

Abstract
Solute carrier family 20 member 2 (SLC20A2) and xenotropic and polytropic retrovirus receptor 1 (XPR1) are transporters with phosphate uptake and efflux functions, respectively. Both are associated with primary familial brain calcification (PFBC), a genetic disease characterized by cerebral calcium-phosphate deposition and associated with neuropsychiatric symptoms. The association of the two transporters with the same disease suggests that they jointly regulate phosphate fluxes and cellular homeostasis, but direct evidence is missing. Here, we found that cross-talk between SLC20A2 and XPR1 regulates phosphate homeostasis, and we identified XPR1 as a key inositol polyphosphate (IP)-dependent regulator of this process. We found that overexpression of WT SLC20A2 increased phosphate uptake, as expected, but also unexpectedly increased phosphate efflux, whereas PFBC-associated SLC20A2 variants did not. Conversely, SLC20A2 depletion decreased phosphate uptake only slightly, most likely compensated for by the related SLC20A1 transporter, but strongly decreased XPR1-mediated phosphate efflux. The SLC20A2-XPR1 axis maintained constant intracellular phosphate and ATP levels, which both increased in XPR1 KO cells. Elevated ATP levels are a hallmark of altered inositol pyrophosphate (PP-IP) synthesis, and basal ATP levels were restored after phosphate efflux rescue with WT XPR1 but not with XPR1 harboring a mutated PP-IP–binding pocket. Accordingly, inositol hexakisphosphate kinase 1-2 (IP6K1-2) gene inactivation or IP6K inhibitor treatment abolished XPR1-mediated phosphate efflux regulation and homeostasis. Our findings unveil an SLC20A2-XPR1 interplay that depends on IPs such as PP-IPs and controls cellular phosphate homeostasis via the efflux route, and alteration of this interplay likely contributes to PFBC.
Keywords: xenotropic and polytropic retrovirus receptor 1 (XPR1), solute carrier family 20 member 2 (SLC20A2), phosphate transporter, primary familial brain calcification (PFBC), phosphate homeostasis, ATP, inositol pyrophosphate, metabolic regulation, SPX domain, efflux transporter, cell metabolism, energy metabolism, anion transport, inositol phosphate
The solute carrier (SLC) SLC20 and SLC53 family genes encode three cell surface multitransmembrane proteins known as PiT1/SLC20A1, PiT2/SLC20A2, and polytropic retrovirus receptor 1 (XPR1)/SLC53A1, which were shown to transport phosphate (1–3) and to serve as retroviral receptors for virus infection (4–8). In contrast to the phosphate transporters of the SLC34 family, which have an expression strictly confined to intestine and kidney cells (9, 10) and a recognized function in blood phosphate homeostasis (11), SLC20A1, SLC20A2, and XPR1 transporters have a broader tissue expression (12), and SLC20 transporters are considered housekeeping players in cellular phosphate homeostasis (13), although direct evidence is missing. A similar role has recently been proposed for XPR1, principally due to its capacity to interact with inositol pyrophosphates (PP-IPs) (14), which are considered intermediates of intracellular phosphate sensing (15). SLC20A1 and SLC20A2 are high-affinity, sodium-dependent transporters of the monovalent H2PO4− form, which control phosphate influx across the plasma membrane (2, 3), while XPR1 ensures phosphate efflux (1, 16) with a yet unknown driving force. It is thought that the rate of transport is dictated by both the intracellular and extracellular phosphate contents, which maintain constant concentrations. The intracellular level of phosphate is tightly regulated, given its central role in many key functions in cells. Thus, phosphate is a fundamental component of nucleic acids and phospholipids and is absolutely required for energy production, intracellular signaling, and acid-base buffering. In mineralizing cells, phosphate is a major component, along with calcium, of the hydroxyapatite mineral found in bones and teeth and acts as a signaling molecule for cell maturation and subsequent mineralization (17). SLC20A1 and SLC20A2 take an active part in these mineralizing processes, both in phosphate sensing, presumably through heterodimerization (18), and in skeleton development (19, 20), and are involved in calcifying disorders (21, 22).
Primary familial brain calcification (PFBC) is a rare genetic disorder characterized by brain calcification in patients with normal phosphate and calcium blood levels (23) and is associated with diverse neuropsychiatric expression and motor symptoms. As seen on computed tomography scans, calcification affects the basal ganglia and other areas of the brain, including the cerebellum, thalamus and caudate nuclei, supratentorial white matter, and the cerebral cortex (24). PFBC is a primarily vascular calcifying disorder, with calcium phosphate deposits observed in mural cells of arterioles and capillaries, namely, vascular smooth muscle cells (VSMC) and pericytes, respectively. Four PFBC causative genes with autosomal dominant inheritance have been identified: SLC20A2 and XPR1 (25, 26), the platelet-derived-growth factor (PDGF) receptor β (PDGFRβ)-encoding gene (PDGFRB) (27), and PDGFB, which encodes PDGF-B, the main ligand of PDGFRβ (28). Very recently, the MYORG and JAM2 genes, which encode the myogenesis-regulating glycosidase and the junctional adhesion molecule 2, respectively, were identified as autosomal recessive causal genes for PFBC (29–31). The deposition of calcium phosphate crystals in the brain and the association of two phosphate transporter genes with PFBC indicate a direct link between phosphate regulation dysfunction and the disease. One clue came from cerebrospinal fluid (CSF) analyses, which unveiled elevated phosphate levels in Slc20a2 KO and heterozygous mice (32, 33), as well as in PFBC patients (34, 35). Remarkably, loss of function of either SLC20A2 or XPR1 results in the same PFBC disorder despite opposite phosphate transport directions.
Here, we assessed for the first time the role of both PFBC-associated phosphate transporters SLC20A2 and XPR1 in intracellular phosphate homeostasis. We found that SLC20A2 and XPR1 regulated the rates of phosphate fluxes in a concerted and PP-IP-dependent manner and controlled intracellular phosphate and ATP levels, providing evidence for a specific SLC20A2, XPR1, and PP-IP interplay in phosphate homeostasis and metabolism, with new insights on the role of phosphate metabolic disorders in PFBC.
Results
PFBC SLC20A2 variants are unable to modulate phosphate uptake and efflux
To date, more than 100 variants have been found in the SLC20A2 gene of PFBC patients (36). It is the major PFBC gene, as variants in this gene account for 66.5% of genetically explained cases. Although a majority of the variants were classified as pathogenic or likely pathogenic based on genetic arguments, very few studies have assessed these variants for phosphate transport (26, 37), and none has been evaluated for expression at the plasma membrane. We tested phosphate uptake ability of SLC20A2 variants harboring four PFBC mutations reported to abolish phosphate transport in a Xenopus oocyte assay (26), c.1492G>A, p.(G498R); c.1723G>A, p.(E575K); c.1784C>T, p.(T595M) and c.1802C>T, p.(S601L) as well as three other variants, c.560A>G, p.(Y187C); c.581A>G, p.(N194S) and p.ΔA145_V205 (24, 38, 39), identified in French PFBC patients. With the exception of SLC20A2 p.ΔA145_V205, which most likely encoded a defective transporter, as it was undetectable by flow cytometry (Fig. 1A) or immunoblotting (Fig. 1B), all of the other SLC20A2 mutants were expressed at the expected size, were present at the plasma membrane as efficiently as WT SLC20A2, and did not affect cell surface expression of endogenous SLC20A1 and XPR1, as observed by flow cytometry using specific retroviral ligands derived from receptor-binding domains (RBDs) of gammaretrovirus envelope (Env) glycoproteins (1, 25, 40, 41). 33P-radiolabeled phosphate flux assays showed that overexpression of human SLC20A2 led to a significant increase in phosphate uptake, as expected, compared with control parental cells (Fig. 1C). In contrast, phosphate uptake remained close to basal levels with the SLC20A2 mutants, suggesting that PFBC variants altered critically phosphate uptake, with no detectable impact on endogenous SLC20A2 expression and phosphate transport. Surprisingly, the increase in phosphate uptake seen for WT SLC20A2 was concomitant with an increase in phosphate efflux (Fig. 1D). Similar observations were obtained with SLC20A1 (Fig. S1). In comparison, phosphate efflux with the SLC20A2 variants remained close to control levels. These results suggested a close interplay between phosphate uptake and efflux. Further ex vivo evaluation of the Y187C and ΔA145_V205 SLC20A2 variants with peripheral blood mononuclear cells (PBMC) isolated from PFBC patients showed a strong decrease in phosphate uptake, compared with PBMC from control healthy individual (Fig. 1E). However, phosphate efflux was similar to controls in patient PBMC (Fig. 1F).
Figure 1.
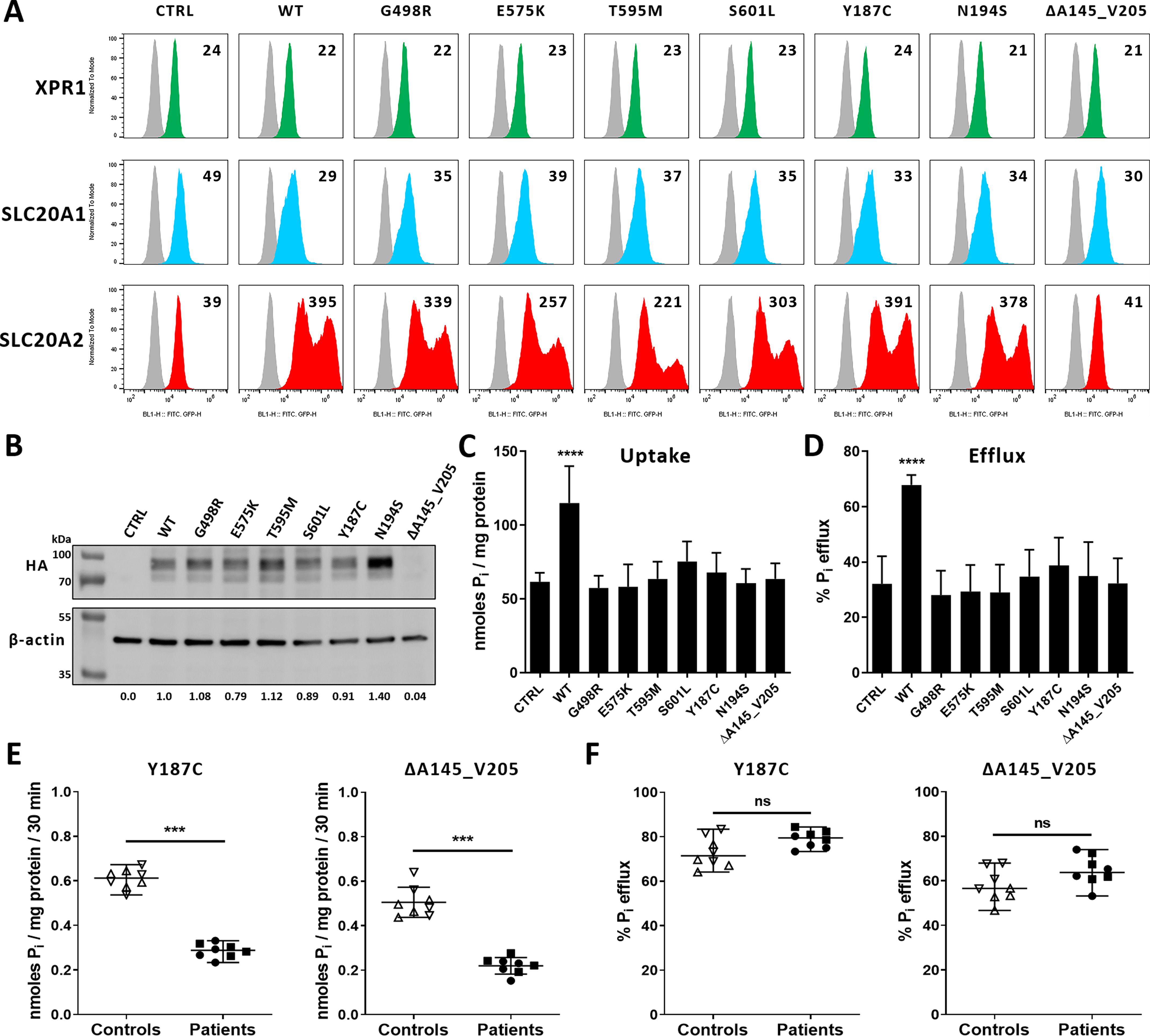
Phosphate transport activity of PFBC SLC20A2 variants. A, cell surface detection of XPR1 (green), SLC20A1 (blue), and SLC20A2 (red) by flow cytometry using cognate retroviral RBD ligands in HEK293T cells transfected with an empty expression vector (control) (CTRL) or encoding either the HA-tagged WT SLC20A2 or the indicated PFBC variants. Numbers indicate the specific change in mean fluorescence intensity of a representative experiment (n = 3), compared with nonspecific staining with the secondary IgG (gray). B, representative immunoblot of HA-tagged SLC20A2 variants in cell lysates from A. SLC20A2 and β-actin bands were quantified by densitometry and expressed as ratios (SLC20A2/β-actin) indicated under the immunoblot. C and D, uptake (C) and efflux (D) assays of [33P]phosphate in HEK293T obtained from A. Data are means ± S.D. from 4 independent experiments. One-way ANOVA with the Holm-Sidak multiple-comparison test; for data in C, F = 21.00 (p < 0.0001); for data in D, F = 20.11 (p < 0.0001). E and F, uptake (E) and efflux (F) of [33P]phosphate in PBMC isolated either from 2 healthy donors (up and down triangles) or from PFBC patients harboring either the p.Y187C or the p.A145_V205 variants (filled squares and circles; 2 patients each). Phosphate fluxes are shown as means ± S.D. from 4 experiments. Two-tailed Student's t tests; ***, p ≤ 0.001, compared with the respective controls; ns, nonsignificant; Pi, inorganic phosphate.
Modulation of both SLC20A2 and XPR1 expression impacts phosphate efflux
The SLC20A2-stimulated phosphate efflux may suggest a cellular adaptation that maintained phosphate homeostasis. Since SLC20A2 and XPR1 are both associated with PFBC and both participate in phosphate fluxes, we first tested whether they balanced each other when overexpressed or down-modulated. To this aim, we used human HAP1 cells in which either SLC20A2 or XPR1 genes were inactivated by genome editing. Knockout of SLC20A2 and XPR1 at the plasma membrane was confirmed by flow cytometry (Fig. 2A) and by cell resistance to infection by enhanced GFP (EGFP) lentiviral vectors pseudotyped with amphotropic murine leukemia virus (A-MLV) or xenotropic murine leukemia virus (X-MLV) Env, respectively (Fig. 2B). Expression of endogenous SLC20A1 and XPR1 in SLC20A2 KO cells remained unchanged (Fig. 2A), although infection assays suggested a slight increase for both transporters (Fig. 2B), while the irrelevant LDL receptor used by the vesicular stomatitis virus G (VSV-G) Env pseudotypes remained unaffected. We also generated SLC20A2-overexpressing HAP1 cells (SLC20A2+ cells) by transduction with a MLV-based retroviral vector carrying the SLC20A2 cDNA. SLC20A2+ cells presented 2.5-fold increases in both binding and infection by A-MLV pseudotype lentiviral vector (Fig. 2, A and B), a small but reproducible decrease in SLC20A1 expression, possibly due to compensation, and no change in XPR1.
Figure 2.
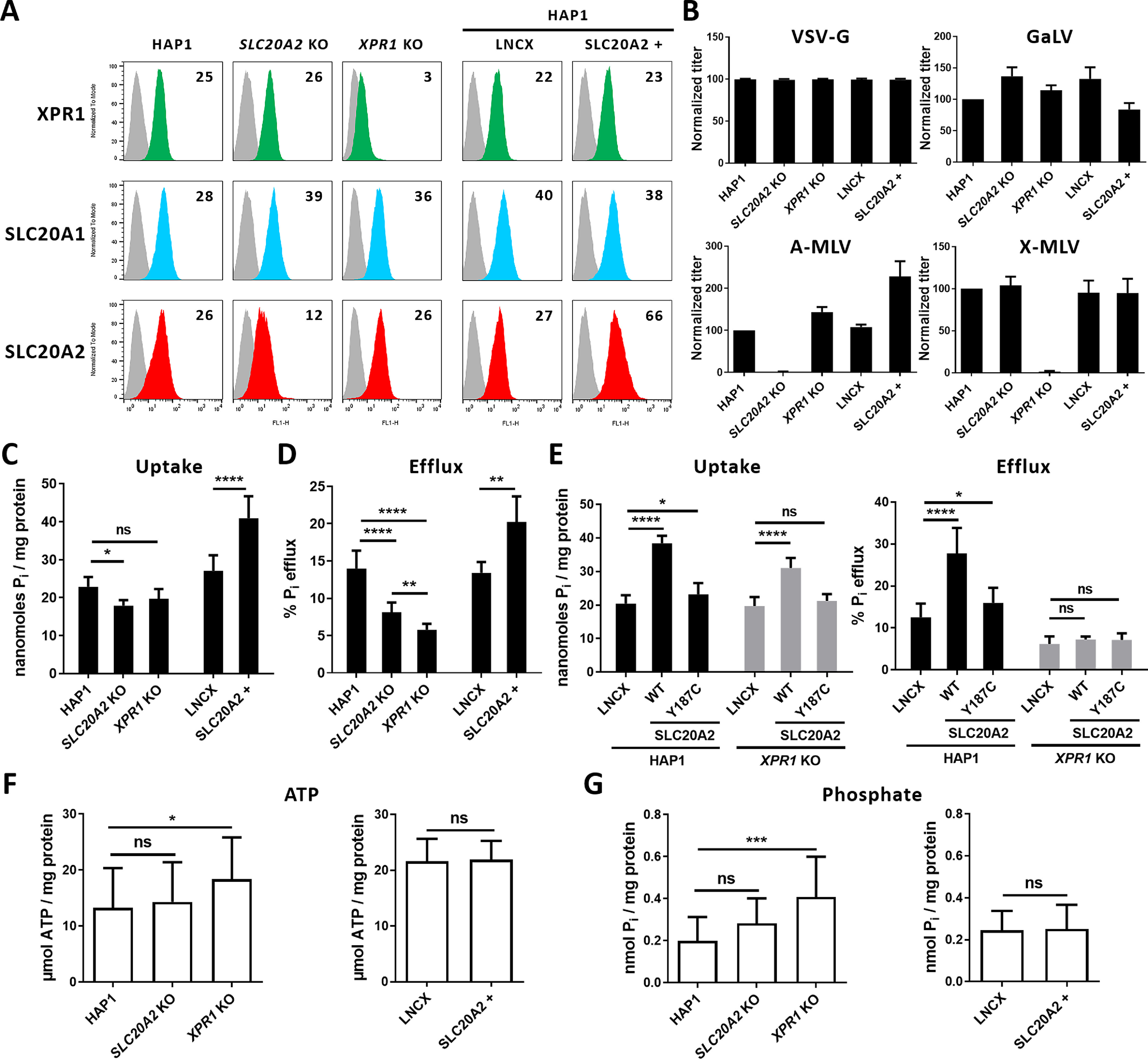
Phosphate efflux, but not uptake, controls phosphate homeostasis and ATP synthesis through XPR1. Human parental, SLC20A2 KO, and XPR1 KO HAP1 cells as well as HAP1 cells stably transduced with retroviral LNCX either empty or carrying the SLC20A2 gene (SLC20A2+) were evaluated. A, cell surface expression of XPR1 (green), SLC20A1 (blue), and SLC20A2 (red) evaluated by flow cytometry using RBD ligands (see text). Numbers indicate the specific change in mean fluorescence intensity of a representative experiment (n = 3). B, sensitivity to retroviral infection by EGFP retroviral vectors pseudotyped with either of the VSV-G protein, the SLC20A1-specific gibbon ape leukemia virus (GaLV) Env, the SLC20A2-specific A-MLV Env, or the X-MLV Env. Graphs represent average values from 3 different wells in 1 representative experiment (n = 3). C and D, uptake (C) and efflux (D) of [33P]phosphate. Data are means ± S.D. from 4 experiments. *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001, one-way ANOVA with the Holm-Sidak multiple-comparison test; for data in C, F = 76.61 (p < 0.0001); for data in D, F = 85.29 (p < 0.0001). E, uptake and efflux of [33P]phosphate in parental HAP1 and XPR1 KO cells stably expressing the LNCX empty vector or encoding either the WT SLC20A2 or the p.Y187C variant. Data are means ± S.D. from 4 experiments. *, p ≤ 0.05; ****, p ≤ 0.0001, two-way ANOVA with Tukey's multiple-comparison test; uptake: F(vector) = 27.95 (p < 0.0001); F(cells) = 212.6 (p < 0.0001); F(interaction) = 10.47 (p = 0.0001); efflux: F(vector) = 228.3 (p < 0.0001); F(cells) = 38.47 (p < 0.0001); F(interaction) = 30.87 (p < 0.0001). F and G, ATP levels (F) and phosphate levels (G) of the HAP1 cells as in A. Results are shown as means ± S.D. from 3 experiments. Left panels: *, p ≤ 0.05; ***, p ≤ 0.001, one-way ANOVA with the Holm-Sidak multiple-comparison test; for data in F, F = 4.186 (p = 0.0184); for data in G, F = 7.905 (p = 0.0012). Right panels: two-tailed Student's t tests. ns, nonsignificant; Pi, inorganic phosphate.
As expected, XPR1 KO cells showed a strong decrease in phosphate efflux and no impact on uptake, which confirmed the role of XPR1 as the main phosphate exporter (Fig. 2, C and D). Moreover, we found that overexpression of human SLC20A2 led to a significant increase in phosphate uptake, compared with parental cells (Fig. 2C), while depletion of SLC20A2 induced only a modest, although statistically significant, decrease of phosphate uptake (p < 0.05), most likely due to the compensatory activity of SLC20A1. Noticeably, the increase in phosphate uptake in SLC20A2+ cells was concomitant with an increase in phosphate efflux (Fig. 2D), although basal levels of XPR1 in these cells were unchanged (Fig. 2A). Consistent with this induced phosphate efflux, lack of SLC20A2 reduced phosphate efflux dramatically in SLC20A2 KO cells without affecting XPR1 expression at the plasma membrane. This SLC20A2-stimulated phosphate efflux depended on XPR1, since it was observed in HAP1 parental cells but not in XPR1 KO cells (Fig. 2E), despite increased phosphate uptake in both cell types. These data provided evidence that SLC20A2, described only as a sodium-dependent phosphate importer, also modulated XPR1-dependent phosphate efflux when overexpressed or depleted. Consistent with a cellular adaptation that maintains phosphate homeostasis, we found that the intracellular levels of both phosphate and ATP were similar in parental HAP1, SLC20A2+, and SLC20A2 KO cells (Fig. 2, F and G). In contrast, phosphate and ATP levels were significantly elevated in XPR1 KO cells, compared with parental cells (p < 0.001), consistent with a pivotal role for XPR1, and not SLC20A2, as a downstream effector of intracellular phosphate sensing required for phosphate homeostasis in HAP1 cells.
Inositol pyrophosphates control XPR1-mediated phosphate efflux and phosphate homeostasis
Fluctuations of extracellular phosphate were shown to directly affect phosphate efflux (1) and to dictate intracellular levels of phosphate, ATP, and PP-IPs, namely, diphosphoinositol pentakisphosphate (IP7) as two isomers, i.e., 1PP-IP5 (1-IP7) and 5PP-IP5 (5-IP7), and diphosphoinositol tetrakisphosphate (IP8) (42). Phosphate, ATP, and PP-IPs are interconnected, since cells with altered PP-IP synthesis show increases in phosphate and ATP levels (43), which is reminiscent to the situation found in XPR1 KO cells (Fig. 2, F and G) and may suggest control of phosphate and ATP concentrations by PP-IPs through XPR1-mediated phosphate export function. To test this hypothesis, we first set up an assay to measure the regulation of phosphate fluxes by extracellular phosphate. We observed that cells responded to elevated extracellular phosphate by increasing phosphate uptake and efflux in a dose-dependent manner (Fig. 3, A and B), which is consistent with the SLC20A2-stimulated phosphate efflux described above. Nevertheless, although loss of SLC20A2 led to a decrease in both fluxes, cells maintained a regulated response to extracellular phosphate. In contrast, XPR1 KO cells were insensitive to extracellular phosphate, since phosphate efflux was maintained at basal levels at 0, 1, and 10 mm phosphate (Fig. 3, B and C), but behaved as parental cells for phosphate uptake (Fig. 3A). Although intracellular phosphate and ATP were at similar levels in control and XPR1 KO HAP1 cells at 0 mm phosphate, their levels increased in the absence of XPR1 at 1 mm phosphate (Fig. 3D), suggesting an active regulating role of XPR1 only in the presence of high phosphate contents. Reintroduction of WT XPR1 in XPR1 KO cells restored the extracellular phosphate dependence of phosphate efflux (Fig. 3C) and decreased the level of cellular phosphate (Fig. 3D) and the level of ATP in a very reproducible manner (Fig. 3E). PP-IPs were shown to interact with the SPX domain of XPR1 in a delineated pocket called the lysine surface cluster (KSC) (14). Expression of a mutated form of XPR1 in the KSC of SPX (XPR1 KSC) did not allow XPR1 KO cells to respond to extracellular phosphate (Fig. 3C) and to lower phosphate and ATP levels (Fig. 3, D and E) despite its measurable cell surface expression (Fig. 3H) and retroviral receptor function (Fig. S2), suggesting that the recognition of SPX by PP-IPs is a mandatory step for the regulation of phosphate efflux and subsequent homeostasis. To assess the presence of PP-IPs in HAP1 cells, inositol polyphosphate (IP) content was analyzed by strong anion exchange (SAX)-HPLC after [3H]inositol labeling. Cells showed significant levels of I(1,3,4,5,6)P5 (referred to as 2OH-IP5), a second IP5 isomer referred to as IP5b, IP6, and IP7, while IP8 levels were under the limit of detection (Fig. 3F). The IP5b peak is likely composed of one or both of the enantiomeric isoforms d/l-I(2,3,4,5,6)P5. Levels of 2OH-IP5, IP5b, and IP7 were significantly increased in XPR1 KO HAP1 cells, compared with parental cells, while a slight increase, statistically nonsignificant, was observed for IP6 (Fig. 3F and Fig. S3). We also found that altering the phosphate concentration could significantly affect the level of IP7. Under 0 mm phosphate conditions, IP7 was reduced by more than 50% (Fig. 3G and Fig. S4), consistent with low phosphate efflux activity of XPR1. Interestingly, and unexpectedly, a smaller decrease in 2OH-IP5 was observed also under phosphate starvation conditions in both cell lines.
Figure 3.
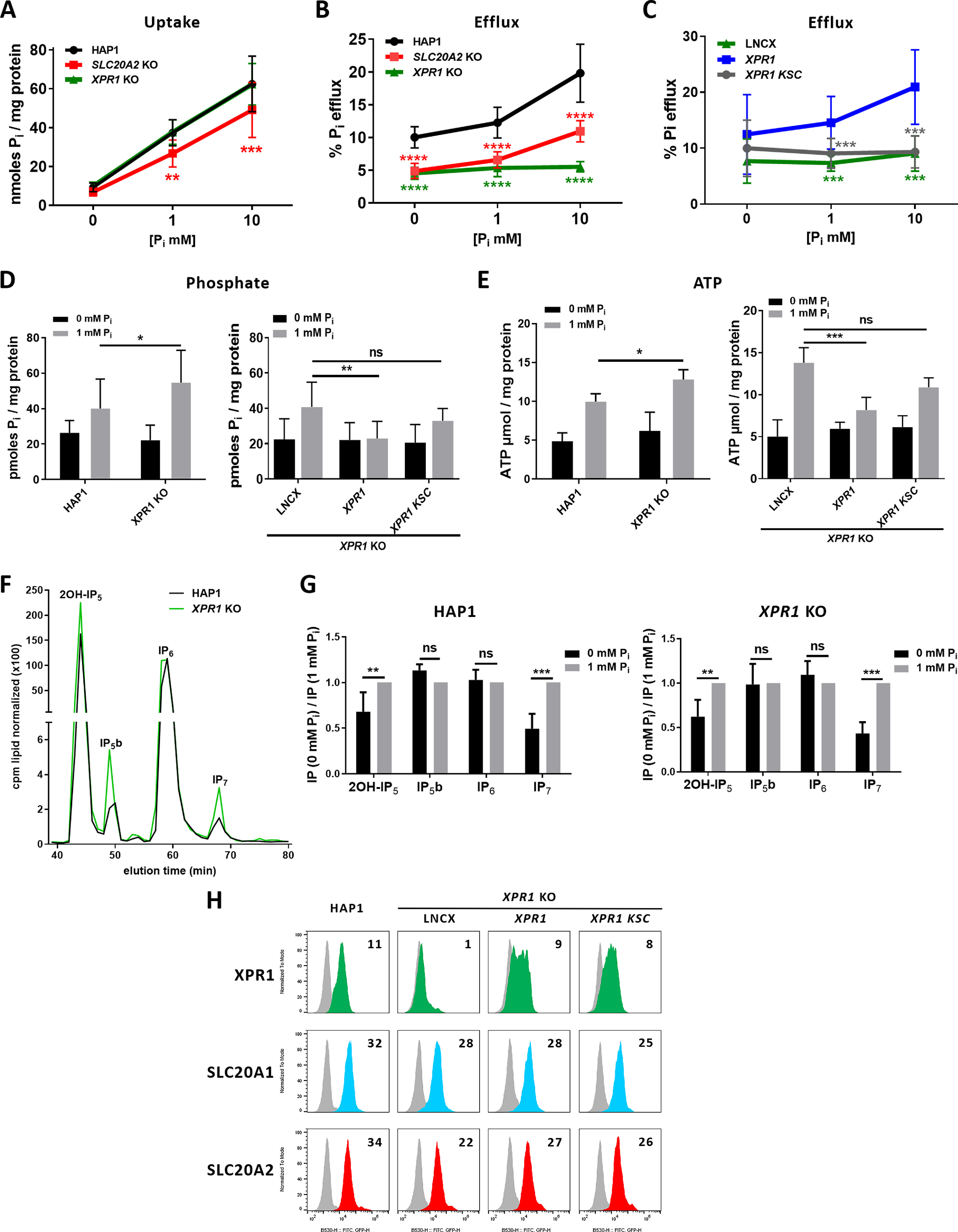
Phosphate efflux is regulated by extracellular phosphate and requires the functional SPX domain of XPR1. A and B, uptake (A) and efflux (B) assays of [33P]phosphate on parental, SLC20A2 KO, and XPR1 KO HAP1 cells at different extracellular phosphate concentrations. Data are shown as means ± S.D. from 4 experiments. **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001, two-way ANOVA with Tukey's multiple-comparison test; uptake: F([Pi]) = 12.99 (p < 0.0001); F(cells) = 296.6 (p < 0.0001); F(interaction) = 1.682 (p = 0.1601); efflux: F([Pi]) = 189.6 (p < 0.0001); F(cells) = 74.82 (p < 0.0001); F(interaction) = 16.65 (p < 0.0001). C, efflux of [33P]phosphate performed in XPR1 KO HAP1 cells stably expressing the LNCX empty vector or carrying either WT (XPR1) or KSC variant XPR1 genes at the indicated extracellular phosphate concentrations. Data are shown as means ± S.D. from 4 experiments; ***, p ≤ 0.001, two-way ANOVA with Tukey's multiple-comparison test: F([Pi]) = 44.99 (p < 0.0001); F(cells) = 6.430 (p = 0.0021); F(interaction) = 4.818 (p = 0.0011). D and E, phosphate intracellular levels (D) and ATP intracellular levels (E) in the same panel of cells as in A and C. Results are shown as means ± S.D. from 6 experiments. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001, two-way ANOVA with Tukey's multiple-comparison test; for data in D, left, F([Pi]) = 9.638 (p = 0.0058); F(cells) = 78.19 (p < 0.0001); F(interaction) = 1.378 (p = 0.2550); right, F([Pi]) = 2.466 (p < 0.0995); F(cells) = 9.987 (p = 0.0032); F(interaction) = 2.313 (p = 0.1139); for data in E, left, F([Pi]) = 0.9621 (p = 0.3340); F(cells) = 18.55 (p = 0.0001); F(interaction) = 3.037 (p = 0.0910); right, F([Pi]) = 6.408 (p = 0.0059); F(cells) = 84.44 (p < 0.0001); F(interaction) = 11.82 (p = 0.0003). No significance was found between 0 mm phosphate-treated cells in D and E. F, SAX-HPLC on parental and XPR1 KO HAP1 cells grown in 1 mm phosphate and labeled for 4 days in [3H]inositol-containing DMEM. Values of IPs are cpm normalized to total lipids. Peaks were annotated based on standard elution times. The HPLC profile is representative of an experiment performed four times. G, comparison of IP levels in WT and XPR1 KO HAP1 cells grown in 0 mm and 1 mm phosphate DMEM. Levels were normalized to 1 mm phosphate in the indicated HAP1 cells. **, p ≤ 0.01; ***, p ≤ 0.001, two-way ANOVA with Tukey's multiple-comparison test; for data in HAP1 cells, F([IPs]) =19.86 (p = 0.0002); F([Pi]) = 15.86 (p < 0.0001); F(interaction) = 15.86 (p < 0.0001); for data in XPR1 KO cells: F(IPs) = 23.17 (p < 0.0001); F([Pi]) = 11.71 (p < 0.0001); F(interaction) = 11.71 (p < 0.0001). H, cell surface detection by flow cytometry of XPR1 (green), SLC20A1 (blue), and SLC20A2 (red) in cells as in C. Numbers indicate the specific change in mean fluorescence intensity of a representative experiment (n = 3), compared with nonspecific staining with the secondary IgG (gray). ns, nonsignificant; Pi, inorganic phosphate; cpm, counts per minute.
We next asked whether the absence of PP-IPs affected phosphate fluxes and associated homeostasis. Since inositol hexakisphosphate kinases (IP6Ks) are responsible for the synthesis of 5-IP7 (the major form of IP7) and IP8, assays were performed by using either HCT116 IP6K1-2 double KO (DKO) cells that were shown to be devoid of PP-IPs (44) or a pan-IP6K inhibitor, N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine (TNP). IP6K1-2 DKO cells contained higher phosphate and ATP levels, compared with parental cells, as recently described (44), a difference also found after TNP treatment of HAP1 cells (Fig. 4, A and B). In the same lines, we found that IP6K1-2 DKO cells had reduced cell surface expression of SLC20A1 and SLC20A2 (Fig. 4F), which correlated with reduced phosphate uptake (Fig. 4C), while TNP treatment decreased uptake without affecting the expression of these transporters (Fig. 4, C and E). This suggested that phosphate uptake can be controlled at multiple levels. As expected, TNP treatment had no effect on IP6K1-2 DKO cells (Fig. S5). We further found that the absence of PP-IPs was accompanied by a dramatic decrease of phosphate efflux and a loss of regulation by extracellular phosphate (Fig. 4D) despite normal levels of cell surface XPR1. These data strongly suggested that phosphate efflux mediated by XPR1 is controlled by IPs like PP-IPs, which thus could participate in phosphate homeostasis in the investigated cells, although we cannot exclude the possibility that nonenzymatically, through scaffolding protein-protein interactions, the IP6Ks may also influence phosphate efflux. The observed decrease in phosphate efflux was not a direct consequence of the decrease in phosphate uptake, since phosphate and ATP levels remained high.
Figure 4.
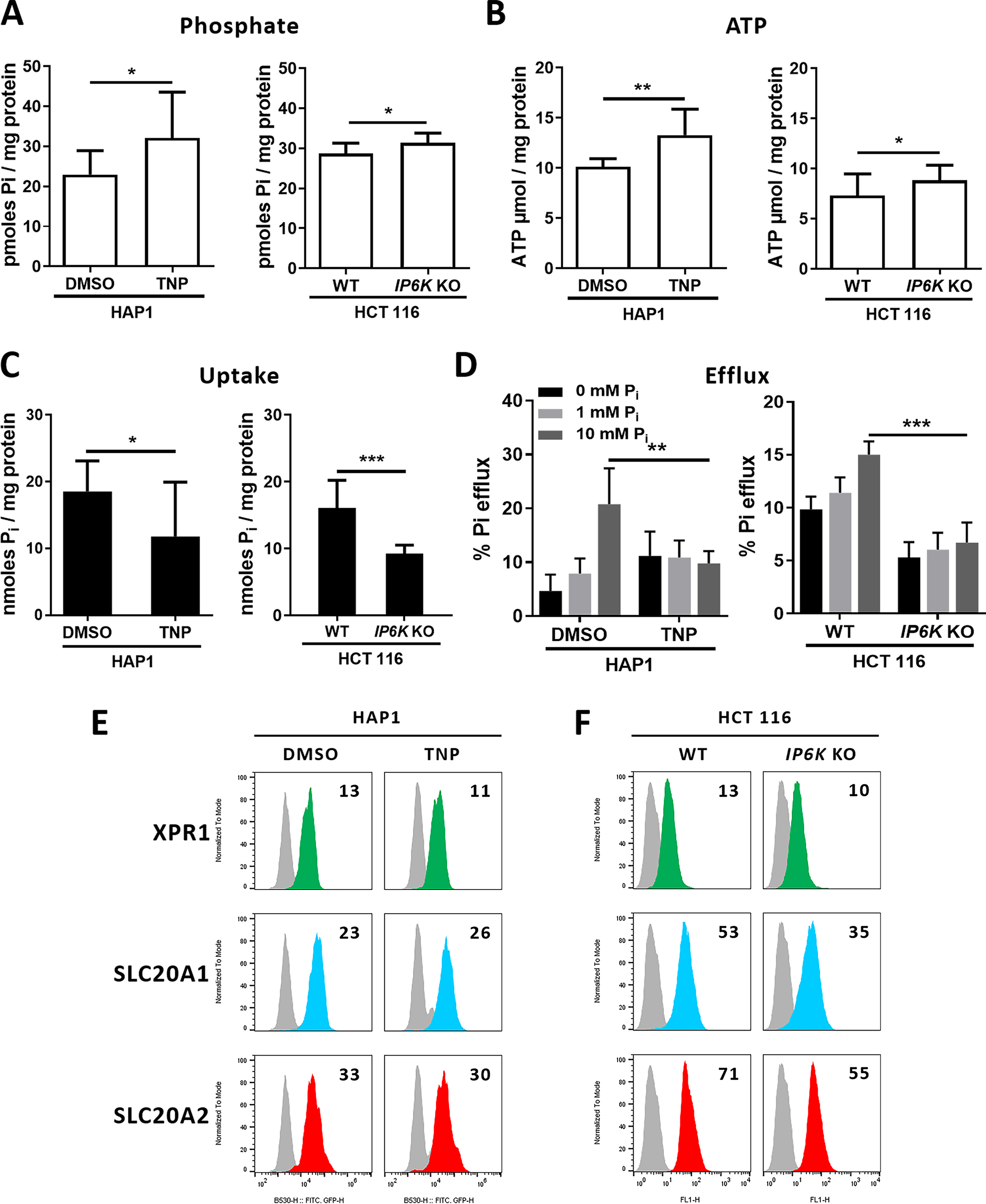
Inactivation of IP6K genes or kinase activity abolishes phosphate efflux regulation. A and B, levels of intracellular phosphate (A) and ATP (B) in HAP1 parental cells treated either with DMSO or with 10 μm TNP for 48 h and in parental or IP6K1-2 DKO HCT116 cells (IP6K KO). Data are means ± S.D. from 6 experiments. *, p ≤ 0.05; **, p ≤ 0.01, two-tailed Student's t tests. C, uptake of [33P]phosphate in HAP1 and HCT116 cells as in A. Data are means ± S.D. from 3 experiments. *, p ≤ 0.05; ***, p ≤ 0.001, two-tailed Student's t tests. D, efflux assays of [33P]phosphate using the same cells as in A with efflux media containing different concentrations of phosphate. Data are shown as means ± S.D. **, p ≤ 0.01; ***, p ≤ 0.001, two-way ANOVA with Tukey's multiple-comparison test; left: F([Pi]) = 19.70 (p < 0.0001); F(cells) = 0.2891 (p = 0.5926); F(interaction) = 27.86 (p < 0.0001); right: F([Pi]) = 385.7 (p < 0.0001); F(cells) = 38.85 (p < 0.0001); F(interaction) = 13.59 (p < 0.0001); ***, p ≤ 0.001. E and F, cell surface detection of XPR1 (green), SLC20A1 (blue), and SLC20A2 (red) in HAP1 parental cells treated with either DMSO or TNP (E) and in HCT116 parental or IP6K1-2 DKO cells (F). Numbers indicate the specific change in mean fluorescence intensity of a representative experiment (n = 3), compared with nonspecific staining with the secondary IgG (gray histograms). ns, nonsignificant; Pi, inorganic phosphate.
Discussion
Although blood phosphate homeostasis is a well-described and accepted regulation process (11), little is known about intracellular phosphate homeostasis. One key condition for such regulation is the presence of an intracellular phosphate-sensing mechanism that would respond to extracellular phosphate variations. We hypothesized that such a response would involve the modulation of phosphate fluxes between the intracellular milieu and the external environment. In this study, we provide evidence for a coordinated interplay between phosphate import and export. Using cells overexpressing or depleted of SLC20A2, we found that increases and decreases of phosphate uptake were concomitant with increases and decreases of efflux, respectively, and that this adjustment of fluxes maintained the levels of phosphate and ATP constant. More surprising was the fact that cells depleted of XPR1 showed a decrease in phosphate efflux without a compensatory adaptation of phosphate entry (Fig. 2C). The immediate consequences of this defect in flux balance were the elevation of intracellular phosphate levels and subsequent enhancement of ATP levels (Fig. 2, F and G). Thus, our results clearly pointed to a role of SLC20A2 and XPR1 in the control of phosphate homeostasis and energy metabolism.
One unanswered question was the nature of the mechanisms and factors that allowed the regulation of phosphate fluxes to occur at the exit level and not at entry. We previously reported that cells incubated in high-phosphate medium have increased phosphate efflux, while efflux was reduced in low-phosphate medium (1) (Fig. 3B). This is likely due to subsequent elevated and low concentrations of intracellular phosphate, respectively, a situation that paralleled the one we observed in the overexpressing SLC20A2+ cells and the SLC20A2 KO cells. If this holds true, phosphate export would constitute a downstream event that follows intracellular phosphate sensing, allowing a readjustment of intracellular phosphate levels in the cytoplasm. Key players of this regulatory process were likely to be PP-IPs, IP7, and IP8, which have been suggested as novel molecules of high energy involved in the regulation of phosphate homeostasis (15). PP-IP levels are considered an indicator of the energetic status of cells, as their synthesis is highly sensitive to fluctuations of intracellular ATP levels and therefore to extracellular phosphate (45, 46). The SPX domain, which is shared by several proteins involved in phosphate metabolism, including XPR1, was an obvious candidate for such a potential sensing, since its 3D crystal structure has revealed key determinants for inositol phosphate and PP-IP interactions (14). Thus, high phosphate extracellular conditions lead cells to acquire high levels of phosphate, ATP, and PP-IPs, which can be transmitted to XPR1 through the interaction of PP-IPs with the SPX domain. As suggested previously (15, 44, 46–49) and as shown here, the SPX domain can function as a sensor for IP7 and IP8 signaling molecules, and the activation of this phosphate-ATP-PP-IPs-XPR1 axis may promote the regulation of XPR1-mediated phosphate exit. Our results are consistent with this putative mechanism, as the absence of IPs, like PP-IPs, or XPR1 or the loss of interaction between PP-IPs and SPX (XPR1 KSC mutant) abolished this regulation despite elevated levels of phosphate and ATP (Fig. 5). We could not exclude the possibility that, beside PP-IPs, other IPs like 2OH-IP5 whose levels are modulated by phosphate starvation (Fig. 3G), could contribute to the regulation of phosphate homeostatic mechanisms. Interestingly, cells depleted of PP-IPs also presented high levels of ATP in yeast (50) and in human cells (43, 44, 49), as we observed in XPR1 KO cells, strongly suggesting that the communication between PP-IPs and XPR1 is an essential step for phosphate exit regulation and intracellular phosphate homeostasis. This communication may modulate the interaction of SPX with cellular partners (51), as shown in plants (47).
Figure 5.

Schematic representation of the interplay between SLC20A2 and XPR1 phosphate transporters In parental cells (WT) (left), phosphate uptake through the SLC20A2 phosphate transporter transiently leads to an increase in the concentrations of intracellular phosphate and consequently ATP. ATP levels are translated into changes in levels of IPs like PP-IPs, IP7, and IP8, through the activity of ATP-dependent IP6K, which, in turn, interact with the SPX domain of XPR1 and stimulate phosphate efflux. The immediate consequence is the return of phosphate, ATP, and PP-IPs to basal levels. In IP6K KO cells (middle), phosphate and ATP accumulate since XPR1-mediated phosphate efflux cannot be stimulated in the absence of PP-IPs. In XPR1 KO cells (right), high ATP levels still stimulate IP6K, leading to the accumulation of phosphate, ATP, PP-IPs, and IPs. Pi, inorganic phosphate.
Little is known about the mechanisms that lead SLC20A2 and XPR1 heterozygous loss of function to PFBC. PFBC is considered a primarily microvascular calcifying disorder, with calcium phosphate deposits in mural cells of basal ganglia capillaries (52). In vitro studies previously designated SLC20A1 as an important contributor to VSMC calcification experimentally induced by high doses of phosphate in culture medium (21, 22), while SLC20A2 has a more protective role (18, 53), most likely for its capacity to counterbalance SLC20A1 transport function (18, 53). The calcification process implies a transition of contractile VSMC into an osteochondrogenic phenotype (54) and the formation of SLC20A1-positive calcifying vesicles in which calcium and phosphate accumulate and precipitate (55). Extracellular phosphate sensing by mineralizing cells is an important step in this process and requires heterodimerization of SLC20A1 and SLC20A2, which in turn activates the ERK1/2 pathway and subsequent expression of mineralization inhibitor-encoded genes (18, 53). It is not known, however, if this heterodimerization-dependent phosphate sensing also controls intracellular phosphate homeostasis, although it clearly does not require phosphate transport activity (18, 53), which is quite in contrast to our defective PFBC SLC20A2 variants, which affected neither phosphate and ATP levels nor phosphate efflux. Our study unveiled an interplay between SLC20A2 and XPR1 with functional transport activities that appeared to control cellular phosphate homeostasis, although only a defect in XPR1 led to increased global phosphate concentrations. Whether loss of function of XPR1, and perhaps SLC20A2, induces elevated phosphate concentrations in mural cells of brain capillaries and subsequent calcification remains to be addressed.
High levels of extracellular phosphate are an important and essential parameter of vascular calcification. In situations of hyperphosphatemia, such as in chronic kidney disease, vascular calcification has severe clinical consequences and is associated with significant mortality and morbidity rates, including significant cerebral complications (56). Although phosphate blood and urinary levels are in the normal ranges in PFBC patients, recent studies have shown that PFBC may be associated with elevated phosphate levels in the CSF (32–35), and thus probably in the glymphatic system in the continuity of the CSF, with which brain capillaries are intimately associated (57). Epithelial cells of the choroid plexus (CP) are the main barrier where phosphate exchanges occur between CSF and blood, and the apical expression of SLC20A2 in the CP is in agreement with a putative role of SLC20A2 in transepithelial phosphate transport in the CSF-to-blood direction, as previously reported (58). As mentioned by others (32), XPR1 is expressed in the CP and may thus participate in transepithelial transport as well, although its localization at the basal side has not yet been assessed. Here, our study clearly shows that altered SLC20A2 expression impacts XPR1-mediated phosphate efflux, reinforcing the hypothesis that PFBC patients carrying heterozygous SLC20A2 loss-of-function variants may have compromised CSF phosphate reabsorption at the apical side of the polarized CP epithelium, leading to a readjustment of intracellular homeostasis and thereby reduced phosphate export to the blood capillary side. The unaltered phosphate import that we observed in XPR1 KO cells despite elevated intracellular phosphate levels is likely to be due to the dual compensatory presence of both SLC20A1 and SLC20A2, a situation that may be very different in CP epithelium, in which SLC20A1 appeared to be absent (57). Further investigations are required in XPR1+/− patients to assess whether elevated phosphate concentration in CSF is a generalized hallmark of PFBC.
Materials and Methods
Cell culture
Human haploid HAP1 cells and derivatives (SLC20A2 and XPR1 KO cells) were obtained from Horizon Discovery and were maintained in Iscove's modified Dulbecco's medium (Gibco) supplemented with 10% FBS (Sigma) and 1% antibiotics (penicillin-streptomycin). HAP1 cells expressing WT SLC20A2 or the Y187C variant were generated by transduction with MLV-based LNCX (60) retroviral vectors pseudotyped with VSV-G protein, followed by G418 selection. HEK293T, HCT116, and IP6K1-2 DKO HCT116 human cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 1% antibiotics, and nonessential amino acids. All cells were cultivated under a humid atmosphere in a 5% CO2 incubator at 37°C. When indicated, HAP1 and HCT116 cells were incubated in phosphate-free DMEM (Gibco) supplemented with 10% dialyzed FBS (Gibco).
Plasmids, viral productions, and infections
cDNAs from WT SLC20A2 or from SLC20A2 variants were generated by site-directed mutagenesis using recombinant PCR and were introduced either in the pCHIX expression vector (59) or in the pLNCX retroviral vector (60). All SLC20A2 variants were fused in-frame at their C-terminal end with two copies of the HA tag. Helper-free LNCX vectors pseudotyped with the VSV-G Env protein were produced by cotransfecting HEK293 cells with the LNCX vectors carrying WT or Y187C SLC20A2 variants, the MLV Gag-Pol expression vector pC57GPBEB (61), and the VSV-G Env expression vector pCSIG (4). Viral supernatants were harvested 48 h after transfection, filtered through 0.45-μm-pore filters, and used for human haploid cell transduction. Lentiviral CSGW vectors expressing the EGFP were produced as described previously (62). CSGW pseudotype vectors bearing Env glycoproteins from VSV, A-MLV, X-MLV, or gibbon ape leukemia virus were used to infect 1 × 104 haploid cells/well (which had been seeded the day before in a 96-well plate) for 48 h. Cells were then resuspended in 50 μl trypsin and 100 μl PBS with 2% FBS (PBA) and analyzed on a NovoCyte flow cytometer (Acea Biosciences, Inc.). Data analyses measuring the percentage of EGFP-positive infected cells were performed using FlowJo software.
ATP and phosphate assays
For measurement of intracellular ATP levels, 25 × 103 cells/well were seeded in a 96-well plate. When indicated, cells were treated with 10 μm TNP for 48 h. The following day, cells were washed with phosphate-free DMEM and incubated for 24 h in either phosphate-free DMEM or DMEM with 1 mm phosphate (NaH2PO4/Na2HPO4 mixture, pH 7.4). The assay was then performed using the ATPlite luminescence assay system (PerkinElmer), according to the manufacturer's instructions. Cells from duplicate wells of a 96-well plate were lysed with 20 μl lysis buffer (1 m Tris HCl, pH 8, 1 m MgCl2, 1% Triton X-100, 5 m NaCl, protease inhibitor mixture (EASYpack tablets; Roche)) before measurement of protein content. For phosphate assays, 2.5 × 105 cells/well were seeded in a 6-well plate. The following day, cells were placed on ice, washed three times with ice-cold buffer (20 mm Hepes, pH 7.5, 150 mm NaCl), and lysed for 15 min at 4°C in 500 μl of washing buffer with 1% Triton X-100; 50-μl samples were used to perform the assay with the malachite green phosphate assay kit (Sigma). ATP and phosphate concentration values were normalized to the protein concentration, as determined with the BCA protein assay kit (Pierce).
Monitoring of cell surface phosphate transporters
Detection of phosphate transporters at the plasma membrane was performed as described previously (1, 25, 40, 41), using soluble RBDs derived from X-MLV, A-MLV (surface unit), and koala retrovirus, fused to a mouse IgG1 Fc domain, and able to bind XPR1, SLC20A2, and SLC20A1, respectively. RBD ligands were produced and used as described previously (1). Briefly, 5 × 104 cells were detached with 1 mm EDTA in PBS, resuspended in 100 μl PBA containing the different ligands, and incubated at 37°C for 30 min, with shaking. Cells were then washed twice with cold PBA and labeled for 20 min on ice with Alexa 488-conjugated anti-mouse IgG1 antibodies (1:200; Invitrogen). Cells were then washed in PBA and analyzed on a NovoCyte flow cytometer (Acea Biosciences, Inc.) or a FACSCalibur system (BD Biosciences). Data analysis was performed using FlowJo software.
Phosphate fluxes
Phosphate uptake and efflux assays in HEK293T, HAP1 and HCT116 cells were carried out as described previously (1). Phosphate uptake was calculated as the ratio of cellular [33P]phosphate to total [33P]phosphate supplemented. The percentage of phosphate efflux was calculated as the ratio of released [33P]phosphate to total cellular [33P]phosphate. Phosphate uptake and efflux assays in PBMC were performed as described previously (25). PBMC of patients carrying the SLC20A2 Y187C and the p.A145_V205del variants were obtained from the patient peripheral venous blood. At the same time, blood samples from an unrelated control were obtained and assayed. Patients and controls provided written informed consent in the context of a study approved by the CPP Ile de France II ethics committee (RBM 02-59).
Immunoblotting
HEK293T cell extracts were separated on a 12% SDS-acrylamide gel under reducing conditions and transferred to PVDF membranes, and protein expression was detected using anti-HA (1:5000, 3F10; Roche) or anti β-actin (1:5000, A5441; Sigma) antibodies, followed by HRP-conjugated anti-mouse or anti-rat IgG1 antibodies, respectively (1:5000; Southern Biotech). Signals were visualized with the Pierce ECL Western blotting substrate (Thermo Scientific) and recorded using the ChemiDoc gel imaging system.
Patients and SLC20A2 variant nomenclature
Patients were recruited in multiple French centers. Patients or their legal representatives provided written informed consent for genetic analyses. This study was approved by the CPP Ile de France II ethics committee (RBM 02-59). This study abided by the Declaration of Helsinki principles. PFBC-associated SLC20A2 variant nomenclature was established using GenBank accession numbers NG_032161.1 and NM_006749.4 as references. G498R refers to c.1492G>A, p.(Gly498Arg); E575K refers to c.1723G>A, p.(Glu575Lys); T595M refers to c.1784C>T, p.(Thr595Met); S601L refers to c.1802C>T, p.(Ser601Leu); Y187C refers to c.560A>G, p.(Tyr187Cys); N194S refers to c.581A>G, p.(Asn194Ser); and ΔA145_V205 refers to the genomic in-frame deletion encompassing exons 4 and 5 of SLC20A2.
Analysis of inositol polyphosphates
IP analyses in control and XPR1 KO HAP1 cell lines were conducted by seeding the cells into 6-well plates in inositol-free DMEM (MP Biomedicals). myo-[3H]Inositol (PerkinElmer) at 5 μCi/ml was added, and the cells were incubated for 4 days. To harvest the IPs, cells were washed once in cold PBS before the addition of cold 1 m perchloric acid. Samples were collected into microcentrifuge tubes after a 10-min incubation on ice. Neutralized extracts were subjected to SAX-HPLC as described previously (63). Results were normalized to radioactivity in the lipid fraction, obtained by incubating the postextraction cells overnight in 0.1 m NaOH, 0.1% SDS. Inositol phosphate peaks were identified using the relevant standards, as described previously (44).
Statistics
Statistical analyses were performed using GraphPad Prism 6 software using t test one-way analysis of variance (ANOVA) or two-way ANOVA with Tukey post hoc test when appropriate. The data were expressed as the mean ± S.D. The results were considered statistically significant at p values of <0.05, <0.01, or <0.001.
Data availability
All data are contained within the manuscript.
Supplementary Material
Acknowledgments
We are grateful to the patients and their families and to collaborators who sent blood samples and medical charts. We acknowledge the MRI facility, a member of the national infrastructure France-BioImaging supported by the French National Research Agency (Grant ANR-10-INBS-04).
This article contains supporting information.
Author contributions—U. L.-S., S. T., M. S., and J.-L. B. conceptualization; U. L.-S., S. T., G. N., M. S. W., V. C., A. S., M. S., and J.-L. B. formal analysis; U. L.-S., S. T., and M. S. W. investigation; U. L.-S., S. T., G. N., M. S., and J.-L. B. writing-original draft; U. L.-S., S. T., G. N., V. C., A. S., M. S., and J.-L. B. writing-review and editing; G. N., M. S., and J.-L. B. supervision; G. N., M. S., and J.-L. B. funding acquisition; G. N., S. J., and X. A. patient selection and supervision of follow-up and clinical patient collection; G. N. supervision of clinical process.
Funding and additional information—U. L.-S. was supported successively by the Labex EpiGenMed (Grant ANR-10-LABX-12-01), Consejo Nacional de Ciencia y Tecnología (CONACYT Grant 243449/440597), the Fondation pour la Recherche Médicale (FRM) (Grant FDT20170436884), and ANR (CALCIPHOS Grant ANR-17-CE14-0008-01); S. T. was supported by ANR (CALCIPHOS Grant ANR-17-CE14-0008-01). This work was supported by grants from FRM grant DBI201312285579 (to M. S.) and NIH grant DK32094 (to M. S.), from ANR (CALCIPHOS Grant ANR-17-CE14-0008 to J.-L. B., G. N., and M. S.), and from Labex GR-Ex (Grant ANR-11-LABX-0051). This study was co-supported by the European Union and Région Normandie, more specifically through the Recherche Innovation Normandie (RIN 2018). Europe gets involved in Normandie with the European Regional Development Fund (ERDF). G. N., M. S., and J.-L. B. are supported by Inserm. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—J.-L. B. and M. S. are inventors on patents describing the use of RBD ligands; M. S. is the co-founder of METAFORA-biosystems, a start-up company that focuses on metabolite transporters under physiological and pathological conditions.
- SLC
- solute carrier
- PFBC
- primary familial brain calcification
- DMEM
- Dulbecco's modified Eagle's medium
- ANOVA
- analysis of variance
- CSF
- cerebrospinal fluid
- CP
- choroid plexus
- EGFP
- enhanced green fluorescent protein
- KSC
- lysine surface cluster
- PBMC
- peripheral blood mononuclear cells
- XPR1
- xenotropic and polytropic receptor 1
- SAX
- strong anion exchange
- DKO
- double knockout
- PDGF
- platelet-derived growth factor receptor β
- PDGFRβ
- platelet-derived growth factor receptor β
- RBD
- receptor-binding domain
- VSV-G
- vesicular stomatitis virus G
- X-MLV
- xenotropic murine leukemia virus
- A-MLV
- amphotropic murine leukemia virus
- VSMC
- vascular smooth muscle cells
- TNP
- N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine
- IP
- inositol polyphosphate
- IP6K
- inositol hexakisphosphate kinase
- IP6
- inositol hexakisphosphate
- IP7
- diphosphoinositol pentakisphosphate
- IP8
- bisdiphosphoinositol tetrakisphosphate
- PP-IP
- inositol pyrophosphate.
References
- 1. Giovannini D., Touhami J., Charnet P., Sitbon M., and Battini J. L. (2013) Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 3, 1866–1873 10.1016/j.celrep.2013.05.035 [DOI] [PubMed] [Google Scholar]
- 2. Kavanaugh M. P., Miller D. G., Zhang W., Law W., Kozak S. L., Kabat D., and Miller A. D. (1994) Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc. Natl. Acad. Sci. U.S.A. 91, 7071–7075 10.1073/pnas.91.15.7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olah Z., Lehel C., Anderson W. B., Eiden M. V., and Wilson C. A. (1994) The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J. Biol. Chem. 269, 25426–25431 [PubMed] [Google Scholar]
- 4. Battini J. L., Rasko J. E., and Miller A. D. (1999) A human cell-surface receptor for xenotropic and polytropic murine leukemia viruses: possible role in G protein-coupled signal transduction. Proc Natl Acad Sci U.S.A. 96, 1385–1390 10.1073/pnas.96.4.1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller D. G., Edwards R. H., and Miller A. D. (1994) Cloning of the cellular receptor for amphotropic murine retroviruses reveals homology to that for gibbon ape leukemia virus. Proc Natl Acad Sci U.S.A. 91, 78–82 10.1073/pnas.91.1.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Hara B., Johann S. V., Klinger H. P., Blair D. G., Rubinson H., Dunn K. J., Sass P., Vitek S. M., and Robins T. (1990) Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ. 1, 119–127 [PubMed] [Google Scholar]
- 7. Tailor C. S., Nouri A., Lee C. G., Kozak C., and Kabat D. (1999) Cloning and characterization of a cell surface receptor for xenotropic and polytropic murine leukemia viruses. Proc Natl Acad Sci U.S.A. 96, 927–932 10.1073/pnas.96.3.927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Y. L., Guo L., Xu S., Holland C. A., Kitamura T., Hunter K., and Cunningham J. M. (1999) Receptors for polytropic and xenotropic mouse leukaemia viruses encoded by a single gene at Rmc1. Nat. Genet. 21, 216–219 10.1038/6005 [DOI] [PubMed] [Google Scholar]
- 9. Hilfiker H., Hattenhauer O., Traebert M., Forster I., Murer H., and Biber J. (1998) Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc Natl Acad Sci U.S.A. 95, 14564–14569 10.1073/pnas.95.24.14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu H., Bai L., Collins J. F., and Ghishan F. K. (1999) Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics 62, 281–284 10.1006/geno.1999.6009 [DOI] [PubMed] [Google Scholar]
- 11. Prié D., and Friedlander G. (2010) Genetic disorders of renal phosphate transport. N. Engl. J. Med. 362, 2399–2409 10.1056/NEJMra0904186 [DOI] [PubMed] [Google Scholar]
- 12. Kavanaugh M. P., and Kabat D. (1996) Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int. 49, 959–963 10.1038/ki.1996.135 [DOI] [PubMed] [Google Scholar]
- 13. Collins J. F., Bai L., and Ghishan F. K. (2004) The SLC20 family of proteins: dual functions as sodium-phosphate cotransporters and viral receptors. Pfluegers Arch. 447, 647–652 10.1007/s00424-003-1088-x [DOI] [PubMed] [Google Scholar]
- 14. Wild R., Gerasimaite R., Jung J. Y., Truffault V., Pavlovic I., Schmidt A., Saiardi A., Jessen H. J., Poirier Y., Hothorn M., and Mayer A. (2016) Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 352, 986–990 10.1126/science.aad9858 [DOI] [PubMed] [Google Scholar]
- 15. Azevedo C., and Saiardi A. (2017) Eukaryotic phosphate homeostasis: the inositol pyrophosphate perspective. Trends Biochem. Sci. 42, 219–231 10.1016/j.tibs.2016.10.008 [DOI] [PubMed] [Google Scholar]
- 16. Wege S., and Poirier Y. (2014) Expression of the mammalian xenotropic polytropic virus receptor 1 (XPR1) in tobacco leaves leads to phosphate export. FEBS Lett. 588, 482–489 10.1016/j.febslet.2013.12.013 [DOI] [PubMed] [Google Scholar]
- 17. Khoshniat S., Bourgine A., Julien M., Weiss P., Guicheux J., and Beck L. (2011) The emergence of phosphate as a specific signaling molecule in bone and other cell types in mammals. Cell. Mol. Life Sci. 68, 205–218 10.1007/s00018-010-0527-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bon N., Couasnay G., Bourgine A., Sourice S., Beck-Cormier S., Guicheux J., and Beck L. (2018) Phosphate (Pi)-regulated heterodimerization of the high-affinity sodium-dependent Pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular Pi sensing independently of Pi uptake. J. Biol. Chem. 293, 2102–2114 10.1074/jbc.M117.807339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beck-Cormier S., Lelliott C. J., Logan J. G., Lafont D. T., Merametdjian L., Leitch V. D., Butterfield N. C., Protheroe H. J., Croucher P. I., Baldock P. A., Gaultier-Lintia A., Maugars Y., Nicolas G., Banse C., Normant S., et al. (2019) Slc20a2, encoding the phosphate transporter PiT2, is an important genetic determinant of bone quality and strength. J. Bone Miner. Res. 34, 1101–1114 10.1002/jbmr.3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki A., Ammann P., Nishiwaki-Yasuda K., Sekiguchi S., Asano S., Nagao S., Kaneko R., Hirabayashi M., Oiso Y., Itoh M., and Caverzasio J. (2010) Effects of transgenic Pit-1 overexpression on calcium phosphate and bone metabolism. J. Bone Miner. Metab. 28, 139–148 10.1007/s00774-009-0121-3 [DOI] [PubMed] [Google Scholar]
- 21. Li X., Yang H. Y., and Giachelli C. M. (2006) Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 98, 905–912 10.1161/01.RES.0000216409.20863.e7 [DOI] [PubMed] [Google Scholar]
- 22. Villa-Bellosta R., Levi M., and Sorribas V. (2009) Vascular smooth muscle cell calcification and SLC20 inorganic phosphate transporters: effects of PDGF, TNF-α, and Pi. Pfluegers Arch 458, 1151–1161 10.1007/s00424-009-0688-5 [DOI] [PubMed] [Google Scholar]
- 23. Ramos E. M., Oliveira J., Sobrido M. J., and Coppola G. (1993) Primary familial brain calcification. in GeneReviews (Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., and Amemiya A., eds.), University of Washington, Seattle, WA. [PubMed] [Google Scholar]
- 24. Nicolas G., Charbonnier C., de Lemos R. R., Richard A. C., Guillin O., Wallon D., Legati A., Geschwind D., Coppola G., Frebourg T., Campion D., de Oliveira J. R., and Hannequin D. (2015) Brain calcification process and phenotypes according to age and sex: lessons from SLC20A2, PDGFB, and PDGFRB mutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 168, 586–594 10.1002/ajmg.b.32336 [DOI] [PubMed] [Google Scholar]
- 25. Legati A., Giovannini D., Nicolas G., Lopez-Sanchez U., Quintans B., Oliveira J. R., Sears R. L., Ramos E. M., Spiteri E., Sobrido M. J., Carracedo A., Castro-Fernandez C., Cubizolle S., Fogel B. L., Goizet C., et al. (2015) Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 47, 579–581 10.1038/ng.3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang C., Li Y., Shi L., Ren J., Patti M., Wang T., de Oliveira J. R., Sobrido M. J., Quintans B., Baquero M., Cui X., Zhang X. Y., Wang L., Xu H., Wang J., et al. (2012) Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 44, 254–256 10.1038/ng.1077 [DOI] [PubMed] [Google Scholar]
- 27. Nicolas G., Pottier C., Maltete D., Coutant S., Rovelet-Lecrux A., Legallic S., Rousseau S., Vaschalde Y., Guyant-Marechal L., Augustin J., Martinaud O., Defebvre L., Krystkowiak P., Pariente J., Clanet M., et al. (2013) Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80, 181–187 10.1212/WNL.0b013e31827ccf34 [DOI] [PubMed] [Google Scholar]
- 28. Keller A., Westenberger A., Sobrido M. J., Garcia-Murias M., Domingo A., Sears R. L., Lemos R. R., Ordonez-Ugalde A., Nicolas G., da Cunha J. E., Rushing E. J., Hugelshofer M., Wurnig M. C., Kaech A., Reimann R., et al. (2013) Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet. 45, 1077–1082 10.1038/ng.2723 [DOI] [PubMed] [Google Scholar]
- 29. Cen Z., Chen Y., Chen S., Wang H., Yang D., Zhang H., Wu H., Wang L., Tang S., Ye J., Shen J., Wang H., Fu F., Chen X., Xie F., et al. (2020) Biallelic loss-of-function mutations in JAM2 cause primary familial brain calcification. Brain 143, 491–502 10.1093/brain/awz392 [DOI] [PubMed] [Google Scholar]
- 30. Grangeon L., Wallon D., Charbonnier C., Quenez O., Richard A. C., Rousseau S., Budowski C., Lebouvier T., Corbille A. G., Vidailhet M., Meneret A., Roze E., Anheim M., Tranchant C., Favrole P., et al. (2019) Biallelic MYORG mutation carriers exhibit primary brain calcification with a distinct phenotype. Brain 142, 1573–1586 10.1093/brain/awz095 [DOI] [PubMed] [Google Scholar]
- 31. Yao X. P., Cheng X., Wang C., Zhao M., Guo X. X., Su H. Z., Lai L. L., Zou X. H., Chen X. J., Zhao Y., Dong E. L., Lu Y. Q., Wu S., Li X., Fan G., et al. (2018) Biallelic mutations in MYORG cause autosomal recessive primary familial brain calcification. Neuron 98, 1116–1123.e1115 10.1016/j.neuron.2018.05.037 [DOI] [PubMed] [Google Scholar]
- 32. Jensen N., Autzen J. K., and Pedersen L. (2016) Slc20a2 is critical for maintaining a physiologic inorganic phosphate level in cerebrospinal fluid. Neurogenetics 17, 125–130 10.1007/s10048-015-0469-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wallingford M. C., Chia J., Leaf E. M., Borgeia S., Chavkin N. W., Sawangmake C., Marro K., Cox T. C., Speer M. Y., and Giachelli C. M. (2016) SLC20A2 deficiency in mice leads to elevated phosphate levels in cerebrospinal fluid and glymphatic pathway-associated arteriolar calcification, and recapitulates human idiopathic basal ganglia calcification. Brain Pathol 27, 64–76 10.1111/bpa.12362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hozumi I., Kurita H., Ozawa K., Furuta N., Inden M., Sekine S. I., Yamada M., Hayashi Y., Kimura A., Inuzuka T., and Seishima M. (2018) Inorganic phosphorus (Pi) in CSF is a biomarker for SLC20A2-associated idiopathic basal ganglia calcification (IBGC1). J. Neurol. Sci. 388, 150–154 10.1016/j.jns.2018.03.014 [DOI] [PubMed] [Google Scholar]
- 35. Paucar M., Almqvist H., Jelic V., Hagman G., Jorneskog G., Holmin S., Bjorkhem I., and Svenningsson P. (2017) A SLC20A2 gene mutation carrier displaying ataxia and increased levels of cerebrospinal fluid phosphate. J. Neurol. Sci. 375, 245–247 10.1016/j.jns.2017.02.007 [DOI] [PubMed] [Google Scholar]
- 36. Ramos E. M., Carecchio M., Lemos R., Ferreira J., Legati A., Sears R. L., Hsu S. C., Panteghini C., Magistrelli L., Salsano E., Esposito S., Taroni F., Richard A. C., Tranchant C., Anheim M., et al. (2018) Primary brain calcification: an international study reporting novel variants and associated phenotypes. Eur. J. Hum. Genet. 26, 1462–1477 10.1038/s41431-018-0185-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Larsen F. T., Jensen N., Autzen J. K., Kongsfelt I. B., and Pedersen L. (2017) Primary brain calcification causal PiT2 transport-knockout variants can exert dominant negative effects on wild-type PiT2 transport function in mammalian cells. J. Mol. Neurosci. 61, 215–220 10.1007/s12031-016-0868-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. David S., Ferreira J., Quenez O., Rovelet-Lecrux A., Richard A. C., Verin M., Jurici S., Le Ber I., Boland A., Deleuze J. F., Frebourg T., Mendes de Oliveira J. R., Hannequin D., Campion D., and Nicolas G. (2016) Identification of partial SLC20A2 deletions in primary brain calcification using whole-exome sequencing. Eur. J. Hum. Genet. 24, 1630–1634 10.1038/ejhg.2016.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicolas G., Pottier C., Charbonnier C., Guyant-Marechal L., Le Ber I., Pariente J., Labauge P., Ayrignac X., Defebvre L., Maltete D., Martinaud O., Lefaucheur R., Guillin O., Wallon D., Chaumette B., et al. (2013) Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 136, 3395–3407 10.1093/brain/awt255 [DOI] [PubMed] [Google Scholar]
- 40. Anheim M., Lopez-Sanchez U., Giovannini D., Richard A. C., Touhami J., N'Guyen L., Rudolf G., Thibault-Stoll A., Frebourg T., Hannequin D., Campion D., Battini J. L., Sitbon M., and Nicolas G. (2016) XPR1 mutations are a rare cause of primary familial brain calcification. J. Neurol. 263, 1559–1564 10.1007/s00415-016-8166-4 [DOI] [PubMed] [Google Scholar]
- 41. Lopez-Sanchez U., Nicolas G., Richard A. C., Maltete D., Charif M., Ayrignac X., Goizet C., Touhami J., Labesse G., Battini J. L., and Sitbon M. (2019) Characterization of XPR1/SLC53A1 variants located outside of the SPX domain in patients with primary familial brain calcification. Sci. Rep. 9, 6776 10.1038/s41598-019-43255-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilson M. S., Livermore T. M., and Saiardi A. (2013) Inositol pyrophosphates: between signalling and metabolism. Biochem. J. 452, 369–379 10.1042/BJ20130118 [DOI] [PubMed] [Google Scholar]
- 43. Gu C., Nguyen H. N., Ganini D., Chen Z., Jessen H. J., Gu Z., Wang H., and Shears S. B. (2017) KO of 5-InsP7 kinase activity transforms the HCT116 colon cancer cell line into a hypermetabolic, growth-inhibited phenotype. Proc Natl Acad Sci U.S.A. 114, 11968–11973 10.1073/pnas.1702370114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilson M. S., Jessen H. J., and Saiardi A. (2019) The inositol hexakisphosphate kinases IP6K1 and -2 regulate human cellular phosphate homeostasis, including XPR1-mediated phosphate export. J. Biol. Chem. 294, 11597–11608 10.1074/jbc.RA119.007848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bennett M., Onnebo S. M., Azevedo C., and Saiardi A. (2006) Inositol pyrophosphates: metabolism and signaling. Cell. Mol. Life Sci. 63, 552–564 10.1007/s00018-005-5446-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gu C., Nguyen H. N., Hofer A., Jessen H. J., Dai X., Wang H., and Shears S. B. (2017) The significance of the bifunctional kinase/phosphatase activities of diphosphoinositol pentakisphosphate kinases (PPIP5Ks) for coupling inositol pyrophosphate cell signaling to cellular phosphate homeostasis. J. Biol. Chem. 292, 4544–4555 10.1074/jbc.M116.765743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dong J., Ma G., Sui L., Wei M., Satheesh V., Zhang R., Ge S., Li J., Zhang T. E., Wittwer C., Jessen H. J., Zhang H., An G. Y., Chao D. Y., Liu D., et al. (2019) Inositol pyrophosphate InsP8 acts as an intracellular phosphate signal in arabidopsis. Mol. Plant 12, 1463–1473 10.1016/j.molp.2019.08.002 [DOI] [PubMed] [Google Scholar]
- 48. Jung J. Y., Ried M. K., Hothorn M., and Poirier Y. (2018) Control of plant phosphate homeostasis by inositol pyrophosphates and the SPX domain. Curr. Opin. Biotechnol. 49, 156–162 10.1016/j.copbio.2017.08.012 [DOI] [PubMed] [Google Scholar]
- 49. Li X., Gu C., Hostachy S., Sahu S., Wittwer C., Jessen H. J., Fiedler D., Wang H., and Shears S. B. (2020) Control of XPR1-dependent cellular phosphate efflux by InsP8 is an exemplar for functionally-exclusive inositol pyrophosphate signaling. Proc. Natl. Acad. Sci. U.S.A. 117, 3568–3574 10.1073/pnas.1908830117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Szijgyarto Z., Garedew A., Azevedo C., and Saiardi A. (2011) Influence of inositol pyrophosphates on cellular energy dynamics. Science 334, 802–805 10.1126/science.1211908 [DOI] [PubMed] [Google Scholar]
- 51. Vaughan A. E., Mendoza R., Aranda R., Battini J. L., and Miller A. D. (2012) Xpr1 is an atypical G-protein-coupled receptor that mediates xenotropic and polytropic murine retrovirus neurotoxicity. J. Virol. 86, 1661–1669 10.1128/JVI.06073-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miklossy J., Mackenzie I. R., Dorovini-Zis K., Calne D. B., Wszolek Z. K., Klegeris A., and McGeer P. L. (2005) Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol. 109, 643–653 10.1007/s00401-005-1007-7 [DOI] [PubMed] [Google Scholar]
- 53. Crouthamel M. H., Lau W. L., Leaf E. M., Chavkin N. W., Wallingford M. C., Peterson D. F., Li X., Liu Y., Chin M. T., Levi M., and Giachelli C. M. (2013) Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: redundant roles for PiT-1 and PiT-2. Arterioscler. Thromb. Vasc. Biol. 33, 2625–2632 10.1161/ATVBAHA.113.302249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Durham A. L., Speer M. Y., Scatena M., Giachelli C. M., and Shanahan C. M. (2018) Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 114, 590–600 10.1093/cvr/cvy010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reynolds J. L., Joannides A. J., Skepper J. N., McNair R., Schurgers L. J., Proudfoot D., Jahnen-Dechent W., Weissberg P. L., and Shanahan C. M. (2004) Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 15, 2857–2867 10.1097/01.ASN.0000141960.01035.28 [DOI] [PubMed] [Google Scholar]
- 56. Bugnicourt J. M., Godefroy O., Chillon J. M., Choukroun G., and Massy Z. A. (2013) Cognitive disorders and dementia in CKD: the neglected kidney-brain axis. J. Am. Soc. Nephrol. 24, 353–363 10.1681/ASN.2012050536 [DOI] [PubMed] [Google Scholar]
- 57. Jessen N. A., Munk A. S., Lundgaard I., and Nedergaard M. (2015) The glymphatic system: a beginner's guide. Neurochem. Res. 40, 2583–2599 10.1007/s11064-015-1581-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guerreiro P. M., Bataille A. M., Parker S. L., and Renfro J. L. (2014) Active removal of inorganic phosphate from cerebrospinal fluid by the choroid plexus. Am. J. Physiol. Renal Physiol. 306, F1275–F1284 10.1152/ajprenal.00458.2013 [DOI] [PubMed] [Google Scholar]
- 59. Manel N., Kim F. J., Kinet S., Taylor N., Sitbon M., and Battini J. L. (2003) The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 115, 449–459 10.1016/S0092-8674(03)00881-X [DOI] [PubMed] [Google Scholar]
- 60. Miller A. D., and Rosman G. J. (1989) Improved retroviral vectors for gene transfer and expression. BioTechniques 7, 980–990 [PMC free article] [PubMed] [Google Scholar]
- 61. Lassaux A., Sitbon M., and Battini J. L. (2005) Residues in the murine leukemia virus capsid that differentially govern resistance to mouse Fv1 and human Ref1 restrictions. J. Virol. 79, 6560–6564 10.1128/JVI.79.10.6560-6564.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mamede J. I., Damond F., Bernardo A., Matheron S., Descamps D., Battini J. L., Sitbon M., and Courgnaud V. (2017) Cyclophilins and nucleoporins are required for infection mediated by capsids from circulating HIV-2 primary isolates. Sci. Rep. 7, 45214 10.1038/srep45214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Azevedo C., and Saiardi A. (2006) Extraction and analysis of soluble inositol polyphosphates from yeast. Nat. Protoc. 1, 2416–2422 10.1038/nprot.2006.337 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.