

Abstract
Angiogenesis-mediated neovascularization in the eye is usually associated with visual complications. Pathological angiogenesis is particularly prominent in the retina in the settings of proliferative diabetic retinopathy, in which it can lead to permanent loss of vision. In this study, by bioinformatics analyses, we provide evidence for elevated expression of actin-binding protein PFN1 (profilin1) in the retinal vascular endothelial cells (VECs) of individuals with proliferative diabetic retinopathy, findings further supported by gene expression analyses for PFN1 in experimentally induced abnormal retinal neovascularization in an oxygen-induced retinopathy murine model. We observed that in a conditional knockout mouse model, postnatal deletion of the Pfn1 gene in VECs leads to defects in tip cell activity (marked by impaired filopodial protrusions) and reduced vascular sprouting, resulting in hypovascularization during developmental angiogenesis in the retina. Consistent with these findings, an investigative small molecule compound targeting the PFN1–actin interaction reduced random motility, proliferation, and cord morphogenesis of retinal VECs in vitro and experimentally induced abnormal retinal neovascularization in vivo. In summary, these findings provide the first direct in vivo evidence that PFN1 is required for formation of actin-based protrusive structures and developmental angiogenesis in the retina. The proof of concept of susceptibility of abnormal angiogenesis to small molecule intervention of PFN1–actin interaction reported here lays a conceptual foundation for targeting PFN1 as a possible strategy in angiogenesis-dependent retinal diseases.
Keywords: Profilin1 (PFN1), angiogenesis, retina, diabetic retinopathy, tip cells, filopodia, oxygen-induced retinopathy (OIR), cytoskeleton, vascularization, vascular endothelial cells (VECs), profilin, actin
Angiogenesis, a process of new blood vessel generation from pre-existing blood vessels, is a fundamental process for vascular expansion during development and vascular regeneration in tissue repair. However, aberrant angiogenesis lies at the heart of a wide range of pathologies including those involving the retina such as proliferative diabetic retinopathy (PDR), wet age-related macular degeneration, and retinopathy of prematurity. In particular, PDR is one of the leading causes of blindness worldwide. Elevated signaling of vascular endothelial growth factor (VEGF) is most often involved in neovascularization-associated eye diseases. Although antiangiogenic therapies targeting VEGF signaling are generally effective in some of these disease contexts, spontaneous or acquired resistance to anti-VEGF therapies continues to be a significant problem in a substantial percentage of the patient population (∼30% in DR and ∼10% in wet age-related macular degeneration) because of involvement of other proangiogenic mediators in disease progression (1–7). Another issue with VEGF signaling inhibition is that VEGF is a trophic factor for vascular endothelial cells (VECs) and neuronal cells (8, 9). Hence, identification of alternative mediators of retinal angiogenesis could potentially pave the way for novel therapeutic avenues in neovascularization-associated ocular diseases.
During angiogenesis, a certain population of VECs are selected in response to proangiogenic growth factors (such as VEGF and fibroblast growth factor) to become tip cells that extend actin-rich filopodial protrusions and assume a highly motile phenotype to invade the stroma and guide the vascular outgrowth. In a highly orchestrated manner, VECs trailing behind (known as stalk cells) proliferate to elongate the vessel sprouts (10). Dynamic regulation of actin cytoskeletal assembly is essential for cell migration. A common structural feature of major actin-assembly factors, such as proteins belonging to the WASP (e.g. N-WASP (Neural Wiskott-Aldrich syndrome protein) and WAVE (WASP-associated verprolin-homology protein)), Mena/VASP (e.g. Mena (mammalian enabled), VASP (vasodilator-stimulated phosphoprotein), and Evl (enabled/VASP-like)), and formin families (we collectively refer to these actin nucleation-promoting and elongating factors as NPEFs from hereon), is the presence of multiple polyproline (PLP) domains. These domains allow NPEFs to interact with G-actin–binding protein PFN1 (profilin1, the abundant isoform of the PFN family of proteins that acts as a major nucleotide exchange factor of G-actin) and in turn enhance their ability to recruit polymerization-competent actin monomers to stimulate actin polymerization (reviewed in Ref. 11). PFN1's associations with N-WASP and WAVE are required for efficient filopodia and actin cluster formations in cultured cells (12, 13). PFN1 is involved in VASP-mediated regulation of actin polymerization at the leading edge of migrating cells, membrane protrusion, and cell motility in vitro (14). WAVE's cooperative action with VASP on Arp2/3-mediated actin assembly in vitro and cell motility during Caenorhabditis elegans embryogenesis requires a functional PLP domain of VASP, further implicating PFN1's role in this process (15). PFN1 also synergizes with formin-family protein Dia2 (diaphanous 2) to promote gastrulation cell movement in zebrafish embryos (16). These actions of PFN1 are consistent with reduced migratory ability and of most normal cells including VECs upon either depletion of the overall expression or disruption of either of two key molecular interactions (actin or PLP ligands) of PFN1 (17–20). Loss of function (LOF) of PFN1 also negatively impacts cell proliferation in vitro and in vivo (17, 21). Collectively, these findings demonstrate that PFN1 is a common denominator of multiple actin-assembly pathways and underscore PFN1 as an important mediator of actin-regulated processes in cells.
We previously demonstrated that depleting PFN1 expression leads to defects in migration, invasion, and morphogenetic and sprouting activities of VECs in vitro and ex vivo (17, 18, 22), but these findings have not been translated in vivo. Other studies established a role of a site-specific post-translational modification of PFN1 in stimulating angiogenesis in vivo (23, 24). Specifically, proangiogenic growth factor (such as VEGF) stimulation of VECs elicits Src-mediated phosphorylation of PFN1 on its Tyr129 residue. This phosphorylation stimulates PFN1–actin interaction and VEC motility, as well as angiocrine factor production through stabilization of hypoxia-inducible factor-1, a major activator of VEGF transcription. Blockade of Tyr129 phosphorylation ability of PFN1 led to reduced angiogenesis associated with tissue repair and tumor progression but interestingly had no discernible effect on developmental angiogenesis in mouse retina (23, 24). However, because abrogating Tyr129 phosphorylation does not inhibit the basal actin interaction or interfere with other molecular interactions (such as those involving PLP ligands or membrane phosphoinositides) of PFN1, it could not rule out PFN1's role in physiological angiogenesis. Therefore, the impact of complete LOF of PFN1 on angiogenesis in vivo, whether physiological or pathological, remains unclear.
In this study, we for the first time provide evidence for PFN1 up-regulation in retinal VECs in PDR patients, PFN1's important role in tip cell activity and vascular sprouting during developmental retinal angiogenesis, and the ability of a small molecule investigative compound targeting the PFN1–actin interaction to attenuate abnormal retinal neovascularization in vivo.
Results
PFN1 expression is elevated in the retina in proliferative diabetic retinopathy in human patients, as well as in experimentally induced abnormal retinal neovascularization in a murine model
Previous studies established PFN1's link with vascular-related pathology in diabetes. Specifically, PFN1 expression is elevated in VECs in diabetes and in response to factors that promote diabetes-associated pathology (e.g. atherosclerosis, advanced glycation end products) in vitro and in vivo (including human patients). PFN1 elevation exacerbates VEC dysfunction in diabetic milieu, and conversely, PFN1 depletion offers protection against oxidative damage, inflammation, obesity-induced glucose tolerance, and VEC dysfunction in diabetic animals in vivo (25–34). Whether PFN1 expression is altered in retinal vasculature in PDR (a later stage of DR that is associated with abnormal retinal neovascularization) is not known. We performed bioinformatics analyses to probe for Pfn1 gene expression in a previously published study that compared transcriptome profiles of CD31+ VECs from fibrovascular membranes from patients with PDR (seven patients) versus four control retinal samples (Gene Expression Omnibus Database data set GSE94019) (35). Although the sample size was small, these analyses revealed a highly significant (false discovery rate = 8.3E-09; p = 1.98E-11) 10.2-fold increase in the average mRNA expression of endothelial PFN1 in the retina of PDR patients relative to the retina of normal subjects (Fig. 1A), suggesting that the Pfn1 gene is transcriptionally up-regulated in retinal VECs in PDR patients.
Figure 1.
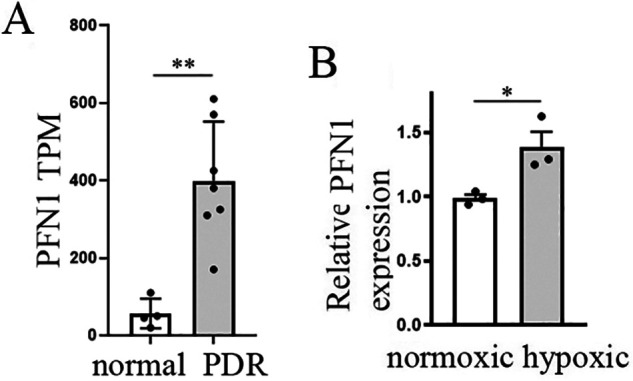
PFN1's association with PDR and abnormal retinal angiogenesis. A, Transcripts per kilobase million (TPM) values for PFN1 in CD31+ ECs from diabetic (n = 7) versus control (n = 4) human retinal samples (based on GEO data set GSE94019) were calculated using Salmon and DESeq2 (p = 1.98E-11; FDR = 8.64E-9; log2FC = 3.35; data are the means ± S.D.). B, quantitative RT-PCR analyses of retinal PFN1 expression relative to the arithmetic mean of three housekeeping genes (GAPDH, Rps26, and Actb) in newborn mice relative to control mice at P17 (data are the means ± S.E. of 3 replicates/mouse; n = 3 mice/group).
We next asked whether the clinical observation of elevated PFN1 expression in retinal VECs in PDR patients is reproducible in animal models. Most genetic and drug-induced mouse models of diabetes reproduce the early stage of DR (basement membrane thickening and vascular leakage) but fail to evoke neo-angiogenesis in the retina as occurs in PDR. Murine oxygen-induced retinopathy (OIR) is a well-established experimental model that captures the hypoxia-induced proliferative phase of abnormal neovascularization in the retina and has been widely used to model abnormal angiogenesis aspects of PDR and retinopathy of prematurity in humans (36–38). In this model, newborn mice are subjected to hyperoxia (75% O2) from P7–P12 and then returned to normoxic condition for an additional 5 days before the animals are sacrificed for retinal evaluation on P17. Exposure to high O2 leads to loss of immature retinal vessels and retards normal vascular development resulting in vaso-obliteration (VO) in the central retina, whereas the hyperoxia-to-normoxic transition creates a relative ischemia triggering both normal vascular regrowth and abnormal neovascularization (NV) (see Fig. 6). Quantitative RT-PCR analyses of P17 mouse retina showed ∼40% higher PFN1 mRNA level in the OIR pups relative to the control pups that were maintained in a normoxic condition throughout (Fig. 1B). These data provide evidence for elevation of retinal PFN1 expression associated with abnormal angiogenesis in vivo, further qualitatively supporting our clinical correlation data in PDR patients (possible reasons underlying the quantitative discrepancy between the order of PFN1 up-regulation in the OIR model versus PDR patients are discussed later).
Figure 6.

Small molecule inhibitor of PFN1–actin interaction attenuates angiogenesis associated with oxygen-induced retinopathy. A, lower (left panels) and higher (right panels) magnification microphotographs of FITC-coupled BS-1 lectin-stained retina of P17-OIR C57BL/6JRj mice after intravitreal injection at P12 and P14 of DMSO (1 µl) (upper panels) or C2 (1 µl of 500 µm solution) (lower panels). VO areas are highlighted in red, and total retinal area is in white. The scale bars in the left and right panels indicate 80 and 50 μm, respectively. B and C, quantification of VO (B) and NV (C) in P17 control versus C2-treated retinal at-mounts). D and E, representative images (D; scale bar, 100 μm) and quantification (E) of vascular density in P17 control versus C2-treated retinal flat mounts in non–vaso-obliterated and tuft-free areas. A total of 11 retinae from two independent experiments in each group were analyzed. *, p < 0.05; NS, not significant (Mann–Whitney t test).
PFN1 is important for tip cell activities and vascular sprouting during developmental retinal angiogenesis
Unlike humans, developmental retinal vascularization in rodents occurs postnatally. During this process, retinal vessels grow radially outward in a reproducible temporal and spatial pattern predominantly through sprouting angiogenesis mechanism involving tip cell invasion and stalk cell proliferation from the center of the retina forming the superficial vascular plexus that reaches the retinal periphery by P8–P10 developmental age. The exact time course of normal vascular development in mice is, however, strain-specific. Subsequent vertical sprouting of superficial capillaries first gives rise to the deep and then intermediate vascular plexi of the retina (36, 38). Our previous study showed that in an ex vivo setting, aortic segments from mice with VEC-specific heterozygous deletion of the Pfn1 gene at the embryonic level display reduced sprouting angiogenesis compared with those isolated from their WT Pfn1 littermates. However, we were unable to obtain live birth of mice with homozygous deletion of the Pfn1 gene, suggesting that the total loss of PFN1 in VECs at the embryonic level might be lethal (22). To circumvent this problem, we generated Pfn1flox/flox:CDH5-CreERT mice to allow for tamoxifen-induced expression of Cre (under the control of VE-cadherin promoter) and excision of the Pfn1 gene selectively in VECs (Cre-negative mice of any PFN1 genotypes served as the control). In our first set of experiments, the pups were subjected to three successive rounds of tamoxifen injection on days P2–P4, and P9 retinae were harvested for staining with FITC-lectin; a subset of retinae of each genotype were also stained with rhodamine-phalloidin to assess the impact of endothelial loss of PFN1 on actin cytoskeleton. Tamoxifen-induced deletion of the Pfn1 gene was confirmed by PCR of genomic DNA prepared from the kidney (a vascular-rich organ) using primer sets 1 and 3, which produces a 700-bp amplicon when the floxed exon1 of Pfn1 is cleaved (Fig. 2A). By P9, the superficial vascular plexus reached the periphery of the retina in 6 of 6 (100%) and 4 of 6 (66%) of Pfn1WT and Pfn1KO retinae, respectively. Although there was considerable zonal variation in the vascular density, the overall vascular density of Pfn1KO retinae was significantly lower (by ∼45 ± 15%) than Pfn1WT retinae (Fig. 2, B and C). These data provided an initial indication of PFN1-dependent changes in retinal vascular architecture. The average fluorescent intensity of phalloidin (labels F-actin) in the lectin-positive regions of the retina was also less intense in Pfn1KO retinae compared with Pfn1WT retinae, indicative of a reduced level of actin polymerization upon the loss of PFN1 in retinal VECs in vivo (Fig. 2, D and E) (note that this average phalloidin fluorescence value is not affected by vascular density). These data suggest that PFN1 promotes actin assembly in retinal VECs in vivo.
Figure 2.

Gene disruption of Pfn1 in VECs affects actin cytoskeleton in retinal vasculature. A, schematic of floxed Pfn1 allele, PCR primer locations for genotyping, and PCR confirmation of cleaved flox-Pfn1 band in CDH5-Cre–positive mice following treatment with tamoxifen. B and C, representative FITC-lectin–stained flat-mounted retina (B) along with quantification of lectin-stained blood vessel density (C) in mice with indicated genotypes (n = 6 retinae/group; scale bar, 100 μm; **, p < 0.01). D and E, representative images of FITC-lectin and rhodamine-phalloidin–stained flat-mounted retina (D) along with quantification of F-actin intensity in lectin-positive retinal blood vessels (E) in mice with the indicated genotypes (n = 6 retinae/group; scale bar, 100 μm; **, p < 0.01). The bottom subpanels in D are the magnified representations of the insets indicated by the box in the middle panels. Avg, average.
P9 retina is developmentally too late to capture the active angiogenesis in the superficial layer of the retina. Therefore, to better capture the effect of LOF of PFN1 on tip cell behavior and sprouting angiogenesis during vascular development, in our subsequent set of experiments, we disrupted Pfn1 gene function by injecting tamoxifen in the pups on days P0–P2 and characterized angiogenesis phenotypes in retinae isolated from Pfn1WT and Pfn1KO mice at an earlier developmental time point (P6). These experiments showed a robust effect of Pfn1 deletion on developmental retinal angiogenesis. Essentially, Pfn1KO retinae exhibited hypovascularization marked by reduced radial outgrowth of blood vessels (suggesting a defect in tip cell invasion) as measured by the overall VEC-covered retinal area (by ∼36 ± 13%), average number of vascular sprouts/field (by ∼62 ± 16%), and vascular density (by ∼55 ± 5%) compared with PFN1WT retinae (Fig. 3, A–E). Because there was no significant difference in the average weight between P6 Pfn1WT (4.07 ± 0.12g) and Pfn1KO (4.12 ± 0.14g) mice, retinal hypovascularization in Pfn1KO mice is not a consequence of global growth defect. Filopodia (a hallmark of tip cells) are crucial for sensing of the environment and guided tip cell invasion at the growing vascular front during sprouting angiogenesis. Filopodia in tip cells can be easily visualized in whole-mount lectin staining of retina, as done previously in a number of studies (39–41). By lectin/phalloidin co-staining of retina, we initially confirmed that lectin-positive protrusive structures in tip cells are also F-actin–positive and are therefore bona fide filopodial structures (Fig. S1). Consistent with reduced ability of PFN1-deficient VECs in filopodial generation in response to acute stimulation of bradykinin (an activator of Cdc42 that is commonly used as an agent to induce filopodia) in a cell-culture setting (Fig. S2), we found filopodial abundance to be dramatically affected by LOF of PFN1 in vivo as marked by a 70% reduction in the average number of filopodia/tip cell in Pfn1KO retinae when compared with Pfn1WT retinae (Fig. 3, F and G). The average length of filopodia in Pfn1KO retinae was also found to be 60% shorter than in Pfn1WT retinae (Fig. 3H). Collectively, these findings demonstrate that PFN1 is a key mediator for filopodial protrusions of tip cells and developmental retinal vascularization.
Figure 3.

PFN1 is an important mediator for tip cell activity and developmental retinal angiogenesis. A, representative images of whole-mount FITC-lectin stained P6 retinae from Pfn1WT and Pfn1KO mice (scale bar, 500 μm). B, quantification of retinal VEC coverage area in P6 Pfn1WT versus Pfn1KO mice (n= 12 retinae/group). C–E, representative close-up images of the growing vascular front in Pfn1WT versus Pfn1KO (C; scale bar, 50 μm) along with association quantifications of sprouts (per 20× field; D) and vascular density (E) in the two groups (n = 12 retinae/group). F–H, representative high-magnification (60×) images of angiogenic front showing tip cells (arrow) and filopodial structures (arrowhead) (F), along with associated quantifications of the filopodial abundance/tip cell (G; scale bar, 10 μm) and the average length of filopodial length (H) in the two groups (60 and 30 filopodia were analyzed for length calculations for Pfn1WT and Pfn1KO retinae, respectively; **, p < 0.01).
Small molecule antagonists of PFN1–actin interaction inhibit retinal angiogenesis in vitro and in vivo
Although PFN1 has been linked to angiogenesis, because of a lack of commercially available inhibitor of PFN1, no studies to date have explored the suitability of targeting PFN1 to mitigate abnormal angiogenesis relevant in disease contexts. Mutagenesis-induced disruption of actin interaction of PFN1 diminishes capillary morphogenesis of VECs in vitro (17), demonstrating the importance of PFN1–actin interaction in angiogenesis. By computationally guided biochemical screen, we recently identified two small molecules with nearly identical structures (C1, 8-(3-hydroxyphenyl)-10-(4-methylphenyl)-2,4,5,6,7,11,12-heptaazatricyclo[7.4.0.03,7]trideca-1(13),3,5,9,11-pentaen-13-ol; and C2, 8-(3-hydroxyphenyl)-10-phenyl-2,4,5,6,7,11,12-heptaazatricyclo[7.4.0.03,7]trideca-1(13),3,5,9,11-pentaen-13-ol) that are capable of reversing PFN1's effect on actin polymerization in vitro and reducing PFN1–actin interaction in cells (42). In this study, we further demonstrated that these first-generation antagonists of PFN1–actin interaction are noncytotoxic (even up to a concentration of 100 µm), and consistent with the effects of depletion of PFN1, treatment of these compounds lead to reduced actin polymerization, migration, proliferation, and angiogenic ability of human dermal microvascular EC in vitro and mouse aortic segments ex vivo. Importantly, the antimigratory effect of these compounds was abrogated in cells when PFN1–actin interaction was genetically disrupted through H119E substitution of PFN1, further suggesting that the mechanism of action of these compounds involved inhibition of PFN1–actin interaction. Whether in vivo angiogenesis is susceptible to any of these investigative compounds is not known. We initially tested the effect of one of these compounds (C2) at two different concentrations (50 and 100 µm) on angiogenic activity of primary microvascular HmRECs in vitro. Depending on the concentration, treatment with C2 resulted in 40–50% reduction in Matrigel-induced cord formation (an in vitro morphogenetic assay that predicts the angiogenic competency of VECs) ability of HmREC relative to the DMSO (vehicle control)–treated condition (Fig. 4, A and B). These data were in qualitative agreement with a dramatic 70% inhibition of cord formation by HmREC in the setting of transient knockdown (by ∼90%) of PFN1 expression (Fig. 4, C–E). Furthermore, C2 had a prominent antimigratory effect in human retinal endothelial cells (HmRECs) as indicated by a ∼60% reduction of the average speed of migration in a random motility assay (Fig. 4F), in addition to causing a profound reduction in cell proliferation (Fig. 4G). To further assess the biological activity of C2 in vivo, we performed Matrigel plug angiogenesis assay and compared new blood vessel formation in subcutaneously implanted Matrigel in Balb/C mice treated with either C2 or DMSO (vehicle control). We found that treatment with C2 resulted in a significant 70% reduction in the number of functional blood vessels (identified by the presence of luminal red blood cells) when compared with DMSO-treated animals (Fig. 5), thus confirming an antiangiogenic activity of C2 in vivo.
Figure 4.

Effect of small molecule inhibitor of PFN1–actin interaction on migration, proliferation, and angiogenic ability of human retinal VECs. A and B, representative images (A) and quantification (B) of Matrigel-induced cord formation of primary HmRECs after overnight treatment with either DMSO or 50 or 100 μm C2 (data summarized from three independent experiments; scale bar, 100 μm). C–E, representative images (D) and quantification (E) of cord formation of control versus PFN1-siRNA transfected primary HmRECs (data summarized from two independent experiments with three technical replicates; scale bar, 100 μm). Immunoblot in C confirms PFN1 knockdown with GAPDH blot serving as the loading control. F, box-and-whisker plot showing the relative migration speed of HmRECs in C2 (50 μm) versus DMSO-treated conditions (based on analyses of ∼50–60 cells/group from two independent experiments). G, line graph showing the effect of 50 μm C2 on proliferation of HmRECs over a period of 7-day treatment (data summarized from three experiments). In all panels, ** represents p < 0.01. cont, control; si, siRNA.
Figure 5.

Antiangiogenic activity of small molecule inhibitor of PFN1–actin interaction in vivo. Shown are representative images (A; 20× magnification) and quantification (B; based on 10× field images) of angiogenesis in Matrigel plugs in Balb/c mice treated with C2 versus DMSO (arrows in A indicate blood vessel formation; **, p < 0.01; n = 4 mice/group; scale bar, 50 μm).
Based on up-regulation of PFN1 mRNA expression in OIR retina versus their room air littermate controls, we hypothesized that increased PFN1 function might contribute to the pathological situation. Therefore, we tested the effect of C2 on retinal neovascularization in the OIR model. In these experiments, C2 was intravitreally administered twice (on days P12 and P14) during the neovascularization phase in OIR pups (control OIR pups were treated with equivalent concentration of DMSO). Several biological parameters contribute to natural variances in the OIR experiments, including include intralitter as well as litter-to-litter variations in weight (and hence size of the retina) of the pups, intrinsic animal-to-animal differences in the degree of vaso-obliteration and neovascular response, and the injection efficiency of any test compound (when applicable). In our experiments, we controlled for the mean weight of the pups to be comparable between the DMSO versus C2-treated groups. Flat-mount staining of P17 retina showed comparable average percentages of vaso-obliteration area between the two treatment settings, whereas the average percentages of neovascularization area in C2-treated pups was ∼35% lower than that of DMSO-treated pups (Fig. 6, A–C). Although development of superficial vascular plexus is complete several days before the time window of hyperoxia-to-normoxia transition and small molecule administration, because neovascular regions originate from the superficial vasculature, we asked whether reduced neovascularization in C2-treated animals could be due to any possible detrimental effect of C2 on the pre-existing vasculature during this time window. To test this, we analyzed the vascular plexus in the outer retina of DMSO- and C2-treated animals, focusing on the regions outside of the tufts (neovascular regions), and found no difference in the vascular density in these regions between the two groups (Fig. 6, D and E). These data likely suggest that C2 administration does not negatively impact pre-existing vasculature and therefore argue against C2-induced inhibition of pathological neovascularization secondary to its effect on precursor blood vessels. Overall, these findings demonstrate a proof of concept for the ability of small molecule antagonist of the PFN1–actin interaction to attenuate pathological retinal angiogenesis in vivo.
Discussion
In this study, we report several novel findings. First, we demonstrated elevated PFN1 expression at the transcriptional level in retinal VECs in PDR patients and experimentally validated changes in retinal PFN1 expression in an animal model that mimics the pathological angiogenesis aspect of PDR. Second, using a novel inducible VEC-specific Pfn1 KO mouse model, we provided the first in vivo evidence of PFN1's requirement for formation of actin-based protrusive structures (such as filopodia) and physiological retinal angiogenesis in a mammalian context. Third, we demonstrated for the first time a proof-of-concept of susceptibility of abnormal retinal angiogenesis to a small molecule antagonist of PFN1–actin interaction.
Although PFN1 up-regulation in the OIR model (increased by 40%) is qualitatively consistent with the clinical finding of elevated PFN1 expression in retinal VECs in PDR patients (by ∼10-fold), the fold changes were very different. This is not unexpected for several reasons. First, mouse models do not always accurately recapitulate the complexity of human diseases. Specifically, in our context, the OIR model only represents the abnormal angiogenesis aspect of the retina and does not recapitulate the entire disease spectrum of DR. Given that PFN1 expression is elevated in VECs in diabetes in general (26), retinal PFN1 up-regulation in the PDR patients would likely be more than what one might encounter in the OIR setting. Second, there might be differences in the biology of mouse versus human retina. Third, methodological differences (transcriptome analyses of whole retina versus VECs isolated from retina) could also account for the quantitative discrepancy between the two systems. In particular, because our analysis is based on whole retina samples, we likely underestimated the extent of PFN1 up-regulation in the neovascular regions. Immunostaining experiments in the future will be necessary to better assess hypoxia-induced changes in PFN1 expression at the protein level in the neoangiogenic regions of the retina.
Although PFN1 is generally regarded as a promoter of actin assembly, the effect of perturbation of PFN1 on actin cytoskeleton has been shown to be context-specific that cannot be readily explained by a single biochemical function of PFN1. For example, PFN1 depletion led to reduced F-actin content in VECs and chondrocytes (18, 21). Consistent with these findings, other studies provided evidence for an increase in the overall F-actin content and/or actin stress-fiber assembly in various cultured cells (Chinese hamster ovary cells, VECs, and breast cancer cells) in response to overexpression of PFN1 (43–45). Contrasting these findings, cultured mouse embryonic fibroblasts displayed an increase in F-actin content upon PFN1 depletion (46), aligning with a similar finding in Dictyostelium amebae when both PFN isoforms (PFN1 and the minor isoform PFN2) were genetically deleted (20). Appreciable reduction in the overall F-actin content (as judged by the phalloidin fluorescence) in retinal VECs in PFN1 KO mice demonstrated in this study is consistent with our previous findings in other types of human VECs (umbilical vein-derived, dermal microvascular) (18, 42) aligning with PFN1's role as a promoter of actin polymerization in vivo. In our previous studies, the most dramatic effect of either depletion of expression or small molecule inhibition of PFN1 on actin cytoskeletal architecture in VECs was the major reduction in actin stress fibers. The actions of two key actin-assembly factor families, which utilize PFN1 to stimulate actin polymerization (diaphanous and Ena/VASP), are involved in the formation of actin stress fibers (47, 48). Therefore, we speculate that LOF of PFN1 negatively impacts actin-assembly pathways associated with these cytoskeletal factors in VECs.
Defects in developmental retinal vascularization in Pfn1 KO mice as demonstrated in our study is in stark contrast to the findings from a previous study where mice harboring VEC-specific expression of nonphosphorylatable Y129F mutant of PFN1 did not appear to exhibit any such phenotype (23). We speculate that the apparent discrepancy is most likely because the Y129F substitution not only does not abrogate the basal actin interaction, but it also has no influence on PLP interactions of PFN1 and therefore does not represent a molecular equivalent of complete LOF of PFN1. Our studies revealed a major inhibitory effect of LOF on PFN1 on vascular branching and sprouting during retinal vascular development. It will be interesting to determine in future studies whether tip cell specification and its underlying signaling pathways (such as Delta/Notch or Robo/slit) are impacted by PFN1 function in VECs.
Reduced radial outgrowth of retinal blood vessels in Pfn1KO mice is suggestive of a defect in tip cell invasion induced by loss of PFN1, although we cannot rule out the additional possibility of PFN1-dependent changes in stalk cell proliferation. Our in vivo observation of filopodial defect in tip cells Pfn1KO retinae is in agreement with the present findings of reduced bradykinin-induced generation of filopodia in VECs in 2D cell culture, as well as our prior findings in 3D cultures of breast cancer cells in the setting of PFN1 knockdown (49). Interestingly, a previous study demonstrated that loss-of-function mutation of chickadee (the Drosophila homolog of PFN1) results in shorter (in agreement with our studies) but more abundant filopodia in cultured primary neurons (50). A recent study has uncovered an unconventional function of autophagy-related protein ATG9 in its ability to interact with and regulate cellular localization of chickadee and Ena/VASP proteins. This study also found increased filopodial abundance in cultured primary neurons derived from ATG9-mutant embryo relative to WT control cells, although a functional link to Pfn underlying this phenotype was not established (51). There could be several possible but not mutually exclusive explanations underlying the filopodial abundance-related difference between our study and the ones seen in cultured Drosophila neurons. First, filopodial abundance is influenced not only by the frequency of initiation but also by the frequency of retraction (determines the stability) of filopodial protrusions. Clearly, the difference in cell-substrate adhesion, a key parameter for the stability of protrusions, between in vivo and in vitro settings could potentially account for such differences. Second, filopodial protrusions are generated by convergent extension of Arp2/3-nucleated actin filaments by the action of formin and Ena/VASP proteins. PFN1 promotes actin polymerization by formin and Ena/VASP proteins but inhibits actin nucleation activity of the Arp2/3 complex (46). Because chickadee is the only isoform of Pfn in Drosophila, LOF of chickadee may promote Arp2/3-mediated actin nucleation and as a result increase the frequency of initiation of filopodial protrusions. However, in a mammalian context, this effect might be less prominent or might even be overcome by a compensatory action of PFN2, the minor isoform of PFN (also expressed in VECs). Third, inherent cell type–specific differences in the expression of actin-nucleating and elongating factors might also contribute to differences in PFN1-related filopodial phenotypes in different experimental systems. Live imaging of filopodial dynamics in VECs in different culture conditions and assessment of localization of other important players of filopodial protrusions in control versus PFN1-depleted condition in future studies will reveal further molecular insights into PFN1's control of filopodial dynamics in tip cells.
An important aspect of this study is the first in vivo demonstration of angiogenesis modulation by small molecule intervention of PFN1–actin interaction, laying a general conceptual foundation for targeting PFN1 as a possible strategy in angiogenesis-dependent diseases. We demonstrated antiangiogenic effect of our compound in both subcutaneously implanted Matrigel plug and OIR angiogenesis assays. The compound proved to be more effective in suppressing angiogenesis in the Matrigel plug (∼70% inhibition) than in the OIR assay (∼35% inhibition). This could be due to differences in the bioavailability and injection frequency (limited by practical constraints) of the compound, the cellular environment (influences the accessibility of the compound to the desirable target cells), and driving force in angiogenesis between the two settings. Although we acknowledge that a 35% inhibition of retinal angiogenesis by C2 is lower than the typical 50–70% inhibition elicited by the gold-standard VEGF-signaling blockers in the OIR setting as reported in other studies (52, 53), these initial results are still promising, given that our compound is only a first-generation inhibitor of the PFN1–actin interaction with likely suboptimal potency (as judged by the requirement for tens of micromolar concentration of the compound for eliciting biological effects in vitro). Our findings justify future endeavors to identify improved classes of PFN1 inhibitors for antiangiogenic purpose, either as a stand-alone agent or in combination with other currently available therapies.
Clinical evidence of PFN1 up-regulation in VECs in diabetes (26) and PDR patients taken together with genetic and pharmacological proofs of concept of PFN1 dependence for retinal angiogenesis in vivo as demonstrated herein suggest that PFN1 could be a potential target in DR. In addition to excessive angiogenesis, another key hallmark change in retinal vasculature in DR is vascular leakage and focal hemorrhage in the retina caused by breakdown of the blood–retinal barrier. We previously showed that PFN1 depletion confers resistance to VEGF-induced junctional disruption in cultured human umbilical vein VECs (18). Furthermore, others have demonstrated that elevating PFN1 expression weakens the barrier function of retinal VECs in vitro as well as in diabetic animals, and conversely, PFN1 depletion improves the barrier function (32). These findings suggest that PFN1 may also be an important regulator of junctional integrity of retinal VECs. Although it is beyond the scope of the present work, it will be also interesting to investigate in the future whether small molecule inhibition of PFN1–actin interaction confers protection against vascular leakage in an actual preclinical model of DR.
Experimental procedures
Cell culture and transfection
HmRECs (ACBRI 191, Cell Systems, Kirkland, WA) were cultured in complete classic medium with serum and culture boost (4Z0-500, Cell Systems) and antibiotics (100 units/ml penicillin and 100 µg/ml streptomycin). For knockdown studies, the cells were transfected with 50 nm of either smart-pool control or PFN1 siRNA for 72 h as before (54). For filopodial induction, VECs, grown sparsely on glass coverslips, were serum-starved overnight and acutely stimulated with 50 ng/ml bradykinin (Sigma) for 30 min before fixation and staining with phalloidin as done before (18).
Cell migration and proliferation assays
For single-cell migration experiments, HmRECs were sparsely plated in a 24-well plate coated with type I collagen (Millipore, Burlington, MA) overnight. The cells were treated overnight with either C2 (50 or 100 µm) or DMSO (vehicle control) prior to imaging. Time-lapse images of randomly migrating cells were collected using a 10× objective for 120 min at 1-min time intervals using MetaMorph (Universal Imaging, Bedford Hills, NY). The centroid of the cell nucleus was tracked using ImageJ, and the average speed of migration was computed on a cell-by-cell basis as before (54). For cell proliferation assay, 10,000 HmRECs were plated in triplicate in a 24-well plate, and proliferation was assessed by time-course analyses of cell count at subsequent days.
Cord formation assay
The cord formation assay was performed as described previously (42). Briefly, HmRECs were plated on polymerized Matrigel and allowed to adhere prior to treatment with either test compound or vehicle control. After 16 h, cord formation was assessed by phase-contrast microscopy. Total cord length was quantified using the angiogenesis plugin of ImageJ.
Protein extraction and immunoblotting
The cell lysates were prepared by a modified radioimmune precipitation assay buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% (v/v) Nonidet P-40, 5% (v/v) glycerol), 1 mm EDTA, 50 mm NaF, 1 mm sodium pervanadate, and protease inhibitors supplemented with 6× sample buffer diluted to 1× with a final SDS concentration in the lysis buffer equivalent to 2%. Conditions for the various antibodies were as follows: monoclonal PFN1 (Abcam, Cambridge, MA; catalog no. ab124904; 1:3000), monoclonal GAPDH (Developmental Studies Hybridoma, University of Iowa; DSHB-hGAPDH-2G7; 1:100).
Animal studies
All animal experiments were conducted in compliance with an approved institutional animal care and use committee protocol according to University of Pittsburgh Division of Laboratory Animal Resources guidelines.
Generation of Pfn1 KO mice
Pfn1flox/flox mice (kindly provided by Dr. Reinhard Fassler, Max Planck Institute,) were crossed with CDH5-Cre-ERT2 mice (kindly provided by Dr. Ralf Adams, Max Planck Institute) to generate Pfn1flox/flox:CDH5-Cre-ERT2 mice. The pups were subjected to daily intragastric injection of 50 µl of 2 mg/ml tamoxifen (to excise PFN1 gene in Cre-positive animals) over a course of 3 days (from either P0–P2 or P2–P4), and retinae (P6 or P9) were collected from age-matched WT and homozygous Pfn1 KO mice for histological staining. PCR-based genotyping of mice was done as previously described (22).
Mouse retinal vasculature labeling
Following sacrificing of pups, the eyes were harvested, fixed in 4% paraformaldehyde at room temperature for 2 h, and then washed with PBS. Following PBS wash, retinae were dissected, fixed again in 4% paraformaldehyde for 30 min at room temperature, and then washed in PBS two more times. The retinae were then blocked in 10% goat serum, 0.4% Triton X-100 in 1 m Tris-HCl (pH 7.4) overnight at 4 °C. The retinae were then washed three times with Pblec buffer (1% Triton X-100 in PBS) and then stained overnight in Pblec with 1:25 FITC-lectin from a stock solution of 5 mg/ml (Sigma, catalog no. L9381). After overnight staining, the retinae were washed three times with PBS-T (0.4% Triton X-100 in PBS). In selected experiments, following PBS-T wash, the retinae were incubated with PBST containing 1:50 rhodamine-phalloidin (Thermo Fisher, catalog no. R415) for 2 h at room temperature and then washed three times with PBS-T. Following PBS-T washes, the retinae were washed three times in PBS, then transferred to glass microscope slide, and further dissected into four petal shapes, flat-mounted, and sealed under a glass coverslip in mounting medium. Fluorescent images were taken using a Nikon A1 confocal microscope, and multiple 20× images fields were stitched to reconstruct the entire retinal images; additional 60× fields were imaged to closely examine filopodia formation. Vascular density was calculated by using the angiogenesis plugin for ImageJ and calculating the total length of all cords. Sprouts per field was determined by counting the number of tip cells per 20× field (a cell was deemed a tip cell if it extended from a vessel and did not connect to another branching vessel). VEC area was determined by estimating the circular area covered by superficial blood vessels in the retina.
Matrigel plug angiogenesis assay
Six-week-old Balb/c mice were anesthetized and injected subcutaneously in the flanks with 0.5 ml of reduced growth factor Matrigel (R&D Systems, 3533-010-02) supplemented with 500 ng of basic fibroblast growth factor and 200 µm of either DMSO or compound C2 (a PFN1–actin inhibitor; described under “Results”) in the contralateral flanks. The animals were subjected to intraplug injection every other day with 25 µl of either 200 µm DMSO or C2 in PBS until they were euthanized on day 7 for extraction of plugs and histological staining of plugs with hematoxylin and eosin. Number of functional blood vessels (red blood cell-positive) were scored from three histological sections at multiple fields per animal.
OIR assay
C57BL/6JRj mouse pups along with their mothers were exposed to 75% oxygen from P7–P12 as previously reported (54). On P12, mice were returned to room air, put with nursing mothers, and injected intravitreally with 0.5% DMSO in PBS as vehicle or C2 (50 μm in 0.5% DMSO). Pups were sacrificed on P17 by CO2 inhalation by CO2 inhalation, and the retinae were dissected from the intact globe and stained with FITC-coupled isolectin B4 (Life Technologies, Inc.). Images were acquired on a DM5500 Leica microscope (Leica). VO and NV areas were calculated on images using MetaMorph software.
Whole retina RT-PCR assay
Retinae from P17 mouse pups grown in normoxic conditions or in hyperoxic conditions from P7 to P12 were dissected in RNase-free conditions (n = 3 in each condition). Total RNA was isolated with Nucleospin RNAII (Macherey Nagel, Duren, Germany). Single-stranded cDNA was synthesized from total RNA using the QuantiTect reverse transcription kit (Qiagen). Real-time PCR was performed by using cDNA and SYBR Green Gene Expression Master Mix (Thermo Fisher Scientific) and the following primers (0.5 pmol/ml; IDT DNA): PFN1 (sense, CTG TCA CCA TGA CTG CCA AG, and antisense, GAT CAA ACC ACC GTG GAC A), GAPDH (sense, GGT GAA GGT CGG TGT GAA CG [sense]; and antisense, CTC GCT CCT GGA AGA TGG TG); Rps26 (sense, AAG TTT GTC ATT CGG AAC ATT; and antisense, AGC TCT GAA TCG TGG TG); β-Actin (Actb) (sense, AAG GCC AAC CGT GAA AAG AT; and antisense, GTG GTA CGA CCA GAG GCA TAC). PCRs were performed in 45 cycles of 15 s at 95 °C and 45s at 60 °C. The data were normalized to the arithmetic mean of three housekeeping genes (GAPDH, Rps26, and Actb) readouts and expressed as relative to the normoxic group values.
Gene expression analyses
Fastq files from SRP097696 archive were downloaded using SRA Toolkit, version 2.9.2. Transcripts from the Ensembl, version 93 transcriptome (hg38) were quantified with Salmon, version 0.14.0. Gene-level expression was calculated using tximport, version 1.12.0, and differential gene expression was calculated with DESeq2, version 1.24.0. Quality control principle component and heat map plots demonstrated two potential outlier samples (SRR5197978 and SRR5197982), which were removed prior to final differential expression analysis. Dot plots of transcripts per kilobase million values were created with ggplot2, version 3.1.1.
Statistics
Statistical tests were performed with either one-way analysis of variance followed by Tukey's post hoc test, Student's t test, or Mann–Whitney test (for small sample size) when appropriate. p values less than 0.05 were considered to be statistically significant.
Data availability
All experimental data are contained in the article. Curated endothelial transcriptome data of PDR patients (GSE94019) are available at the Gene Expression Omnibus Database.
Supplementary Material
Acknowledgments
We acknowledge the Center for Biological Imaging at the University of Pittsburgh for the use of the Nikon confocal microscope purchased with provided through National Institute of Health Grant S10OD019973.
This article contains supporting information.
Author contributions—D. G., L. V., A. A., Z. G., and D. B. data curation; D. G. validation; D. G., L. V., A. A., X. G., and P. R. investigation; D. G ., L. V., and A. A. methodology; D. G., D. B., X. G., and P. R. writing-original draft; Z. G., D. B., D. K., and X. G. formal analysis; J. S., D. K., X. G., and P. R. conceptualization; J. S. and D. K. resources; X. G. and P. R. supervision; X. G. and P. R. writing-review and editing; P. R. funding acquisition; P. R. project administration.
Funding and additional information—This work was supported by National Institute of Health Grants R01CA108607 (to P. R.) and R01GM108340 (to D. K.), by a developmental pilot grant from the Hillman Cancer Center (University of Pittsburgh), by INSERM, and by LabEx Lifesenses Grant ANR-10-LABEX-65 (to X. G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PDR
- proliferative diabetic retinopathy
- DR
- diabetic retinopathy
- VEC
- vascular endothelial cell
- OIR
- oxygen-induced retinopathy
- VEGF
- vascular endothelial growth factor
- PLP
- polyproline
- LOF
- loss of function
- Pn
- postnatal day n
- VO
- vaso-obliteration
- NV
- neovascularization
- HmREC
- human retinal endothelial cell
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Paulus Y. M., and Sodhi A. (2017) Anti-angiogenic therapy for retinal disease. Handb. Exp. Pharmacol. 242, 271–307 10.1007/164_2016_78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown D. M., and Regillo C. D. (2007) Anti-VEGF agents in the treatment of neovascular age-related macular degeneration: applying clinical trial results to the treatment of everyday patients. Am. J. Ophthalmol. 144, 627–637 10.1016/j.ajo.2007.06.039 [DOI] [PubMed] [Google Scholar]
- 3. Eghøj M. S., and Sørensen T. L. (2012) Tachyphylaxis during treatment of exudative age-related macular degeneration with ranibizumab. Br. J. Ophthalmol. 96, 21–23 10.1136/bjo.2011.203893 [DOI] [PubMed] [Google Scholar]
- 4. Forooghian F., Cukras C., Meyerle C. B., Chew E. Y., and Wong W. T. (2009) Tachyphylaxis after intravitreal bevacizumab for exudative age-related macular degeneration. Retina 29, 723–731 10.1097/IAE.0b013e3181a2c1c3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McAuley A. K., Sanfilippo P. G., Hewitt A. W., Liang H., Lamoureux E., Wang J. J., and Connell P. P. (2014) Vitreous biomarkers in diabetic retinopathy: a systematic review and meta-analysis. J Diabetes Complications 28, 419–425 10.1016/j.jdiacomp.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 6. Yoshimura T., Sonoda K. H., Sugahara M., Mochizuki Y., Enaida H., Oshima Y., Ueno A., Hata Y., Yoshida H., and Ishibashi T. (2009) Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS One 4, e8158 10.1371/journal.pone.0008158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ellis M. P., Lent-Schochet D., Lo T., and Yiu G. (2019) Emerging concepts in the treatment of diabetic retinopathy. Curr. Diab. Rep. 19, 137 10.1007/s11892-019-1276-5 [DOI] [PubMed] [Google Scholar]
- 8. Kurihara T., Westenskow P. D., Bravo S., Aguilar E., and Friedlander M. (2012) Targeted deletion of Vegfa in adult mice induces vision loss. J. Clin. Invest. 122, 4213–4217 10.1172/JCI65157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saint-Geniez M., Maharaj A. S. R., Walshe T. E., Tucker B. A., Sekiyama E., Kurihara T., Darland D. C., Young M. J., and D'Amore P. A. (2008) Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. PLoS One 3, e3554 10.1371/journal.pone.0003554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stapor P., Wang X., Goveia J., Moens S., and Carmeliet P. (2014) Angiogenesis revisited: role and therapeutic potential of targeting endothelial metabolism. J. Cell Sci. 127, 4331–4341 10.1242/jcs.153908 [DOI] [PubMed] [Google Scholar]
- 11. Ding Z., Bae Y. H., and Roy P. (2012) Molecular insights on context-specific role of profilin-1 in cell migration. Cell Adh. Migr. 6, 442–449 10.4161/cam.21832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miki H., Suetsugu S., and Takenawa T. (1998) WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 17, 6932–6941 10.1093/emboj/17.23.6932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suetsugu S., Miki H., and Takenawa T. (1998) The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 17, 6516–6526 10.1093/emboj/17.22.6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gau D., Veon W., Shroff S. G., and Roy P. (2019) The VASP-profilin1 (PFN1) interaction is critical for efficient cell migration and is regulated by cell-substrate adhesion in a PKA-dependent manner. J. Biol. Chem. 294, 6972–6985 10.1074/jbc.RA118.005255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Havrylenko S., Noguera P., Abou-Ghali M., Manzi J., Faqir F., Lamora A., Guérin C., Blanchoin L., and Plastino J. (2015) WAVE binds Ena/VASP for enhanced Arp2/3 complex-based actin assembly. Mol. Biol. Cell 26, 55–65 10.1091/mbc.E14-07-1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai S. L., Chan T. H., Lin M. J., Huang W. P., Lou S. W., and Lee S. J. (2008) Diaphanous-related formin 2 and profilin I are required for gastrulation cell movements. PLoS One 3, e3439 10.1371/journal.pone.0003439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding Z., Gau D., Deasy B., Wells A., and Roy P. (2009) Both actin and polyproline interactions of profilin-1 are required for migration, invasion and capillary morphogenesis of vascular endothelial cells. Exp. Cell Res. 315, 2963–2973 10.1016/j.yexcr.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding Z., Lambrechts A., Parepally M., and Roy P. (2006) Silencing profilin-1 inhibits endothelial cell proliferation, migration and cord morphogenesis. J. Cell Sci. 119, 4127–4137 10.1242/jcs.03178 [DOI] [PubMed] [Google Scholar]
- 19. Kullmann J. A., Neumeyer A., Gurniak C. B., Friauf E., Witke W., and Rust M. B. (2011) Profilin1 is required for glial cell adhesion and radial migration of cerebellar granule neurons. EMBO Rep. 13, 75–82 10.1038/embor.2011.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haugwitz M., Noegel A. A., Karakesisoglou J., and Schleicher M. (1994) Dictyostelium amoebae that lack G-actin-sequestering profilins show defects in F-actin content, cytokinesis, and development. Cell 79, 303–314 10.1016/0092-8674(94)90199-6 [DOI] [PubMed] [Google Scholar]
- 21. Böttcher R. T., Wiesner S., Braun A., Wimmer R., Berna A., Elad N., Medalia O., Pfeifer A., Aszódi A., Costell M., and Fässler R. (2009) Profilin 1 is required for abscission during late cytokinesis of chondrocytes. EMBO J. 28, 1157–1169 10.1038/emboj.2009.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gau D., Veon W., Capasso T. L., Bottcher R., Shroff S., Roman B. L., and Roy P. (2017) Pharmacological intervention of MKL/SRF signaling by CCG-1423 impedes endothelial cell migration and angiogenesis. Angiogenesis 20, 663–672 10.1007/s10456-017-9560-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fan Y., Arif A., Gong Y., Jia J., Eswarappa S. M., Willard B., Horowitz A., Graham L. M., Penn M. S., and Fox P. L. (2012) Stimulus-dependent phosphorylation of profilin-1 in angiogenesis. Nat. Cell Biol. 14, 1046–1056 10.1038/ncb2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fan Y., Potdar A. A., Gong Y., Eswarappa S. M., Donnola S., Lathia J. D., Hambardzumyan D., Rich J. N., and Fox P. L. (2014) Profilin-1 phosphorylation directs angiocrine expression and glioblastoma progression through HIF-1α accumulation. Nat. Cell Biol. 16, 445–456 10.1038/ncb2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pae M., and Romeo G. R. (2014) The multifaceted role of profilin-1 in adipose tissue inflammation and glucose homeostasis. Adipocyte 3, 69–74 10.4161/adip.26965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romeo G. R., Pae M., Eberlé D., Lee J., and Shoelson S. E. (2013) Profilin-1 haploinsufficiency protects against obesity-associated glucose intolerance and preserves adipose tissue immune homeostasis. Diabetes 62, 3718–3726 10.2337/db13-0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caglayan E., Romeo G. R., Kappert K., Odenthal M., Südkamp M., Body S. C., Shernan S. K., Hackbusch D., Vantler M., Kazlauskas A., and Rosenkranz S. (2010) Profilin-1 is expressed in human atherosclerotic plaques and induces atherogenic effects on vascular smooth muscle cells. PLoS One 5, e13608 10.1371/journal.pone.0013608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Romeo G. R., and Kazlauskas A. (2008) Oxysterol and diabetes activate STAT3 and control endothelial expression of profilin-1 via OSBP1. J. Biol. Chem. 283, 9595–9605 10.1074/jbc.M710092200 [DOI] [PubMed] [Google Scholar]
- 29. Romeo G. R., Moulton K. S., and Kazlauskas A. (2007) Attenuated expression of profilin-1 confers protection from atherosclerosis in the LDL receptor null mouse. Circ. Res. 101, 357–367 10.1161/CIRCRESAHA.107.151399 [DOI] [PubMed] [Google Scholar]
- 30. Romeo G., Frangioni J. V., and Kazlauskas A. (2004) Profilin acts downstream of LDL to mediate diabetic endothelial cell dysfunction. FASEB J. 18, 725–727 10.1096/fj.03-0841fje [DOI] [PubMed] [Google Scholar]
- 31. Li Z., Zhong Q., Yang T., Xie X., and Chen M. (2013) The role of profilin-1 in endothelial cell injury induced by advanced glycation end products (AGEs). Cardiovasc. Diabetol. 12, 141 10.1186/1475-2840-12-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding H., Chen B., Lu Q., and Wang J. (2018) Profilin-1 mediates endothelial dysfunction in diabetic retinopathy through HIF1-α dependent pathway. Int. J. Clin. Exp. Pathol. 11, 1247–1255 [PMC free article] [PubMed] [Google Scholar]
- 33. Lu Q., Lu L., Chen W., Chen H., Xu X., and Zheng Z. (2015) RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 253, 669–680 10.1007/s00417-015-2985-3 [DOI] [PubMed] [Google Scholar]
- 34. Lu Q., Lu P., Chen W., Lu L., and Zheng Z. (2018) ANGPTL-4 induces diabetic retinal inflammation by activating Profilin-1. Exp. Eye Res. 166, 140–150 10.1016/j.exer.2017.10.009 [DOI] [PubMed] [Google Scholar]
- 35. Lam J. D., Oh D. J., Wong L. L., Amarnani D., Park-Windhol C., Sanchez A. V., Cardona-Velez J., McGuone D., Stemmer-Rachamimov A. O., Eliott D., Bielenberg D. R., van Zyl T., Shen L., Gai X., D'Amore P. A., et al. (2017) Identification of RUNX1 as a mediator of aberrant retinal angiogenesis. Diabetes 66, 1950–1956 10.2337/db16-1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Selvam S., Kumar T., and Fruttiger M. (2018) Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 63, 1–19 10.1016/j.preteyeres.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 37. Connor K. M., SanGiovanni J. P., Lofqvist C., Aderman C. M., Chen J., Higuchi A., Hong S., Pravda E. A., Majchrzak S., Carper D., Hellstrom A., Kang J. X., Chew E. Y., Salem N. Jr., Serhan C. N., et al. (2007) Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 13, 868–873 10.1038/nm1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stahl A., Connor K. M., Sapieha P., Chen J., Dennison R. J., Krah N. M., Seaward M. R., Willett K. L., Aderman C. M., Guerin K. I., Hua J., Löfqvist C., Hellström A., and Smith L. E. (2010) The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51, 2813–2826 10.1167/iovs.10-5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sakabe M., Fan J., Odaka Y., Liu N., Hassan A., Duan X., Stump P., Byerly L., Donaldson M., Hao J., Fruttiger M., Lu Q. R., Zheng Y., Lang R. A., and Xin M. (2017) YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. U.S.A. 114, 10918–10923 10.1073/pnas.1704030114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weinl C., Riehle H., Park D., Stritt C., Beck S., Huber G., Wolburg H., Olson E. N., Seeliger M. W., Adams R. H., and Nordheim A. (2013) Endothelial SRF/MRTF ablation causes vascular disease phenotypes in murine retinae. J. Clin. Invest. 123, 2193–2206 10.1172/JCI64201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stenzel D., Franco C. A., Estrach S., Mettouchi A., Sauvaget D., Rosewell I., Schertel A., Armer H., Domogatskaya A., Rodin S., Tryggvason K., Collinson L., Sorokin L., and Gerhardt H. (2011) Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 12, 1135–1143 10.1038/embor.2011.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gau D., Lewis T., McDermott L., Wipf P., Koes D., and Roy P. (2018) Structure-based virtual screening identifies a small-molecule inhibitor of the profilin 1-actin interaction. J. Biol. Chem. 293, 2606–2616 10.1074/jbc.M117.809137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zou L., Jaramillo M., Whaley D., Wells A., Panchapakesa V., Das T., and Roy P. (2007) Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br. J. Cancer 97, 1361–1371 10.1038/sj.bjc.6604038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finkel T., Theriot J. A., Dise K. R., Tomaselli G. F., and Goldschmidt-Clermont P. J. (1994) Dynamic actin structures stabilized by profilin. Proc. Natl. Acad. Sci. U.S.A. 91, 1510–1514 10.1073/pnas.91.4.1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moldovan N. I., Milliken E. E., Irani K., Chen J., Sohn R. H., Finkel T., and Goldschmidt-Clermont P. J. (1997) Regulation of endothelial cell adhesion by profilin. Curr. Biol. 7, 24–30 10.1016/S0960-9822(06)00024-8 [DOI] [PubMed] [Google Scholar]
- 46. Rotty J. D., Wu C., Haynes E. M., Suarez C., Winkelman J. D., Johnson H. E., Haugh J. M., Kovar D. R., and Bear J. E. (2015) Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell 32, 54–67 10.1016/j.devcel.2014.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Price C. J., and Brindle N. P. J. (2000) Vasodilator-stimulated phosphoprotein is involved stress-fiber and membrane ruffle formation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 20, 2051–2056 10.1161/01.atv.20.9.2051 [DOI] [PubMed] [Google Scholar]
- 48. Krebs A., Rothkegel M., Klar M., and Jockusch B. M. (2001) Characterization of functional domains of mDia1, a link between the small GTPase Rho and the actin cytoskeleton. J. Cell Sci. 114, 3663–3672 [DOI] [PubMed] [Google Scholar]
- 49. Chakraborty S., Jiang C., Gau D., Oddo M., Ding Z., Vollmer L., Joy M., Schiemann W., Stolz D. B., Vogt A., Ghosh S., and Roy P. (2018) Profilin-1 deficiency leads to SMAD3 upregulation and impaired 3D outgrowth of breast cancer cells. Br. J. Cancer 119, 1106–1117 10.1038/s41416-018-0284-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gonçalves-Pimentel C., Gombos R., Mihály J., Sánchez-Soriano N., and Prokop A. (2011) Dissecting regulatory networks of filopodia formation in a Drosophila growth cone model. PLoS One 6, e18340 10.1371/journal.pone.0018340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kiss V., Jipa A., Varga K., Takáts S., Maruzs T., Lörincz P., Simon-Vecsei Z., Szikora S., Földi I., Bajusz C., Tóth D., Vilmos P., Gáspár I., Ronchi P., Mihály J., et al. (2020) Drosophila Atg9 regulates the actin cytoskeleton via interactions with profilin and Ena. Cell Death Differ. 27, 1677–1692 10.1038/s41418-019-0452-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tokunaga C. C., Mitton K. P., Dailey W., Massoll C., Roumayah K., Guzman E., Tarabishy N., Cheng M., and Drenser K. A. (2014) Effects of anti-VEGF treatment on the recovery of the developing retina following oxygen-induced retinopathy. Invest. Ophthalmol. Vis. Sci. 55, 1884–1892 10.1167/iovs.13-13397 [DOI] [PubMed] [Google Scholar]
- 53. Montassar F., Darche M., Blaizot A., Augustin S., Conart J. B., Millet A., Elayeb M., Sahel J. A., Reaux-Le Goazigo A., Sennlaub F., Marrakchi N., Messadi E., and Guillonneau X. (2017) Lebecetin, a C-type lectin, inhibits choroidal and retinal neovascularization. FASEB J. 31, 1107–1119 10.1096/fj.201600351R [DOI] [PubMed] [Google Scholar]
- 54. Joy M., Gau D., Castellucci N., Prywes R., and Roy P. (2017) The myocardin-related transcription factor MKL co-regulates the cellular levels of two profilin isoforms. J. Biol. Chem. 292, 11777–11791 10.1074/jbc.M117.781104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data are contained in the article. Curated endothelial transcriptome data of PDR patients (GSE94019) are available at the Gene Expression Omnibus Database.