrifuged at 400×g for 5 minutes, supernatant was removed by aspiration, and the pellet was resuspended in 20 μL of room-temperature P3 electroporation buffer (Lonza) per reaction. 20 μL of cell suspension was then gently mixed with each RNP and aliquoted into a 96-well electroporation cuvette for nucleofection with the 4D 96-well shuttle unit (Lonza) using pulse code EH-115. Immediately after electroporation, 100 μL of pre-warmed media without IL-2 was added to each well and cells were allowed to rest for 30 minutes in a 37°C cell culture incubator. Cells were subsequently moved to 96-well flat-bottomed culture plates pre-filled with 100 μL warm complete media with IL-2 at 40 U/mL (for a final concentration of 20 U/mL) and anti-CD3/anti-CD2/anti-CD28 beads (T cell Activation and Stimulation Kit, Miltenyi) at a 1:1 bead:cell ratio. Cells were cultured at 37°C / 5% CO2 in a dark, humidified cell culture incubator for 4 days to allow for gene knock-out and protein clearance, with additional media added on day 2. To check knock-out efficiency, 50 μL of mixed culture was removed to a centrifuge tube. Cells were pelleted, supernatant was removed, and pellets were resuspended in 100 μL 2.5x Laemmli Sample Buffer. Protein lysates were heated to 98°C for 20 min before storage at −20°C until western blotting.
Detection of Reverse Transcripts, 2-LTR circles, and integration in CD4+ T cells
Total DNA was extracted from 1 × 106 infected cells. Samples were processed at 12, 36, and 66 h post-infection using QIAmp DNA micro kit (QIAGEN). Viral DNA forms of wild-type and mutant HIV-1 were amplified using real-time PCR (Butler et al., 2001; Valle-Casuso et al., 2012). β-actin amplification was used for normalization. ERT, which refers to the early reverse transcripts (prior the minus strand transfer), and LRT, which refers to the whole synthesis of DNA by HIV-1, were measured at 12 h post-infection. 2-LTR circles were measured at 36 h post-infection to assess nuclear import. Provirus integration in cellular genome was measured at 66 h post-infection using Alu-PCR. Reactions were performed in 1X FastStart Universal Probe Master Mix (Rox) 2 × (Roche) in 20 μl volume. The PCR reaction consisted of the following steps: initial annealing (50°C 2 min), denaturation step (95°C 15 min), 40 cycles of amplification (95°C 15 s, 58°C 30 s, 72°C 30 s). Primer or probe sequences are as follows: ERT hRU5-F2: 5′-GCCTCAA TAAAGCTTGCCTTGA-3′; hRU5-R: 5′-TGACTAAAAGGGTCTGAGGGATCT-3′; ERT-Probe: 5′-(FAM)-AGAGTCACACAACAGACGGGCACACACTA-(TAMRA)-3′. LRTMH531: 5′-TGTGTGCCCGTCTGTTGTGT-3′; MH532: 5′-GAGTCCTGCGTCGAGAGAGC-3′; LRT-Probe: 5′-(FAM)-CAGTGGCGCCCGAACAGGGA-(TAMRA)-3′. 2-LTR circles: MH535: 5′-AACTAGGGAACCCACTGCTTAAG-3′; MH536: 5′-TCCACAGATCAAGGATATCTTGTC-3′; 2-LTR probe: MH603: 5′-(FAM)-ACACTACTTGAAGCACTCAAG-GCAAGCTTT-(TAMRA)-3′. Alu-PCR 1st round: Alu 1: 5′-TCCCAGCTACTCGGGAGGCTGAGG-3′; Alu 2: 5′-GCCTCCCAAAGTGCTGGGATTACAG-3′; Lambda U3: 5′-ATGCCACGTAAGCGAAACTTTCCGCTGGGGACTTTCCAGGG-3′. Alu quantitative PCR 2nd round: Lambda: (5′-ATGCCACGTAAGCGAAACT-3′); U5: (5′-CTGACTAAAAGGGTCTGAGG-3′; Probe: 5′-(FAM)- TTAAGCCTCAATAAAGCTTGCCTT GAGTGC-(TAMRA)-3′. β-actin: FX: 5′-AACACCCCAGCCATGTACGT-3′, RX: 5-CGGTGAGGATCTTCATGAGGTAGT-3′, Actin-Probe: (6FAM)-CCAGCCAGGTCCAGACGCAGGA-(BBQ)-3′.
Western blot analysis
Cells were lysed in lysis buffer (50 mM Tris [pH 8.0], 280 mM NaCl, 0.5% IGEPAL 40, 10% glycerol, 5 mM MgCl2). Proteins were detected by western blot using anti-FLAG (1:1,000 dilution; Sigma), anti-hemagglutinin (HA) (1:1,000 dilution; Sigma), anti-CypA (1:5,000 dilution; catalog number ab58144; Abcam), anti-TRIM5αhu (1:1,000 dilution; catalog number 14326S; Cell Signaling Technology), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5,000 dilution; Invitrogen), or anti-p24 (1:1,000 dilution, catalog number 3637; NIH) antibodies. Secondary antibodies against rabbit and mouse IgG conjugated to IRDye 680LT or IRDye 800CW were obtained from Li-Cor (1:10,000 dilution). Bands were detected by scanning blots using the Li-Cor Odyssey imaging system in the 700-nm or 800-nm channels.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using unpaired t test. Information on sample number, amount of repeats and p value can be found in the corresponding figure legend. Quantification of the intensity of western blot bands was performed using ImageJ. For all experiments the mean and standard deviation values were calculated using GraphPad Prism 7.0c.
DATA AND CODE AVAILABILITY
This study did not generate any datasets or code.
Supplementary Material
Highlights.
Depletion of cyclophilin A (CypA) expression in CD4+ T cells blocks HIV-1 infection
CypA binding to the core prevents HIV-1 restriction by TRIM5a during T cell infection
Endogenously expressed TRIM5α binds to the HIV-1 core and blocks reverse transcription
Human TRIM5α binds to HIV-1 cores but not to cores bearing capsid changes A92E/G94D
ACKNOWLEDGMENTS
We thank the NIH AIDS repository for important reagents. We are very grateful to Kyusik Kim and Jeremy Luban for reagents and helpful discussions. M.P. and C.B. acknowledge support from National Institutes of Health grant T32 AI07501. F.D.-G. is supported NIH NIAID grant R01 AI087390. J.F.H. is supported by amfAR grant 109504-61-RKRL, with funds raised by generation-CURE, the Gilead Sciences Research Scholars Program in HIV, NIH grant K22 AI136691, a supplement from the NIH-supported Third Coast CFAR P30 AI117943, and a supplement from the NIH-sponsored HARC Center P50 GM082250. V.S. and F.D.N. were funded by ANRS (Agence Nationale de Recherche sur le SIDA) grant ECTZ4469.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.100.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Berthoux L, Sebastian S, Sokolskaja E, and Luban J (2005). Cyclophilin A is required for TRIM5αlpha-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. USA 102, 14849–14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaten D, Franke EK, and Luban J (1996). Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol 70, 3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaten D, and Luban J (2001). Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 20, 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, and Elledge SJ (2008). Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926. [DOI] [PubMed] [Google Scholar]
- Brennan G, Kozyrev Y, and Hu SL (2008). TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. USA 105, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffone C, Martinez-Lopez A, Fricke T, Opp S, Severgnini M, Cifola I, Petiti L, Frabetti S, Skorupka K, Zadrozny KK, et al. (2018). Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol 92, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick RC, Delviks-Frankenberry KA, Chen J, Janaka SK, Sastri J, Hu WS, and Pathak VK (2017). Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 13, e1006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burse M, Shi J, and Aiken C (2017). Cyclophilin A potentiates TRIM5α inhibition of HIV-1 nuclear import without promoting TRIM5α binding to the viral capsid. PLoS ONE 12, e0182298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, and Bushman FD (2001). A quantitative assay for HIV DNA integration in vivo. Nat. Med 7, 631–634. [DOI] [PubMed] [Google Scholar]
- Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, and Debyser Z (2008). Transportin-SR2 imports HIV into the nucleus. Curr. Biol 18, 1192–1202. [DOI] [PubMed] [Google Scholar]
- Di Nunzio F, Danckaert A, Fricke T, Perez P, Fernandez J, Perret E, Roux P, Shorte S, Charneau P, Diaz-Griffero F, and Arhel NJ (2012). Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS ONE 7, e46037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nunzio F, Fricke T, Miccio A, Valle-Casuso JC, Perez P, Souque P, Rizzi E, Severgnini M, Mavilio F, Charneau P, and Diaz-Griffero F (2013). Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 440, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F, Perron M, McGee-Estrada K, Hanna R, Maillard PV, Trono D, and Sodroski J (2008). A human TRIM5αlpha B30.2/SPRY domain mutant gains the ability to restrict and prematurely uncoat B-tropic murine leukemia virus. Virology 378, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, and Sodroski J (2009). A B-box 2 surface patch important for TRIM5αlpha self-association, capsid binding avidity, and retrovirus restriction. J. Virol 83, 10737–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F, Vandegraaff N, Li Y, McGee-Estrada K, Stremlau M, Welikala S, Si Z, Engelman A, and Sodroski J (2006). Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351, 404–419. [DOI] [PubMed] [Google Scholar]
- Fernandez J, Machado AK, Lyonnais S, Chamontin C, Gärtner K, Léger T, Henriquet C, Garcia C, Portilho DM, Pugnière M, et al. (2019). Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol 4, 1840–1850. [DOI] [PubMed] [Google Scholar]
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, and Hill CP (1996). Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, and Yeager M (2011). Hexagonal assembly of a restricting TRIM5αlpha protein. Proc. Natl. Acad. Sci. USA 108, 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T, Perez-Caballero D, Cowan S, and Bieniasz PD (2005). Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol 79, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Hiatt J, Schumann K, McGregor MJ, Roth TL, Haas P, Doudna JA, Marson A, and Krogan NJ (2019). CRISPR-Cas9 genome engineering of primary CD4(+) T cells for the interrogation of HIV-host factor interactions. Nat. Protoc 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Schumann K, Woo JM, Manganaro L, McGregor MJ, Doudna J, Simon V, Krogan NJ, and Marson A (2016). A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 17, 1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idica A, Sevrioukov EA, Zisoulis DG, Hamdorf M, Daugaard I, Kadan-dale P, and Pedersen IM (2017). MicroRNA miR-128 represses LINE-1 (L1) retrotransposition by down-regulating the nuclear import factor TNPO1. J. Biol. Chem 292, 20494–20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Guardeño JM, Apolonia L, Betancor G, and Malim MH (2019). Immunoproteasome activation enables human TRIM5α restriction of HIV-1. Nat. Microbiol 4, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keckesova Z, Ylinen LM, and Towers GJ (2006). Cyclophilin A renders human immunodeficiency virus type 1 sensitive to Old World monkey but not human TRIM5 alpha antiviral activity. J. Virol 80, 4683–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Dauphin A, Komurlu S, McCauley SM, Yurkovetskiy L, Carbone C, Diehl WE, Strambio-De-Castillia C, Campbell EM, and Luban J (2019). Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol 4, 2044–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al. (2008). Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, and Engelman A (2010). The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J. Virol 84, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, et al. (2010). Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A, Hayouka Z, Friedler A, and Loyter A (2010). Transportin 3 and importin a are required for effective nuclear import of HIV-1 integrase in virus-infected cells. Nucleus 1, 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kar AK, and Sodroski J (2009). Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol 83, 10951–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li X, Stremlau M, Lee M, and Sodroski J (2006). Removal of arginine 332 allows human TRIM5αlpha to bind human immunodeficiency virus capsids and to restrict infection. J. Virol 80, 6738–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yeung DF, Fiegen AM, and Sodroski J (2011). Determinants of the higher order association of the restriction factor TRIM5αlpha and other tripartite motif (TRIM) proteins. J. Biol. Chem 286, 27959–27970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienlaf M, Hayashi F, Di Nunzio F, Tochio N, Kigawa T, Yokoyama S, and Diaz-Griffero F (2011). Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5αlpha(rh): structure of the RING domain of TRIM5alpha. J. Virol 85, 8725–8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, and Emerman M (2008). Determinants of cyclophilin A-dependent TRIM5 alpha restriction against HIV-1. Virology 379, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luban J, Bossolt KL, Franke EK, Kalpana GV, and Goff SP (1993). Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Matreyek KA, and Engelman A (2011). The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol 85, 7818–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Yücel SS, Li X, and Engelman A (2013). Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 9, e1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbisa JL, Delviks-Frankenberry KA, Thomas JA, Gorelick RJ, and Pathak VK (2009). Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol. Biol 485, 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan AM, Saenz DT, Guevera R, Morrison JH, Peretz M, Fadel HJ, Hamada M, van Deursen J, and Poeschla EM (2014). A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection. PLoS Pathog. 10, e1003969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O’Neil SP, and Johnson W (2008). Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog. 4, e1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, et al. (2011). HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 7, e1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhAinle M, Helms L, Vermeire J, Roesch F, Humes D, Basom R, Delrow JJ, Overbaugh J, and Emerman M (2018). A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, and Sodroski J (2007). The human TRIM5αlpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol 81, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham QT, Bouchard A, Grütter MG, and Berthoux L (2010). Generation of human TRIM5αlpha mutants with high HIV-1 restriction activity. Gene Ther. 17, 859–871. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, and Yeager M (2009). X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, Bobardt MD, de Béthune MP, Neyts J, De Clercq E, et al. (2008). Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob. Agents Chemother 52, 1302–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, and Luban J (2004). Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430, 569–573. [DOI] [PubMed] [Google Scholar]
- Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hué S, Fletcher AJ, Lee K, KewalRamani VN, et al. (2011). HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyutina A, Bulnes-Ramos A, and Diaz-Griffero F (2018). Binding of host factors to stabilized HIV-1 capsid tubes. Virology 523, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry B, Zybarth G, Alfano M, Dubrovsky L, Mitchell R, Rich D, Ulrich P, Bucala R, Cerami A, and Bukrinsky M (1998). Role of cyclophilin A in the uptake of HIV-1 by macrophages and T lymphocytes. Proc. Natl. Acad. Sci. USA 95, 1758–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolskaja E, Sayah DM, and Luban J (2004). Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol 78, 12800–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, and Sodroski J (2006a). Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5αlpha restriction factor. Proc. Natl. Acad. Sci. USA 103, 5514–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Song B, Javanbakht H, Perron M, and Sodroski J (2006b). Cyclophilin A: an auxiliary but not necessary cofactor for TRIM5αlpha restriction of HIV-1. Virology 351, 112–120. [DOI] [PubMed] [Google Scholar]
- Sundquist WI, and Hill CP (2007). How to assemble a capsid. Cell 131, 17–19. [DOI] [PubMed] [Google Scholar]
- Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, and Göttlinger HG (1994). Functional association of cyclophilin A with HIV-1 virions. Nature 372, 363–365. [DOI] [PubMed] [Google Scholar]
- Thys W, De Houwer S, Demeulemeester J, Taltynov O, Vancraenenbroeck R, Gérard M, De Rijck J, Gijsbers R, Christ F, and Debyser Z (2011). Interplay between HIV entry and transportin-SR2 dependency. Retrovirology 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, and Bieniasz PD (2003). Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med 9, 1138–1143. [DOI] [PubMed] [Google Scholar]
- Valle-Casuso JC, Di Nunzio F, Yang Y, Reszka N, Lienlaf M, Arhel N, Perez P, Brass AL, and Diaz-Griffero F (2012). TNPO3 is Required for HIV-1 Replication After Nuclear Import but Prior to Integration and Binds the HIV-1 Core. J Virol 86, 5931–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg MA, Dascal A, Blain N, Fitz-Gibbon L, Boulerice F, Numazaki K, and Tremblay M (1988). The effect of cyclosporine A on infection of susceptible cells by human immunodeficiency virus type 1. Blood 72, 1904–1910. [PubMed] [Google Scholar]
- Yap MW, Nisole S, and Stoye JP (2005). A single amino acid change in the SPRY domain of human TRIM5αlpha leads to HIV-1 restriction. Curr. Biol 15, 73–78. [DOI] [PubMed] [Google Scholar]
- Yin L, Braaten D, and Luban J (1998). Human immunodeficiency virus type 1 replication is modulated by host cyclophilin A expression levels. J. Virol 72, 6430–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, and Espeseth AS (2008). Genomescale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets or code.