

Abstract
Bacteriophage T7 encodes its own DNA polymerase, the product of gene 5 (gp5). In isolation, gp5 is a DNA polymerase of low processivity. However, gp5 becomes highly processive upon formation of a complex with Escherichia coli thioredoxin, the product of the trxA gene. Expression of a gp5 variant in which aspartate residues in the metal-binding site of the polymerase domain were replaced by alanine is highly toxic to E. coli cells. This toxicity depends on the presence of a functional E. coli trxA allele and T7 RNA polymerase-driven expression but is independent of the exonuclease activity of gp5. In vitro, the purified gp5 variant is devoid of any detectable polymerase activity and inhibited DNA synthesis by the replisomes of E. coli and T7 in the presence of thioredoxin by forming a stable complex with DNA that prevents replication. On the other hand, the highly homologous Klenow fragment of DNA polymerase I containing an engineered gp5 thioredoxin-binding domain did not exhibit toxicity. We conclude that gp5 alleles encoding inactive polymerases, in combination with thioredoxin, could be useful as a shutoff mechanism in the design of a bacterial cell-growth system.
Keywords: DNA polymerase, DNA replication, enzyme mutation, bacteriophage, bacterial genetics, gene 5 (gp5), thioredoxin, processivity, replisome
The minimal replication machinery of bacteriophage T7 provides a superb model system to dissect the molecular mechanisms of DNA replication. Only four proteins are required to reconstitute coordinated leading- and lagging-strand DNA synthesis in vitro (1–3). The primase-helicase, gp4, unwinds dsDNA via its C-terminal helicase domain, providing the templates for DNA synthesis; the N-terminal primase domain of gp4 synthesizes tetraribonucleotides to prime lagging-strand synthesis. T7 DNA polymerase holoenzyme, a complex of the phage-encoded DNA polymerase, gp5, and its host-encoded processivity factor, thioredoxin (Trx), is responsible for the synthesis of leading and lagging strands. Finally, the T7 ssDNA-binding protein, gp2.5, interacts with ssDNA, T7 DNA polymerase, and primase-helicase gp4 to coordinate the enzymatic events between the leading and lagging strands (2–4).
All members of the DNA polymerase superfamily employ two invariant carboxylic acid-containing side chains to coordinate catalytic metal ions in the palm domain of the polymerase active site (5, 6). The A family of polymerases includes T7 gp5, the eukaryotic mitochondrial polymerase γ, and Escherichia coli DNA Pol I. In E. coli Pol I, the carboxylates are present as aspartate 705 and aspartate 882. Mutation of either of these residues to alanine results in undetectable polymerase activity (7). Mechanistic experiments show that these residues function in steps prior to the chemical step (8). Aspartate 882 functions in the finger-closing transition, whereas aspartate 705 functions to facilitate entry of a metal associated with the dNTP delivered into the active site. Although no similar mutants for gp5 have been identified, it seems likely that metal-coordinating aspartate residues in gp5 will play an analogous role as asp 705 and 882 in DNA Pol I because of the conservation of the nucleotidyl transfer step among all DNA polymerases (9, 10).
While investigating the transfer of primers from T7 DNA primase to T7 DNA polymerase (11), we constructed a gp5 variant that lacks polymerase activity. The alteration consisted of replacing the two aspartate residues, corresponding to those described above in the metal-binding site for E. coli DNA Pol I, with alanine. (8, 12). Expression of this inactive polymerase (gp5-D475A/D654A) was deleterious to E. coli, as evidenced by a lack of growth of E. coli containing gene 5 expression plasmids. This toxic effect depended on the co-expression of thioredoxin by E. coli. In the present study we show that both in vivo and in vitro, the inactive T7 gp5 and thioredoxin form a complex that inhibits DNA synthesis mediated by the E. coli and T7 replisomes.
Results
Polymerase-inactive gp5 is toxic to E. coli in a thioredoxin-dependent manner
The X-ray crystallographic structure of T7 DNA polymerase in complex with a primer/template shows that aspartate residues at position 475 and 654 in gp5 coordinate the two Mg2+ ions observable in the polymerase domain (Fig. 1) (12). We constructed gp5 variants with aspartate-to-alanine substitution at positions 475 and 654 and examined their ability to complement a T7 phage with a deletion in gene 5 (T7Δ5). None of the gp5 alleles we constructed containing alanine substitutions of the invariant Mg2+-binding active site aspartates were able to complement T7Δ5 (Table 1), suggesting that the mutations disrupted the enzymatic activity of gp5.
Figure 1.

The polymerase active site of T7 DNA polymerase in the crystal structure. Aspartates 475 and 654 contact the two stably bound magnesium ions, MgA and MgB (yellow circles), respectively. MgA mediates abstraction of a proton from the primer 3′-OH, whereas MgB coordinates with the β- and γ-phosphates of the incoming dNTP and stabilizes the pyrophosphate leaving group. A recently postulated third magnesium ion, MgC (10), is not observed in this X-ray crystallographic structure (12).
Table 1.
Effect of T7 DNA polymerase (gp5) expression on E. coli and T7 growth
Substitution of the catalytic aspartates essential for gp5 polymerase activity is lethal for E. coli as well as T7 phage. Plasmids expressing WT gp5, gp5-D475A/D654A, gp5-D475A, or gp5-D654A alleles were transformed into E. coli BL21 (DE3) (encodes Trx and T7 RNA polymerase), BL21 (DE3) ΔtrxA (encodes T7 RNA polymerase), or BL21 (encodes Trx). For each cell line, the number of transformants resulting from transformation of plasmids expressing mutant gp5 was normalized against the number of transformants obtained with the plasmid expressing WT gp5. Plasmids expressing WT, D475A/D654A, D475A, or D654A gp5 were also transformed into E. coli DH5α cells, and transformants were infected with either WT T7 or T7Δ5. In the bottom two rows, no inhibition of E.coli growth is observed when E. coli are transformed with plasmids encoding Klenow fragment containing the TBD of T7 DNA polymerase or a polymerase-inactive Klenow fragment (D705A/D882A)-TBD. + and – indicate phage plaques detected and not detected, respectively.
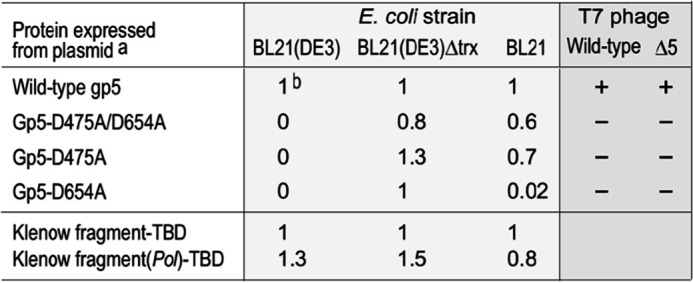
a T7 gene 5 coding both exonuclease and polymerase is located downstream of T7 promoter.
b Efficiency of transformation. All values are normalized against number of colonies obtained with WT gene 5 protein.
To our surprise we were unable to transform the E. coli expression strain BL21 (DE3) with plasmids containing mutant gene 5 alleles under the control of the T7 promoter. Interestingly, transformation was successful with E. coli A307 (DE3) (data not shown), a protein overexpression strain that lacks the gene encoding thioredoxin (trxA), the processivity factor for T7 DNA polymerase, and that is routinely used in our laboratory to purify gp5 devoid of thioredoxin contamination. This result suggests that mutations in the polymerase active site of gp5 result in toxicity for E. coli growth, but this toxicity only occurs in the presence of thioredoxin.
To examine the effect of thioredoxin on transformation efficiency in the same genetic background as E. coli BL21(DE3), we constructed an E. coli BL21(DE3) strain lacking gene trxA but that still retains the λDE3 lysogen designated E. coli BL21(DE3) ΔtrxA. The λDE3 lysogen integrated into this strain's genome encodes T7 RNA polymerase under the control of the lactose-inducible lacUV5 promoter and is equally capable of expressing plasmid-encoded genes under the control of a T7 promoter as the parental BL21 (DE3) (data not shown). Plasmids harboring polymerase-inactive gp5 alleles can be transformed into E. coli BL21(DE3) ΔtrxA as efficiently as the plasmid encoding WT gp5 (Table 1). This result confirms the necessity of thioredoxin in E. coli for inhibition of E. coli growth by expression of polymerase-inactive gp5 (gp5-D475A/D654A) (Table 1). Transformation efficiency in E. coli BL21, a strain that lacks the gene for T7 RNA polymerase in its chromosome, was also examined. This strain would be unable to express the plasmid-encoded gene 5, and comparison of transformation efficiency shows that the lack of expression of gp5-D475A/D654A or gp5-D475A does not produce a toxic effect, suggesting that the toxicity requires expression of mutant gp5. However, we observed a lower transformation efficiency of BL21 with a plasmid encoding the single gp5-D654A mutation (Table 1).
The expression levels resulting from leaky basal expression of T7 RNA polymerase in (DE3) host cells are sufficient to drive the production of gp5-D475A/D654A to toxic levels, because isopropyl β-d-thiogalactopyranoside was not present to induce high levels of expression. Toxicity is observed only when host cells containing both the trxA gene and a λDE3 lysogen are transformed with plasmids encoding gp5-D475A/D654A (Table 1). The genetic requirement for trxA for toxicity and the strong interaction between the two proteins (13) suggest that gp5-D475A/D654A binds E. coli trx in vivo to form a protein with a deleterious effect on E. coli growth. Additionally, a single substitution of alanine for aspartate at positions 475 or 654 in gp5 is sufficient for inhibition of E. coli growth (Table 1). The 3′–5′ exonuclease activity of gp5 is not required for the toxic effect, because gene 5 alleles expressing gp5 with or without exonuclease activity exhibit toxicity (data not shown).
Further, no toxicity is observed in control experiments when E. coli is transformed by a polymerase-inactive version of E. coli Klenow fragment (KF-D705A/D882A-TBD) bearing an insertion of the gp5 thioredoxin-binding domain (TBD), which we previously showed confers increased processivity (Table 1) (14). The observation that KF-D705A/D882A-TBD did not exhibit toxicity could be because although this chimeric protein has enhanced processivity in the presence of thioredoxin compared with WT Klenow fragment, it binds thioredoxin with a 10-fold lower affinity, its polymerase activity is only stimulated ∼8-fold by thioredoxin (compared with >2,500-fold for gp5), and it shows a 3-fold lower specific activity than gp5 in the presence of thioredoxin (14).
In addition we found that gp5-D475A/D654A is similarly toxic for T7 phage growth. WT T7 phage is unable to grow in E. coli cells containing an expression plasmid bearing alleles encoding polymerase-inactive variants of gp5, whereas the presence of an expression plasmid with WT gp5 shows no inhibition of growth (Table 1).
gp5-D475A/D654A is devoid of polymerase activity and inhibits DNA synthesis mediated by T7 and E. coli replisomes
We expressed and purified gp5-D475A/D654A as well as an exonuclease-deficient variant (gp5-D5A/D65A/D475A/D654A) in E. coli A307 (DE3), an expression strain in which the endogenous trxA gene is deleted. Substitution of Asp475 and Asp654 to Ala selectively inactivated the polymerase activity of gp5. Both purified gp5-D475A/D654A versions have no detectable polymerase activity (<0.2%) as compared with WT T7 DNA polymerase using primed M13 (Fig. 2A). The selectivity of the mutations in targeting polymerase activity is underscored by the 3′–5′ exonuclease activity displayed by purified exonuclease-competent gp5-D473A/D654A (Fig. 2B).
Figure 2.

Substitution of catalytic aspartate residues at positions 475 and 654 by alanine results in the selective inactivation of gp5 polymerase activity. A, primed M13 DNA polymerase assay. Incorporation of radioactively labeled dGMP was measured as a function of the concentration of WT gp5 (red), gp5 exo− (blue), gp5-D475A/D654A exo+ (orange), and gp5-D475A/D654A exo− (green). Trx was present at a 10-fold higher concentration over gp5 concentration. B, substitution of catalytic aspartate residues 475 and 654 does not affect exonuclease activity of gp5. The rate of release of acid-soluble deoxyribonucleotides from an internally 32P-labeled dsDNA substrate was measured as a function of the concentration of WT gp5 (red), gp5 exo− (blue), gp5-D475A/D654A exo+ (orange), and gp5-D475A/D654A exo− (green). Trx was present at a 10-fold higher concentration over gp5 concentration.
A plausible explanation for the inhibition of E. coli growth by the presence of an inactive T7 DNA polymerase is that the inhibition results from an interference of the E. coli replisome by the inactive polymerase. To determine whether this is the case, the effect of T7 gp5-D475A/D654A on an in vitro E. coli replication system was examined. This reconstituted system results in coordinated synthesis of leading and lagging strands, with rates comparable with those determined for E. coli DNA replication in vivo (15). The addition of a complex of gp5-D475A/D654A and Trx into DNA replication reactions catalyzed by the E. coli replisome led to a greater than 95% reduction in DNA synthesis (Fig. 3A). We conclude that gp5-D475A/D654A inhibits E. coli growth by interfering directly with DNA synthesis mediated by the bacterial replisome.
Figure 3.
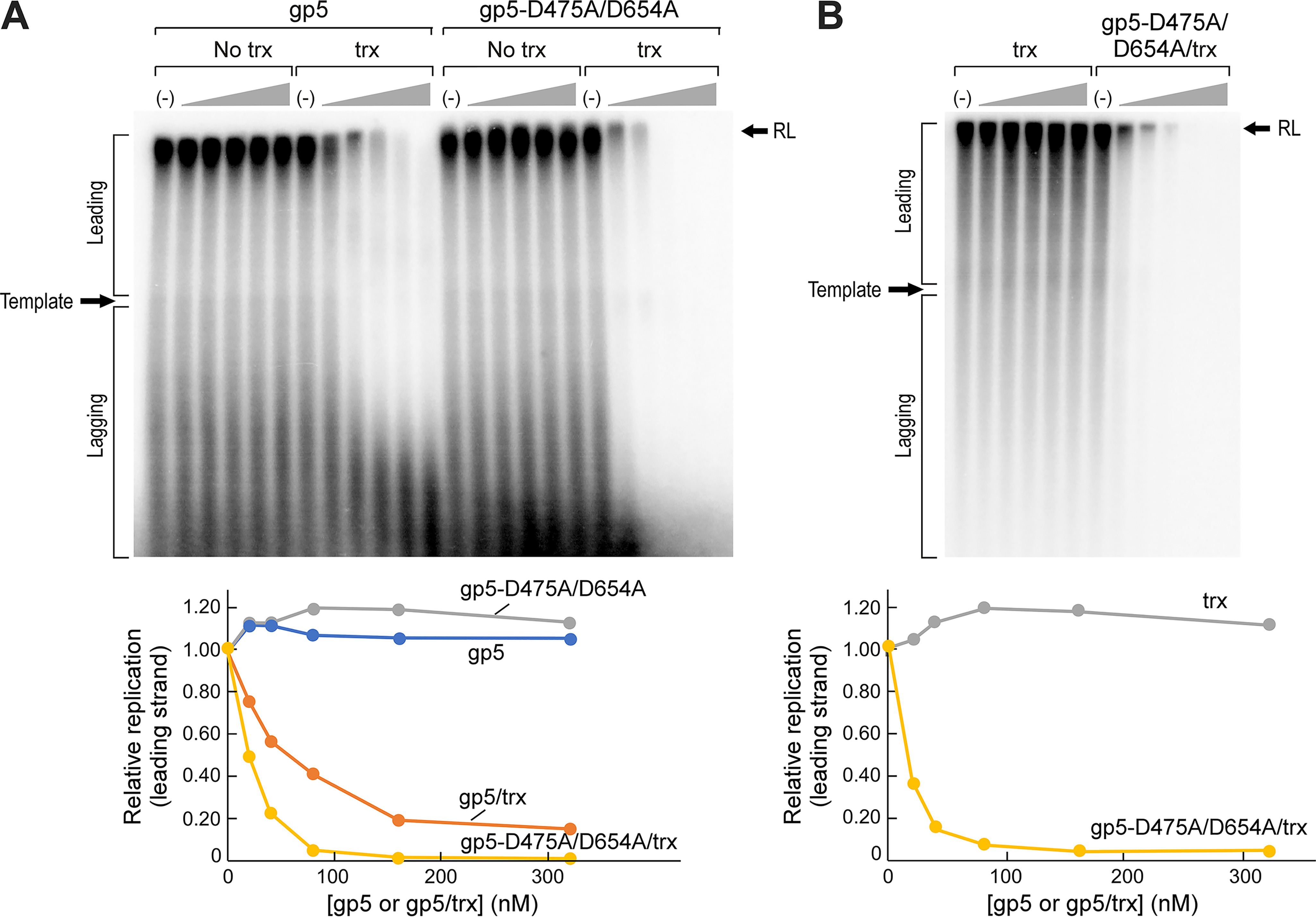
gp5-D475A/D654A inhibits DNA synthesis by the reconstituted E. coli replisome in a thioredoxin-dependent manner. A, DNA synthesis by the E. coli replisome is inhibited by the presence of gp5-D475A/D654A/Trx. Denaturing agarose gel showing replication by the E. coli replisome in the presence of various concentrations of gp5 (+/− Trx) and gp5-D475A/D654A (+/− Trx). The E. coli replisome was assembled onto a rolling circle DNA substrate, and DNA synthesis was initiated as described under “Experimental procedures.” 10 s after addition of the initiation mixture containing [α-32P]dATP, the reaction was supplemented with various concentrations of WT gp5 or gp5-D475A/D654A in the absence and presence of Trx. Quenched samples were loaded on a denaturing agarose gel, and the radiolabel incorporated into DNA was detected by phosphorimaging. Bottom panel, the intensity of DNA replication products was plotted as a function of the concentration of gp5 variant added. Intensities were normalized to the average intensity of the replication product in no addition controls. B, inhibition of the E. coli replisome is not mediated by thioredoxin. Denaturing agarose gel showing replication by E. coli replisome in the presence of various concentrations of thioredoxin alone (Trx) and gp5-D475A/D654A (+/−) Trx. Bottom panel, the intensity of DNA replication products was plotted as a function of the concentration of Trx or gp5-D475A/D654A/Trx added. Intensities were normalized to the average intensity of the replication product in no addition controls. RL, Resolution Limit.
The inhibition of DNA synthesis by gp5-D475A/D654A depends on the presence of thioredoxin. The addition of gp5-D475A/D654A in the absence of thioredoxin to DNA synthesis reactions mediated by the E. coli replisome, up to a concentration of 320 nm, does not result in any significant decrease in DNA synthesis (Fig. 3). Likewise, the presence of thioredoxin alone fails to inhibit E. coli Pol III–dependent DNA synthesis (Fig. 3B). Although WT gp5/Trx was able to attenuate E. coli replication at high concentrations, it did so with markedly lower potency than gp5-D75A/D654A/Trx. At concentrations greater than 80 nm, gp5-D475A/D654A/Trx essentially eliminated DNA synthesis by E. coli Pol III, whereas significant levels of DNA synthesis by Pol III persisted even at the highest concentration of WT gp5/Trx tested (Fig. 3A). The decrease in DNA synthesis by WT gp5/Trx may arise from an indirect effect on DNA synthesis (see “Discussion”).
Given that polymerase-inactive gp5 alleles are also toxic for T7 phage growth (Table 1), we examined the effect of gp5-D475A/D654A on the T7 DNA replication system. The presence of gp5-D475A/D654A partially inhibits the activity of WT T7 DNA polymerase and the T7 replisome. We first tested the effect of gp5-D475A/D654A on primer extension by T7 DNA polymerase. There is significant inhibition of the extension of a primer annealed to M13 ssDNA by WT T7 DNA polymerase only at gp-5D475A/D654A concentrations equivalent or higher than the concentration of WT polymerase present in the reaction (Fig. 4A). In the absence of gp5-D475A/D654A, partially extended intermediates that arise from polymerase pausing at regions of secondary structure accumulate at earlier time points but diminish as synthesis progresses through the structured region (Fig. 4A, second and fourth lanes, black arrow) At concentrations of gp5-D475A/D654A approximately equimolar or in excess to WT gp5, these intermediates persist even at the longest time points, with a concomitant decrease in fully extended products (Fig. 4A, eighth through sixteenth lanes, white arrow).
Figure 4.

Inhibition of WT T7 DNA polymerase by gp5-D475A/D654A. A, gp5-D475A/D654A inhibits primer extension by T7 DNA polymerase. A 32P-5′-end-labeled primer (50 nm) was annealed to M13 ssDNA (10 nm) and incubated with 100 nm WT T7 DNA polymerase, in the presence of 0.3 mm dNTPs, and increasing concentrations (0.03, 0.13 0.5, or 2 μm) of gp5-D475A/D654A. Samples were removed at 10, 30, and 90 s after addition of MgCl2 and separated on a denaturing polyacrylamide gel. Examples of extension intermediates accumulating (black arrow) or decreasing (white arrow) as the concentration of gp5-D475A/D654A increases. B, inhibition of strand-displacement DNA synthesis by gp5-D475A/D654A. Strand-displacement reactions rely on the helicase activity of gp4 to displace dsDNA encountered during DNA synthesis by T7 DNA polymerase. The reaction mixtures were supplemented with gp5-D475A/D654A (0, 1.9, 3.8, 7.5, 15, 30, or 120 nm). The samples were incubated for 5 min at 30 °C after initiating the reaction by addition of MgCl2 and quenched with 50 mm EDTA, and the incorporation of dGMP was measured by liquid scintillation counting. C, gp5-D475A/D654A inhibits coordinated leading and lagging-strand DNA synthesis. gp5-D475A/D654A (0, 5, 20, 80, or 320 nm) was incubated in a standard minicircle reaction (see “Experimental Procedures”) for 5 min prior to addition of MgCl2. The reactions were incubated at 30 °C for 5 min and quenched with 50 mm EDTA, and incorporation of dGMP (for leading strand synthesis) or dCMP (for lagging-strand synthesis) was measured by liquid scintillation counting.
gp5-D475A/D654A inhibits leading strand DNA synthesis, catalyzed by T7 DNA polymerase and the T7 helicase gp4 (Fig. 4B). During leading strand DNA synthesis, gp4 unwinds upstream dsDNA, generating the single-stranded template for nucleotide polymerization by T7 DNA polymerase. Together, gp4 and T7 DNA polymerase display a processivity of >17 kb (3, 4). The presence of gp5-D475A/D654A leads to a dose-dependent decrease in deoxyribonucleotide incorporation. At a concentration of 30 nm, gp5-D475A/D654A inhibits DNA synthesis by ∼50%. However, at the highest concentration of gp5-D475A/D654A tested (320 nm), synthesis of the leading strand still occurs at a value of ∼20% of control reactions (Fig. 4C).
The effect of gp5-D475A/D654A was also examined under conditions where leading and lagging strands are synthesized in a coordinated manner (Fig. 4C). The presence of gp5-D475A/D654A leads to a similar degree of inhibition of DNA synthesis of leading and lagging strands (Fig. 4C). Similar to the results observed for leading strand synthesis, significant DNA synthesis persists even at high concentrations of gp5-D475A/D654A. In conclusion, gp5-D475A/D654A also inhibits T7 DNA replication in vitro, but to a lesser extent than the inhibition observed for the E. coli replisome in vitro.
gp5-D475A/D654A/trx displays a more rapid association to primer/template DNA
T7 DNA polymerase has a high affinity for a primer template, with a Kd of 20 nm (9, 16). One possibility for the inhibitory effect of gp5-D475A/D654A/Trx is that it retains its ability to bind to a primer template but is locked in position, because it is unable to covalently link the incoming nucleotide to the primer. We compared the ability of WT gp5 and gp5-D475A/D654A to bind to a primer template and to ssDNA using an electrophoretic mobility shift assay (EMSA), in both the presence and the absence of Trx (Fig. 5A). gp5-D475A/D654A exhibits the same binding characteristics as WT gp5. In the case of both gp5 variants, no significant binding is observed to either ssDNA or primer/template in the absence of Trx (Fig. 5A, lanes 2–5 and 15–18). Like WT gp5, gp5-D4754A/D654A binds the primer/template stably only in the presence of thioredoxin (Fig. 5A, lanes 19–26). This strong binding is specific for a primer/template configuration because only weak binding to ssDNA is observed (Fig. 5A, lanes 2–5 and 15–18). The presence of thioredoxin slightly increases the affinity of gp5-D475A/D654A for ssDNA (Fig. 5A, lanes 4, 5, and 10–13). However, the polymerase–DNA complex formed is unstable under these electrophoretic conditions. Nevertheless, a decrease in the signal for free, labeled ssDNA and a smear representing labile protein–DNA complexes during electrophoresis is observed (Fig. 5A, lanes 2–13).
Figure 5.

gp5-D475A/D654A/Trx shows increased DNA-binding stability. A, EMSA gp5 or gp5-D475A/D654A in the presence and absence of thioredoxin. Left panel, lanes 1–13, 5′-32P-end-labeled ssDNA (19 nt in length). Right panel, lanes 14–26, ds 5′-32P-end-labeled primer/template (19-nt primer and 26-nt template). The samples were incubated as described under “Experimental procedures” and loaded on a native 0.5 × TBE (45 mm Tris, 45 mM Borate, 0.5 mm EDTA, pH 8.0) gel. After drying, the gel was exposed to a phosphorimaging screen. B, sensograms obtained using a biotinylated primer/template coupled to streptavidin chip under flow of solutions containing WT gp5 (red) or gp5-D475A/D654A (dark blue) in the absence of nucleotides. C, kinetic and binding constants for the interaction of WT gp5 or gp5-D475A/D654A to an immobilized primer/template ligand determined by surface plasmon resonance.
We used surface plasmon resonance to obtain a more quantitative description of the DNA-binding characteristics of gp5-D475A/D654. We used a series of binding experiments in which a concentration series of either WT gp5/Trx or gp5-475A/D654A/Trx was flowed over a immobilized primer template in the absence of deoxyribonucleotides and determined the kinetic parameters for their interaction (Fig. 5B and C). The results show that gp5-D475A/D654A/Trx binds a primer/template DNA with a 4-fold greater affinity than WT gp5/Trx. The increased affinity of gp5-D475A/D654A/Trx for primer/template DNA is solely due to enhanced association kinetics. As suggested in the experiment presented in Fig. 5B, the dissociation rate constant (koff) is identical between both gp5 variants. However, the association rate constant (kon) of gp5-D475A/D654A/Trx for primer/template DNA is roughly four times greater than for WT gp5/Trx. These results suggest that although the active site in gp5-D475/D654A/Trx cannot incorporate a nucleotide because it lacks the catalytic aspartates for polymerization, it associates with primer/template DNA faster, resulting in a higher affinity compared with WT gp5/Trx (17, 18).
Discussion
DNA polymerases employ two carboxylic acid-containing residues to coordinate a catalytic metal cation, usually Mg2+, in the active site (5, 10, 17). In the case of DNA polymerases from family A, two conserved aspartate residues are responsible for the coordination of metal ligands and are indispensable for catalysis (7, 12). From the crystal structure of T7 DNA polymerase (12), we identified Asp475 and Asp654 as the metal-coordinating residues in gp5 equivalent to those previously identified in the Klenow fragment of E. coli DNA polymerase I (7, 8). We targeted these postulated catalytic residues by replacing each or both aspartates with alanine.
We were surprised when we were unable to obtain transformants of singly or doubly substituted aspartate-to-alanine gene 5 alleles using common E. coli expression strains. We find that expression of polymerase-inactive gp5 is toxic if the host cells contain a functional allele for thioredoxin; no toxicity is observed in cells lacking thioredoxin. In addition, we find that the toxic effect is not observed in E. coli strains lacking the (DE3) lysogen, which encodes T7 RNA polymerase. This finding suggests that the levels of gp5-D475A/D654A protein that accumulate as a result of “leaky expression” of T7 RNA polymerase from the lacUV5 promoter in the (DE3) lysogen are sufficient to inhibit E. coli growth. In other words, our genetic analysis indicates that the toxic effect is manifest with very low expression of the altered polymerase. Combined, these results show that a complex of polymerase-inactive gene 5 DNA polymerase with thioredoxin is toxic for E. coli growth. We do not observe a toxic effect with WT gp5 or other polymerase active site gp5 variants. Toxicity depends on the presence of thioredoxin and is specific for gp5; no toxicity is observed in trxA− strains. Furthermore, a pol− Klenow fragment bearing homologous mutations corresponding to the magnesium-coordinating residues in the polymerase active site of gp5 and containing the TBD of gp5 did not exhibit toxicity. This latter result is not too surprising, considering that the chimeric protein shows a marginal level of stimulation in the presence of thioredoxin compared with gp5, has a 10-fold lower affinity for Trx than does T7 gp5, and is 3-fold less active than gp5/Trx, and its complex with Trx has a lower processivity compared with gp5/Trx (14).
Thioredoxin is essential for the T7 DNA replication machinery. It binds tightly to T7 gp5 and confers processivity to the polymerization reaction (16, 19, 20). T7 gp5 alone has a low processivity of ∼5–20 nucleotides per binding cycle, and the acquisition of thioredoxin increases the processivity to ∼800 nucleotides per binding event (19). This increase in processivity arises from an increased affinity of gp5/Trx for a primer/template: The Kd of gp5/Trx for a primer/template determined by incorporation assays is 20 nm, whereas it is at least 20-fold higher in the case of gp5 alone (9, 16). Thioredoxin also provides gp5 with contacts for the T7 gene 4 helicase-primase and to the gene 2.5 ssDNA-binding protein (13, 21). Binding of thioredoxin to gp5 leads to the formation of two basically charged unstructured regions in gp5 that interact with the negatively charged unstructured C-terminal domains of both the ssDNA-binding protein (gp2.5) and helicase-primase (gp4) of phage T7 (22). Of these two functions, it seems most likely that the toxic effect arises from the increases affinity for DNA, because it is unlikely that proteins in the E. coli replisome have specific contacts with thioredoxin. However, we cannot exclude the possibility that gp5/Trx might interact with E. coli SSB, because this protein has an acidic C-terminal tail with similar length, charge distribution, and a C-terminal phenylalanine, as do the acidic N-terminal tails of T7 gp2.5 and gp4. In fact, a chimeric gp2.5 containing the C terminus of E. coli SSB supports T7 growth and interacts with T7 DNA polymerase. However, a chimeric SSB protein containing the C-terminal tail of gp2.5 does not support T7 growth (23).
The toxic effect of gp5-D475A/D654A/Trx appears to be directly related to its effect on E. coli DNA replication. The inactive gp5/Trx complex inhibits DNA synthesis by reconstituted E. coli replisomes, and this inhibition depends on the presence of the polymerase-inactive gp5/Trx complex: either protein alone has no effect on the efficiency of DNA synthesis. Intriguingly, WT gp5 also inhibits the reconstituted E. coli replisome, albeit to a much lesser extent. In vitro, the E. coli replisome retains significant activity in the presence of high concentrations of WT gp5/Trx, but similar concentrations of gp5-D475A/D654A/Trx essentially abolish replication activity. Consistent with these results, expression of gp5-D475A/D654A/Trx in vivo is toxic, whereas expression of WT gp5/Trx is not toxic. The residual level of replication activity in the presence of high concentrations of WT gp5/Trx is likely sufficient to support cell viability. Whether the inhibition of E. coli replication by WT gp5/Trx reflects a viral mechanism to further shut off residual host DNA replication, whereby excess amounts of WT gp5/Trx might interfere with the assembly of functional E. coli replisomes, remains to be investigated. Polymerase-inactive gp5/Trx also inhibits the activities of the T7 replication system in vitro, and gene 5 alleles bearing mutations in the catalytic aspartates are dominant-lethal in vivo for growth of WT T7. Similarly, in a previous study (24), a histidine-to-alanine mutation of the C-terminal residue of gp5 resulted in a dominant-negative phenotype for T7 growth, and the purified mutant protein also displayed strikingly decreased polymerase activity.
The molecular mechanism for the toxicity of gp5-D475A/D654A/Trx on E. coli DNA replication is difficult to dissect. The simplest mechanism is that the high affinity of gp5/Trx for a primer template inhibits movement of the E. coli replisome on DNA by direct competition with assembly of E. coli replisome components onto DNA, perhaps interfering with the ability of Pol III, the replicative DNA polymerase of E. coli for binding to the primer/template junction. An inactive DNA polymerase bound to DNA could be locked into place as a result of its inability to add the next incoming nucleotide. Such a scenario is similar to the stable polymerase complex created by the incorporation of a dideoxy-nucleotide, where the absence of condensation of the next nucleotide locks the polymerase in a polymerizing mode (12). An alternative, nonmutually exclusive, possibility is that the toxic effect exerted by polymerase-inactive gp5/Trx could be due to the generation or propagation of toxic DNA damage. DNA replication machineries encounter multiple barriers during genomic replication, resulting in stalled or abandoned replication forks that, if left unchecked, result in highly deleterious dsDNA breaks (25). The reactivation of replication forks is a major pathway in the maintenance of genomic integrity (26). Polymerase-inactive gp5/Trx could hinder the assembly of replication fork restart pathways, resulting in catastrophic genomic damage and triggering cell death, whereas any inhibition by WT gp5/Trx would be likely less potent (27, 28).
We envision that pol− gene 5 alleles could have applications in synthetic biology or industrial settings as a cell host shutoff mechanism. Bacterial cellular toxicity triggered through inhibition of DNA synthesis can form the basis for the removal of selected cells in a population in a specific manner by designing a regulatory circuit that depends on mutant gene 5 or trxA co-expression in a temporally controlled manner.
Experimental Procedures
Construction of plasmids and protein purification
Mutations were simultaneously introduced into codons for aspartate 475 and 654 of T7 gene 5 in plasmids encoding WT gene 5 (pGP5-3) and exonuclease-deficient gene 5 (D5A, D65A) (pGP5-3 exo−) using Gibson assembly (29). Site-directed mutagenesis was used to introduce single mutations into T7 gene 5. Similarly, we used Gibson assembly to construct a double substitution (D705A/D882A) variant of Klenow fragment containing an insertion of the T7 gp5 TBD using pKLN-TBD as template (14). BL21 (DE3) ΔtrxA was constructed by replacing trxA with a gene encoding kanamycin resistance using the λ RED system (30). T7 gp5 variants were expressed in E. coli A307 (DE3), which lacks endogenous thioredoxin and the overexpressed gene 5 proteins were purified as previously described (19). WT E. coli thioredoxin was prepared as previously described (31). E. coli clamp loader complex (τ3dd′χy), β2 clamp, DnaG, DnaB, and ssDNA-binding protein (SSB) were purified as previously described (32, 33). αεθ, αεQθ, and αεLθ were provided by Nicholas Dixon (University of Wollongong).
Genetic assays
E. coli BL21 (DE3), BL21, and E. coli BL21 (DE3) ΔtrxA were transformed with plasmids containing the genes for WT or variant T7 gene 5 or the Klenow fragment of E. coli DNA polymerase I containing the T7 gp5 TBD according to standard methods. Transformation mixtures were plated onto LB agar plates containing 100 µg/ml ampicillin and grown overnight at 37 °C. The number of colonies after incubation were counted. For phage growth and complementation assays, E. coli DH5α transformed with gene 5–expressing plasmid were grown in LB to exponential phase. Cell culture (0.3 ml) was mixed with 3 ml of soft agar (LB broth + 0.7% agar) and plated onto LB-agar/ampicillin plates. Aliquots of serially diluted WT or gene 5-deletion (Δ5) T7 phage were spotted onto the plates to determine phage titer.
DNA polymerase assays
Primed M13 ssDNA (7 nm) was incubated with increasing concentrations (3–2,000 nm) of purified WT gp5 or gp5-D475A/D654A in the presence of a 10-fold molar excess of thioredoxin in a buffer consisting of 40 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 10 mm MgCl2, 5 mm DTT, and 0.3 mm dTTP, dATP, dCTP, and [α-32P]dGTP at 25 °C for 2 min. The reactions were quenched with EDTA and spotted onto DE81 filters, and the amount of nucleotides incorporated into DNA was determined as previously described (1).
3′–5′ exonuclease activity of gp5 variants was measured using an internally 32P-labeled 3-kb dsDNA generated by PCR in the presence of [α-32P]dGTP from a plasmid template. Reaction mixtures contained 40 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 5 mm DTT, 50 mm potassium glutamate, 3 μm uniformly labeled dsDNA (expressed as nucleotides) and varying concentrations of gp5 variants. Thioredoxin was included at a 10-fold molar excess over gp5 concentrations in the assay. Samples were taken after incubation at 37 °C and quenched with 18% (w/v) TCA and 2 mg/ml BSA. The acid-soluble radioactivity in the supernatant was measured by liquid scintillation counting.
The primer template for rolling-circle DNA synthesis mediated by the E. coli replication proteins was prepared and purified as described (32). A 73-mer tailed oligonucleotide (T36GAATTTCGCAGCCGTCCACAGGTAGCACTGAATCATG) was annealed to a modified M13mp7(L2) circular closed ssDNA and extended with T7 DNA polymerase (New England Biolabs) at 37 °C for 20 min. The filled M13mp7(L2) was purified by phenol-chloroform extraction. The E. coli replisome was assembled by mixing 1 nm template, 20 nm τ3δδ′χy, 20 nm β2 clamp dimer, 50 nm DnaB hexamer, 60 nm Pol III core complex (αεθ), 50 μm ATPγS, and 60 μm dCTP and dGTP in 50 mm Hepes-KOH, pH 7.9, 12 mm Mg (OAc)2, 0.1 mg/ml BSA, 10 mm DTT on ice and incubating the mixture in a water bath at 37 °C for 6 min Replication was initiated by adding prewarmed 10× initiation mixture to the assembly reactions. The 10× initiation mixture contained 1 mm ATP, 250 μm rNTPs, 60 μm dTTP, and [α-32P]dATP, 500 nm SSB tetramer, and 600 nm DnaG. All concentrations are the final concentrations in the reactions. 10 s after initiation, the indicated concentrations of either WT T7 gp5 or its variants were added to the replication reactions. Replication reactions took place in a 37 °C water bath. Replication reactions were quenched by adding EDTA to a final concentration of 25 mm. Quenched reactions were applied to a 0.6% alkaline denaturing agarose gel, and replication products were resolved by running the gel in an alkaline electrophoresis buffer (50 mm NaOH, 2 mm EDTA) at 21 V for 15 h. Replication products were visualized by autoradiography (32).
To assess the effect of gp5-D475A/D654A on the activity of WT gp5, we incubated various concentrations of gp5. D475A/D654A(exo−)/Trx along with 100 nm gp5(exo−)/Trx in the presence of M13 DNA annealed to a 5′-32P-labeled primer in 40 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 5 mm DTT, and 0.3 mm dNTPs. DNA synthesis was initiated by adding MgCl2 to a final concentration of 10 mm. At 10, 30, or 90 s, the reactions were stopped by the addition of an equal volume of formamide loading dye (93% formamide, 50 mm EDTA, 0.01% xylene cyanol, and 0.01% bromphenol blue). The samples were heated to 95 °C for 5 min and loaded onto a denaturing 5% polyacrylamide gel. The gel was dried and exposed to a phosphorimaging screen.
Leading-strand DNA synthesis was measured in a reaction containing 10 nm of a DNA minicircle bearing a replication fork (1), 50 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 5 mm DTT, 0.6 mm dATP, dCTP, dTTP, and [α-32P]dGTP, 60 nm gp4 (monomer), 80 nm WT gp5/Trx, and varying concentrations of gp5-D475A/D654A/Trx. The reaction was initiated by adding MgCl2 to a final concentration of 10 mm, incubated at 25 °C for 5 min before quenching with EDTA, and processed as described (34).
Coordinated leading- and lagging-strand DNA synthesis reactions were assembled in the presence of varying concentrations of gp5-D475A/D654A/Trx. The reaction mixtures contained 40 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, mm DTT, 10 mm MgCl2, 0.6 mm dNTPs (with either [α-32P]dGTP to measure synthesis of the leading strand or [α-32P]dCTP to measure lagging-strand synthesis), 60 nm gp4 (monomer), 80 nm WT T7 DNA polymerase, 3 μm gp2.5, and 10 nm minicircle DNA. The sequence bias of minicircle DNA allows for monitoring of synthesis of the leading and lagging strands by measuring the incorporation of dGMP or dCMP, respectively (1).
DNA-binding assays
An EMSA was used to measure binding of DNA polymerase to both ssDNA and a primer/template DNA duplex. A 19-nt ssDNA (5′-TGCCGACGATAAACGACCC-3′) was 5′-end-labeled with T4 polynucleotide kinase and [γ-32P]ATP and served as the primer strand. A dsDNA containing a 5′ overhang representing a primer/template duplex was formed by annealing the labeled primer strand with a 26-nt ssDNA (5′-CAGTGACGGGTCGTTTATCGTCGGCA-3′). Binding reactions contained 3 nm 5′-32P primer or primer/template in 40 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 5 mm DTT, 10 mm MgCl2, and various concentrations of gp5 exo− or gp5-D475A/D654A exo− in the presence or absence of Trx. The samples were incubated at room temperature for 10 min and loaded onto a native 10% polyacrylamide gel with 0.5× TBE (45 mm Tris, 45 mm Borate, 0.5 mm EDTA, pH 8.0) as the electrophoretic buffer. The gel was run in a cold room, dried, and visualized by phosphorimaging.
In surface plasmon resonance assays, the 19-nt primer used for EMSA was annealed to the 26-nt template strand containing a biotin attached to the 3′-end. dsDNA was coupled to a streptavidin chip at a concentration of 0.25 μm in 10 mm Hepes, pH 7.4, 150 mm NaCl, 0.005% Tween 20 (vol./vol.) at a flow rate of 10 μl/min. Free streptavidin on the flow cells was then blocked by washing with biotin. Binding studies were carried out by flowing 200 nm gp5 exo−/Trx or gp5-D475A/D654A exo−/Trx over 600 RU immobilized primer/template in 20 mm Hepes, pH 7.5, 5 mm MgCl2, 2.5 mm DTT, 200 mm potassium glutamate, 1% glycerol (w/v) at a flow rate of 10 μl/min. For determination of kinetic parameters, 50 RU of primer/template was immobilized and various concentrations (12.5, 25, 50, 75, and 100 nm) of gp5 exo−/Trx or gp5-D475A/D654A exo−/Trx were flowed over the chip. Kinetic data were obtained by fitting binding sensorgrams using BIA-Evaluation software. All experiments were repeated at least four times, with the exception of those whose data are shown in Figs. 3B and 5C, which were done in duplicate.
Data availability
All of the data presented are contained in the manuscript and are available upon request from Alfredo J. Hernandez, Harvard Medical School, alfredo_hernandez@hms.harvard.edu.
Acknowledgments
We thank Stephen C. Blacklow for helpful discussions, Stanley Tabor for the gift of pKLN-TBD, Slobodan Jergic and Nicholas Dixon (University of Wollongong) for the generous gift of E. coli replisome proteins, Steve Moskowitz for preparation of figures, and Kelly Arnett at the Center for Macromolecular Interactions at Harvard Medical School for the use of Biacore equipment.
Author contributions—A. J. H. conceptualization; A. J. H., S.-J. L., S. C., J. J. L., and C. C. R. formal analysis; A. J. H. and S.-J. L. validation; A. J. H., S.-J. L., S. C., and J. A. L. investigation; A. J. H. and S.-J. L. methodology; A. J. H. writing-original draft; A. J. H., S.-J. L., S. C., J. J. L., and C. C. R. writing-review and editing; C. C. R. supervision; C. C. R. and J. J. L. funding acquisition; C. C. R. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grant R01 GM114065 (to J. J. L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- Trx
- thioredoxin
- Pol
- polymerase
- EMSA
- electrophoretic mobility shift assay.
References
- 1. Lee J., Chastain P. D. 2nd, Kusakabe T., Griffith J. D., and Richardson C. C. (1998) Coordinated leading and lagging strand DNA synthesis on a minicircular template. Mol. Cell 1, 1001–1010 10.1016/S1097-2765(00)80100-8 [DOI] [PubMed] [Google Scholar]
- 2. Hamdan S. M., and Richardson C. C. (2009) Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 78, 205–243 10.1146/annurev.biochem.78.072407.103248 [DOI] [PubMed] [Google Scholar]
- 3. Kulczyk A. W., and Richardson C. C. (2016) The replication system of bacteriophage T7. Enzymes 39, 89–136 10.1016/bs.enz.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 4. Lee S. J., and Richardson C. C. (2011) Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 15, 580–586 10.1016/j.cbpa.2011.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brautigam C. A., and Steitz T. A. (1998) Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 8, 54–63 10.1016/S0959-440X(98)80010-9 [DOI] [PubMed] [Google Scholar]
- 6. Wu S., Beard W. A., Pedersen L. G., and Wilson S. H. (2014) Structural comparison of DNA polymerase architecture suggests a nucleotide gateway to the polymerase active site. Chem. Rev. 114, 2759–2774 10.1021/cr3005179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Polesky A. H., Dahlberg M. E., Benkovic S. J., Grindley N. D., and Joyce C. M. (1992) Side chains involved in catalysis of the polymerase reaction of DNA polymerase I from Escherichia coli. J. Biol. Chem. 267, 8417–8428 [PubMed] [Google Scholar]
- 8. Bermek O., Grindley N. D., and Joyce C. M. (2011) Distinct roles of the active-site Mg2+ ligands, Asp882 and Asp705, of DNA polymerase I (Klenow fragment) during the prechemistry conformational transitions. J. Biol. Chem. 286, 3755–3766 10.1074/jbc.M110.167593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel S. S., Wong I., and Johnson K. A. (1991) Pre–steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 10.1021/bi00216a029 [DOI] [PubMed] [Google Scholar]
- 10. Raper A. T., Reed A. J., and Suo Z. (2018) Kinetic mechanism of DNA polymerases: contributions of conformational dynamics and a third divalent metal ion. Chem. Rev. 118, 6000–6025 10.1021/acs.chemrev.7b00685 [DOI] [PubMed] [Google Scholar]
- 11. Hernandez A. J., Lee S. J., and Richardson C. C. (2016) Primer release is the rate-limiting event in lagging-strand synthesis mediated by the T7 replisome. Proc. Natl. Acad. Sci. U.S.A. 113, 5916–5921 10.1073/pnas.1604894113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doublié S., Tabor S., Long A. M., Richardson C. C., and Ellenberger T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 391, 251–258 10.1038/34593 [DOI] [PubMed] [Google Scholar]
- 13. Hamdan S. M., Marintcheva B., Cook T., Lee S. J., Tabor S., and Richardson C. C. (2005) A unique loop in T7 DNA polymerase mediates the binding of helicase-primase, DNA binding protein, and processivity factor. Proc. Natl. Acad. Sci. U.S.A. 102, 5096–5101 10.1073/pnas.0501637102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bedford E., Tabor S., and Richardson C. C. (1997) The thioredoxin binding domain of bacteriophage T7 DNA polymerase confers processivity on Escherichia coli DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 94, 479–484 10.1073/pnas.94.2.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu C. A., Zechner E. L., Hughes A. J. Jr., Franden M. A., McHenry C. S., and Marians K. J. (1992) Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork: IV. Reconstitution of an asymmetric, dimeric DNA polymerase III holoenzyme. J. Biol. Chem. 267, 4064–4073 [PubMed] [Google Scholar]
- 16. Huber H. E., Tabor S., and Richardson C. C. (1987) Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J. Biol. Chem. 262, 16224–16232 [PubMed] [Google Scholar]
- 17. Florián J., Goodman M. F., and Warshel A. (2003) Computer simulation studies of the fidelity of DNA polymerases. Biopolymers 68, 286–299 10.1002/bip.10244 [DOI] [PubMed] [Google Scholar]
- 18. Florián J., Goodman M. F., and Warshel A. (2003) Computer simulation of the chemical catalysis of DNA polymerases: discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 125, 8163–8177 10.1021/ja028997o [DOI] [PubMed] [Google Scholar]
- 19. Tabor S., Huber H. E., and Richardson C. C. (1987) Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 262, 16212–16223 [PubMed] [Google Scholar]
- 20. Etson C. M., Hamdan S. M., Richardson C. C., and van Oijen A. M. (2010) Thioredoxin suppresses microscopic hopping of T7 DNA polymerase on duplex DNA. Proc. Natl. Acad. Sci. U.S.A. 107, 1900–1905 10.1073/pnas.0912664107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamdan S. M., Johnson D. E., Tanner N. A., Lee J. B., Qimron U., Tabor S., van Oijen A. M., and Richardson C. C. (2007) Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 27, 539–549 10.1016/j.molcel.2007.06.020 [DOI] [PubMed] [Google Scholar]
- 22. Zhang H., Lee S. J., Zhu B., Tran N. Q., Tabor S., and Richardson C. C. (2011) Helicase–DNA polymerase interaction is critical to initiate leading-strand DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 9372–9377 10.1073/pnas.1106678108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kong D., and Richardson C. C. (1998) Role of the acidic carboxyl-terminal domain of the single-stranded DNA-binding protein of bacteriophage T7 in specific protein-protein interactions. J. Biol. Chem. 273, 6556–6564 10.1074/jbc.273.11.6556 [DOI] [PubMed] [Google Scholar]
- 24. Kumar J. K., Tabor S., and Richardson C. C. (2001) Role of the C-terminal residue of the DNA polymerase of bacteriophage T7. J. Biol. Chem. 276, 34905–34912 10.1074/jbc.M104151200 [DOI] [PubMed] [Google Scholar]
- 25. Windgassen T. A., Wessel S. R., Bhattacharyya B., and Keck J. L. (2018) Mechanisms of bacterial DNA replication restart. Nucleic Acids Res. 46, 504–519 10.1093/nar/gkx1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., and Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 10.1038/35003501 [DOI] [PubMed] [Google Scholar]
- 27. Bayles K. W. (2014) Bacterial programmed cell death: making sense of a paradox. Nat. Rev. Microbiol. 12, 63–69 10.1038/nrmicro3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanouchi Y., Lee A. J., Meredith H., and You L. (2013) Programmed cell death in bacteria and implications for antibiotic therapy. Trends Microbiol. 21, 265–270 10.1016/j.tim.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 30. Yu D., Ellis H. M., Lee E. C., Jenkins N. A., Copeland N. G., and Court D. L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 97, 5978–5983 10.1073/pnas.100127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee S. J., Tran N. Q., Lee J., and Richardson C. C. (2018) Hydrophobic residue in Escherichia coli thioredoxin critical for the processivity of T7 DNA polymerase. Biochemistry 57, 5807–5817 10.1021/acs.biochem.8b00341 [DOI] [PubMed] [Google Scholar]
- 32. Kath J. E., Chang S., Scotland M. K., Wilbertz J. H., Jergic S., Dixon N. E., Sutton M. D., and Loparo J. J. (2016) Exchange between Escherichia coli polymerases II and III on a processivity clamp. Nucleic Acids Res. 44, 1681–1690 10.1093/nar/gkv1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jergic S., Horan N. P., Elshenawy M. M., Mason C. E., Urathamakul T., Ozawa K., Robinson A., Goudsmits J. M., Wang Y., Pan X., Beck J. L., van Oijen A. M., Huber T., Hamdan S. M., and Dixon N. E. (2013) A direct proofreader-clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 32, 1322–1333 10.1038/emboj.2012.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghosh S., Marintcheva B., Takahashi M., and Richardson C. C. (2009) C-terminal phenylalanine of bacteriophage T7 single-stranded DNA-binding protein is essential for strand displacement synthesis by T7 DNA polymerase at a nick in DNA. J. Biol. Chem. 284, 30339–30349 10.1074/jbc.M109.024059 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the data presented are contained in the manuscript and are available upon request from Alfredo J. Hernandez, Harvard Medical School, alfredo_hernandez@hms.harvard.edu.