

Abstract
Natural killer (NK) cells are key innate immunity effectors that combat viral infections and control several cancer types. For their immune function, human NK cells rely largely on five different cytotoxic proteases, called granzymes (A/B/H/K/M). Granzyme B (GrB) initiates at least three distinct cell death pathways, but key aspects of its function remain unexplored because selective probes that detect its activity are currently lacking. In this study, we used a set of unnatural amino acids to fully map the substrate preferences of GrB, demonstrating previously unknown GrB substrate preferences. We then used these preferences to design substrate-based inhibitors and a GrB-activatable activity-based fluorogenic probe. We show that our GrB probes do not significantly react with caspases, making them ideal for in-depth analyses of GrB localization and function in cells. Using our quenched fluorescence substrate, we observed GrB within the cytotoxic granules of human YT cells. When used as cytotoxic effectors, YT cells loaded with GrB attacked MDA-MB-231 target cells, and active GrB influenced its target cell-killing efficiency. In summary, we have developed a set of molecular tools for investigating GrB function in NK cells and demonstrate noninvasive visual detection of GrB with an enzyme-activated fluorescent substrate.
Keywords: amino acid, granzyme B, serine protease, natural killer cells (NK cells), activity-based probe, innate immune system, quenched fluorescence assay, substrate specificity, peptide, protease
In the last two decades, significant advances in the understanding of natural killer (NK) cells have been made (1). These cytotoxic lymphocytes are key effectors of innate immunity and are involved in viral infection responses as well as controlling several types of tumors. The activation of these cells is initiated by major compatibility complex (MHC) class I protein loss in compromised cells (1). NK cell activation changes the balance between the activating and inhibiting receptors on cell surfaces. Activated cells rapidly and quickly secrete cytokines, tumor necrosis factor α and γ interferon, leading to subsequent stimulation of the immune system. Reciprocal interactions with dendritic cells, macrophage T cells, and endothelial cells also enhance the immune system response. To prevent autoimmune damage, the NK cell inhibitory receptors recognize MHC class I proteins and protect healthy host cells (2).
Among the various weapons of NK cells and cytotoxic T lymphocytes (CTLs), the most important, located in the cytotoxic granules, are perforin and granzymes (granule-associated enzymes, or Grs). Grs are the family of homologous serine proteases and the five different human granzymes (A/B/H/K/M); the most studied and abundant are granzymes A and B (3, 4). These enzymes are not only stored within the cytotoxic granules of immune killer cells but also detected in primary breast carcinoma and in chondrocytes of articular cartilage (5). Upon the activation of NK cells and binding to the target cell, the granule membrane fuses with the plasma membrane of the NK cell, and perforins (proteins that form pores in cell membranes) and granzymes are released from the cytotoxic effector cell into the intermembrane space (6, 7). The mechanism of GrB entering the target cell through perforin-formed pores in the plasma membrane is still unclear (8). GrB is transported into the target cell to carry out its effect. Within the cell, GrB initiates at least three distinct pathways of programmed cell death, namely, 1) the activation of caspase 3, which triggers apoptosis, 2) GrB caspase-3 substrate hydrolysis with the inhibitor of caspase-activated DNase (ICAD) BID, or 3) the direct hydrolysis of lamin B.
The detection of individual granzymes with antibody-related techniques is challenging due to their structural similarities; for example, GrB antibody cross-reacts with GrA and GrH (3). For this reason, the detailed analysis of individual Grs is challenging. To overcome this obstacle, chemical approaches for developing enzymatic inhibitors and activity-based probes as efficient tools for granzyme exploration have been attempted. However, one problem is that the substrate specificity of GrB is similar to that of some caspases (caspase-8) (9), cleaving the same substrates at the same cleavage site (synthetic, IEPD tetrapeptide [10]; natural, caspase-3 or BID [11]); therefore, the reliable detection of individual Grs with either chemical or antibody-related techniques remains challenging (10, 12).
To date, only a few groups have succeeded in making functional activity-based probes for granzymes, but the major issues in these studies are the lack of selectivity of these probes toward individual enzymes and the low kinetic parameters (10, 13). For example, an activity-based probe for GrB developed using combinatorial chemistry resulted in an inactivation rate constant for the target activity-based probes (GrB) equal to kobs/I = 460 m−1 s−1 (13).
Here, we design and characterize a set of selective chemical probes, including a quenched fluorescence substrate for GrB imaging in live cells. The development of new chemical tools for investigating Grs allows the examination of the unexplored functions of these proteases and their localization.
Results
Search for the optimal chain length for the substrate peptide
Our objective was to develop selective and potent substrates and probes for human granzyme B (GrB). Although the available GrB substrates are tetrapeptides, it was previously reported that lengthening the peptide substrate improves the substrate hydrolysis rate for some proteases (14). To define the optimal peptide length for which GrB is most active, six fluorescent substrates were designed based on literature data. The S1 binding pocket of GrB has an unusual preference for aspartic acid (10, 15); therefore, we incorporated Asp at P1, and we elongated the peptide up to six amino acids. The investigated peptides were Ac-AAIEPD-ACC, Ac-AIEPD-ACC, Ac-IEPD-ACC, Ac-EPD-ACC, Ac-PD-ACC, and Ac-D-ACC. We measured the activity of GrB against each of the new substrates (at equal concentrations) and observed that neither the tripeptide, the dipeptide, nor the single-amino-acid-based substrates were hydrolyzed. The longer tetra-, penta-, and hexapeptides were cleaved by GrB. The pentapeptide and hexapeptides were more efficiently cleaved by GrB than was the classic tetrapeptide substrate (Fig. 1A). Because there was no significant difference in the efficiency of the cleavage of the penta- and hexapeptides, we selected the pentapeptides as the most appropriate chain length for activity-based probes and substrates.
Figure 1.
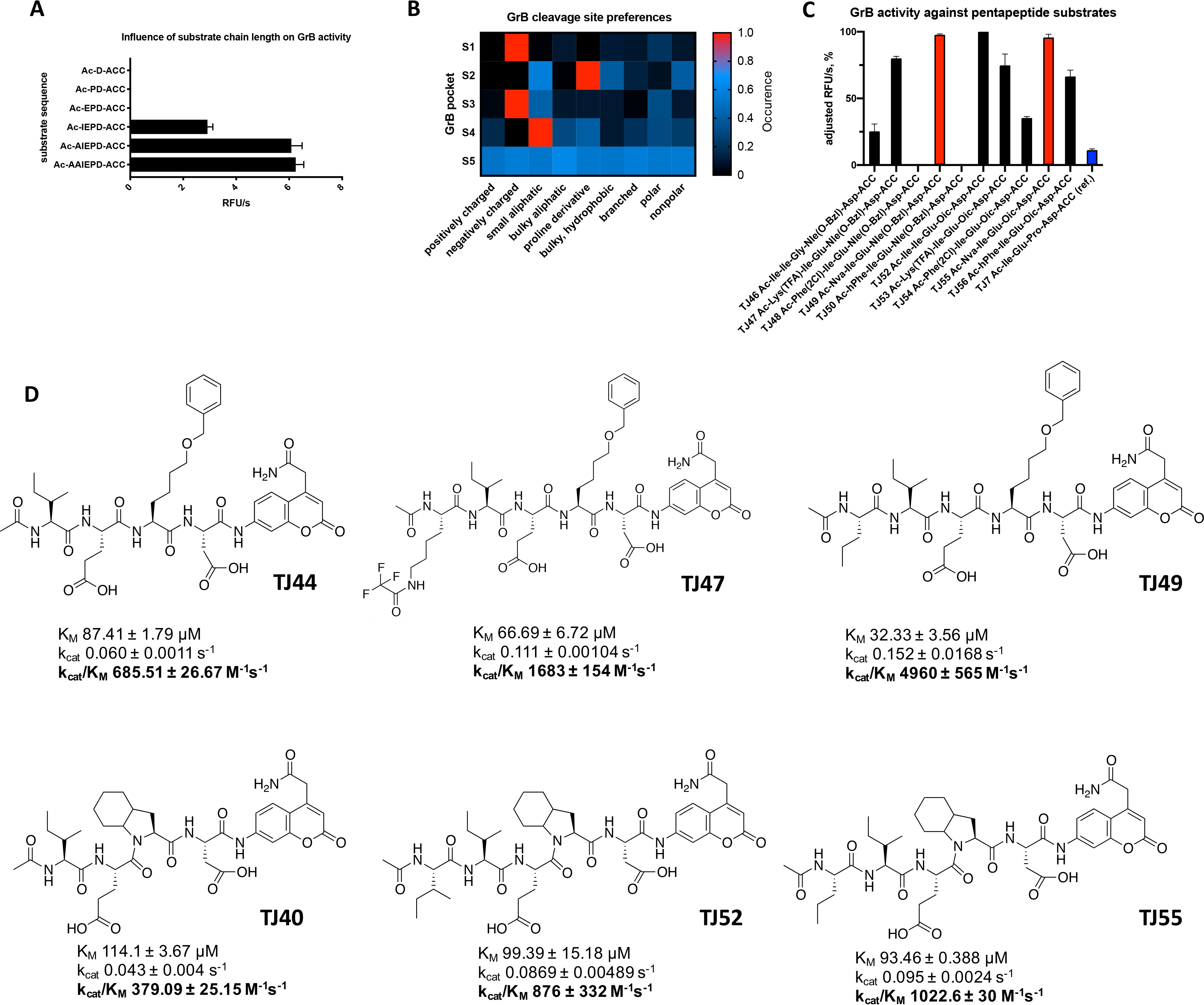
A, influence of substrate chain length on GrB activity. The substrates (at the same concentrations) were treated with GrB. The data in the column diagram are presented as RFU/s values (relative fluorescence units/second). B, GrB substrate cleavage site preferences. The GrB specificity for the S4–S2 pockets was tested against Asp-HyCoSuL (Hybrid Combinatorial Substrate Library), while the GrB S5 pocket catalytic preferences were tested using the pentapeptides in the P5 library (Ac-P5-X-Glu-X-Asp-ACC) and the P1 library (Ac-Ile-Ser-Pro-P1-ACC). The results, shown as the relative fluorescent units per second (RFU/s), were divided into 9 groups according to the amino acid character and are presented as a heat map. The average value of RFU/s for each group was normalized with the highest value from all groups being equal to 1. C and D, screening, structures of selected pentapeptide substrates, and kinetic parameters. Data are presented as the mean ± standard deviation and represent at least 2 independent experiments. The substrate hydrolysis rate (A–D) was measured with a plate reader with 355/460-nm excitation/emission wavelengths. All diagrams and kcat/Km values were prepared and calculated using GraphPad Prism software.
Catalytic preferences in nonprime S1–S5 enzyme pockets of GrB
To further explore the substrate specificity of GrB and allow better optimization of GrB substrates, we analyzed the S1–S5 pocket preferences using both combinatorial chemistry methods and the screening of defined peptides possessing natural and diverse unnatural amino acids (16, 17).
The GrB S1 pocket almost exclusively recognizes aspartic acid, and this feature is also seen in cysteine protease caspases (9, 10, 17). To reduce substrate cross-reactivity, especially with caspases, we tested whether any acidic and nonacidic modifications of natural amino acid residues are accommodated by the GrB S1 pocket. For this purpose, we designed and synthesized (using a solid-phase peptide synthesis method, see Fig. S1) a library of 95 defined peptides that share the same leading sequence and differ only by one amino acid residue at P1 (for the structures, see Fig. S2). ACC (7-amino-4-carbamoylmethylcoumarin) was applied as a fluorescent leaving group, allowing the substrate hydrolysis rate to be determined based on the increase in fluorescence. In the positions P4–P2, defined natural amino acid residues were incorporated based on literature data (Ac-Ile-Ser-Pro-P1-ACC), while the N termini of the peptide substrates were acetylated.
We tested the activity of GrB against the new P1 library and determined that GrB almost exclusively requires aspartic acid in the S1 pocket (l-Asp, 100%); it can accommodate the methylated derivative (l-Asp [O-Me], 30%) and, less potently, tyrosine (l-Tyr) and its derivatives (<10%) (Fig. 1B and Fig. S3A). This confirms the literature data where, similar to caspases (10, 17), the GrB S1 pocket is essentially restricted to aspartic acid due to its interaction with the positively charged guanidine group of Arg-226. Additionally, the carboxyl group of Asp forms hydrogen bonds with three water molecules within the S1 subsite of GrB (18). Not only the charge but also the shape of this pocket dictates amino acid binding, since only Asp or uncharged Asp derivatives with minimal modifications (Asp [O-Me]) can occupy this pocket, despite the lack of a negative charge, while more sizable Asp derivatives (Asp [O-Chx] and Asp [O-Bzl]) are not recognized by GrB, revealing the limited capacity of the S1 subsite and the involvement of additional interactions in peptide binding. The shape of Asp allows it to fit perfectly in the S1 pocket, and this is supported by the interaction between the guanidine group and the COOH group (for the P1 library screening, see Fig. S3).
Although P1-Asp specificity distinguishes GrB from other granzymes, many GrB substrates are also recognized by caspases (10), so conventional strategies for enzyme activity analysis cannot be used to selectively monitor GrB activity in cells. Therefore, we sought a substrate sequence that distinguishes GrB activity from other proteases. To this end, we next determined the extended catalytic preferences of GrB at S4–S2 using a well-established HyCoSuL strategy (Hybrid Combinatorial Substrate Library) incorporating a wide range of different nonproteinogenic amino acids (16, 17, 19) (Fig. S3). We observed that bulky hydrophobic proline derivatives, such as octahydroindolecarboxylic acid (l-Oic), 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (Tic), and O-benzyl-l-hydroxyproline (l-Hyp [Bzl]) are strongly preferred by GrB in P2, while substrates with hydroxyproline (l-Hyp) bearing unprotected hydroxyl groups are not hydrolyzed. Additionally, bulky hydrophobic 6-benzyloxy-l-norleucine (l-Nle [O-Bzl]), benzyl-l-histidine (l-His [Bzl]), and benzyl-l-serine (l-Ser [Bzl]) were tolerated in the S2 pocket, indicating that amino acids with benzyl groups can be accommodated within this pocket (Fig. 1B and Fig. S3) and revealing that the S2 pocket is very capacious. We speculate that this pocket, filled by the flexible cyclohexane group of proline derivative l-Oic or the benzyl groups of l-Nle (O-Bzl) or l-Ser (Bzl), enables the amino acid residues to perfectly fill the S2 pocket.
The HyCoSuL strategy also revealed that the S3 pocket of GrB has a broad substrate scope; however, it has strong preferences for glutamic acid (l-Glu, 100%), hydrophobic tyrosine bearing a benzyl group (l-Tyr [Bzl], 72%), and mono-oxidized methionine l-Met (O) (Fig. 1B and Fig. S3). The strong preference for an acidic amino acid is due to Asn-218 and Lys-192 in the S3 pocket of GrB, which interact with the side chain of l-Glu and stabilize the positively charged side chain of lysine (18). The S3 pockets of all caspases (10) share GrB's preferences for glutamic acid, confirming the structural similarity of these enzymes (17).
Our results demonstrated that the S4 pocket also possesses broad substrate preferences. It can accommodate branched amino acids, such as isoleucine (l-Ile, 72%), valine (l-Val, 36%), or linear hydroxyl-l-norvaline (l-Hnv, 82%), as well as a proline derivative (l-Tic, 100%) and benzyloxymethyl-l-histidine (l-His [3-Bom], 82%) and some basic amino acids, such as 2,4-diaminobutyrylic acid (l-Dab, 39%), citrulline (l-Cit, 10%), and 1,3-diaminopropionic acid (l-Dap, 10%) (Fig. 1B and Fig. S3). According to Rotonda et al., this GrB pocket is a “shallow hydrophobic depression formed by aromatic rings” (Tyr-174 and Tyr-215) and the side chain of Leu-172 (18); therefore, there is not enough space for phenylalanine within this pocket. Our data from the HyCoSuL screening revealed that a bulkier amino acid (l-His [3-Bom]) was hydrolyzed by GrB (Fig. S3). To find the optimal amino acid for P5, we synthesized a combinatorial library of pentapeptides. For this purpose, based on the literature data and our results related to GrB specificity, we designed a library consisting of 1) defined amino acids at P1 (Asp) and P3 (Glu), 2) equimolar mixtures (X) at P2 and P4 (a mixture of natural amino acids with l-Nle replacing l-Met and l-Cys), and 3) one of the 174 defined amino acids at P5 (Ac-P5-X-Glu-X-Asp-ACC) (for the structures, see Fig. S2). GrB was tested against the library, and we observed that it displays no S5 substrate specificity and is capable of hydrolyzing most substrates regardless of the residue at this position; however, the addition of an extra amino acid to the substrate (P5) clearly leads to a dramatic increase in substrate hydrolysis (Fig. 1B–D).
Design, synthesis, and kinetic analysis of pentapeptide substrates for GrB
To validate the results of the HyCoSuL screening, we selected the most promising amino acid residues for the S4–S2 positions (P4, l-Tic, l-His [3-Bom], and l-Ile; P3, l-Glu and l-Tyr [Bzl]; and P2, l-His [Bzl], l-Oic, l-Tic, and l-Hyp [Bzl]) and synthesized eighteen different fluorogenic tetrapeptides using a previously described method (20). We then tested the activity of GrB on the new substrates and observed that peptides with Ile at P4 were exclusively hydrolyzed by GrB (TJ40–44), while substrates with other selected amino acids, such as Tic (TJ2, TJ4, TJ6, and TJ35–39) and l-His (3-Bom) (TJ3 and TJ30–34), were not recognized by GrB under these assay conditions. Additionally, we confirmed that sequences containing l-Nle (O-Bzl) or l-Oic at S2 were preferred by GrB, confirming the large size of S2 (Fig. S4A).
With this in mind, we performed a detailed kinetic analysis of the most hydrolyzed and promising substrates, namely, TJ40 (Ac-Ile-Glu-Oic-Asp-ACC), TJ41 (Ac-Ile-Glu-Hyp [Bzl]-Asp-ACC), TJ42 (Ac-Ile-Glu-Tic-Asp-ACC), TJ43 (Ac-Ile-Glu-His [Bzl]-Asp-ACC), and TJ44 (Ac-Ile-Glu-Nle [O-Bzl]-Asp-ACC) (Fig. S4), and we compared those results with those of reference substrate TJ7 (Ac-Ile-Glu-Pro-Asp-ACC). Substrates TJ40, TJ43, and TJ44 were cleaved more rapidly by GrB than was TJ7, and their kinetic constants were kcat/Km = 379.09 m−1 s−1, 267.89 m−1 s−1, and 685.51 m−1 s−1, respectively, and that of the reference is kcat/Km = 76.95 m−1 s−1 (Fig. S4B). This was in agreement with our initial substrate screening (Fig. S4A). Since l-Oic and other proline derivatives are poorly or not recognized by caspases (10, 17, 21), we decided to use the sequence of substrate TJ40 as the core for our future substrates and probes. The optimization of the P4–P1 GrB peptide sequence resulted in a champion substrate that is well recognized by GrB and is less likely to show cross-reactivity with other granzymes and caspases. From the broad range of investigated amino acids, we selected the most promising structures (l-Lys [TFA], l-Nva, l-Ile, l-Phe [2-Cl], and l-hPhe), and we synthesized ten defined pentapeptide substrates, incorporating the sequences previously selected for P4–P1 (l-Ile-l-Glu-l-Oic/l-Nle [O-Bzl]-l-Asp). After an initial screening of GrB activities against the new substrates (TJ46–TJ56) (Fig. 1C), we selected substrates TJ47, TJ49, TJ52, and TJ55 as being hydrolyzed with the highest efficiency, and we studied their kinetics in detail (Fig. 1D). Substrate TJ49 possesses the lowest Km value (32.33 μm) and, at the same time, the highest kcat parameter (0.152 s−1), with 40% higher activity (kcat/Km of 4960 m−1 s−1) than that of the optimal tetrapeptide. Exchanging l-Nle (O-Bzl) at P2 (in TJ49) for l-Oic (affording TJ55) caused the enzyme activity to decrease by approximately a factor of four; however, we used this sequence in further analyses to minimize the cross-reactivity with the majority of caspases (17).
Granzyme B activity-based probe design, synthesis, and evaluation
The most valuable tools for GrB investigations are activity-based probes, both inhibitor- and substrate-based. However, because GrB and caspases share similar substrate specificity (10), the development of potent and selective GrB probes is challenging. To address this limitation, we first designed an inhibitor-like molecule (activity-based probe) that is selective for GrB with low cross-reactivity with caspases.
We designed our probe to be built from three main functional segments. First, we used a reactive functional group that covalently binds to the active site of the enzyme. We selected diphenyl phosphonate, which mimics natural amino acids and potently reacts with serine proteases (22, 23), thereby reducing the possibility of the probe binding with caspases. Second, we attached a peptide sequence to the warhead based on the optimal GrB catalytic preferences to ensure the best fitting of the probe within the active site of GrB. For this purpose, we selected our champion substrate (TJ55) (Fig. 1D), since GrB showed its highest activity with this substrate, and Oic at P2 disfavors binding with caspases. Third, we attached an affinity (biotin) (TJ55.Bt) or a fluorescent (Cy5) (TJ55.5) tag at the N terminus for membrane and in-gel detection, respectively (Fig. 2A). The probes were synthesized using a mix of classic solid-phase and solution-phase peptide synthesis methods. Briefly, the peptide sequence with Cy5 or biotin was attached to the warhead in solution following a procedure described elsewhere (Fig. S5). Using the same method, we obtained a covalent inhibitor of GrB with the general structure of Ac-Nva-Ile-Glu-Oic-AspP (OPh)2. As we noticed from the calculation of the inhibition kinetic constants, GrB strongly prefers biotin as a tag relative to the Cy5 derivative, and it binds with TJ55.Bt almost 70 times more rapidly than it does to TJ55.5 (Fig. 2A). We speculate that this substantial difference in enzyme binding to biotinylated versus fluorescent probes is due to the presence of a biotin-binding exosite in the GrB structure, and biotin binding is preferred due to the three-dimensional enzyme structure and probe interactions.
Figure 2.

Specific granzyme B detection in cell lysates. A, covalent GrB inhibitor, biotinylated and fluorescent activity-based probe structures, and kobs/I values for GrB calculated using GraphPad Prism software. Data are reported as the mean ± standard deviation and represent at least 2 independent experiments. B and C, optimization of the recombinant enzyme incubation time with TJ55.Bt (red) (A) or TJ55.5 (red) (B). GrB was incubated with TJ55.Bt or TJ55.5 for the indicated times (only the probe or only the enzyme was used in the controls). Samples then were analyzed by SDS-PAGE, followed by transfer to the membrane and streptavidin conjugate (red) and antibody labeling (green). D and E, YT cell lysates corresponding to 1 × 107 cells/ml were treated with TJ55.Bt (red) for the indicated time (D), or samples were incubated with various concentrations of GrB probe (from 5–500 nm) for 60 min (E). Samples then were analyzed by SDS-PAGE, followed by transfer to the nitrocellulose membrane, streptavidin conjugate labeling (red), and immunoblotting using anti-GrB (green). As a control, lysates were pretreated with a competitive inhibitor prior to probe addition, or the probe and the lysates were run separately. The data reflect at least three separate biological replicates.
We tested the binding of our new probes to purified GrB and showed that both probes were capable of GrB labeling (TJ55.Bt [Fig. 2B] and TJ55.5 [Fig. 2C, red bands]). First, we optimized the enzyme-probe incubation time for detecting GrB. After 5 min of incubation, we noticed a strongly labeled species between 35 and 40 kDa, corresponding to the size of purified GrB (Fig. 2B, lane 2, and C, lane 2, red bands). As a control, to test whether the probe covalently binds in the GrB active site, we preinhibited GrB with a covalent inhibitor (Ac-Nva-Ile-Glu-Oic-AspP [OPh]2) prior to probe addition (Fig. 2B, lane 8, and C, lane 8). The inhibitor prevented probe binding; therefore, we concluded that TJ55.Bt and TJ55.5 bind to the active site of GrB. Additionally, TJ55.5 binds less potently than TJ55.Bt, which is consistent with their inhibition kinetic constants (Fig. 1A). In addition, to confirm that the 35- and 40-kDa species represent GrB, we applied an anti-GrB mAb, and the signals from the probes and antibodies overlapped exactly (Fig. 2B and C, antibody [green bands] and overlay [yellow bands]), confirming that our probe labels GrB. Furthermore, to test whether the sample autofluoresces under our assay conditions, we ran one sample without an activity-based probe and only detected a signal from the antibody (Fig. 2B, lane 7, and C, lane 7, green bands). Thus, TJ55.Bt and TJ55.5 1) bind GrB; 2) bind to the GrB active site; and 3) can be utilized in future active GrB detection assays.
Active GrB detection in cell lysates
Since our activity-based probes allow efficient GrB labeling, in the next step, we verified their specificity in a complex system of cell lysates. We selected the human NK cell line YT, since it constitutively expresses and releases GrB (24), to observe if the probe binds with enzymes or cellular components other than the target enzyme. First, we optimized the concentration of the probe for efficient GrB labeling (Fig. 2C, red bands) and found that 5 nm is sufficient for GrB detection in complex systems. As indicated, even at a very high probe concentration (500 nm) and a long incubation time (60 min) (Fig. 2C, lane 7, red band), the probe binds almost exclusively with GrB; a signal from an additional band between 70 and 100 kDa was observed, but it does not have proteolytic activity (25). Additionally, we observed that the TJ55.Bt probe binds with GrB from YT cell lysates rapidly, and the complex was detectable after 1 min of incubation (Fig. 2D, lane 2, red band). Pretreatment of the cell lysates with a competitive inhibitor of GrB activity prevented probe from binding (Fig. 2D, lane 9). (The ∼100-kDa species seen in Fig. 2D and E [red bands] is a nonspecific product of the streptavidin reagent, as it appears in samples not containing TJ55.Bt [lane 7]). To verify whether the observed species corresponds to the target enzyme, we performed additional staining using an anti-GrB mAb, and we noted that the signals from the probe and antibody overlapped, confirming probe binding with GrB. We also observed an additional species at 70 kDa when using the antibody (Fig. 2D–E, green bands). We speculate that this represents a complex between GrB and the cytosolic serpin PI-9 (Serpinb9), which forms postlysis and is detected by the mAb of GrB. Since our probe is specific to GrB even in a complex system of YT cell lysates, we speculated that it can be applied for GrB detection in other cells, especially cancer cell lines. To test that hypothesis, we screened GrB activity in different cell lines, such as YT (as a control cell line), MDA-MB-231, Su-DHL-4, Jurkat-T, NK92, MG63, SEMK2, REM, and NALM-6, using classic SDS-PAGE, and we observed that GrB is present in its active form mainly in the NK-like YT and NK92 cells. Interestingly, there was far less GrB detected in NK92 cells (below the detection level of the antibody), while minimal labeling with the probe was detected, emphasizing the sensitivity of this active-site probe.
GrB was also detected in leukemia cell lines SEMK2, REM, and NALM but only in an inactive form, as it was strongly labeled with the antibody and not labeled with the activity-based probe (Fig. 3, probe [red] and antibody [green]). We think that this is due to the presence of inactive GrB within these cells or nonspecific antibody binding.
Figure 3.
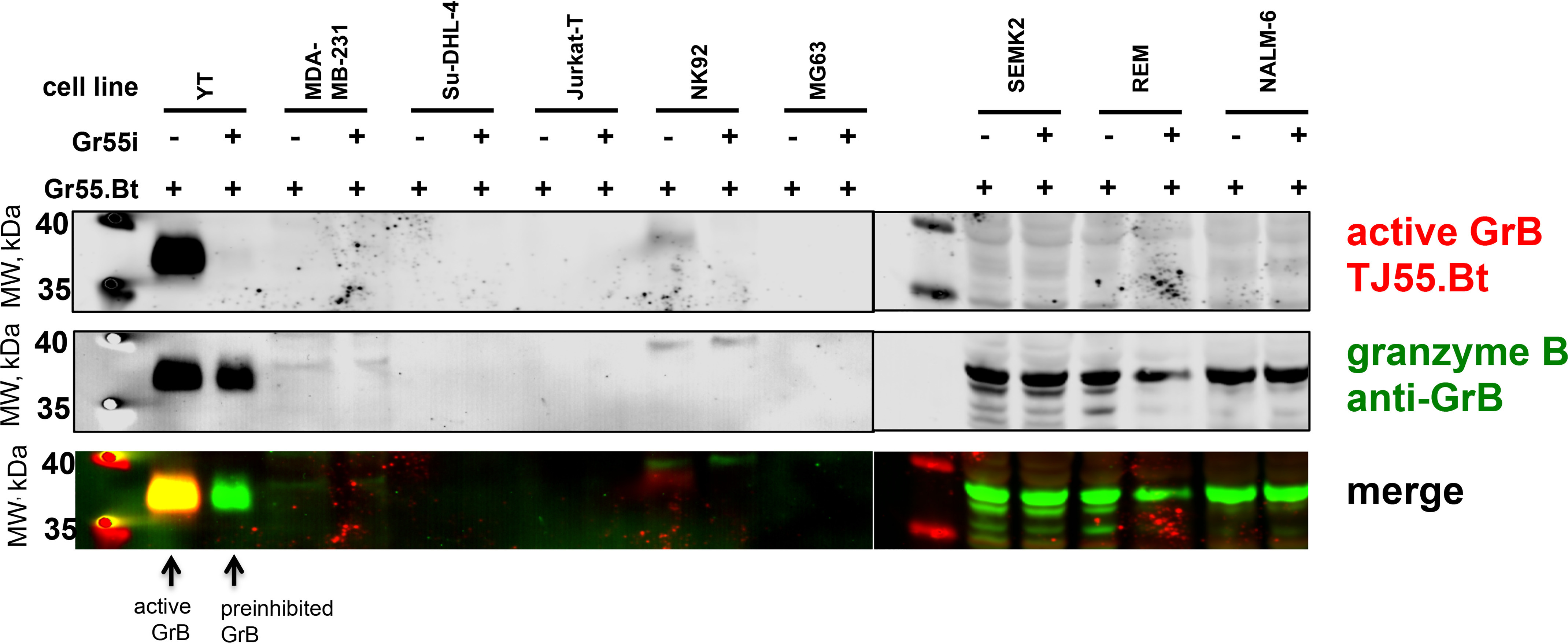
Detection of GrB in cell lines. Cells were harvested from liquid cultures and suspended to the same initial cell concentration (1 × 107 cells/ml) in lysis buffer and then subjected to freeze-thaw cycles and sonication. The same volume of each sample then was treated with the same concentration of TJ55.Bt (250 nm, red) for 30 min. The samples were assessed by SDS-PAGE and transferred to a nitrocellulose membrane, followed by immunoblotting using anti-GrB (green). The presented data are representative of at least two biological replicates.
The design of a quenched fluorescence substrate probe for GrB
Biotinylated and fluorescent probes are valuable tools, but this type of inhibitor-like active-site probe presents problems for enzyme monitoring in real time due to the inhibition of the enzyme activity, potentially leading to changes in its function. In addition, classic inhibitor-like probes are frequently equipped with unquenched fluorescent tags, which emit a signal regardless of binding with the enzyme; therefore, they are called “always on” probes. This may cause false-positive results in biological assays, since the cells, by pinocytosis or another mechanism, may take up the probe, and the fluorescent signal will be detected even if probe will not bind with the enzyme. Therefore, to avoid these limitations, we designed another type of molecule, a quenched fluorescent substrate, that emits fluorescence only after hydrolysis by the targeted enzyme. In addition, this substrate will not inhibit the enzyme activity and, therefore, will not modify the biological function of the enzyme. To generate this probe, we first optimized its leading sequence. For this purpose, we used two previously optimized amino acid sequences for the P5–P1 positions based on TJ49 (Nva-Ile-Glu-Nle [O-Bzl]-Asp) and TJ55 (Nva-Ile-Glu-Oic-Asp), and we utilized variations of the P1′–P3′ sequence (Phe-Gly-Arg or Gly-Gly-Gly). Additionally, at the P6 position, we attached a PEG(4) linker (or not) to increase the distance between a recognition sequence and the fluorophore. To identify the most active sequence for GrB, we utilized ACC and Lys (Dnp) as the fluorescence donor-acceptor pair. After synthesizing the twelve substrates (Table 1), we tested their hydrolysis rates using GrB, and we observed that in this type of GrB substrate, the predicted Oic (instead of the Nle [O-Bzl] group that was indicated in the HyCoSuL screening) is crucial at P2 for GrB substrate detection; moreover, the S1′–S3′ pockets are significant for GrB activity. We selected two of the most promising sequences, TJ65, H2N-ACC-PEG(4)-Nva-Ile-Glu-Oic-Asp-Phe-Gly-Arg-Lys (Dnp)-CO (NH2), and TJ71, H2N-ACC-Nva-Ile-Glu-Oic-Asp-Phe-Gly-Arg-Lys (Dnp)-CO (NH2), and we calculated their exact kinetic parameters. Table 1 shows the structures and kinetic constants for all substrates. Regardless of the presence of a linker at P6, the substrates possess high kcat/Km kinetic rates (TJ65 and TJ71). We also noticed a 33% reduction in Km if PEG(4) was incorporated at P6. Surprisingly, the kcat/Km for GrB on internally quenched fluorescent (IQF) substrates was dramatically higher than what was observed with shorter, classic substrates (TJ49 and TJ55). To test the selectivity of the most promising sequence (TJ71), we tested its activity with caspases, since these enzymes share similar substrate specificity (10), and we noticed robust GrB activity but only minimal hydrolysis of TJ71 by caspase-6 and caspase-8 (Fig. 4B and Table 2); other caspases did not lead to fluorescence increases. Thus, the peptide sequence TJ71 was selected as a scaffold for future synthesis.
Table 1.
Structures of the GrB IQF substrates and calculated kinetic constants for the hydrolyzed substratesa
| Code, sequence for GrB IQF substrates | Km (μm) | kcat (s−1) | kcat/Km (m−1 s−1) |
|---|---|---|---|
| TJ65, H2N-ACC-PEG(4)-Nva-Ile-Glu-Oic-Asp-Phe-Gly-Arg-Lys (Dnp)-CO (NH2) | 5.9 ± 1.37 | 0.7 ± 0.06 | 1.2 × 105 ± 1.8 × 103 |
| TJ66, H2N-ACC-PEG(4)-Nva-Ile-Glu-Oic-Asp-Gly-Gly-Gly-Lys (Dnp)-CO (NH2) | NDb | ND | ND |
| TJ67, H2N-ACC-PEG (4)-Nva-Ile-Glu-Oic-Asp-PEG(4)-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ68, H2N-ACC-PEG(4)-Nva-Ile-Glu-Nle (O-Bzl)-Asp-Phe-Gly-Arg-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ69, H2N-ACC-PEG(4)-Nva-Ile-Glu-Nle (O-Bzl)-Asp-Gly-Gly-Gly-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ70, H2N-ACC-PEG(4)-Nva-Ile-Glu-Nle (O-Bzl)-Asp-PEG(4)-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ71, H2N-ACC-Nva-Ile-Glu-Oic-Asp-Phe-Gly-Arg-Lys (Dnp)-CO (NH2) | 7.65 ± 0.59 | 0.8 ± 0.07 | 1 × 105 ± 1.1 × 103 |
| TJ72, H2N-ACC-Nva-Ile-Glu-Oic-Asp-Gly-Gly-Gly-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ73, H2N-ACC-Nva-Ile-Glu-Oic-Asp-PEG(4)-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ74, H2N-ACC-Nva-Ile-Glu-Nle (O-Bzl)-Asp-Phe-Gly-Arg-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ75, H2N-ACC-Nva-Ile-Glu-Nle (O-Bzl)-Asp-Gly-Gly-Gly-Lys (Dnp)-CO (NH2) | ND | ND | ND |
| TJ76, H2N-ACC-Nva-Ile-Glu-Nle (O-Bzl)-Asp-PEG(4)-Lys (Dnp)-CO (NH2) | ND | ND | ND |
a The kcat/Km values were calculated using GraphPad Prism software. Data are the mean ± standard deviation and represent at least 2 independent experiments.
bND, no activity detected.
Figure 4.
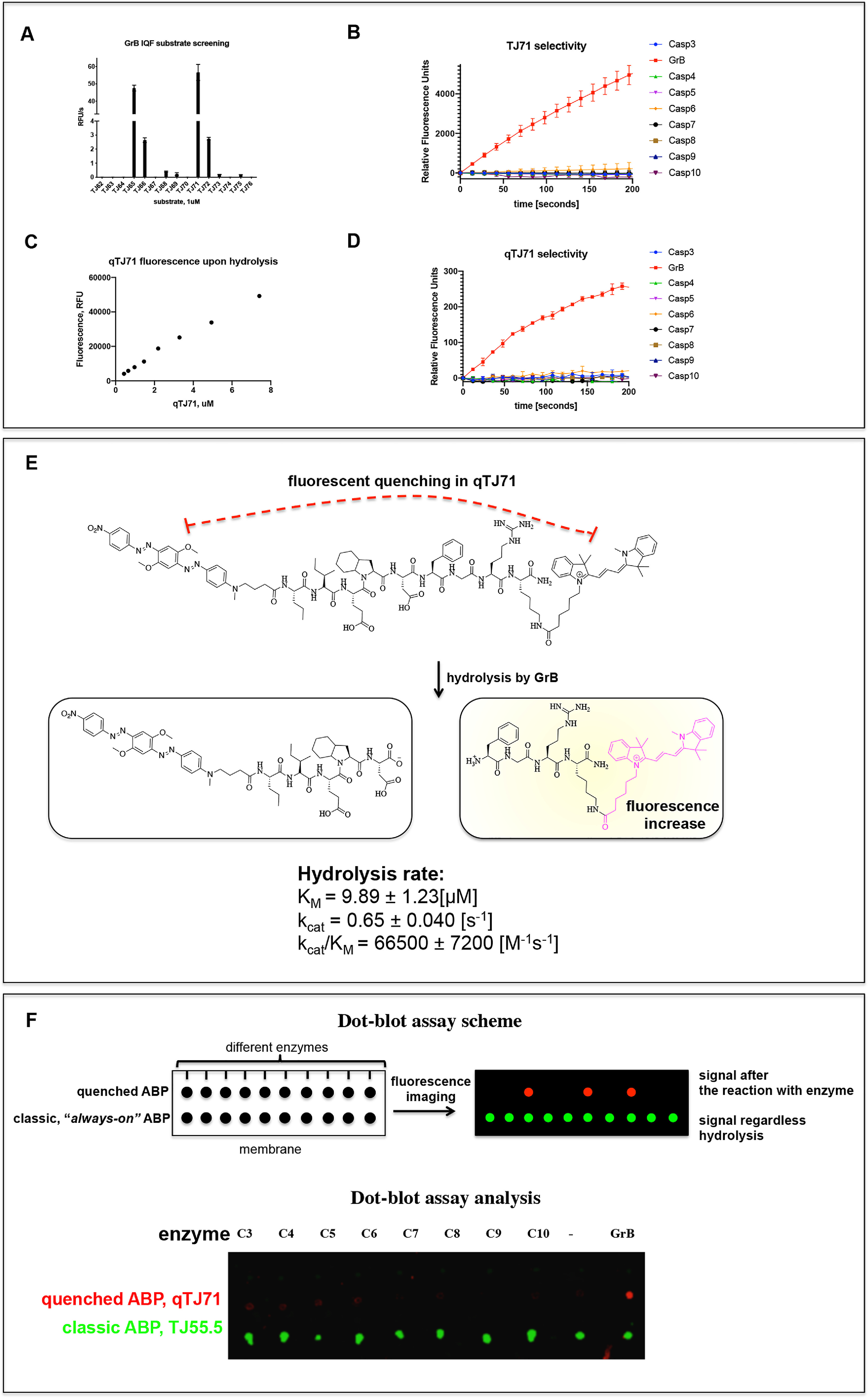
A, hydrolysis rate of GrB IQF substrates. The increase in fluorescence over time was measured using a spectrofluorometer and analyzed in GraphPad Prism. B, TJ71 selectivity. Experiments were performed using GrB and caspases at equal concentrations. The increase in fluorescence over time was measured and analyzed in GraphPad Prism. C, the fluorescence increase upon hydrolysis of qTJ71 is proportional to the substrate concentration. D, qTJ71 selectivity. The experiments were performed with GrB and caspases at equal concentrations. The increase in fluorescence over time was measured and analyzed in GraphPad Prism. E, a scheme of the qTJ71 cleavage site and kinetic parameters of the GrB hydrolysis of qTJ71. qTJ71 exhibits no fluorescent signal until it interacts with GrB. F, dot-blot analysis of quenched qTJ71 (red dots) compared with unquenched TJ55.5 (green dots). The upper panel shows a scheme of the experiment demonstrating that the two types of chemical markers exhibit significantly different fluorescence properties upon interaction with the investigated enzymes. The lower panel shows the test of qTJ71 utility in dot-blot analysis (top lane) and its specificity determination. The strong signal from substrate hydrolysis was noticed only with GrB, demonstrating the specificity of qTJ71. The classic inhibitor-like activity-based probe (TJ55.5, bottom lane) with the unquenched fluorescent moiety (always on) released fluorescence regardless of binding with the enzymes (green dots). All data (A–F) are the mean of at least two independent experiments performed in duplicate, and the standard error of the mean is provided.
Table 2.
TJ71 specificitya
| Enzyme | Km (μm) | kcat (s−1) | kcat/Km (m−1 s−1) |
|---|---|---|---|
| GrB | 7.65 ± 0.59 | 0.8 ± 0.07 | 101,700 ±1100 |
| Casp-3 | 36 ± 0.32 | 0.204 ± 0.002 | 5590 ± 111 |
| Casp-6 | 15 ± 3.4 | 0.178 ± 0.150 | 11,800 ± 1240 |
| Casp-8 | 3.48 ± 0.35 | 0.0105 ± 0.00095 | 3050 ± 43 |
a kcat/Km values were calculated using GraphPad prism software. Data are the mean ± standard deviation from at least 2 independent experiments.
The GrB quenched fluorescent substrate is activated by recombinant human GrB
The ACC moiety is not a suitable fluorophore for the live imaging of enzymes within cells since its fluorescence emission is close to the natural autofluorescence of cells, which may cause a false-positive result. Additionally, ACC possesses a poor quantum yield compared with those of other fluorophores; therefore, to use our quenched fluorescent substrate for in-cell GrB investigation, we exchanged ACC with the cyanine derivative Cy3, which is more stable and convenient for cell-based analysis. To prevent false-positive signals and minimize the background fluorescence from the fluorophore of classic always-on probes, we applied a nonfluorescent quencher in the fluorescent substrate sequence that efficiently silences the fluorescence signal of the unhydrolyzed substrate. After hydrolysis, due to the separation of the donor-acceptor pair, Cy3 fluorescence is activated by the enzyme, and the signal is released. We applied Black Hole Quencher® 2 (BHQ2) as the quencher, since it is characterized by a high quenching yield of Cy3, and we speculated that it can be utilized in cell-based analyses. The lack of measurable activity toward caspases made the TJ71 sequence an ideal candidate for the recognition sequence of the quenched fluorescent substrate. Importantly, the fluorophore was attached to the C terminus of the substrate and the quencher was attached to the N terminus, since, upon substrate hydrolysis by an enzyme, the product containing Cy3 will be amplified in place of hydrolysis (Fig. 4E). The qTJ71 quenched fluorescent substrate was synthesized using a mixture of solid-phase and solution-phase synthesis techniques (Fig. S6).
We tested the activation of qTJ71 by GrB, utilizing kinetic analysis and as indicated in Fig. 4C. The total relative fluorescence units (RFU) from the entire peptide hydrolysis increased proportionally to the substrate concentration, confirming our hypothesis that qTJ71 is activated by GrB and that the fluorescence quenched by BHQ2 is released upon hydrolysis. Since caspases have also been reported to recognize Asp at the P1 position, we evaluated the hydrolysis of our substrate with these enzymes. For this purpose, we monitored the number of RFU as a function of time upon qTJ71 hydrolysis with GrB and caspases (at equal concentrations). As indicated in Fig. 4D, qTJ71 is hydrolyzed exclusively by GrB. Therefore, in the next step, we calculated kcat/Km parameters for the hydrolysis of qTJ71 by GrB to be as high as 66,500 m−1 s−1. Next, we tested the cleavage site of qTJ71 by GrB and observed that, as expected, the cleavage site was after the Asp residue (Fig. 4E and Fig. S7). qTJ71 was designed to be utilized in cell culture-based assays; however, first, we tested the utility of both quenched fluorescent substrate qTJ71 and unquenched TJ55.5 probe in a simple dot-blot assay, and we also tested the cross-reactivity of qTJ61 with caspases. Therefore, we added GrB or caspases-3, -4, -5, -6, -7, -8, -9, and -10 (separately) to qTJ71 or TJ55.5, and then we performed dot-blot analysis. We observed the signal increased only in the samples containing GrB and qTJ71 (Fig. 4F, red dot). As a control, we utilized an untreated quenched fluorescent substrate in an assay buffer and did not observe fluorescence. In the same test using unquenched TJ55.5, we observed a strong false-positive signal in every sample from the always-on activity-based probe (Fig. 4F, green dots). Therefore, we concluded that our qTJ71 is selective for GrB and can be utilized in cell culture assays for real-time imaging of GrB within cells.
qTJ71 quenched fluorescent substrate cellular uptake
qTJ71 possesses selectivity toward GrB and is not recognized by active caspases; therefore, in the next step, we used it to monitor GrB activity in YT cells. First, as a pilot experiment, we tested qTJ71 cellular uptake over time (Fig. S8). We performed live-cell imaging of GrB in YT cells (in growth medium) treated with 500 nm of qTJ71 and noticed that the quenched fluorescent substrate allows the detection of GrB immediately after addition; however, the optimal labeling time for living cells was 15 min. Next, we addressed the question of whether the fluorescence from qTJ71 cleavage is GrB specific and can be prevented by covalent inhibition of GrB. In order to test this, YT cells were pretreated with TJ55i and subsequently labeled with qTJ71. We observed a strong fluorescence increase in YT cells labeled with qTJ71 but no fluorescence in cells pretreated with TJ55i. Therefore, we concluded that the qTJ71 compound is selective to GrB and can be utilized for future GrB monitoring in living cells.
GrB activity monitoring in direct NK cell recognition of tumor-targeted cells
GrB is considered to be involved in target cell killing, acting in concert with perforin, and by entering the target cell, among other things, it can hydrolyze caspase substrates and cause target cell death. Therefore, our next goal was to observe the accumulation of active GrB in the breast cancer cell line MDA-MB-231 (target cells, T) after the addition of YT cells (effector cells, E). As indicated previously using the unquenched TJ55.5 probe (Fig. 3), active GrB is not usually present in MDA-MB-231 cells, while YT cells produce a substantial amount of this enzyme.
However, to test the utility of qTJ71 in GrB assays in living cells, we performed two controls. First, GrB was inactivated using a covalent GrB inhibitor prior to the addition of the qTJ71 quenched fluorescent substrate. We observed no fluorescence, but when we used qTJ71 alone, fluorescence was detected. This demonstrates that other components in the cell do not hydrolyze our quenched fluorescent substrate; therefore, it can be used in cell-based assays. Second, to test the selectivity of qTJ71, we treated both MDA-MB-231 and YT cell lines separately with the quenched fluorescent substrate and analyzed the samples by flow cytometry. We also analyzed naive cells and cells stained with anti-GrB, followed by a fluorescently labeled secondary antibody. We observed robust fluorescence in YT cells treated with qTJ71, and, in contrast, MDA-MB-231 cells showed no fluorescence. This was confirmed with antibody staining, which indicated the absence of GrB in MDA-MB-231 and its presence in YT (Fig. 5B). These experiments demonstrated that our quenched fluorescent substrate can detect active GrB within cells using simple and straightforward assays and classic methods, such as FACS and microscopy (Fig. 5A and B).
Figure 5.
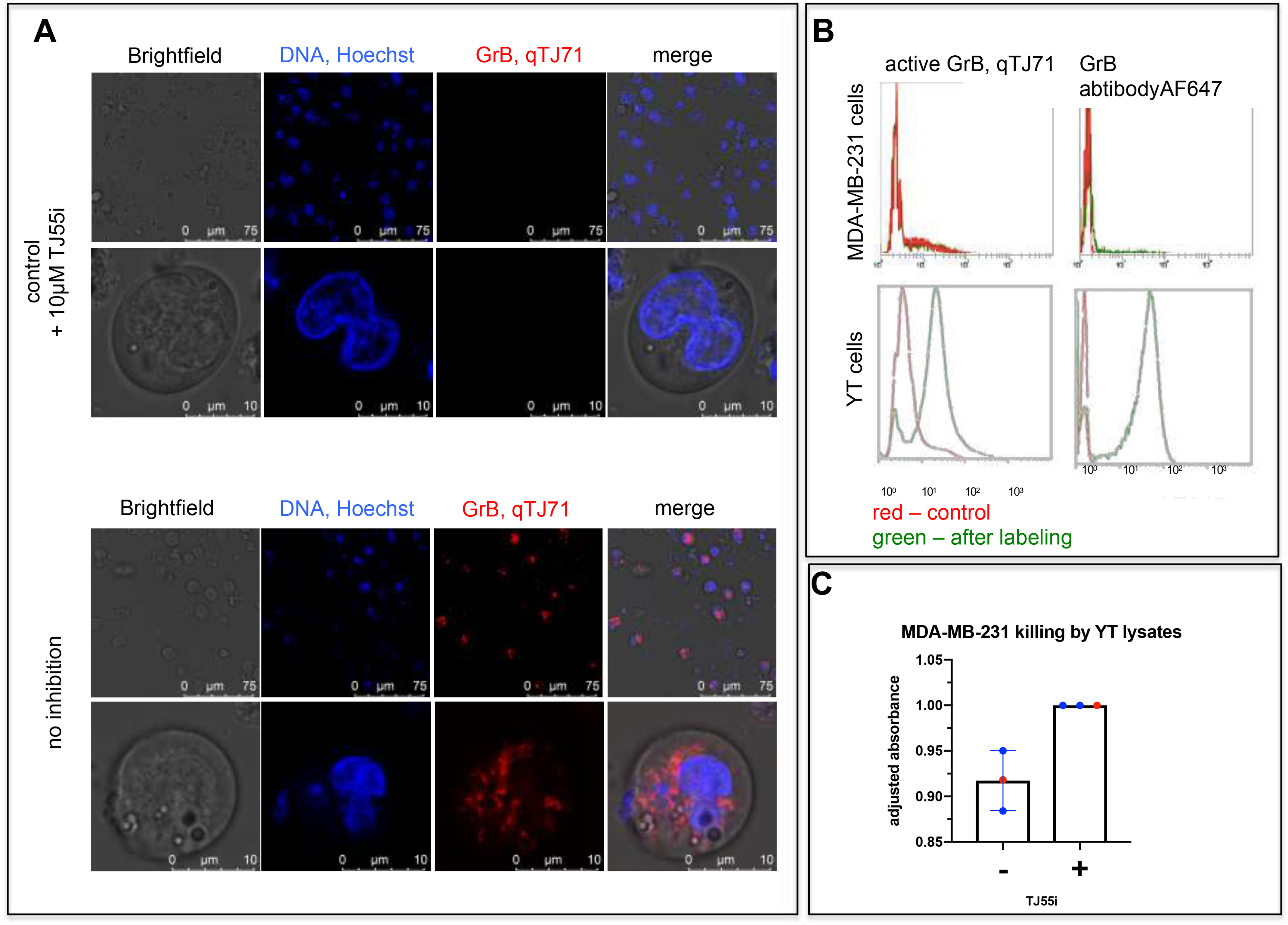
A, active GrB detection in YT cells with qTJ71 by confocal microscopy. YT cells were treated with qTJ71 for 5 min and with Hoechst 33342 for 5 min, followed by live imaging using a Leica TCS SP8 confocal microscope. As a control, cells were pretreated with GrB inhibitor prior to quenched fluorescent substrate addition. B, GrB detection in YT cells and the absence of active GrB in MDA-MB-231 cells labeled with qTJ71. YT and MDA-MB-231 cells were stained with qTJ71, fixed with PFA, and then analyzed at Sysmex CyFlow Cube. As a control, unlabeled cells and anti-GrB-labeled cells were analyzed. C, quenched fluorescent substrate. MDA-MB-231 cells were treated with YT cell lysates for 4 h, and an MTS assay was performed. As a control, lysates were pretreated with TJ55i. Data are from 3 independent biological replicates.
GrB is considered one of the most important enzymes involved in target cell killing. Therefore, with a specific GrB inhibitor, we tested whether the inhibition of GrB in YT cell lysates influences its target cell killing efficiency. To do so, we measured MDA-MB-231 cell viability in the presence of YT cell lysates or YT cell lysates pretreated with GrB inhibitor (TJ55i). As indicated in Fig. 5C, the inhibition of GrB led to a 10% increase in cell viability but did not prevent target cell killing. With this in mind, we speculate that GrB is important but is not the only YT cell component that can cause target cell death.
Discussion
Granzyme B, a serine protease involved in programmed cell death through the cleavage of caspase substrates (BID and ICAM), is one of the key factors within NK cells and CTLs leading to cell death (10, 12, 15). GrB enters the target cell via perforin, and, by hydrolyzing cellular proteins, it activates caspases, which are pivotal enzymes in cell death. The mechanism of GrB entrance into target cells using perforins remains unclear; however, a recent discovery led to the hypothesis that perforin forms a pore within the target cell that allows GrB to enter (26). Within the cell, GrB is capable of cleaving different substrates, and it can induce a caspase-independent or caspase-dependent cell death cascade.
GrB, in order to perform its functions, has a relatively broad substrate specificity and can hydrolyze a wide range of peptidic epitopes. The classic artificial substrates of GrB described previously are tetrapeptide derivatives (13). Interestingly, the peptide chain elongation and incorporation of one or two additional amino acid residues increased GrB activity against the substrates, while shorter substrates (tri- or dipeptides) are not hydrolyzed by GrB, as we show in Fig. 1A.
It is worth noting that granzyme B is the only known serine protease that hydrolyzes substrates after aspartic acid, sharing this feature with caspases. The shape and chemical properties of the S1 pocket are controlled by the presence of a positively charged arginine 226 moiety oriented into the S1 cavity by the cis-proline conformation in the AA221–Pro-224–Pro-225–Arg226 motif, which is conserved between human and mouse GrB (18). Therefore, GrB interacts almost exclusively with negatively charged Asp, and surprisingly, as we observed for its methylated derivative (Asp [O-Me]), GrB does not cleave glutamic acid, which has one additional methylene group, and this is probably due to the depth of the cavity. On the other hand, in the S2 pocket the specificity to proline and its derivatives is characteristic of a majority of serine proteases, including the granzyme family (10), neutrophil elastase (27), cathepsin G (27), proteinase 3 (27), chymotrypsin, and blood coagulation factor II. The crystal structure of GrB complex with an inhibitor with proline at P2 (Ac-IEPD-CO), Phe-99, forms a wall in the active site of GrB, and as demonstrated previously, the proline residue forms no specific interactions with the S2 pocket, but above the proline ring an additional cavity was detected (18), and we speculate that it can accommodate the proline extension in nonproteinogenic amino acids. Therefore, in our GrB substrates, we applied several proline derivatives at P2, and we observed that Oic dramatically improves substrate binding. Oic is a proline derivative modified with a cyclohexane ring that can adopt a three-dimensional structure, known as chair conformation, giving it flexibility. Interestingly, bulky hydrophobic Nle (O-Bzl) at P2 was also recognized, confirming the presence of an additional cavity around the S2 subsite. In our opinion, the S2 pocket is crucial for distinguishing between granzyme B and caspases, since it is the only position in which these enzymes possess distinct specificity. With our HyCoSuL screening of GrB, we confirmed that it recognizes glutamic acid at P3 and shares this feature with caspases (10, 17, 21, 28). The carboxyl group of glutamic acid interacts with Lys192 and Asn218 in the GrB S3 pocket, consistent with GrB specificity (18). These results lead us to an improved, selective, and potent leading sequence for our GrB reagents.
NK cells and CTLs are loaded with GrB and deliver it to the targeted cancer cells during invasion. However, little is known about GrB presence in other cells, especially in cancer cell lines; therefore, we aimed to investigate it with covalent activity-based probes that are the method of choice for the specific detection of protease activity. Previously, Mahrus et al. synthesized a biotinylated activity-based probe that allowed GrB detection in NK cell lysates. In our work, we improved the kinetic parameters of activity-based probes for in-gel detection of GrB and synthesized two activity-based probes 1) with biotin (TJ55.Bt) or 2) with cyanine derivative Cy5 (TJ55.5), and we demonstrated the utility of these probes in selective GrB detection in both pure enzyme solutions and cellular environment (cell lysates) (Fig. 2B and C). These probes allow us to test for GrB presence in selected cell lines (YT, MDA-MB-231, Su-DHL-4, Jurkat-T, NK92, MG63, SEMK2, REM, and NALM-6), and we demonstrated that active GrB is almost exclusively present in the NK cell line YT, and a substantial amount of GrB can be detected in the NK92 cell line (Fig. 3).
Covalent activity-based probes are valuable tools for protease detection in cells and can be used to analyze enzyme functions in cells. However, because of their inhibitory activity, the utility of such probes is limited, and this type of molecule cannot be applied in noninvasive live-cell imaging. To circumvent this, we designed a quenched fluorescent substrate that fluoresces only after hydrolysis by GrB. To do that, we utilized a peptide structure based on our results, and we applied a Cy3/BHQ2 pair as a fluorophore/quencher. This new structure turned out to be specific to GrB and is barely recognized by caspase-6 and caspase-8.
The activity of GrB depends on the pH, and GrB stored in low-pH granules is not enzymatically active. However, after the treatment of YT cells with our qTJ71, we detected a strong fluorescence, which was confirmed by FACS analysis (Fig. 5A and B). Signal was dismissed by the presence of GrB inhibitor, confirming that hydrolysis by GrB indeed occurs. These data suggest that YT cells constitutively release active GrB via granule exocytosis, consistent with a previous study (24). We speculate that the treatment of YT with the granule destabilizer LLOMe should markedly increase GrB binding (29). We believe that our quenched fluorescent substrate will allow the monitoring of GrB delivery from NK cells to target cancer cell lines, which, as observed in MDA-MB-231 cells, do not have active GrB (Fig. 5B), and the observed fluorescence increase is associated with the substrate interaction with GrB.
In summary, in our work, we designed and obtained a set of specific and potent tools for GrB investigation: 1) substrates, 2) an inhibitor, 3) inhibitor-based, activity-based probes, and 4) an enzyme-activated fluorescent substrate. With this, we were able to detect active GrB in lysates from different cell lines, and we noted the presence of this enzyme in NK-like cell lines YT and NK92. Our enzyme-activated fluorescent substrate allowed the noninvasive optical detection of GrB in NK cells.
Materials and methods
Chemicals
All chemicals were purchased from commercial suppliers and were used without further purification. All Fmoc amino acids (purity of >99%) used for libraries and individual substrate synthesis were purchased from Iris Biotech GmbH, Combi-blocks, QMBIO, CreoSalus, Bachem, and APExBIO. Fmoc-Rink amide (AM) polystyrene resin (loading, 0.74 mmol/g), DICI (diisopropylcarbodiimide, peptide grade), HBTU (O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate, peptide grade), HATU [2-(1-H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium, peptide grade], and TFA (purity, 99%) were purchased from Iris Biotech GmbH, Germany. HOBt (N-hydroxybenzotriazole, purity of >98%) was purchased from CreoSalus, USA. DCM (dichloromethane, analytically pure), MeOH (methanol, analytically pure), Et2O (diethyl ether, analytically pure), and AcOH (acetic acid, purity of 99%) were purchased from POCh. DIPEA (N,N-diisopropylethylamine, peptide grade) was purchased from VWR International, Poland. Pip (piperidine, purity of 99%), collidine (2,4,6-trimethylpyridine, peptide grade), and TIPS (triisopropylsilane, purity of 99%) were purchased from Sigma Aldrich sp. z o.o., Poland. DMF (N,N′-dimethylformamide, peptide grade) and ACN (acetonitrile, HPLC grade) were purchased from Avantor, Poland. Triphenyl phosphite was purchased from Sigma Aldrich sp. z.o.o., Poland. BHQ2 and BHQ3 succinimidyl esters were purchased from Future Synthesis, Poland. Cyanine5 NHS ester was purchased from Lumiprobe, Germany.
All individual substrates were purified by HPLC (Waters M600 solvent delivery module, Waters M2489 detector system) using a C8 Supelco Discovery Bio Wide column (Sigma Aldrich). The solvents were the following: phase A (95% water, 0.1% TFA) and phase B (5% acetonitrile, 0.1% TFA). The purity of the substrates was determined by analytical HPLC (C8 Supelco Discovery Bio Wide analytical column, Sigma Aldrich). Finally, molecular weights were confirmed with a high-resolution mass spectrometer (Waters LCT Premier XE high-resolution mass spectrometer, electrospray ionization, and TOF detector) and LC-MS QDa (Waters e2695,2489 UV-vis detector, Acquity QDa detector).
Kinetic assays
All kinetic experiments were performed using a spectrofluorometer (SpectraMax Gemini XPS plate reader, Molecular Devices) and analyzed using SoftMax® software (Molecular Devices), Microsoft Excel®, and GraphPad Prism®.
The granzyme B assay buffer contained 50 mm Tris-base, 100 mm NaCl, 25 mm CaCl2, 0.1% Tween, pH 7.4. The buffer was prepared at room temperature, and the enzyme kinetic studies were performed at 37 °C.
HyCoSuL screening was utilized to define the substrate specificity of granzyme B
P4, P3, and P2 fluorogenic substratesublibraries (33 μm) with P1-Asp, with the general structure Ac-P4-X-X-Asp-ACC, Ac-X-P3-X-Asp-ACC, and Ac-X-X-P2-Asp-ACC (where P4-2 is a defined amino acid and X is an equimolar mixture of natural amino acids with Nle replacing Met and Cys) were scanned with GrB (48 nm). The final volume of the reaction mixture was 30 μL. The substrate hydrolysis (fluorescence release in the presence of enzyme) was monitored in real time at λex= 355 nm and λem= 460 nm for a minimum of 30 min, but for each substrate, only the linear part of the curve was used to calculate the reaction rate (RFU/s, relative fluorescent units per second). Each sublibrary was screened twice, and the average value was calculated. For each sublibrary, the amino acid with the highest number of RFU/s was set to 100%, and the other amino acids were adjusted accordingly. The results are presented as a bar diagram (P4–P2) (Fig. S3). Additionally, the adjusted values for amino acids sharing similar chemical properties were added and then divided by the number of events, showing the GrB preferences for a particular molecular feature. Data are presented as a heatmap (Fig. 1A). In the same manner, the P5 library (Ac-P5-X-E-X-D-ACC, 166 μm final concentration) was screened using 12 nm of GrB and analyzed as above. Data are presented as a violin diagram of GrB amino acid preferences.
GrB activity against classic substrates (mono-, di-, tri-, tetra-, penta-, and hexapeptides)
The substrates TJ2–7 and TJ30–44 (166 μm) were placed in a well of a 384-well plate (Corning®, opaque) and treated with 29 μL of 12 nm GrB. 100 μm Ac-D-ACC, Ac-PD-ACC, Ac-EPD-ACC, Ac-IEPD-ACC, Ac-AIEPD-ACC, and Ac-AAIEPD-ACC were treated with 28 nm GrB, and 166 μm TJ46-56 substrate was placed in a well of a 384-well plate (Corning®, opaque) and treated with 29 μL of 12 nm GrB. Substrate hydrolysis (fluorescence release in the presence of enzyme) was monitored in real time at λex= 355 nm and λem= 460 nm for a minimum of 30 min. The linear region of the curve was used to calculate the reaction rate. RFU/s values were then adjusted to 100%, and the data are presented as bar graphs, with each value being the average from 3 replicates. For selected substrates, kcat/Km values were calculated using substrates in a range of 666–5 μm for TJ7, TJ40, TJ43, and TJ44 or 333–2.6 μm for TJ47, TJ49, TJ52, and TJ55 with the Michaelis-Menten equation in GraphPad Prism. Data are presented as the mean with standard deviation and represent at least 2 independent experiments.
Quenched fluorescent substrate sequence optimization and kcat/Km value determination
IQF substrates TJ65–76 (6.6 μm) were screened with GrB (2.1 nm final concentration) in 96-well Corning® opaque plates with λex= 355 nm and λem= 460 nm for a minimum of 30 min. Data were analyzed as above (“GrB activity against classic substrates”) and are presented as a bar chart. To calculate the kcat/Km value, serial dilutions of TJ65 or TJ71 in a range from 14.81 μm to 0.86 μm were placed into a 96-well Corning® black plate, and the enzyme at 12 nm in assay buffer was added. The fluorescence increase was monitored as a function of time for at least 30 min. The linear region of the curve was used to calculate the reaction rate using the Michaelis-Menten equation in GraphPad Prism. Data are presented as the mean with standard deviation and represent at least 2 independent experiments.
GrB activity against qTJ71
The optimal excitation/emission wavelengths were determined to be λex= 540 nm and λem= 580 nm. Serial dilutions of qTJ71 from 33.3 μm to 1.95 μm were placed in a 96-well Corning® black plate, and 12 nm enzyme was added. The fluorescence increase over time was monitored for 30 min. The linear region of the curve was analyzed to calculate the reaction rate and analyzed using the Michaelis-Menten equation in GraphPad Prism. Data are presented as the mean with standard deviation and represent at least 2 independent experiments.
qTJ71 specificity
Kinetic measurements were performed using black Corning® plates with λex= 540 nm and λem= 580 nm in assay buffers optimized for particular enzymes: 1) 50 mm Tris base, 100 mm NaCl, 25 mm CaCl2, 0.1% Tween, pH 7.4, for granzyme B; 2) 10 mm PIPES, 100 mm NaCl, 1 mm EDTA, 10% sucrose, pH 7.4, for caspases 3–7; and 3) 10 mm sodium citrate, 10 mm PIPES, 100 mm NaCl, 1 mm EDTA, 10% sucrose, pH 7.4, for caspases 8–10. qTJ71 (1 μm) was placed into 96-well Corning® black plates, and after the addition of the enzyme (120 nm), the fluorescence increase over time was monitored for 30 min. Only the linear regions of the curves were used for calculations. The kcat and Km values were calculated using GraphPad Prism and Microsoft Excel software. All measurements were repeated three times, and the data presented are averages from these replicates.
Synthesis of P1 fluorogenic substrate library
To synthesize peptides with the sequence Ac amino acids ACC, we used the method described previously (16, 20), with small modifications. A total of 100 mg of Rink AM resin (0.74 mmol/g) was added to a glass reaction vessel containing DCM, and the mixture was stirred gently once every 10 min for 30 min. The mixture then was filtered and washed 3 times with DMF. The Fmoc-protecting group was then removed by treatment with 20% (v/v) piperidine in DMF for 5 min, 5 min, and then 25 min. After each cycle, the resin was washed with DMF and then filtered, and after the final deprotection, it was washed carefully six times with DMF. Afterward, 2.5 eq of Fmoc-ACC-OH was preactivated with 2.5 eq of HOBt and 2.5 eq of DICI in DMF for 5 min, and that mixture was poured into the resin. The reaction was gently stirred for 24 h at room temperature. The resin was then washed three times with DMF, and the Fmoc-ACC-OH coupling reaction was repeated using 1.5 eq of the above reagents to improve the yield of the coupling reaction. After this, the reaction mixture was removed, and the resin was washed five times with DMF, affording ACC-resin (compound 1). As previously described, to remove the Fmoc-protecting group, 20% (v/v) piperidine in DMF was added to the reaction vessel, and three deprotection cycles were conducted (5 min, 5 min, and 25 min). The mixture was filtered and washed with DMF after each cycle to afford H2N-ACC-resin (compound 2). Next, 2.5 eq of Fmoc-P1-OH was preactivated with 2.5 eq of HATU and 2.5 eq of 2,4,6-trimethylpiridine in DMF, and this mixture was added to the reaction vessel with 2. The reaction was carried out overnight at room temperature with gentle stirring. The resin then was washed with DMF three times, and the Fmoc-P1-OH coupling reaction was repeated using 1.5 eq of the reagents to increase the reaction yield and to obtain Fmoc-P1-ACC-resin (compound 3). The Fmoc-protecting group was removed as described above, and the peptide chain elongation was continued using 2.5 eq of Fmoc-aa-OH (aa, amino acid), 2.5 eq of HOBt, and 2.5 eq of DICI until the desired peptide length was obtained. Each time, the coupling reaction or deprotection efficiency was tested using the ninhydrin test. At the end of this process, the N terminus was protected with an acetyl group using 5 eq of AcOH, 5 eq of HBTU, and 5 eq of DIPEA in DMF with gentle stirring for 60 min at room temperature to afford Ac-peptide-ACC-resin (compound 4). The resin then was washed five times with DMF, three times with DCM, and twice with MeOH and dried over P2O5 overnight. Next, peptide cleavage from the dry resin was performed with a mixture of cold TFA–TIPS–H2O (v/v/v, 95:2.5:2.5) for an hour at room temperature with gentle stirring once every 10 min. The peptide then was precipitated from cold Et2O for an hour and centrifuged. The supernatant was removed, while the pellet was recrystallized from an additional portion of cold Et2O and centrifuged. The product, as a light-yellow pellet, was dried overnight at room temperature, purified by HPLC, and then lyophilized. Product purity was confirmed by analytical HPLC and high-resolution mass spectrometry (HRMS) analysis. All substrates were stored as 20 mm solutions at −80 °C until use.
Synthesis of internally quenched substrates
A total of 100 mg of Rink AM resin was added to a glass reaction vessel or 48-well reaction vessel containing DCM and stirred gently once every 10 min for 30 min, and then the mixture was filtered and washed 3 times with DMF. The Fmoc-protecting group was then removed by treatment with 20% (v/v) piperidine in DMF for 5 min, 5 min, and 25 min. After each cycle, the piperidine was removed and the resin was washed with DMF three times. Prior to transfer to the glass reaction vessel, 2.5 eq of Fmoc-Lys (Dnp)-OH was preactivated for 5 min by mixing with 2.5 eq of HOBt and 2.5 eq of DICI in DMF. The reaction then was stirred for 12 h at room temperature. The reaction mixture was filtered and the resin was washed three times with DMF, and the Fmoc-protecting group was removed from Fmoc-Lys (Dnp)-resin (5) as above using 20% (v/v) piperidine in DMF to afford H2N-Lys (Dnp)-resin (6). Next, 2.5 eq of Fmoc-AA-OH (P3′ position) was preactivated with 2.5 eq HOBt and 2.5 eq DICI in DMF for 5 min and added to a reaction vessel with H2N-Lys (Dnp)-resin. The reaction was carried out for three hours with gentle stirring. Fmoc-AA-Lys (Dnp)-resin (7) then was filtered and washed with DMF three times. Afterward, the Fmoc-protecting group was removed as above, and the peptide chain elongation was continued until the peptide with the desired length was obtained (using 2.5 eq of HOBt and 2.5 eq of DICI as the coupling reagents). Next, 2.5 eq of Fmoc-ACC-OH was attached to H2N-peptide-Lys (Dnp)-resin (8) using 2.5 eq of HOBt and 2.5 eq of DICI, and the mixture was stirred for 24 h at room temperature. Resin was then washed three times with DMF, and the reaction was repeated using 1.5 eq of the above reagents to improve the yield of the coupling of Fmoc-ACC-OH to (8). The final product, H2N-ACC-peptide-Lys (Dnp)-resin (9), was cleaved from the Fmoc-ACC-peptide-Lys (Dnp)-resin-protecting group and precipitated from Et2O as above. The product, a bright-yellow pellet, was dried overnight at room temperature, purified by HPLC, and lyophilized. Product purity was confirmed by HPLC and HRMS analysis. All substrates were stored as 20 mm solutions at −80 °C until use.
Fluorogenic and biotinylated activity-based probe and inhibitor synthesis with TJ55.5, Cy5-Gly-Nva-Ile-Glu-Oic-AspP (OPh)2, TJ55.Bt, Biot-Nva-Ile-Glu-Oic- AspP (OPh)2, and TJ55i, Ac-Nva-Ile-Glu-Oic- AspP (OPh)2
In the first step, H2N-Nva-Ile-Glu-Oic resin was synthesized using solid-phase synthesis on 2-chlorotityl chloride (CTC) resin. CTC resin (100 mg) was activated with 5 ml of anhydrous DCM and gently stirred once every 5 min for 30 min. The mixture then was filtered, and the filtrate was washed three times with DCM. 3 eq of Fmoc-Oic-OH (P2 position) then was preactivated with 5 eq of DIPEA in anhydrous DCM and added to the glass reaction vessel with the CTC resin. The reaction was stirred gently in an argon atmosphere for 12 h at room temperature. After that, the reaction mixture was filtered, and the resin was washed three times with DCM and two times with DMF to afford Fmoc-Oic-CTC (10). The Fmoc-protecting group was then removed from 10 using 20% (v/v) piperidine in DMF for 5, 5, and 25 min, as described above, to afford H2N-Oic-CTC (11). Peptide elongation with other amino acids (2.5 eq of P3, Fmoc-Glu (O-tBu)-OH; P4, Fmoc-Ile-OH; and P5, Fmoc-Nva-OH) was achieved with a series of coupling (with 2.5 eq of HOBt and 2.5 eq of DICI) and deprotection (20% PIP–DMF) reactions to obtain H2N-Nva-Ile-Glu (O-tBu)-Oic-CTC (11). Next, Boc-Gly-OH was introduced using 2.5 eq of HOBt and 2.5 eq of DICI (for TJ55.5) (12) or D-biotin was introduced using 3 eq of DIPEA (for TJ55.Bt) (13). After the coupling of the last amino acid, the resin was washed five times with 2-ml portions of DMF, five times with 2-ml portions of DCM, and three times with 2-ml portions of MeOH and dried over P2O5 overnight. The crude Boc-Gly-Nva-Ile-Glu (O-tBu)-Oic-COOH (12) and Biot-Nva-Ile-Glu (O-tBu)-Oic-COOH (13) peptides were cleaved from the resin using a mixture of TFE–AcOH–DCM (v/v/v, 1:1:8) for one hour. The supernatant was collected in a round-bottom flask, a portion of hexane was added, and the volatile products were evaporated under reduced pressure. Obtained compound 13 was dissolved in a mixture of ACN–H2O (v/v, 3:1), frozen, and lyophilized. The purities of 12 and 13 were confirmed with analytical HPLC, and the molecular weights were determined with HRMS.
The aspartic acid phosphonate warhead (14) was synthesized according to Mahrus et al., with slight modifications (13). In a round-bottom flask, 8.64 g of Meldrum's acid was mixed with 28 ml of triethyl orthoformate and stirred under reflux at 80 °C. After three hours, the mixture was concentrated under reduced pressure to obtain a crude compound as a brown oil, which was stored at +4 °C for 24 h (it solidified at that temperature). The solid material (2.37 g) was vigorously stirred for one hour in 2 m HCl (34 ml). The suspension was then partitioned between diethyl ether and brine (3 × 100 ml). The organic phase was collected and dried over MgSO4, and the volatile components were evaporated under reduced pressure to give the product as a yellow solid. Next, to a 50-ml round-bottom flask was added formyl Meldrum's acid (1.3 g, 7.5 mmol), p-nitrobenzyl alcohol (1.2 g, 7.5 mmol), and toluene (15 ml). The reaction mixture was stirred under reflux in an oil bath for 30 min, and afterward, the solvent was removed under reduced pressure. The obtained crude oil (1.5 g) was used in the next step without purification. In the next step, in a 50-ml round-bottom flask, 1.5 g of p-nitrobenzyl formylacetate, 0.9 g of benzyl carbamate, 2.1 ml of triphenyl phosphite, and 2.1 ml of glacial acetic acid were stirred for 1 h under reflux at 80 °C, and then the solvent was removed under reduced pressure. The obtained product (15) was purified over SiO2 (2:3 ethyl acetate–hexane), and 1.2 g of pure compound was obtained. Afterward, 15 was treated with 30% HBr in AcOH (10 ml) with stirring for 1 h at room temperature. The solvent was then evaporated under reduced pressure, and the material was purified by reverse-phase HPLC. Compound 16 was analyzed by HRMS and stored at −80 °C until use.
In the next step, 2.5 eq (25 mg) of the aspartic acid phosphonate warhead [H2N-AspP (OPh)2; 16] was coupled with 12 or 13 using 2.5 eq of HATU and 2.5 eq of collidine in DMF. Each reaction was carried out for 2 h at room temperature. Afterward, each mixture was diluted in ethyl acetate and washed 2 times with 5% NaHCO3 solution, 2 times with 5% citric acid solution, and 2 times with brine. The organic phase was collected and dried over MgSO4, and the volatile components were evaporated under reduced pressure. Next, the deprotection of the aspartic acid carboxyl group from the warhead was performed by hydrogenolysis (H2, Pd/C, and MeOH), and global deprotection was achieved in TFA–DCM (1:1, v/v; with 3% TIPS for 30 min) to afford H2N-Gly-Nva-Ile-Glu-Oic-Asp-AspP (OPh)2 (17) and Biot-Nva-Ile-Glu-Oic-Asp-AspP (OPh)2 (18). Crude 17 was purified by HPLC, analyzed using HRMS, and lyophilized. In the next step, 1 eq of Cy5-NHS was dissolved in DMF, 5 eq of DIPEA was added, and the mixture was stirred occasionally. This mixture was then added to pure, dry 17. The reaction was carried out at room temperature for four hours with gentle stirring. The reaction progress was monitored by analytical HPLC. The final products, Cy5-Gly-Nva-Ile-Glu-Oic-AspP (OPh)2 (18) and Biot-Nva-Ile-Glu-Oic-AspP (OPh)2 (19), were purified on HPLC, analyzed using HRMS, lyophilized, dissolved to a concentration of 20 mm in DMSO, and stored at −80 °C until use.
To obtain the inhibitor, the amine group of 11 was acetylated with 5 eq of AcOH, 5 eq of DIPEA, and 5 eq of HBTU in DMF. The reaction was carried out at room temperature for one hour. The remaining steps were analogous to the synthesis of the activity-based probe.
qTJ71 synthesis (BHQ2-Nva-Ile-Glu-Oic-Asp-Phe-Gly-Arg-Lys-Cy3)
In the first step, a peptide derivative was obtained using solid-phase synthesis with Rink AM resin in the same manner as that described above; however, a quencher was coupled to the N terminus. For that coupling, 12.5 mg of BHQ2 (BHQ-2000S) was preactivated with 2.5 eq of DIPEA in DMF and added to a glass reaction vessel containing H2N-Nva-Ile-Glu (O-tBu)-Oic-Asp (O-tBu)-Phe-Gly-Arg (Pbf)-Lys (Boc)-resin. The reaction was carried out at room temperature overnight, and the product was cleaved from the resin and purified as above. The purity of the obtained BHQ2-Nva-Ile-Glu (O-tBu)-Oic-Asp (O-tBu)-Phe-Gly-Arg (Pbf)-Lys (Boc)-NH2 (19) was confirmed by analytical HPLC, and the molecular weight of the compound was determined by HRMS. The protecting groups on the side chains of the amino acids in (19) were removed with TFA–DCM (1:1, v/v; supplemented with 3% TIPS) in 30 min, and after deprotection, the volatile products were evaporated. 1 eq of Cy3-NHS then was attached to the side chain of Lys with 2.5 eq of DIPEA in DMF. The final product was purified with HPLC to afford BHQ2-Nva-Ile-Glu-Oic-Asp-Phe-Gly-Arg-Lys-Cy3 (20). The compound was analyzed by HRMS and stored as a 20 mm solution at −80 °C until use.
Inhibition kinetic assay
The inhibitory constants of TJ55i, TJ55.5, and TJ55.Bt were measured using Opaque Corning® plates (Corning) with a spectrofluorometer (Spectramax Gemini XPS, Molecular Devices) and analyzed using SoftMax software (Molecular Devices) and Microsoft Excel®. The measurements were performed in an assay buffer containing 50 mm Tris base, 100 mm NaCl, 25 mm CaCl2, 0.1% Tween, pH 7.4, at 37 °C with excitation and emission wavelengths of 355 and 460, respectively, with a cutoff of 455 nm. To each reaction well was added 20 μl of an inhibitor at various concentrations (375 nm–32 nm for TJ55i, 2 μm–175 nm for TJ55.Bt, and 166 μm–10 μm for TJ55.5), followed by 20 μl of substrate (TJ71, 24 μm) and then 60 μl of GrB (130 nm). The inhibitory efficiency and potency were calculated using kobs (app)/I (apparent second-order rate constant for inhibition) under pseudo-first-order conditions. kobs/I values were calculated taking into account the Km value for the assay substrates (S) using the equation kobs/I = kobs (app)/I × [1+ ([S]/Km)].
Dot-blot analysis of quenched fluorescent substrate and activity-based probe fluorescence and selectivity
Recombinant caspase-3, -4, -5, -6, -7, -8, -9, or -10, or granzyme B (120 nm), each was incubated with 1 μm the quenched fluorescent substrate probe (qTJ71) or classic activity-based probe (TJ55.5) in an assay buffer (50 mm Tris base, 100 mm NaCl, 25 mm CaCl2, 0.1% Tween, pH 7.4, for granzyme B; 10 mm PIPES, 100 mm NaCl, 1 mm EDTA, 10% sucrose, pH 7.4, for caspases 3–7; 1 m sodium citrate, 10 mm PIPES, 100 mm NaCl, 1 mm EDTA, 10% sucrose, pH 7.4, for caspases 8–10) for 20 min at 37 °C. 10 μl of each solution then was dotted onto a dry nitrocellulose membrane (Bio-Rad, 0.2 μm) and allowed to dry for 5 min The membrane then was scanned with a SapphireTM biomolecular imager with two lasers dedicated to Cy5 (658 nm) and Cy3 (520 nm), and the data were analyzed using Azure Biosystems software. The experiment was repeated 3 times.
Cell culturing
SU-DHL1, YT, NALM-6, and Jurkat-T cells were cultured in 75-cm3 flasks in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin; SEMK2 and MG63 were cultured on culture plates in Eagle's minimum essential medium (EMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin; and MDA-MB-231 cells were cultured on culture plates in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin under incubating conditions at 37 °C, 90% relative humidity, and 5% CO2. The primary concentration for optimal growth was 1 × 105 cells/ml, and the media were changed every other day. For all further experiments, cells with a low passage number (up to 30) were taken ∼24 h after the last seeding.
Western blot analysis of the cell lysates
To prepare the cell lysates, 1 × 107 cells/ml were lysed with 1 ml of cold lysis buffer containing 15 mm KCl, 5 mm MgCl2, 10 mm Tris-HCl, 0.5% (v/v) Triton X-100. The cells then were sonicated (2.0 kJ for 10 s) and immediately treated with an activity-based probe (TJ55.5 or TJ55.Bt) for the indicated time (0 min to 1 h) at 37 °C. The reaction was stopped by the addition of 30 μl of 3× SDS–DTT to 60 μl of sample (lysate plus probe), followed by boiling at 95 °C for 5 min. 30 μl of sample then was loaded on 4–12% Bis-Tris Plus gel (Life Technologies); electrophoresis was performed at 200 V for 30 min, followed by transfer to a nitrocellulose membrane (0.2 μm, Bio-Rad, 1620112) for 60 min at 10 V. The membrane then was blocked with 2% BSA in TBS-T (TBS with 0.1% [v/v] Tween-20) for 60 min at room temperature, and when TJ55.Bt was used, the membranes were treated with fluorescent streptavidin conjugate (Streptavidin, Alexa FluorTM 647 conjugate, no. S21374, Invitrogen) (1:10,000) for 1 h at room temperature, followed by rabbit recombinant monoclonal granzyme B antibody (Abcam, ab208586), and incubated overnight at 4 °C. The membrane then was incubated with the secondary antibody (Alexa Fluor® 532 goat anti-rabbit IgG [H + L], Invitrogen, A11009) for 30 min at room temperature. The fluorescence was scanned at wavelengths of 649 nm for AF647 or Cy5 and 554 nm for AF532 using a SapphireTM biomolecular imager and Azure Biosystems software. The blots were then analyzed using Image Studio software.
Flow cytometry
All experiments were performed using CyFlow Cube6 (Sysmex) and analyzed in Sysmex software. MDA-MB-231 cells (1 × 105 cells/ml) in culturing media were incubated with (or without, as a control) 250 nm of qTJ71 for 1 h. YT cells (1 × 105 cells/ml) were incubated in cell culture media with (or without, as a control) 250 nm of qTJ71 for 1 h. All samples then were spun down, fixed with 4% PFA for 20 min, and washed twice with DPBS. The cells then were treated with 10% BSA in DPBS for 30 min, and the samples were spun down and treated with anti-GrB primary antibody (rabbit recombinant monoclonal granzyme B antibody, Abcam, ab208586) and incubated overnight at 4 °C. After washing, DPBS secondary antibody (Alexa Fluor® 532 goat anti-rabbit IgG [H + L], Invitrogen, A11009) was added, and the mixture was incubated for 1 h at 37 °C. The samples then were washed twice with DPBS, resuspended in 200 μl of DPBS, and analyzed with CyView® software (Sysmex) using blue and red lasers and FL2 (580-nm) and FL4 (675-nm) filters. The experiment was repeated three times.
Data availability
The data used and/or analyzed during the current study are contained within the manuscript and supporting information. The raw data will be available from the corresponding author upon reasonable request.
Supplementary Material
This article contains supporting information.
Author contributions—T. J., S. K., and P. K. data curation; T. J. and P. K. formal analysis; T. J., S. K., P. I. B., and P. K. validation; T. J., S. K., and P. K. investigation; T. J., S. K., G. S., M. D., and P. K. methodology; T. J. and P. K. writing-original draft; T. J., S. K., D. K., S. J. S., S. L., J. K., J. S., N. B., G. S., M. D., P. I. B., and P. K. writing-review and editing; D. K., S. J. S., J. K., J. S., N. B., G. S., M. D., P. I. B., and P. K. resources; P. K. conceptualization; P. K. supervision; P. K. funding acquisition; P. K. visualization; P. K. project administration; D. K., S. L., and N. B. contributed enzymes; S. J. S. and J. K. acquisition of data; J. S. contributed cell lines, data analysis; M. D. contributed HyCoSuL library; P. I. B. contributed enzymes.
Funding and additional information—This work was supported by the HOMING Programme, a Grant Project of the Foundation for Polish Science, funded by the European Union under agreement no. 2016-3/24. The publication fee was covered with the Statutory Funds of Wroclaw University of Science and Technology (subvention number: 8201003902). P. K. is a beneficiary of L'Oreal Poland and Polish Ministry of Science and Higher Education scholarships.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- NK
- natural killer cells
- CTLs
- cytotoxic T lymphocytes
- Grs
- granule-associated enzymes
- GrB
- granzyme B
- ACC
- 7-amino-4-carbamoylmethylcoumarin
- HyCoSuL
- Hybrid Combinatorial Substrate Library
- RFU
- relative fluorescence units
- AM
- amide
- DICI
- diisopropylcarbodiimide
- HBTU
- O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HATU
- 2-(1-H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium
- HOBt
- N-hydroxybenzotriazole
- DCM
- dichloromethane
- MeOH
- methanol
- Et2O
- diethyl ether
- AcOH
- acetic acid
- DIPEA
- N,N-diisopropylethylamine
- TIPS
- triisopropylsilane
- DMF
- N,N′-dimethylformamide
- HRMS
- high-resolution mass spectrometry
- CTC
- 2-chlorotityl chloride.
References
- 1. Caligiuri M. A. (2008) Human natural killer cells. Blood 112, 461–469 10.1182/blood-2007-09-077438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vivier E., Tomasello E., Baratin M., Walzer T., and Ugolini S. (2008) Functions of natural killer cells. Nat. Immunol. 9, 503–510 10.1038/ni1582 [DOI] [PubMed] [Google Scholar]
- 3. Jenne D. E., and Tschopp J. (1989) Granzymes: a family of serine proteases in granules of cytolytic T lymphocytes. Curr. Top. Microbiol. Immunol. 140, 33–47 10.1007/978-3-642-73911-8_4 [DOI] [PubMed] [Google Scholar]
- 4. Masson D., and Tschopp J. (1987) A family of serine esterases in lytic granules of cytolytic T lymphocytes. Cell 49, 679–685 10.1016/0092-8674(87)90544-7 [DOI] [PubMed] [Google Scholar]
- 5. Horiuchi K., Saito S., Sasaki R., Tomatsu T., and Toyama Y. (2003) Expression of granzyme B in human articular chondrocytes. J. Rheumatol. 30, 1799–1810 [PubMed] [Google Scholar]
- 6. Jans D. A., Jans P., Briggs L. J., Sutton V., and Trapani J. A. (1996) Nuclear transport of granzyme B (fragmentin-2). Dependence of perforin in vivo and cytosolic factors in vitro. J. Biol. Chem. 271, 30781–30789 10.1074/jbc.271.48.30781 [DOI] [PubMed] [Google Scholar]
- 7. Pinkoski M. J., Hobman M., Heibein J. A., Tomaselli K., Li F., Seth P., Froelich C. J., and Bleackley R. C. (1998) Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood 92, 1044–1054 10.1182/blood.V92.3.1044.415k12_1044_1054 [DOI] [PubMed] [Google Scholar]
- 8. Voskoboinik I., Whisstock J. C., and Trapani J. A. (2015) Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 10.1038/nri3839 [DOI] [PubMed] [Google Scholar]
- 9. Poreba M., Strozyk A., Salvesen G. S., and Drag M. (2013) Caspase substrates and inhibitors. Cold Spring Harb. Perspect. Biol. 5, a008680 10.1101/cshperspect.a008680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., and Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 10.1074/jbc.272.29.17907 [DOI] [PubMed] [Google Scholar]
- 11. Andrade F., Roy S., Nicholson D., Thornberry N., Rosen A., and Casciola-Rosen L. (1998) Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity 8, 451–460 10.1016/S1074-7613(00)80550-6 [DOI] [PubMed] [Google Scholar]
- 12. Zhang D., Beresford P. J., Greenberg A. H., and Lieberman J. (2001) Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc. Natl. Acad. Sci. U S A 98, 5746–5751 10.1073/pnas.101329598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahrus S., and Craik C. S. (2005) Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell-mediated lysis of target cells. Chem. Biol. 12, 567–577 10.1016/j.chembiol.2005.03.006 [DOI] [PubMed] [Google Scholar]
- 14. Rut W., and Drag M. (2016) Human 20S proteasome activity towards fluorogenic peptides of various chain lengths. Biol. Chem. 397, 921–926 10.1515/hsz-2016-0176 [DOI] [PubMed] [Google Scholar]
- 15. Van de Craen M., Van den Brande I., Declercq W., Irmler M., Beyaert R., Tschopp J., Fiers W., and Vandenabeele P. (1997) Cleavage of caspase family members by granzyme B: a comparative study in vitro. Eur. J. Immunol. 27, 1296–1299 10.1002/eji.1830270535 [DOI] [PubMed] [Google Scholar]
- 16. Kasperkiewicz P., Poreba M., Snipas S. J., Parker H., Winterbourn C. C., Salvesen G. S., and Drag M. (2014) Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. U S A 111, 2518–2523 10.1073/pnas.1318548111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poreba M., Kasperkiewicz P., Snipas S. J., Fasci D., Salvesen G. S., and Drag M. (2014) Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ. 21, 1482–1492 10.1038/cdd.2014.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rotonda J., Garcia-Calvo M., Bull H. G., Geissler W. M., McKeever B. M., Willoughby C. A., Thornberry N. A., and Becker J. W. (2001) The three-dimensional structure of human granzyme B compared to caspase-3, key mediators of cell death with cleavage specificity for aspartic acid in P1. Chem. Biol. 8, 357–368 10.1016/S1074-5521(01)00018-7 [DOI] [PubMed] [Google Scholar]
- 19. Poreba M., Salvesen G. S., and Drag M. (2017) Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat. Protoc. 12, 2189–2214 10.1038/nprot.2017.091 [DOI] [PubMed] [Google Scholar]
- 20. Maly D. J., Leonetti F., Backes B. J., Dauber D. S., Harris J. L., Craik C. S., and Ellman J. A. (2002) Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J. Org. Chem. 67, 910–915 10.1021/jo016140o [DOI] [PubMed] [Google Scholar]
- 21. Poreba M., Rut W., Groborz K., Snipas S. J., Salvesen G. S., and Drag M. (2019) Potent and selective caspase-2 inhibitor prevents MDM-2 cleavage in reversine-treated colon cancer cells. Cell Death Differ. 26, 2695–2709 10.1038/s41418-019-0329-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oleksyszyn J., and Powers J. C. (1994) Amino acid and peptide phosphonate derivatives as specific inhibitors of serine peptidases. Methods Enzymol. 244, 423–441 [DOI] [PubMed] [Google Scholar]
- 23. Oleksyszyn J., and Powers J. C. (1991) Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry 30, 485–493 10.1021/bi00216a026 [DOI] [PubMed] [Google Scholar]
- 24. Prakash M. D., Bird C. H., and Bird P. I. (2009) Active and zymogen forms of granzyme B are constitutively released from cytotoxic lymphocytes in the absence of target cell engagement. Immunol. Cell Biol. 87, 249–254 10.1038/icb.2008.98 [DOI] [PubMed] [Google Scholar]
- 25. Rut W., Poręba M., Kasperkiewicz P., Snipas S. J., and Drąg M. (2018) Selective substrates and activity-based probes for imaging of the human constitutive 20S proteasome in cells and blood samples. J. Med. Chem. 61, 5222–5234 10.1021/acs.jmedchem.8b00026 [DOI] [PubMed] [Google Scholar]
- 26. Law R. H. P., Lukoyanova N., Voskoboinik I., Caradoc-Davies T. T., Baran K., Dunstone M. A., D'Angelo M. E., Orlova E. V., Coulibaly F., Verschoor S., Browne K. A., Ciccone A., Kuiper M. J., Bird P. I., Trapani J. A., et al. (2010) The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 468, 447–451 10.1038/nature09518 [DOI] [PubMed] [Google Scholar]
- 27. Kasperkiewicz P., Altman Y., D'Angelo M., Salvesen G. S., and Drag M. (2017) Toolbox of fluorescent probes for parallel imaging reveals uneven location of serine proteases in neutrophils. J. Am. Chem. Soc. 139, 10115–10125 10.1021/jacs.7b04394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poreba M., Szalek A., Kasperkiewicz P., Rut W., Salvesen G. S., and Drag M. (2015) Small molecule active site directed tools for studying human caspases. Chem. Rev. 115, 12546–12629 10.1021/acs.chemrev.5b00434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bird C. H., Christensen M. E., Mangan M. S., Prakash M. D., Sedelies K. A., Smyth M. J., Harper I., Waterhouse N. J., and Bird P. I. (2014) The granzyme B-Serpinb9 axis controls the fate of lymphocytes after lysosomal stress. Cell Death Differ. 21, 876–887 10.1038/cdd.2014.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used and/or analyzed during the current study are contained within the manuscript and supporting information. The raw data will be available from the corresponding author upon reasonable request.