

Abstract
Unimolecular dual agonists of the glucagon (GCG) receptor (GCGR) and glucagon-like peptide-1 receptor (GLP-1R) are a new class of drugs that are potentially superior to GLP-1R–specific agonists for the management of metabolic disease. The dual-agonist, peptide 15 (P15), is a glutamic acid 16 analog of GCG with GLP-1 peptide substitutions between amino acids 17 and 24 that has potency equivalent to those of the cognate peptide agonists at the GCGR and GLP-1R. Here, we have used cryo-EM to solve the structure of an active P15-GCGR-Gs complex and compared this structure to our recently published structure of the GCGR-Gs complex bound to GCG. This comparison revealed that P15 has a reduced interaction with the first extracellular loop (ECL1) and the top of transmembrane segment 1 (TM1) such that there is increased mobility of the GCGR extracellular domain and at the C terminus of the peptide compared with the GCG-bound receptor. We also observed a distinct conformation of ECL3 and could infer increased mobility of the far N-terminal His-1 residue in the P15-bound structure. These regions of conformational variance in the two peptide-bound GCGR structures were also regions that were distinct between GCGR structures and previously published peptide-bound structures of the GLP-1R, suggesting that greater conformational dynamics may contribute to the increased efficacy of P15 in activation of the GLP-1R compared with GCG. The variable domains in this receptor have previously been implicated in biased agonism at the GLP-1R and could result in altered signaling of P15 at the GCGR compared with GCG.
Keywords: glucagon receptor, glucagon-like peptide-1 receptor, dual agonist, cryo-electron microscopy, single particle analysis, structure-function, structural biology, peptide 15 (P15), metabolic disorder, G protein–coupled receptor (GPCR), glucagon, GLP-1 receptor
The G protein–coupled receptor (GPCR) superfamily is one of the largest membrane protein families, and GPCRs are widely distributed in the human body, where they are involved in most physiological activities (1). GPCRs can be divided into subclasses based on sequence homology and evolution, and among these the B1 class encompasses receptors for many important peptide hormones (1, 2). There are 15 class B1 GPCRs, including the glucagon receptor subfamily that comprises receptors for glucagon (GCG), glucagon-like peptide-1 (GLP-1), glucagon-like peptide-2 (GLP-2), and gastric inhibitory peptide (GIP). GCG, GLP-1, and GIP are key metabolic hormones that are essential for glucose homeostasis and caloric intake and disposal, with the peptides having both complementary and opposing actions (3).
There has been much interest in receptors for these metabolic hormones as potential therapeutic targets, with GLP-1 receptor (GLP-1R) agonists now established as highly effective drugs for treatment of type 2 diabetes and obesity (4). In a drug-specific manner, the GLP-1R agonists may also have cardiovascular and weight loss benefits in overweight and obese patients (5). To date, there are no drugs that target the GCG receptor (GCGR) and this is primarily because of the complexity of glucagon-regulated physiological effects. Glucagon is a key regulator of carbohydrate, lipid, and amino acid metabolism that is recognized for its counter-regulatory role to the actions of insulin, and is responsible for glycogen and glycolipid decomposition resulting in elevation of blood glucose (6). However, glucagon also increases energy expenditure and can attenuate food intake leading to interest in combinatory effects with GLP-1R agonists for treatment of obese and diabetic patients. Recently, novel unimolecular agonists that target multiple receptors of the GCGR family have been developed and have shown high efficacy and promising safety profiles in clinical trials (7). Among them, dual agonists of GLP-1R and GCGR have attracted much attention as they were more effective than single drug or multi-drug combination (8–10). Here, we report the cryo-EM structure of GCGR in complex with heterotrimeric Gs protein and a dual agonist, peptide 15 (P15) (11), at a global resolution of 3.4 Å. This structure, combined with mutagenesis and pharmacological studies, reveals molecular details of ligand binding and activation of the GCGR. The results provide valuable information for the design and development of future drugs.
Results
P15 is a modified form of the GCG peptide containing the following residues from GLP-1 (numbered from the GCG sequence), Gln-17, Ala-18, Lys-20, Glu-21, Ile-23, and Ala-24, with an additional Ser to Glu substitution at position 16 (Fig. S1A) (11). The peptide had approximately equal potency to GLP-1(7-36)NH2 in CHO-K1 cells expressing the WT human GLP-1R (Fig. S1B and Table S1), and to GCG in CHO-K1 cells expressing WT human GCGR (Fig. S1C and Table S1). However, it had ∼10-fold lower affinity than the cognate peptides for each receptor in competition binding assays in these cells (Fig. S1, D and E and Table S2).
Structure determination
To understand molecular details of P15 binding to GCGR, we determined a cryo-EM structure of the active P15-GCGR-Gs complex. To achieve this, we utilized the GCGR expression construct (HA-GCGR-HPC4) previously used for determination of the active GCG-GCGR structure (12), with the complex stabilized by use of a dominant negative form of Gαs (13, 14), and nanobody 35 (Nb35) that binds across the Gαs-Gβ interface (15). Although the HA-GCGR-HPC4 construct had lower expression than the WT GCGR in CHO-K1 cells, the relative pharmacology of P15 and GCG was equivalent for both receptors (Fig. S1, C and E and Tables S1 and S2). Purified complex was resolved as a monodisperse peak on size size exclusion chromatography (SEC) (Fig. 1A), with all components of the complex identified in Coomassie Blue–stained SDS-PAGE of the SEC peak (Fig. 1B). Although there was significant orientation preference of particles (Fig. 1C), cryo-EM imaging of the sample yielded 2D class averages with well-resolved secondary structure (Fig. 1D) that was reconstructed into a consensus 3D density map with a global resolution at gold standard FSC 0.143 of 3.4 Å, with local resolution ranging from 3.2 to 5.8 Å (Fig. 1, E–H). Highest resolution was observed in the G protein and at the interface between the G protein and receptor, with lower resolution at the extracellular face of the receptor and only limited resolution for the receptor ECD (Fig. 1G), indicative of higher mobility of this domain. The α-helical domain of the Gαs subunit was poorly resolved and masked out during the final refinement. The map resolution enabled modeling of side chains of most amino acids in the receptor core and G protein, albeit that assignment of rotamers was often ambiguous (Fig. S2). Density for parts of ECL1 (Gln-204–Ser-213), ICL3 (His-340–Asp-342), and ECL3 (Val-368–Gly-375) was ambiguous and these segments were not modeled. There was no density for Ala-26ECD or C-terminal residues beyond Trp-4188.53 and these were not modeled. Where there was insufficient density to model the α-carbon of side chains, these residues were stubbed, including many residues in the ECD, along with Leu-3546.45 (superscript numbers refer to the Wootten et al. (16) class B GPCR numbering scheme), Thr-3767.33, and Leu-3777.34 (Fig. S3). The N terminus of P15 was reasonably resolved, but there was more limited density for the C-terminal half of the peptide (Fig. S2), consistent with the notion of reduced stability of interactions between the receptor ECD and peptide C terminus.
Figure 1.
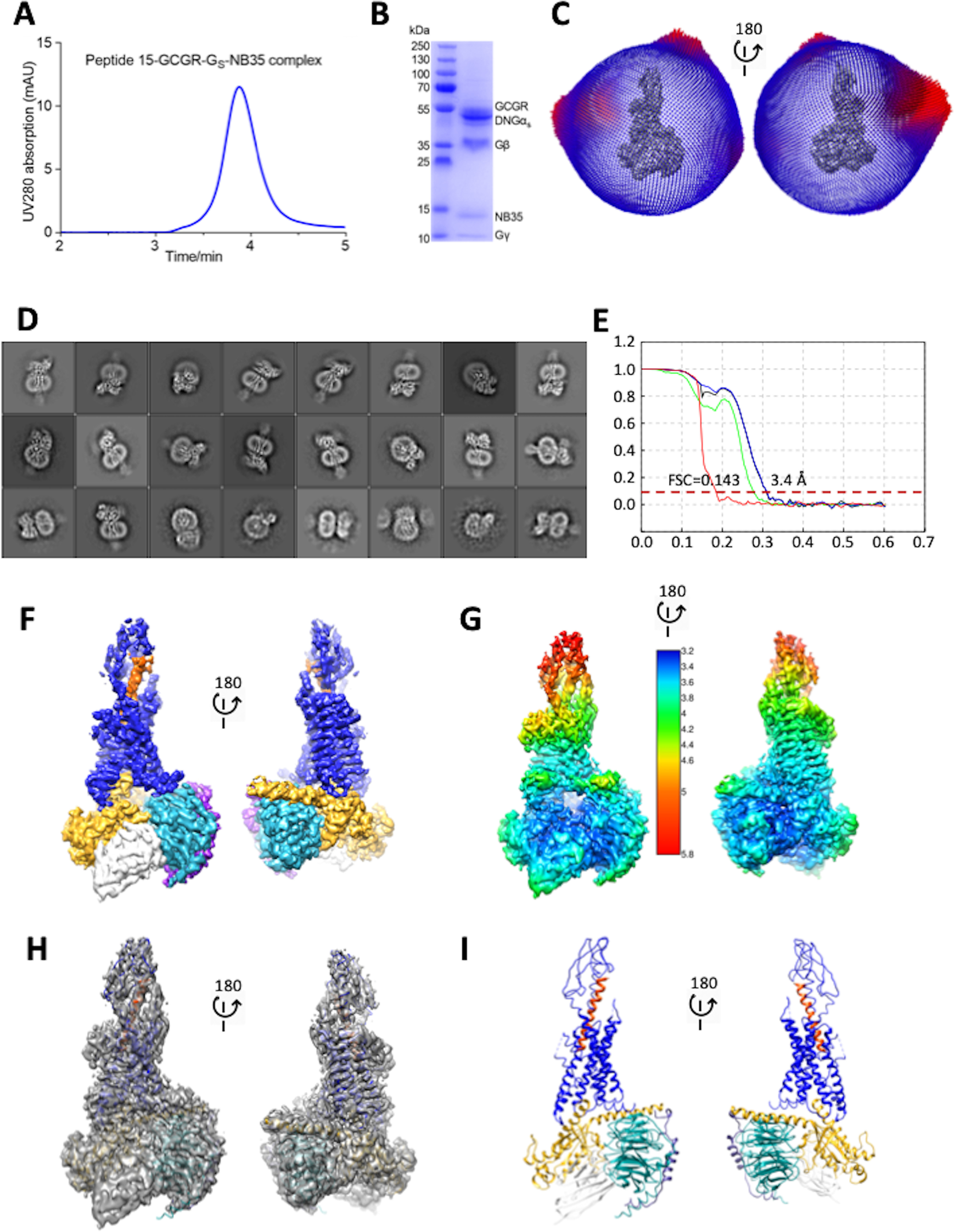
Cryo-EM structure of the P15-GCGR-GsDN-Nb35 complex. A, analytical SEC trace of the purified complex. B, Coomassie blue–stained SDS-PAGE of the purified complex. C, 3D histogram representation of the Euler angle distribution of all the particles used in the reconstruction overlaid on the density map drawn on the same coordinate axis. D, 2D class averages of cryo-EM projections of the receptor complex. E, gold standard Fourier shell correlation (FSC) curves for the final map and map validation from half maps, showing the overall nominal resolution of 3.4 Å. F, surface representation of the final map colored by protein segment. G, local resolution-filtered EM map displaying local resolution (Å) colored from highest resolution (dark blue) to lowest resolution (red). H, full map containing the backbone model of the complex in ribbon format. I, backbone model of the receptor complex in ribbon format. Blue, GCGR; orange, P15; gold, Gαs; cyan, Gβ1; purple, Gγ2; white, Nb35.
Peptide–receptor interactions
P15 formed an extended α-helix, exiting the receptor core with an ∼30-degree angle from perpendicular (Fig. 1I) and displayed polar and hydrophobic interactions with both the ECD and the receptor core (Fig. 2). The deepest residue was His-1P15 that was located above the conserved central polar network of class B GPCRs but that interacted principally with residues in TM5, Trp-3045.36, Arg-3085.40, and Val-3115.43 (Fig. 2B and Fig. 3A). However, there was alternative density for His-1P15 in lower contoured maps that extended toward the central polar network (Fig. 3A) that was not observed in equivalent low contour maps of the GCG-bound GCGR (Fig. 3B), suggesting that this residue in P15 is more dynamic. Gln-3P15 was within H-bonding distance of the hydroxyl of Tyr-1491.47 that may stabilize the peptide N terminus in the receptor core (Fig. 2, A–C). Additional polar interactions were predicted between Ser-8P15 and the backbone of Asn-298ECL2, Ser-11P15, and both Thr-296ECL2 and Ser-297ECL2, whereas Asp-15P15 interacted with both Gln-293ECL2 and residues of the far N terminus, Gln-27ECD and Met-29ECD (Fig. 2). These interactions were supported by density in the EM map (Fig. 2C). An extended network of hydrophobic interactions also occurred between the peptide and TM1 (Val-1341.32, Ala-1351.33, Tyr-1381.36, and Tyr-1451.43), and TM7 (Asp-3857.42, Leu-3867.43), as well as with parts of TM2/ECL1 (Leu-1982.71, Tyr-2022.75). Alanine mutation of residues within the binding pocket provided support for many of the key interactions identified in the active structure (Tables 1 and 2 and Figs. S4 and S5), including Tyr-1451.43, Tyr-1491.47, Tyr-2022.75, Arg-3085.40, Asp-3857.42, and Leu-3867.43. There was also a range of effects observed that likely relates to impact of alanine mutation on the secondary structure or dynamics of the receptor. For example, whereas the H-bond between Ser-8P15 and Asn-298ECL2 was with the amino acid backbone, the N298A mutation likely disrupts the potential polar interaction of the side chain of Asn-298ECL2 and the backbone of Asn-300ECL2 that helps maintain the fold of ECL2. Likewise, the large impact of the T295ECL2A mutant is likely because of disruption to the active ECL2 conformation. In the map, the tops of TM6/TM7/ECL3 were poorly defined, suggesting that this segment has higher mobility. The mutational analysis also revealed >10-fold loss of potency for residues at the top of TM7/ECL3 (R3787.35A, L3827.39A), suggesting that they may play a role via either transient interactions with the peptide or in influencing the mobility of ECL3. Not surprisingly, we also observed marked loss of potency with mutation of key residues of the central polar network, Lys-1872.60 and Glu-3626.53, consistent with the key role of this network in signal propagation of many class B GPCRs (17–19).
Figure 2.
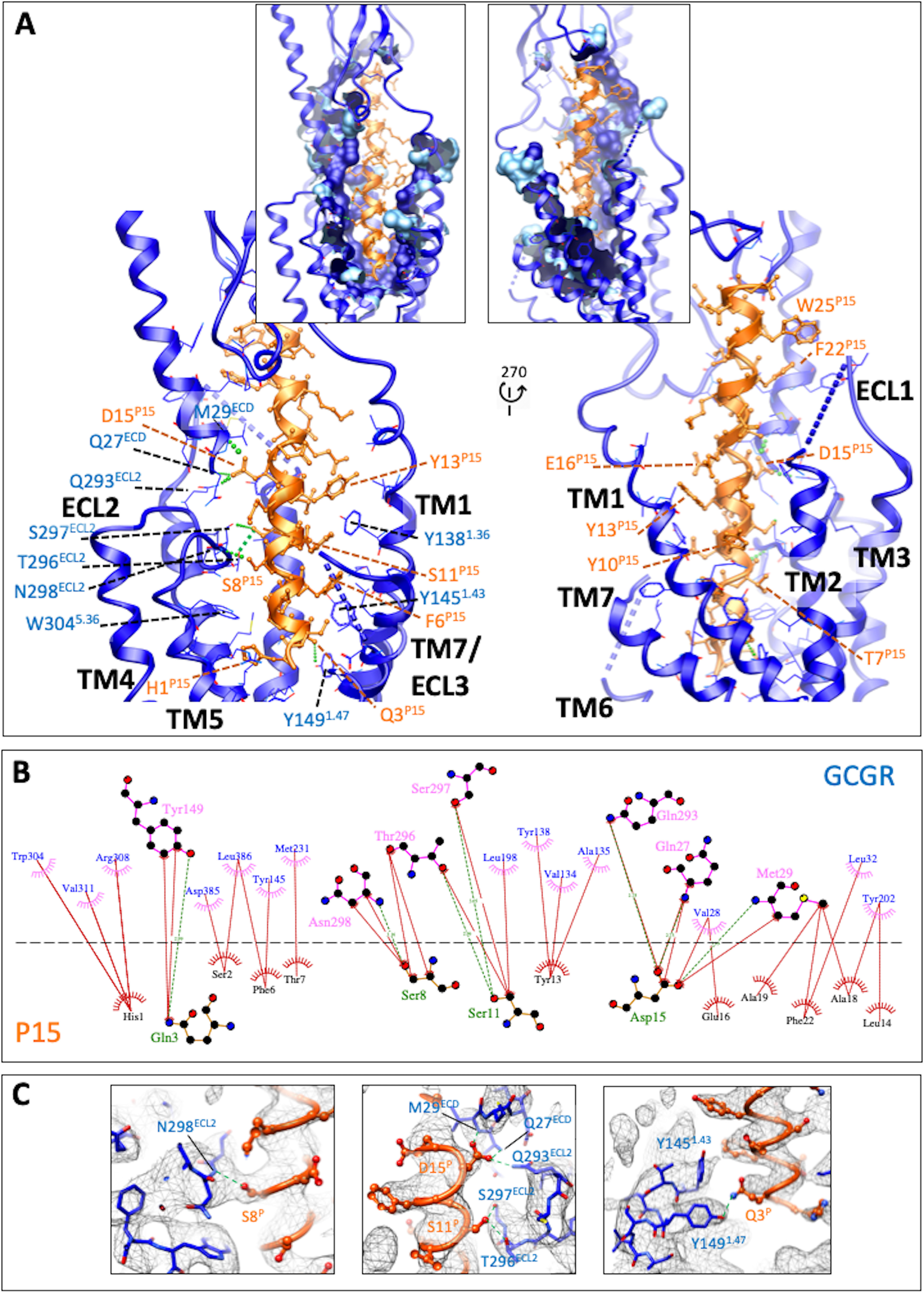
The peptide 15-binding interface with GCGR. A, GCGR (blue) residues within 5 Å of P15 are displayed in wire format with the GCGR protein backbone in ribbon format. Blue dashed lines represent segments of GCGR that could not be modeled because of ambiguous density. P15 is colored orange with amino acid side chains in x-stick format and backbone shown as ribbon display. H-bonds are displayed as dashed green lines. The insets include surface display of interacting GCGR residues with the dark blue–colored surface illustrating the proportion of the side chain within 5 Å. B, interactions were determined using LigPlot+. GCGR residues are located above the dashed black line, and P15 residues below the line. Hydrophobic interactions are illustrated by red (P15) or pink (GCGR) arcs, and interacting residues are joined by a red line. Amino acids involved in H-bonds are shown in atomic detail with H-bonds shown as dashed green lines. C, map to model densities for side chains involved in H-bond (green dashed lines) formation.
Figure 3.

A and B, map to model figures for the P15-GCGR-GsDN-Nb35 (A) and GCG-GCGR-GsDN-Nb35 (PDB:6LMK; EMD-0917) (B) structures at different levels of contour. In the higher contour map for the P15 complex, there is incomplete density for His-1P15 (A, left panel, white arrow); however, density for the full side chain is present at lower contour, along with density that would support an alternate location for this residue (A, right panel; solid red arrow). In contrast, a single continuous density is observed for His-1GCG in both higher contour and lower contour maps (B, red circles). GCGR residues in the P15 complex are displayed in blue wire format, and purple wire format for the GCG complex. P15 is displayed in orange, protein worm, and x-stick format. GCG is displayed in light blue, protein worm and x-stick format.
Table 1.
Quantitative analysis of peptide competition for 125I-glucagon binding and cell surface expression of WT and mutant GCGRs
| Mutant | Glucagon pIC50 ± S.E.a | Peptide 15 pIC50 ± S.E.a | Expression (% of WT)b |
|---|---|---|---|
| WT-GCGR | 7.47 ± 0.03 | 6.52 ± 0.03 | 100 ± 0 |
| Q1421.40bA | 7.03 ± 0.07c ** | 6.29 ± 0.07 | 80 ± 5 |
| Y1451.43bA | Nbd | Nb | 38 ± 4*** |
| Y1491.47bA | Nb | Nb | 103 ± 3 |
| K1872.60bA | 7.98 ± 0.09*** | 7.11 ± 0.05*** | 78 ± 8 |
| V1912.64bA | Nb | Nb | 119 ± 11 |
| I1942.67bA | 7.57 ± 0.05 | 6.78 ± 0.06* | 94 ± 8 |
| Y2022.75bA | Nb | Nb | 74 ± 4 |
| M2313.36bA | 7.60 ± 0.05 | 6.58 ± 0.06 | 95 ± 5 |
| Q2323.37bA | 7.02 ± 0.09*** | 6.20 ± 0.11* | 83 ± 4 |
| I2353.40bA | 7.34 ± 0.09 | 6.34 ± 0.10 | 71 ± 6* |
| N2383.43bA | 7.22 ± 0.09 | 6.42 ± 0.09 | 41 ± 2*** |
| W295 ECL2A | Nb | Nb | 68 ± 6* |
| S297 ECL2A | 7.59 ± 0.06 | 6.65 ± 0.06 | 62 ± 6* |
| N298 ECL2A | 6.90 ± 0.13*** | Nb | 68 ± 14* |
| R3085.40bA | Nb | Nb | 71 ± 7* |
| N3185.50bA | Nb | Nb | 80 ± 3 |
| H3616.52bA | 7.11 ± 0.08* | 6.60 ± 0.07 | 85 ± 3 |
| E3626.53bA | 6.98 ± 0.15*** | Nb | 67 ± 8* |
| R3787.35bA | 7.17 ± 0.08* | 6.79 ± 0.07* | 115 ± 5 |
| L3827.39bA | Nb | Nb | 84 ± 5 |
| D3857.42bA | 7.65 ± 0.06 | 6.76 ± 0.06 | 65 ± 4* |
| L3867.43bA | Nb | Nb | 86 ± 16 |
| S3897.46bA | 7.28 ± 0.08 | 6.38 ± 0.09 | 90 ± 8 |
| Q3927.49bA | 7.01 ± 0.07*** | 6.25 ± 0.13* | 70 ± 8* |
a pIC50 values are shown as means ± S.E. of at least three independent experiments.
b Cell surface expression was detected by flow cytometry and is reported as percentage of WT-GCGR.
c Data from Fig. S5 were fit to a three-parameter logistic equation. Statistical evaluation was performed using one-way analysis of variance and Dunnett's post test, *, p < 0.05; **, p < 0.001; ***, p < 0.0001 compared with WT control.
d Nb, insufficiently robust binding to enable a curve to be fitted.
Table 2.
Quantitative analysis of peptide-mediated cAMP accumulation in cells stably expressing WT or mutant GCGRs
| Mutants | Glucagon |
Peptide 15 |
||
|---|---|---|---|---|
| pEC50 ± S.E.Ma | Emaxb (% of WT) | pEC50 ± S.E.M | Emax (% of WT) | |
| WT-GCGR | 10.99 ± 0.03 | 100 ± 1.3 | 10.65 ± 0.04 | 100 ± 1.5 |
| Q1421.40bA | 10.44 ± 0.06c*** | 100 ± 2.2 | 9.84 ± 0.06*** | 100 ± 2.4 |
| Y1451.43bA | 8.93 ± 0.05*** | 97 ± 2.2 | 8.27 ± 0.04*** | 95 ± 1.7 |
| Y1491.47bA | 9.20 ± 0.06*** | 92 ± 2.5 | 8.57 ± 0.04*** | 93 ± 1.7 |
| K1872.60bA | 9.34 ± 0.08*** | 99 ± 3.4 | 8.81 ± 0.03*** | 97 ± 1.1 |
| V1912.64bA | 8.69 ± 0.04*** | 100 ± 2.0 | 7.93 ± 0.05*** | 100 ± 2.7 |
| I1942.67bA | 10.52 ± 0.05*** | 95 ± 1.7 | 10.39 ± 0.06* | 94 ± 2.5 |
| Y2022.75bA | 9.65 ± 0.05*** | 102 ± 2.0 | 9.21 ± 0.05*** | 103 ± 2.2 |
| M2313.36bA | 10.74 ± 0.04* | 100 ± 1.8 | 10.22 ± 0.05*** | 102 ± 2.2 |
| Q2323.37bA | 10.49 ± 0.04*** | 101 ± 1.7 | 9.62 ± 0.05*** | 103 ± 2.0 |
| I2353.40bA | 10.97 ± 0.04 | 99 ± 1.8 | 10.37 ± 0.05* | 100 ± 2.1 |
| N2383.43bA | 10.67 ± 0.08*** | 97 ± 2.9 | 9.93 ± 0.05*** | 99 ± 2.1 |
| W295 ECL2A | 7.30 ± 0.05*** | 101 ± 3.0 | 6.33 ± 0.04*** | 105 ± 2.6 |
| S297 ECL2A | 10.83 ± 0.05 | 96 ± 1.9 | 10.66 ± 0.05 | 98 ± 2.0 |
| N298 ECL2A | 9.26 ± 0.04*** | 101 ± 1.9 | 8.23 ± 0.06*** | 100 ± 2.7 |
| R3085.40bA | 8.14 ± 0.04*** | 104 ± 2.4 | 7.21 ± 0.05*** | 93 ± 2.3 |
| N3185.50bA | 9.33 ± 0.14*** | 56 ± 3.4*** | 8.45 ± 0.16*** | 56 ± 3.7*** |
| H3616.52bA | 10.57 ± 0.06*** | 98 ± 2.2 | 9.74 ± 0.09*** | 98 ± 3.6 |
| E3626.53bA | 9.34 ± 0.04*** | 100 ± 1.7 | 8.49 ± 0.05*** | 99 ± 2.1 |
| R3787.35bA | 7.72 ± 0.02*** | 106 ± 1.3 | 6.90 ± 0.06*** | 94 ± 3.1 |
| L3827.39bA | 9.62 ± 0.05*** | 97 ± 2.1 | 8.78 ± 0.05*** | 96 ± 2.1 |
| D3857.42bA | 9.31 ± 0.05*** | 100 ± 2.1 | 8.60 ± 0.08*** | 98 ± 3.0 |
| L3867.43bA | 8.86 ± 0.03*** | 103 ± 1.5 | 7.93 ± 0.06*** | 101 ± 2.9 |
| S3897.46bA | 10.94 ± 0.05 | 100 ± 2.2 | 10.25 ± 0.08*** | 99 ± 3.4 |
| Q3927.49bA | 10.85 ± 0.05 | 101 ± 2.2 | 10.00 ± 0.07*** | 101 ± 3.1 |
a pEC50 values are shown as means ± S.E. of at least three independent experiments.
b Emax means the maximal response measured relative to that of WT GCGR. Data from Fig. S4 were fit to a three-parameter logistic equation.
c Statistical evaluation was performed using one-way analysis of variance and Dunnett's post test, *, p < 0.05; **, p < 0.001; ***, p < 0.0001 compared with WT control.
G protein interactions
The P15-bound GCGR formed a series of polar and nonpolar interactions with the Gαs subunit, particularly with the αH5 helix and the proximal segment of the αN helix (Fig. 4, A–C). Polar interactions occurred between Arg-38αN and Thr-257ICL2 and also the backbone of Pro-259ICL2, together with side chain to receptor backbone interactions for His-41αN and Ala-256ICL2, Gln-384αH5 and Leu-2533.58, Arg-385αH5 and Lys-3325.64, His-387αH5 and Leu-2523.57, and between Glu-392αH5 and Lys-405H8.40 (Fig. 4, B and C). ICL2 of GCGR formed additional hydrophobic interactions with both GαN and GαH5, while the GαH5 had extended interactions with TM2, TM3, TM5, TM6, and TM7 (Fig. 4). There were also limited interactions between the Gβ subunit (Phe-335β, Arg-52β) and ICL1 (Ser-167ICL1, Leu-169ICL1, His-170ICL1) and between Gβ (Ala-309β, Asp-312β) and helix 8 of the receptor (Arg-4178.52, Arg-4138.48), with the potential for an H-bond or salt bridge to be formed with Asp-312β; however, there was limited density in the EM map, indicating that this was likely to be transient if it were formed.
Figure 4.
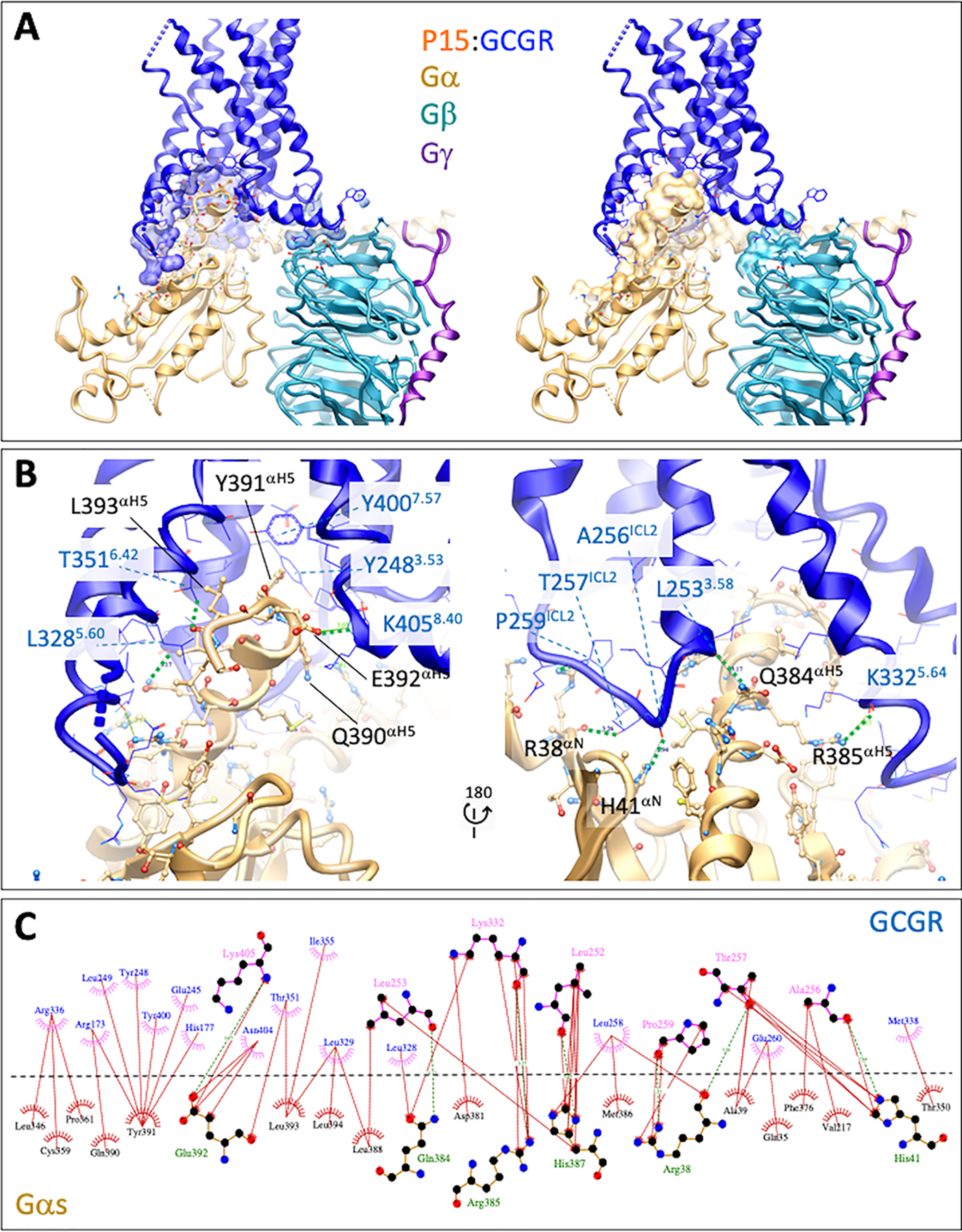
The GCGR-Gs protein interface of the active P15-bound complex. A, overview of the interface with protein backbones displayed in ribbon format and side chains within 5 Å displayed in either x-stick (G protein) or wire (GCGR) format. Left panel includes surface representation of the interacting GCGR residues. Right panel includes surface representation of the interacting G protein residues. GCGR, blue; Gαs, gold; Gβ1, cyan; Gγ2, purple. B, close up of the Gαs-GCGR interface. Predicted H-bonds are displayed as green dashed lines. C, GCGR-Gαs interface. Interactions were determined using LigPlot+. GCGR residues are located above the dashed black line, and Gαs residues below the line. Hydrophobic interactions are illustrated by red (Gαs) or pink (GCGR) arcs, and interacting residues are joined by a red line. Amino acids involved in H-bonds are shown in atomic detail with H-bonds shown as dashed green lines.
Comparison of P15-bound and GCG-bound GCGR and GLP-1–bound GLP-1R structures
Overall, there was a high degree of conservation in the structure of the GCGR bound to GCG (12) versus P15, including the location of the ECD; most TM helices; and in the conformation of ECL2, ICL2, and ICL3 (Fig. 5A). However, key distinctions were observed, most notably in the resolution and secondary structure of ECL1, ECL3, the upper segment of TM1 on the extracellular face of the receptor, and ICL1 on the intracellular face (Fig. 5A), with these differences clearly observed in comparisons of the cryo-EM maps for the two structures (Fig. 6, A–D). P15 differs from GCG across residues 16-24 where 6/8 amino acids are distinct (Fig. S1A). These differences alter the interaction of the peptides with ECL1 and the top of the TM1 helix where GCG makes more extensive interactions (Fig. 7, A and B) that in turn stabilize the secondary structure of the receptor in these regions (Fig. 7B). In the EM density map, there is limited density for the P15 substituted residues, indicative of weak or transient interactions and greater mobility of the peptide C terminus that likely also contributes to the limited resolution of the ECD (Fig. 6, A and B). Despite the conservation in the N terminus of P15 and GCG, there was marked difference in the conformation of ECL3 in the consensus map with substantively reduced interaction between the top of TM6/TM7/ECL3 for P15 (Fig. 6, C and D). Although the GCGR-Gs heterotrimer interface was very similar for the P15 and GCG-bound structures, there were distinct conformations of ICL1 that altered the interface with the Gβ subunit (Fig. 8, A–C). This was correlated with a small local shift in the orientation of the G protein (Fig. 8A).
Figure 5.

A and B, comparison of the active P15-GCGR-GsDN-Nb35 complex with GCG-GCGR-GsDN-Nb35 (A, PDB: 6LMK) and GLP-1-GLP1R-GsDN-Nb35 (B, PDB:,5VAI) complexes. Protein backbone is shown in ribbon format. Only the receptor and peptides are displayed for clarity. GCGR is blue and P15 orange in the P15-GCGR complex. GCGR is purple and GCG light blue in the GCG-GCGR complex. GLP-1R is gray and GLP-1 pink in the GLP-1–GLP-1R complex. White arrows depict areas of conformational divergence between the complexes. The solid red arrow highlights the differential positioning of the L2 loop of the GCGR and GLP-1R ECDs relative to the C terminus of peptides.
Figure 6.

EM density maps for divergent regions of GCGR in the P15- versus GCG-bound active structures. A, highlights differences in peptide interactions with the upper segment of TM1 that extends and connects the receptor ECD and transmembrane domain bundle. Left panel, the P15 (orange)-GCGR (blue) active complex. Right panel, the GCG (light blue)-GCGR (purple) active structure. GCGR is in wire format. Peptides are shown in protein worm and x-stick format. B, highlights differences in peptide interactions with GCGR ECL1. Left panel, the P15 (orange)-GCGR (blue) active complex. Right panel, the GCG (light blue)-GCGR (purple) active structure. GCGR is in wire format. Peptides are shown in protein worm and x-stick format. C and D, differences in the conformation of ECL3 between the P15 complex (C) and GCG complex (D). Proteins are displayed in ribbon format with colors as in A and B.
Figure 7.

Peptide 15 and glucagon make distinct interactions with GCGR. A, P15–GCGR interface with the divergent peptide amino acids from GCG displayed in x-stick format and conserved residues in wire format. GCGR residues that interact with the nonconserved peptide amino acids are displayed in wire and surface representation. GCGR is blue and P15 orange. The receptor backbone is shown in protein worm representation. B, GCG–GCGR interface with the divergent peptide amino acids from P15 displayed in x-stick format and conserved residues in wire format. GCGR residues that interact with the nonconserved peptide amino acids are displayed in wire and surface representation. GCGR is purple and GCG light blue. The receptor backbone is shown in protein worm representation. Lower panels include cpk representation of the nonconserved peptide residues. The red circle highlights the distinction in interaction surfaces between the top of TM1 and the two peptides. The black circle highlights the distinction in interaction surfaces between ECL1 and the two peptides.
Figure 8.

Distinctions in the conformation of ICL1 between the P15-bound and GCG-bound GCGR structures alter the orientation of the Gs protein interface. A, ribbon representation of the protein backbone of GCGR and G protein subunits. GCGR in the P15 bound complex is blue and in the GCG bound complex, purple. In the P15 complex structure, Gαs is gold; Gβ1, cyan; Gγ2, dark purple. All subunits are colored gray in the GCG-bound complex. B, EM density map (contour 0.01) to model for GCGR ICL1 in the P15-bound complex. GCGR, blue; Gαs, gold; Gβ1, cyan. C, EM density map (contour 0.037) to model for GCGR ICL1 in the GCG bound complex. GCGR, purple; Gαs, gold; Gβ1, cyan.
Intriguingly, the areas of distinction in structure of the peptide-binding domain of GCGR bound to P15 versus GCG were also the major sites of difference in conformation between the active GLP-1–bound GLP-1R (20) and the GCGR structures (Fig. 5, B versus A). A notable difference to the GCGR structures, in the GLP-1–GLP-1R structure, and also the published ExP5-bound GLP-1R structure (13), is disordering of the top of the TM1 helix and the stalk connecting TM1 to the ECD (Figs. 5–7). The ECD orientation between the related receptors is remarkably similar, and this is likely driven by the overlap in the angle that the peptides exit the receptor (Fig. 5), with the minor differences related to the extent of interaction between the L2 loop of the ECD and the C termini of the peptides (Fig. 5B).
Discussion
Diabetes and obesity are major health burdens, and there has been much interest in harnessing physiological systems that regulate metabolism for treatment of these diseases. Among the key glucoregulatory hormones are GLP-1 and GCG that have opposing actions on regulation of plasma glucose but complementary actions in regulation of food intake, energy expenditure, and other metabolic events (3, 6). Oxyntomodulin is a related peptide that, like GLP-1, is secreted from the intestine following meal ingestion that is a dual agonist of GLP-1R and GCGR but with lower potency in canonical cAMP signaling assays than the “cognate” agonists of these receptors (21). Preclinical studies provided early evidence that chronic oxyntomodulin administration could induce greater weight loss than GLP-1 selective agonists with equivalent glycemic control (22–24), suggesting that dual agonists of GLP-1R and GCGR could provide additional benefits above that of GLP-1R agonists in treatment of metabolic disease. This has driven interest in development of stable, high-potency unimolecular dual agonists of GCGR and GLP-1R, and also tri-agonists that also target GIP receptor (7, 8, 11, 25–28), and several of these have now entered clinical trials (9, 10, 26, 27).
The principal approach to engendering GCGR/GLP-1R dual agonism with high potency has been substitution of nonconserved residues in the mid and C-terminal segments of the peptides. An exemplar is the dual agonist P15, first described by Day and colleagues (11). It is a substituted analog of GCG that retains equivalent potency to the parental peptide at the GCGR but has equal potency in cAMP assay to GLP-1 at the GLP-1R. We recently published the first active structure of the GCGR bound to GCG (12) and in the current study sought to understand the impact of the P15 modifications on the engagement of this peptide with the receptor.
Multiple structures have now been solved for active-state, G protein–coupled class B GPCRs, including the calcitonin receptor (29, 30), calcitonin gene-related peptide (31) and adrenomedullin receptors (32), parathyroid hormone-1 receptor (33), corticotrophin releasing factor-1 and -2 receptors (34, 35), pituitary adenylate cyclase-activating peptide receptor (35–37), as well as the GCG-bound GCGR (12) and multiple GLP-1R complexes (13, 20, 38). Comparison of the P15-bound GCGR to other class B GPCR active structures reveals that it most closely resembles the peptide-bound structures of the GCGR receptor subfamily (Fig. 5) (12, 13, 20), particularly with respect to the consensus orientation of the ECD that is a major area of divergence within this broader receptor class (discussed in detail in Ref. 35). However, the P15-GCGR-Gs structure exhibits the classic hallmarks of activated class B GPCRs compared with inactive class B GPCR structures exemplified by the GCGR (39, 40) that include the pronounced kink in TM6 around the conserved Pro6.47-X-X-Gly6.50 motif, and the intracellular reorganization of TMs 5 and 6 that allows engagement with the G protein; class-dependent details of the global changes that occur in activated receptors are described in detail elsewhere (12, 34, 35). Although there are receptor-specific differences in interactions with Gs protein, there is general conservation in the receptor regions involved in binding, which have been discussed previously (41, 42), that are also conserved for the P15-bound GCGR. As such, we have concentrated the following discussion on the distinctions between GCG and P15-bound GCGR that may give insight into the nature of dual agonism that is a property of P15.
The N-terminal 15 residues of GCG and P15 are identical and, unsurprisingly, the two active GCGR structures had a high degree of overlap in the transmembrane domain bundle and in the receptor–G protein interface, with mutations in the TM-binding pocket having similar impact on both P15- and GCG-induced cAMP production (Table 2). Nonetheless, interesting differences in the structures were observed, even in proximity to the conserved sequence of the peptides. Of particular note, there was marked difference in the conformation of ECL3, with this loop folded toward and forming additional interactions with GCG compared with P15 (Fig. 7). In contrast, in the P15-bound structure, ECL3 is poorly resolved and oriented away from the peptide. Within the deep binding pocket, the density for His-1P15 is less well resolved, although it best supports modeling of the side chain in an equivalent orientation to that observed for GCG (12) (Fig. 3). At lower contour of the EM maps, as noted above, there is additional density that could support alternate modeling of His-1P15 whereas no equivalent density is observed in lower contoured maps of the GCG-bound GCGR, and this may relate to the more extended interactions of this peptide with ECL3 that could limit conformational dynamics within the core. In the GLP-1–GLP-1R active complex, ECL3 is folded in toward the peptide, similar to GCG-GCGR (Fig. 4), however, ECL3 is among the least conserved, conformationally, across different structures of active GLP-1R complexes (13, 20, 38). Moreover, mutational analysis of GLP-1R ECL3/TM7 has revealed that this domain is critically important in the biased agonism of GLP-1R agonists, including the dual agonist oxyntomodulin (43), and ExP5 (13). Later work suggested that TM6/ECL3/TM7 is functionally linked to TM1 in controlling GLP-1R signaling (44). This is notable as the top of TM1 was also structurally different in GCGR bound to P15 versus GCG. In the GCG-bound structure, TM1 forms an extended α-helix that is stabilized by direct interaction with GCG residues that are distinct in P15 (Fig. 5). In the native GCG sequence, residue 18 is Arg but Ala in P15 (and GLP-1). Arg-18GCG extends toward and is tightly packed with residues in ECL1 and this loop is highly structured in the GCG-GCGR complex (12). In contrast, there is much more limited interaction between P15 and ECL1 and the loop is disordered and less well resolved. Thus, there is reduced interaction between P15 and the receptor as the peptide exits the GCGR core and this in turn leads to greater mobility of the peptide and receptor ECD that is reflected in the reduced resolution of this domain in the EM density map, compared with GCG-GCGR. This likely also contributes to the stability of interaction of the peptide and ECL3 and the resolution of this domain.
With the exception of ECL3, the areas of difference between the P15- and GCG-bound GCGR are the major areas of structural distinction observed between peptide-bound structures of GLP-1R and GCGR, and indeed areas that are less well resolved in GLP-1R versus the GCG-GCGR EM density maps. While highly speculative, flexibility of the receptor ECD and interaction of this domain with peptide agonists may be a requirement for efficacious engagement. As such, one of the mechanisms underlying increased potency of P15 at the GLP-1R may be related to enhanced conformational dynamics of this peptide versus GCG when binding to GLP-1R. In support of this hypothesis, ECL1 plays a more important role in the cAMP response to oxyntomodulin at the GLP-1R compared with selective GLP-1R agonists (43), consistent with greater interaction of oxyntomodulin with this domain than the selective agonists.
As noted above, at the GLP-1R, mutagenesis studies have provided evidence that residues in TM1 and ECL3/TM7 give rise to divergent signaling profiles in a peptide-specific manner (43, 44). These regions were among the most distinct in conformation between the P15- versus GCG-bound GCGR and, by analogy, would suggest that these two peptides would have altered signaling profiles and that P15 could be a biased agonist of the GCGR.
In conclusion, our data reveal conformational differences in the active, Gs-complexed structure of GCGR when bound to the dual agonist, P15 versus GCG. The distinct residues in the mid/C-terminal region of the peptide reduce engagement of the peptide with receptor as it exits the receptor core, leading to loss of GCGR secondary structure in these regions and overall increased mobility of the peptide and ECD. These alterations in GCGR structure occur in regions that are also different between GCGR and known structures of GLP-1R and likely contribute to the increased efficacy of P15 at the GLP-1R, but may also alter the signaling profile of P15 relative to the native peptides at both receptors.
Experimental Procedures
The human GCGR gene was cloned into pFastBac1 vector with GP64 promoter to enhance the protein yield. Forty-five residues (His-433–Phe-477) were truncated at the C terminus to improve the thermostability, and the affinity tag hpc4 was added at the C terminus (GP64-HA-GCGR-GSGS linker-HPC4).
The modified GCGR had decreased expression and subsequent peptide-induced cAMP accumulation when expressed in CHO-K1 cells (Fig. S1 and Table S1) but equivalent pharmacology to the WT receptor in HEK-293 cells (12). Gαs was modified to become dominant negative (DN) with eight mutations (S54N, G226A, E268A, N271K, K274D, R280K, T284D, and I285T) thereby stabilizing the interaction with βγ subunits (13, 14). Nb35 was expressed and purified as described previously (12).
Insect cell expression
Human GCGR construct, DNGαs, His6-tagged Gβ1 and Gγ2 were co-expressed in HighFive insect cells (Invitrogen), which were infected with three separate baculoviruses at a ratio of 4:1:1. Cells were grown to a density of 3 million cells/ml and cultured for 48 h thereafter before collection by centrifugation (1500 rpm, 15 min). The pellets were stored at −80°C for subsequent protein purification.
Complex purification
Cell pellets were resuspended in 20 mm HEPES, pH 7.4, 50 mm NaCl, 2 mm MgCl2 with protease inhibitor cOmplete mixture tablets (Roche), 5 μm P15 (GL Biochem), 10 μg/ml Nb35 and 25 milliunits/milliliter apyrase (New England Biolabs). The suspension was incubated for 1 h at room temperature to promote the formation of complexes. Membranes were collected by centrifugation at 30,000 rpm for 30 min and solubilized in 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG) (Anatrace), 0.03% (w/v) cholesteryl hemisuccinate (CHS) (Anatrace), 5 μm P15, and 25 milliunits/milliliter apyrase (New England Biolabs) for 2 h at 4°C. Supernatant was collected by centrifugation at 30,000 rpm for 30 min. GCGR complex was immobilized by anti–hpc4 affinity resin in the presence of 2 mm CaCl2 overnight, washed with 20 column volumes of 20 mm HEPES, pH 7.4, 100 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 5 μm P15, 0.01% (w/v) LMNG, and 0.006% (w/v) CHS, and eluted with 5 column volumes of buffer by adding 6 mm EDTA and 5 μm P15. The complexes were concentrated using a 100-kDa molecular weight cutoff concentrator (Millipore) and then further separated by SEC on a Superdex 200 Increase 10/300 column (GE Healthcare) pre-equilibrated with 20 mm HEPES, pH 7.4, 100 mm NaCl, 2 mm MgCl2, 0.01% (w/v) LMNG, 0.006% (w/v) CHS, and 5 μm P15 was employed to yield the final product. The purified complex was subsequently concentrated to 3-5 mg/ml with a 100-kDa molecular weight cutoff concentrator, and analyzed by SDS-PAGE and analytical SEC. Negative stain transmission EM was performed on the final purified complex as previously described (29).
Cryo-EM
Cryo-EM samples were prepared by plunge vitrification in liquid ethane on a Vitrobot Mark IV (Thermo Fisher Scientific) with blotting chamber set to 4°C and 100% humidity. A 3-μl sample solution was applied on Quantifoil R1.2/1.3 Cu 200 mesh (Quantifoil) glow-discharged grids and blotted for 10 s before plunging. Data were collected on a Titan Krios G3i (Thermo Fisher Scientific) 300 kV electron microscope equipped with a Gatan BioQuantum energy filter and K3 direct electron detector (Gatan). The microscope was set to zero-loss EFTEM NanoProbe mode at × 105,000 indicated magnification with 25 eV energy selection slit, 50 μm condenser aperture, 100 μm objective aperture, spot 4, beam diameter 1.85 μm. The equivalent pixel size on the detector was 0.83 Å/pixel in non-superresolution counting mode. The exposure parameters were as follows: exposure rate 9.9 e−/pixel/s = 14.4 e−/Å2/s, exposure time 4.015 s, total exposure 57.8 e−/Å2, number of frames 67. Multiframe movies were automatically acquired with SerialEM software (45) in non–gain-normalized compressed TIFF format with a 9-position beam-image shift data acquisition scheme and target defocus range of 0.8 to 1.5 μm. In total, 5571 movies were acquired in 22.5 h with an average throughput of 255 movies/h.
Data processing
Movies were motion-corrected, dose-weighted, and integrated using UCSF MotionCor2 (46, 47). This was followed by CTF estimation using the GCTF (48) software package. Particles were picked from the micrographs using the automated procedure in the crYOLO software package (49). Particle extraction and reference-free 2D classification was carried out in RELION (version 3.0.7) (50). CryoSPARC (version 2.7) (51) was used to generate an ab initio model of the GCGR complex, which was used in RELION for 3D classification. A homogeneous subset of particles was then subjected to cycles of Bayesian particle polishing and CTF refinement as implemented in RELION. This homogeneous subset of polished particles was used for a 3D refinement in RELION and was further classified into 3D classes with a fine grain angular sampling only allowing for local Euler angle searches. Particles belonging to the 3D class were further refined in RELION (version 3.1), where their higher order CTF parameters were re-refined, taking into account particles belonging to each image shift group. Further 3D refinements where the α-helical domain of the Gαs protein and the detergent micelle were masked and a final 3D refinement was carried out in RELION (version 3.1), yielding consensus maps of the complex at a global resolution (FSC = 0.143) of 3.4 Å.
Atomic model refinement
The model of GCGR-Gs complex bound to GCG (PDB: 6LMK) (12) was used as the initial template and was fitted in the cryo-EM density map in Chimera (UCSF), followed by molecular dynamics flexible fitting simulation with nanoscale molecular dynamics (52). The initial model was then subjected to real-space refinement, as implemented in the PHENIX (53). GCGR transmembrane domain, Gs protein and Nb35 were further refined by manual model building in COOT (54). After iterative refinement and manual adjustments, comprehensive validation implemented in PHENIX was performed to assess the model quality as presented in Table S3. No electron density was observed for N-terminal residues before Ala-26ECD or C-terminal residues beyond Trp-4188.53 of the GCGR, N terminus of Gβ (Arg-8) and Gγ (Leu-15). The extracellular domain (ECD) was less resolved and was modeled at a backbone level beyond Leu-50ECD. Density of ECL1 (Gln-204ECL1–Ser-213ECL1), ICL3 (His-340ICL3–Asp-342ICL3), and ECL3 (Val-368ECL3–Gly-375ECL3) was discontinuous and these sequences were deleted from the final model. Side chains of Leu-3546.45, Thr-3767.33, and Leu-3777.34 were omitted because of limited density. Structure statistics are detailed in Table S3.
Receptor mutagenesis and mammalian cell culture
WT GCGR was subcloned into pDONR201 plasmids. Alanine mutagenesis was achieved using the Muta-direct Site-Directed Mutagenesis Kit according to the manufacturer's instructions (SBS Genetech). The target gene was transferred to pEF5/FRT/V5-DEST destination vector by LR recombination reaction and FlpIn-Chinese hamster ovary (CHO) cells stably expressing WT or mutant GCGR were established using Gateway technology (Invitrogen). The cells were selected and maintained in F12 medium supplemented with 10% FBS, 600 μg/ml hygromycin B and incubated in a humidified atmosphere at 37°C in 5% CO2. Cells were routinely tested for mycoplasma contamination.
cAMP accumulation assay
cAMP accumulation was measured using a TR-FRET cAMP kit (PerkinElmer) according to manufacturer's instructions. Briefly, cells were seeded into 384-well culture plates (1.2 × 104 cells/well) and incubated overnight at 37°C in 5% CO2. Upon removal of the culture medium, stimulating solution (5 μl/well containing 0.5 mm IBMX) and 5 μl/well ligands of different concentrations were added and incubated for 40 min at room temperature. Eu-cAMP tracer (5 μl) and 5 μl ULight–anti-cAMP were introduced followed by 1 h incubation at room temperature. Signal was detected thereafter with an Envision Multilabel Plate Reader (PerkinElmer). Data were converted to absolute concentration of cAMP using a cAMP standard curve generated in parallel. For analysis, data were normalized to the response of WT receptor and analyzed using a three-parameter logistic equation in GraphPad Prism (v8.0; GraphPad Software Inc.).
Whole cell radioligand binding assay
Cells were seeded into 96-well culture plates (5 × 104 cells/well) and incubated for 24 h at 37°C in 5% CO2. They were then washed twice using F12 with 0.1% BSA and 33 mm HEPES and incubated for 2 h at 37°C. The medium was removed and 125I–GLP-1 or 125I-glucagon (40 picomolar) (PerkinElmer) as well as increasing concentrations of unlabeled ligand were added. After overnight incubation at 4°C, cells were washed three times with ice-cold PBS and lysed in PBS with 1% Triton X-100 and 20 mm Tris-HCl. After addition of scintillation mixture (PerkinElmer), radioactivity (counts per minute) was counted on a MicroBeta2 microplate counter (PerkinElmer). Data were normalized to the response of WT and analyzed using a three-parameter logistic equation.
Receptor expression
The cell surface expression level of GCGR was determined by flow cytometry with an anti-GCGR antibody (Abcam). Cells were seeded at a density of 2 × 104 cells/well into 6-well culture plates and incubated overnight at 37°C in 5% CO2. They were washed three times in PBS and Hanks' Balanced Salt Solution with 0.02% EDTA was added until cells were dispersed. They were then washed three times in PBS, counted, and resuspended in 1% BSA at a density of 4 × 106 cells/tube. Cell suspension (50 μL) was transferred to a new centrifuge tube and blocked by 5% BSA for 15 min at room temperature. The primary antibody (1:100) was then introduced followed by 1 h incubation at room temperature. After three washes with 1% BSA, anti-rabbit Alexa Fluor 488–conjugated secondary antibody (1:1000, Cell Signaling Technology) was added for 1 h at 4°C in the dark. Cells were resuspended in 200 μl PBS containing 1% BSA after three washes and fluorescence signals were detected by NovoCyte flow cytometer (ACEA Biosciences) using laser excitation and emission wavelengths of 488 nm and 519 nm, respectively. For each data point, ∼20,000 cellular events were collected, and the total fluorescence intensity of positive expression cell population was calculated. Data were normalized to the WT GCGR.
Statistical analysis
Data were analyzed using GraphPad Prism (v8.0). Statistic evaluation was performed using one-way analysis of variance and Dunnett's post test. Significance was accepted at p < 0.05.
Data availability
Cryo-EM maps and atomic models are deposited in the PDB and EMDB databases with the following codes: 6WHC, EMD-21671.
Supplementary Material
Acknowledgments
We thank the cryo-EM facility at Shanghai Institute of Materia Medica, Chinese Academy of Sciences, the Monash University Ramaciotti Center for cryo-electron microscopy, and the Monash University MASSIVE high-performance computing facility.
This article contains supporting information.
Author contributions—R. C., X. Z., M. J. B., S. H., D. Y., R. D., M-W. W., D. W., B. W., and P. M. S. formal analysis; R. C., X. Z., A. Q., A. D., M. J. B., Q. T., L. S., L. Z., G. L., Y-L. L., L. M., and R. D. investigation; R. C., S. H., D. Y., D. W., and P. M. S. visualization; R. C., D. Y., M-W. W., D. W., B. W., and P. M. S. writing-original draft; R. C., X. Z., A. Q., A. D., M. J. B., Q. T., L. S., L. Z., G. L., Y.-L. L., L. M., S. H., D. Y., R. D., M.-W. W., D. W., B. W., and P. M. S. writing-review and editing; X. Z. and M. J. B. data curation; X. Z. and D. Y. validation; X. Z., M. J. B., and R. D. methodology; Y.-L. L., D. Y., M-W. W., D. W., B. W., and P. M. S. supervision; D. Y., R. D., M-W. W., D. W., B. W., and P. M. S. funding acquisition; M-W. W., D. W., B. W., and P. M. S. conceptualization; M.-W. W., D. W., B. W., and P. M. S. project administration.
Funding and additional information—This work was supported by the National Natural Science Foundation of China Grants 81872915 (to M.-W. W.), 31825010 (to B. W.), 81973373 (to D. Y.), and 81773792 (to D. Y.); the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China, Grants 2018ZX09735-001 (to M.-W. W.), 2018ZX09711002-002-005 (to D. Y.), and 2018ZX09711002-001 (to L. M.); the National Key R&D Program of China Grant 2018YFA0507000 (to M.-W. W. and B. W.); Novo Nordisk-CAS Research Fund NNCAS-2017-1-CC (to D. Y.); the National Health and Medical Research Council of Australia (NHMRC) Project Grants 1184726 and 1126857 (to D. W.) and Program Grant 1150083 (to P. M. S.); Takeda Science Foundation 2019 Medical Research Grant (to R. D.); and Japan Science and Technology Agency (JST) PRESTO 18069571 (to R. D.). P. M. S. is a Senior Principal Research Fellow and D. W. a Senior Research Fellow of the NHMRC.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein–coupled receptor
- GCG
- glucagon
- GIP
- gastric inhibitory peptide
- Nb
- nanobody
- SEC
- exclusion chromatography
- FSC
- Fourier shell correlation
- ECD
- extracellular domain
- LMNG
- lauryl maltose neopentyl glycol
- CHS
- cholesteryl hemisuccinate
- CTF
- contrast transfer function
- CHO
- Chinese hamster ovary.
References
- 1. Alexander S. P. H., Christopoulos A., Davenport A. P., Kelly E., Mathie A., Peters J. A., Veale E. L., Armstrong J. F., Faccenda E., Harding S. D., Pawson A. J., Sharman J. L., Southan C., and Davies J. A, CGTP Collaborators (2019) The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 176, Suppl. 1, S21–S141 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wootten D., Miller L. J., Koole C., Christopoulos A., and Sexton P. M. (2017) Allostery and biased agonism at class B G protein-coupled receptors. Chem. Rev. 117, 111–138 10.1021/acs.chemrev.6b00049 [DOI] [PubMed] [Google Scholar]
- 3. Sekar R., Singh K., Arokiaraj A. W., and Chow B. K. (2016) Pharmacological actions of glucagon-like peptide-1, gastric inhibitory polypeptide, and glucagon. Int. Rev. Cell. Mol. Biol. 326, 279–341 10.1016/bs.ircmb.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 4. Aroda V. R. (2018) A review of GLP-1 receptor agonists: Evolution and advancement, through the lens of randomised controlled trials. Diabetes Obes. Metab. 20, Suppl. 1, 22–33 10.1111/dom.13162 [DOI] [PubMed] [Google Scholar]
- 5. Chudleigh R. A., Platts J., and Bain S. C. (2020) Comparative effectiveness of long-acting GLP-1 receptor agonists in Type 2 diabetes: A short review on the emerging data. Diabetes Metab. Syndr. Obes. 13, 433–438 10.2147/DMSO.S193693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Campbell J. E., and Drucker D. J. (2015) Islet α-cells and glucagon-critical regulators of energy homeostasis. Nat. Rev. Endocrinol. 11, 329–338 10.1038/nrendo.2015.51 [DOI] [PubMed] [Google Scholar]
- 7. Knerr P. J., Mowery S. A., Finan B., Perez-Tilve D., Tschöp M. H., and DiMarchi R. D. (2020) Selection and progression of unimolecular agonists at the GIP, GLP-1, and glucagon receptors as drug candidates. Peptides 125, 170225 10.1016/j.peptides.2019.170225 [DOI] [PubMed] [Google Scholar]
- 8. Sánchez-Garrido M. A., Brandt S. J., Clemmensen C., Müller T. D., DiMarchi R. D., and Tschöp M. H. (2017) GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 60, 1851–1861 10.1007/s00125-017-4354-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ambery P., Parker V. E., Stumvoll M., Posch M. G., Heise T., Plum-Moerschel L., Tsai L. F., Robertson D., Jain M., Petrone M., Rondinone C., Hirshberg B., and Jermutus L. (2018) MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: A randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet 391, 2607–2618 10.1016/S0140-6736(18)30726-8 [DOI] [PubMed] [Google Scholar]
- 10. Tillner J., Posch M. G., Wagner F., Teichert L., Hijazi Y., Einig C., Keil S., Haack T., Wagner M., Bossart M., and Larsen P. J. (2019) A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: Results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes. Metab. 21, 120–128 10.1111/dom.13494 [DOI] [PubMed] [Google Scholar]
- 11. Day J. W., Ottaway N., Patterson J. T., Gelfanov V., Smiley D., Gidda J., Findeisen H., Bruemmer D., Drucker D. J., Chaudhary N., Holland J., Hembree J., Abplanalp W., Grant E., Ruehl J., et al. (2009) A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 5, 749–757 10.1038/nchembio.209 [DOI] [PubMed] [Google Scholar]
- 12. Qiao A., Han S., Li X., Li Z., Zhao P., Dai A., Chang R., Tai L., Tan Q., Chu X., Ma L., Thorsen T. S., Reedtz-Runge S., Yang D., Wang M. W., et al. (2020) Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 367, 1346–1352 10.1126/science.aaz5346 [DOI] [PubMed] [Google Scholar]
- 13. Liang Y. L., Khoshouei M., Glukhova A., Furness S. G. B., Zhao P., Clydesdale L., Koole C., Truong T. T., Thal D. M., Lei S., Radjainia M., Danev R., Baumeister W., Wang M. W., Miller L. J., et al. (2018) Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 10.1038/nature25773 [DOI] [PubMed] [Google Scholar]
- 14. Liang Y. L., Zhao P., Draper-Joyce C., Baltos J. A., Glukhova A., Truong T. T., May L. T., Christopoulos A., Wootten D., Sexton P. M., and Furness S. G. B. (2018) Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 1, 12–20 10.1021/acsptsci.8b00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wootten D., Simms J., Miller L. J., Christopoulos A., and Sexton P. M. (2013) Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. U. S. A. 110, 5211–5216 10.1073/pnas.1221585110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wootten D., Reynolds C. A., Koole C., Smith K. J., Mobarec J. C., Simms J., Quon T., Coudrat T., Furness S. G., Miller L. J., Christopoulos A., and Sexton P. M. (2016) A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: Lessons from class B crystal structures. Mol. Pharmacol. 89, 335–347 10.1124/mol.115.101246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chugunov A. O., Simms J., Poyner D. R., Dehouck Y., Rooman M., Gilis D., and Langer I. (2010) Evidence that interaction between conserved residues in transmembrane helices 2, 3, and 7 are crucial for human VPAC1 receptor activation. Mol. Pharmacol. 78, 394–401 10.1124/mol.110.063578 [DOI] [PubMed] [Google Scholar]
- 19. Vertongen P., Solano R. M., Perret J., Langer I., Robberecht P., and Waelbroeck M. (2001) Mutational analysis of the human vasoactive intestinal peptide receptor subtype VPAC(2): Role of basic residues in the second transmembrane helix. Br. J. Pharmacol. 133, 1249–1254 10.1038/sj.bjp.0704195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Sun B., Feng D., Hu H., Chu M., Qu Q., Tarrasch J. T., Li S., Kobilka T. S., Kobilka B. K., and Skiniotis G. (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 10.1038/nature22394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pocai A. (2014) Action and therapeutic potential of oxyntomodulin. Mol. Metab. 3, 241–251 10.1016/j.molmet.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Du X., Kosinski J. R., Lao J., Shen X., Petrov A., Chicchi G. G., Eiermann G. J., and Pocai A. (2012) Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 303, E265–E271 10.1152/ajpendo.00142.2012 [DOI] [PubMed] [Google Scholar]
- 23. Kosinski J. R., Hubert J., Carrington P. E., Chicchi G. G., Mu J., Miller C., Cao J., Bianchi E., Pessi A., Sinharoy R., Marsh D. J., and Pocai A. (2012) The glucagon receptor is involved in mediating the body weight lowering effects of oxyntomodulin. Obesity 20, 1566–1571 10.1038/oby.2012.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santoprete A., Capitò E., Carrington P. E., Pocai A., Finotto M., Langella A., Ingallinella P., Zytko K., Bufali S., Cianetti S., Veneziano M., Bonelli F., Zhu L., Monteagudo E., Marsh D. J., et al. (2011) DPP-IV-resistant, long-acting oxyntomodulin derivatives. J. Pept. Sci. 17, 270–280 10.1002/psc.1328 [DOI] [PubMed] [Google Scholar]
- 25. Finan B., Yang B., Ottaway N., Smiley D. L., Ma T., Clemmensen C., Chabenne J., Zhang L., Habegger K. M., Fischer K., Campbell J. E., Sandoval D., Seeley R. J., Bleicher K., Uhles S., et al. (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 21, 27–36 10.1038/nm.3761 [DOI] [PubMed] [Google Scholar]
- 26. Visentin R., Schiavon M., Göbel B., Riz M., Cobelli C., Klabunde T., and Dalla Man C. (2020) Dual glucagon-like peptide-1 receptor/glucagon receptor agonist SAR425899 improves beta-cell function in type 2 diabetes. Diabetes Obes. Metab. 22, 640–647 10.1111/dom.13939 [DOI] [PubMed] [Google Scholar]
- 27. Parker V. E. R., Robertson D., Wang T., Hornigold D. C., Petrone M., Cooper A. T., Posch M. G., Heise T., Plum-Moerschel L., Schlichthaar H., Klaus B., Ambery P. D., Meier J. J., and Hirshberg B. (2020) Efficacy, safety, and mechanistic insights of cotadutide, a dual receptor glucagon-like peptide-1 and glucagon agonist. J. Clin. Endocrinol. Metab. 105, dgz047 10.1210/clinem/dgz047 [DOI] [PubMed] [Google Scholar]
- 28. Capozzi M. E., DiMarchi R. D., Tschöp M. H., Finan B., and Campbell J. E. (2018) Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocr. Rev. 39, 719–738 10.1210/er.2018-00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liang Y. L., Khoshouei M., Radjainia M., Zhang Y., Glukhova A., Tarrasch J., Thal D. M., Furness S. G. B., Christopoulos G., Coudrat T., Danev R., Baumeister W., Miller L. J., Christopoulos A., Kobilka B. K., et al. (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 10.1038/nature22327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dal Maso E., Glukhova A., Zhu Y., Garcia-Nafria J., Tate C. G., Atanasio S., Reynolds C. A., Ramírez-Aportela E., Carazo J. M., Hick C. A., Furness S. G. B., Hay D. L., Liang Y. L., Miller L. J., Christopoulos A., et al. (2019) The molecular control of calcitonin receptor signaling. ACS Pharmacol. Transl. Sci. 2, 31–51 10.1021/acsptsci.8b00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang Y. L., Khoshouei M., Deganutti G., Glukhova A., Koole C., Peat T. S., Radjainia M., Plitzko J. M., Baumeister W., Miller L. J., Hay D. L., Christopoulos A., Reynolds C. A., Wootten D., and Sexton P. M. (2018) Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 10.1038/s41586-018-0535-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang Y. L., Belousoff M. J., Fletcher M. M., Zhang X., Khoshouei M., Deganutti G., Koole C., Furness S. G. B., Miller L. J., Hay D. L., Christopoulos A., Reynolds C. A., Danev R., Wootten D., and Sexton P. M. (2020) Structure and dynamics of adrenomedullin receptors AM1 and AM2 reveal key mechanisms in the control of receptor phenotype by receptor activity-modifying proteins. ACS Pharmacol. Transl. Sci. 3, 263–284 10.1021/acsptsci.9b00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao L. H., Ma S., Sutkeviciute I., Shen D. D., Zhou X. E., de Waal P. W., Li C. Y., Kang Y., Clark L. J., Jean-Alphonse F. G., White A. D., Yang D., Dai A., Cai X., Chen J., et al. (2019) Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 10.1126/science.aav7942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma S., Shen Q., Zhao L. H., Mao C., Zhou X. E., Shen D. D., de Waal P. W., Bi P., Li C., Jiang Y., Wang M. W., Sexton P. M., Wootten D., Melcher K., Zhang Y., et al. (2020) Molecular basis for hormone recognition and activation of corticotropin-releasing factor receptors. Mol. Cell. 77, 669–680.e4 10.1016/j.molcel.2020.01.013 [DOI] [PubMed] [Google Scholar]
- 35. Liang Y. L., Belousoff M. J., Zhao P., Koole C., Fletcher M. M., Truong T. T., Julita V., Christopoulos G., Xu H. E., Zhang Y., Khoshouei M., Christopoulos A., Danev R., Sexton P. M., and Wootten D. (2020) Toward a structural understanding of class B GPCR peptide binding and activation. Mol. Cell. 77, 656–668.e5 10.1016/j.molcel.2020.01.012 [DOI] [PubMed] [Google Scholar]
- 36. Kobayashi K., Shihoya W., Nishizawa T., Kadji F. M. N., Aoki J., Inoue A., and Nureki O. (2020) Cryo-EM structure of the human PAC1 receptor coupled to an engineered heterotrimeric G protein. Nat. Struct. Mol. Biol. 27, 274–280 10.1038/s41594-020-0386-8 [DOI] [PubMed] [Google Scholar]
- 37. Wang J., Song X., Zhang D., Chen X., Li X., Sun Y., Li C., Song Y., Ding Y., Ren R., Harrington E. H., Hu L. A., Zhong W., Xu C., Huang X., et al. (2020) Cryo-EM structures of PAC1 receptor reveal ligand binding mechanism. Cell Res. 30, 436–445 10.1038/s41422-020-0280-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao P., Liang Y. L., Belousoff M. J., Deganutti G., Fletcher M. M., Willard F. S., Bell M. G., Christe M. E., Sloop K. W., Inoue A., Truong T. T., Clydesdale L., Furness S. G. B., Christopoulos A., Wang M. W., et al. (2020) Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 577, 432–436 10.1038/s41586-019-1902-z [DOI] [PubMed] [Google Scholar]
- 39. Siu F. Y., He M., de Graaf C., Han G. W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J. S., Liu W., Lau J., Cherezov V., Katritch V., Wang M. W., et al. (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 10.1038/nature12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang H., Qiao A., Yang D., Yang L., Dai A., de Graaf C., Reedtz-Runge S., Dharmarajan V., Zhang H., Han G. W., Grant T. D., Sierra R. G., Weierstall U., Nelson G., Liu W., et al. (2017) Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264 10.1038/nature22363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Glukhova A., Draper-Joyce C. J., Sunahara R. K., Christopoulos A., Wootten D., and Sexton P. M. (2018) Rules of engagement: GPCRs and G proteins. ACS Pharmacol. Transl. Sci. 1, 73–83 10.1021/acsptsci.8b00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. García-Nafría J., and Tate C. G. (2019) Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol. Cell. Endocrinol. 488, 1–13 10.1016/j.mce.2019.02.006 [DOI] [PubMed] [Google Scholar]
- 43. Wootten D., Reynolds C. A., Smith K. J., Mobarec J. C., Koole C., Savage E. E., Pabreja K., Simms J., Sridhar R., Furness S. G. B., Liu M., Thompson P. E., Miller L. J., Christopoulos A., and Sexton P. M. (2016) The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell 165, 1632–1643 10.1016/j.cell.2016.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lei S., Clydesdale L., Dai A., Cai X., Feng Y., Yang D., Liang Y. L., Koole C., Zhao P., Coudrat T., Christopoulos A., Wang M. W., Wootten D., and Sexton P. M. (2018) Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. J. Biol. Chem. 293, 9370–9387 10.1074/jbc.RA118.003278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schorb M., Haberbosch I., Hagen W. J. H., Schwab Y., and Mastronarde D. N. (2019) Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 10.1038/s41592-019-0396-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., and Cheng Y. (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 10.1038/nmeth.2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., and Agard D. A. (2017) MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang K. (2016) Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wagner T., Merino F., Stabrin M., Moriya T., Antoni C., Apelbaum A., Hagel P., Sitsel O., Raisch T., Prumbaum D., Quentin D., Roderer D., Tacke S., Siebolds B., Schubert E., et al. (2019) SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 21810 10.1038/s42003-019-0437-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J., Lindahl E., and Scheres S. H. (2018) New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Punjani A., Rubinstein J. L., Fleet D. J., and Brubaker M. A. (2017) cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 10.1038/nmeth.4169 [DOI] [PubMed] [Google Scholar]
- 52. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kale L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comp. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liebschner D., Afonine P. V., Baker M. L., Bunkóczi G., Chen V. B., Croll T. I., Hintze B., Hung L.-W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., et al. (2019) Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in PHENIX. Acta Crystallogr. D Struct. Biol. 75, 861–877 10.1107/S2059798319011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps and atomic models are deposited in the PDB and EMDB databases with the following codes: 6WHC, EMD-21671.