

Abstract
Alzheimer's disease (AD) is a very common neurodegenerative disorder, chiefly caused by increased production of neurotoxic β-amyloid (Aβ) peptide generated from proteolytic cleavage of β-amyloid protein precursor (APP). Except for familial AD arising from mutations in the APP and presenilin (PSEN) genes, the molecular mechanisms regulating the amyloidogenic processing of APP are largely unclear. Alcadein α/calsyntenin1 (ALCα/CLSTN1) is a neuronal type I transmembrane protein that forms a complex with APP, mediated by the neuronal adaptor protein X11-like (X11L or MINT2). Formation of the ALCα–X11L–APP tripartite complex suppresses Aβ generation in vitro, and X11L-deficient mice exhibit enhanced amyloidogenic processing of endogenous APP. However, the role of ALCα in APP metabolism in vivo remains unclear. Here, by generating ALCα-deficient mice and using immunohistochemistry, immunoblotting, and co-immunoprecipitation analyses, we verified the role of ALCα in the suppression of amyloidogenic processing of endogenous APP in vivo. We observed that ALCα deficiency attenuates the association of X11L with APP, significantly enhances amyloidogenic β-site cleavage of APP, especially in endosomes, and increases the generation of endogenous Aβ in the brain. Furthermore, we noted amyloid plaque formation in the brains of human APP-transgenic mice in an ALCα-deficient background. These results unveil a potential role of ALCα in protecting cerebral neurons from Aβ-dependent pathogenicity in AD.
Keywords: amyloid β (Aβ), alcadein, calsyntenin, Alzheimer's disease, amyloid β protein precursor (APP), membrane protein, metabolism, X11-like, Mint2, neurodegeneration
Alzheimer's disease (AD) is the most common neurodegenerative disorder, primarily caused by augmented production of neurotoxic β-amyloid (Aβ) peptide generated from one of the alternative proteolytic cleavages of β-amyloid protein precursor (APP). One of the factors that could affect the onset of AD is the alteration of Aβ generation in quality and quantity.
In familial AD cases associated with causative mutations in PSEN genes, neurotoxic longer Aβ species such as Aβ42 increase along with attenuated γ-secretase activity (1). Similar altered γ-secretase activity is also observed in brains of sporadic AD subjects (2). In another familial AD case associated with causative mutations in the APP gene, Aβ generation altered in quality and quantity is induced (3–6). Mutations in Aβ sequence also alter the aggregative state of Aβ (7). Individuals with Down syndrome carry an extra copy of chromosome 21, where the APP gene resides, and are prone to develop AD in their 50s or 60s (reviewed in Ref. 8), suggesting that even a relatively moderate increase in Aβ generation may affect the onset of AD. Although the primary causes of sporadic AD may be various and it is still controversial whether altered Aβ generation in quality and quantity contributes to the pathology of sporadic AD (9–11), there are reports that Aβ generation can be altered qualitatively and/or quantitatively in the absence of pathogenic mutations on causative genes (12, 13). These observations underscore the relevance of elucidating modulatory factors involved in amyloidogenic processing of endogenous APP in vivo.
One such factor is a submembrane scaffolding protein, X11-like (X11L), encoded by the APBA2 gene. X11L was isolated as a binding partner of APP and shown to suppress Aβ generation in vitro (14). Although overexpressed X11L suppressed overall metabolism of APP in vitro (14, 15), loss of X11L protein preferentially enhanced the amyloidogenic cleavage of APP in the brains of X11L-deficient mice (16, 17). Furthermore, amyloid plaque formation in the brains of human APP-transgenic mice was facilitated in an X11L-deficient background (18). Aβ generation in the brain was suppressed in transgenic mice producing increased amounts of X11L (19). These results indicate that X11L is involved in the suppression of amyloidogenic processing of APP; however, it is unclear whether other molecules associated with X11L affect APP metabolism.
Alcadein α (Alcα) was isolated as a single-pass transmembrane protein, and it binds to X11L through its cytoplasmic region (20). Alcα colocalizes with APP in dystrophic neurites and senile plaques of AD patients' postmortem brains, suggesting that the Alcα may be involved in AD pathogenesis (20). This notion is further supported by reduced expression of Alcα in AD patients' brains (21). Alcα is one of three closely related protein family members (Alcα, Alcβ, and Alcγ). It was also identified as a postsynaptic Ca2+-binding protein calsyntenin1 (Clstn1) (20, 22). Binding of APP to X11L in vitro is strengthened in the presence of Alcα by the formation of a tripartite complex comprising APP, X11L, and Alcα (20, 23). APP within the tripartite complex is subject to greater suppression of proteolytic processing compared with APP with X11L alone in transient expression studies (20, 23). These results suggest that Alcα may play a role in APP metabolism. In addition, Alcs are subject to proteolytic processing as is APP, the quality and quantity of which may correlate with a pathogenic processing of APP (24, 25).
APP generates a p3 fragment by sequential cleavages with α-secretase (mainly ADAM10/17) and γ-secretases. We have shown that Alcα is also cleaved by these proteases to generate a p3-Alcα fragment (26). APP is alternatively cleaved by a combination of β-secretase (BACE1) and γ-secretases to generate Aβ peptide (reviewed in Ref. 27). Alcs are not cleaved by BACE1; however, the generation of p3-Alcα from Alcα likely correlates with pathobiology in Alzheimer's disease. We have shown that the quantity and quality of p3-Alcα peptide in cerebrospinal fluid and blood are altered and correlate with AD pathogenesis (28–32). Another in vitro study showed that siRNA-mediated reduction of Alcα, in cultured neurons, enhanced amyloidogenic processing of APP (21). These observations collectively suggest a functional link between Alcα and APP metabolism; however, the physiological significance of Alcα in APP metabolism in vivo remains unclear.
Here, we explored the role of Alcα in APP metabolism, in vivo, by generating Alcα-deficient mice. The β-site cleavage of endogenous APP and generation of Aβ were enhanced in the brains of Alcα-deficient mice, and amyloid plaque formation was facilitated in the brains of human APP-producing transgenic mice in an Alcα-deficient background. These observations indicate that Alcα plays a physiologically relevant role in APP metabolism to ameliorate AD pathogenesis.
Results
Generation of Alcα-deficient mice
To evaluate the role of Alcα in endogenous APP metabolism, Alcα-deficient mice were generated using a standard gene knockout method with a targeting vector in which the coding sequence of exon 1 was replaced with the LacZ-pA-PGK-Neo-pA cassette (Fig. 1A). Mutant mice carrying the allele without pGKNeo were backcrossed with C57BL/6 mice over 10 generations. The mutation was confirmed by Southern blotting and PCR analysis (Fig. 1, B and C). The absence of Alcα protein was confirmed by immunoblotting and immunostaining (Fig. 1, D and E) of brain extracts and slices with specific antibodies, as Alcα is predominantly expressed in brain tissue (20). Two bands representing full-length Alcα and Alcα C-terminal fragment (CTF), which is the cytoplasmic CTF generated following the cleavage of Alcα by APP α-secretase (ADAM10/17), were detected with antibodies that recognized the carboxyl cytoplasmic region of Alcα in the WT (+/+) mouse brains (20, 23, 26). Neither band was detected in the brains of knockout (−/−) mice (Fig. 1D). Alcα was expressed in whole-brain tissue with a stronger expression in the hippocampal neurons of WT (+/+) mice, based on immunohistochemical staining; these signals were not detected in knockout (−/−) mice (Fig. 1E). These results indicate that homozygous mutant mice expressed neither the full-length Alcα nor the truncated/processed fragments of Alcα.
Figure 1.

Generation of Alcα-deficient mice. A, gene-targeting procedure. Shown is a schematic of the partial gene structure of the Alcα allele, including exon 1, the targeting construct, and targeted allele after crossing with FLPe-Tg mice. B, Southern blotting analysis. Probes indicated in A were used to detect WT (20 kbp) and the targeted (8.5 kbp) fragments. C, PCR products specific to the WT allele (+/+) generated with primers i plus ii (416 bp) and to the targeted allele (−/−) generated with primers ii plus iii (1,224 bp) were analyzed by agarose gel electrophoresis. D, immunoblot analysis of Alcα and Alcα CTF. Whole-brain lysates (20 μg of protein) of WT (+/+) and homozygous mutant (−/−) mice were analyzed in 8% resolving gel with an anti-Alcα antibody and anti-actin antibody. *, nonspecific product. E, immunostaining of sagittal sections of WT (+/+) and homozygous mutant (−/−) mouse (2–3 months old) brains with the anti-Alcα antibody. Scale bar, 1 mm.
Alcα deficiency enhances cleavages of APP by β-secretase but not α-secretase
APP is primarily cleaved by either α- or β-secretase to generate APP CTFs. The α-cleavage of APP generates C83/CTFα by cleaving the peptide bond between Lys-687 and Leu-688 (sequence numbering refers to the APP770 isoform) within the Aβ sequence. Thus, this cleavage is amyloidolytic. Alternatively, β-cleavage of APP generates C99/CTFβ by cleaving the peptide bond between Met-671 and Asp-672 and generates C89/CTFβ′ by cleaving the peptide bond between Tyr-681 and Gln-682 (27, 33). We examined whether defects in Alcα would influence the primary cleavages of APP in vivo (Fig. 2). The cerebral cortex and hippocampus membrane fractions, prepared from WT (+/+) and Alcα-KO (−/−) mice brains (3 months old), were analyzed for Alcα expression and cleavages of APP by immunoblotting. Identical protein amounts were loaded per lane, as indicated in the figure legends, and the densities of bands were quantified followed by normalization using the integral membrane protein flotillin-1. Among the three major APP isoforms (amino acid numbers 770, 751, and 695), brain neurons exclusively expressed the APP695 isoform and showed three protein bands. These comprised two mature forms with different O-glycosylation with N-glycosylation and one immature form with N-glycosylation alone on an immunoblotted membrane (reviewed in Ref. 34). Maturation of APP was largely identical and a slight alteration of APP protein level was observed, however not statistically significant, between the WT (+/+) and knockout (−/−) mouse brains (Fig. 2 (A and C) and Fig. S1). This suggests that defects in Alcα expression did not significantly affect the expression or posttranslational modification of the APP.
Figure 2.

Enhanced β-site cleavages of APP in the brains of Alcα-deficient mice. A and B, immunoblot analysis of APP (A) and APP CTFs (B). Membrane fraction (5 µg (A) or 15 µg (B) of protein) of the hippocampus and cerebral cortex of WT (+/+) and homozygous mutant (−/−) mice (3 months old) were analyzed in 8% (A) or 15% (B) resolving gel with anti-APP, anti-Alcα, and anti-flotillin-1 antibodies. APP, mature (N- and O-glycosylated APP695) and immature (N-glycosylated APP695) forms. Alcα CTF exhibits double bands under these electrophoresis conditions. C, band densities of APP and APP CTFs for WT (open columns) and Alcα-deficient (filled columns) mice were standardized to the density of flotillin-1, and the value of WT was assigned as a reference value of 1.0. m, mature APP (top two bands); im, immature APP (bottom band); t, total APP (mature plus immature APP); C99, CTFβ; C89, CTFβ′; C83, CTFα of APP CTFs (unpaired t test; *, p < 0.05; n = 3 mice/group). Error bars, S.E. D, endogenous mouse Aβ40 and Aβ42 in the hippocampus and cerebral cortex of WT (open columns) and Alcα-deficient (filled columns) mice at the indicated ages (2, 6, and 12 months old) were quantified using sandwich ELISA. The Aβ40 and Aβ42 concentrations were normalized to tissue weight (unpaired t test; **, p < 0.01; ***, p < 0.001; n = 5 mice/group). Error bars, S.E.
We next examined APP CTF levels in the brains of these mice. Three CTF fragments (C99, C89, and C83) were detected and measured by immunoblotting phosphatase-treated membrane fractions prepared from the cerebral cortex and hippocampus of the indicated mice, with antibodies raised against the APP cytoplasmic region (see Fig. S1). Levels of C99 and C89, generated by β-cleavage of APP, were significantly increased in Alcα-KO mouse brains compared with WT mice. Levels of C83, generated by α-cleavage of APP, were not altered among the brains of WT and Alcα-deficient mice (Fig. 2, B and C). These results indicate that the β-site cleavage of APP was enhanced in the brains of Alcα-deficient mice. We further examined age-specific alterations of the enhanced β-site cleavage in Alcα-KO mice. Selective enhancement of β-site cleavage in Alcα-KO mice was observed from 2-month-old adult mice (Fig. S1). The ratio of β-site cleavage enhancement was virtually unaltered with age. The observed enhancement of β-site cleavage in Alcα-KO mice was probably not due to the augmented expression of BACE1, as no statistically significant alteration of BACE1 expression was observed between the WT and Alcα-deficient mouse brains (see Fig. 4A).
Figure 4.

Alcα-, but not Alcβ-deficient mice showed significant alterations in amyloidogenic processing of APP. A, immunoblot analysis of APP CTFs in Alcα- or Alcβ-deficient mice. A total of 15 µg of membrane fraction of the hippocampus and cerebral cortex of WT (+/+) and homozygous mutant (−/−) mice (6 months old) were analyzed in 15% resolving gel with anti-APP and anti-synaptophysin (SYP) antibodies. The same fractions were also analyzed in an 8% resolving gel with anti-Alcα, anti-Alcβ, anti-BACE1, and anti-flotillin-1 antibodies. B, band densities of APP CTFs for WT (black columns) and Alcα- or Alcβ-deficient (colored columns as indicated) were standardized to the density of synaptophysin, and the value of WT was assigned a reference value of 1.0. C99, CTFβ; C89, CTFβ′; C83, CTFα of APP CTFs (n = 4 mice/group; two-way ANOVA; Dunnett's post hoc test compared with WT; *, p < 0.05; **, p < 0.01). Band densities of APP (m, mature APP; im, immature APP; t, total APP) and BACE1 were also quantified and standardized to the density of flotillin-1, and the value of WT was assigned a reference value of 1.0. Error bars, S.E. C, endogenous mouse Aβ40 or Aβ42 in the hippocampus and cerebral cortex of WT (open column) and Alcα- or Alcβ-deficient (colored columns as indicated) mice were quantified using sandwich ELISA. The Aβ40 and Aβ42 concentrations were normalized to tissue weight (n = 4 mice/group; one-way ANOVA; Dunnett's post hoc test compared with WT; *, p < 0.05; ***, p < 0.001). Error bars, S.E.
Enhanced β-site cleavage of APP should induce increased Aβ generation as APP CTFs are subject to a secondary cleavage by γ-secretase that generates Aβ and p3 peptides. We next quantified the endogenous Aβ levels in the brains of WT and Alcα-deficient mice (Fig. 2D). Mouse Aβ is largely recovered into a TBS-insoluble fraction, regardless of its nonaggregative nature (17). We thus examined Aβ40 and Aβ42 levels in TBS-insoluble fractions of the hippocampus and cerebral cortex of WT and Alcα-deficient mice at ages 2, 6, and 12 months. Endogenous mouse Aβ40 and Aβ42 levels were significantly increased in the brains of Alcα-deficient mice (filled columns) compared with the WT mice (open columns). The enhancements were roughly comparable with those of the enhanced production of Aβ peptides at the corresponding ages. As stated earlier, endogenous mouse Aβ is less aggregation-prone than the human Aβ, and it is rather natural to not observe significant age-related accumulation of mouse Aβ in these mice. Taken together, these results indicate that Alcα may functionally suppress amyloidogenic β-cleavage of APP in the brain.
Mouse Aβ does not form amyloid plaques due to its nonaggregative nature (33), and the mouse endogenous Aβ peptides did not accumulate in their brains with age, as shown in Fig. 2D. Hence, it is unclear whether Alcα deficiency would be sufficient to contribute to plaque formation that characterizes AD pathology. Therefore, we crossed Alcα-deficient mice with APP23 human APP-transgenic mice to generate APP23/Alcα-deficient mice and examined brain amyloid plaque formation (Fig. 3). Brain slices (10 35-µm-thick slices with 350-µm intervals, −2.8 to +0.7 mm to bregma) of these mice (12 months old) were immunostained with an anti-human Aβ-specific antibody (Fig. 3B). APP23/Alcα-deficient mice appeared to form more plaques, including cortex and hippocampus, without readily recognizable morphological alteration at this age (Fig. 3A). The numbers and area of amyloid plaques in APP23 and APP23/Alcα-deficient mice were quantified, including the cerebral cortex, hippocampus, and entorhinal cortex. The numbers of plaques and proportion of plaque area were significantly higher in APP23/Alcα-deficient mice than in APP23 mice (Fig. 3C), implying that Alcα ameliorates AD pathogenesis and that Alcα deficiency augments amyloid plaque formation.
Figure 3.

Quantification of amyloid plaques in APP23 mouse brain in the presence or absence of Alcα. A, immunostaining of coronal sections of brain regions including the cerebral cortex and hippocampus of APP23 (top) and APP23/Alcα-deficient mice (bottom) at 12 months of age. The brain sections were stained with anti-human Aβ antibody. Arrowheads, typical amyloid plaques. A magnified view of the area around the hippocampus and cortex is shown on the right with magnified images of plaques (squares in red and green). Scale bar, 300 μm (sections) or 50 μm (plaques). B and C, 10 35-µm-thick sections with 315-µm intervals were examined in one mouse. The total plaque numbers in 10 sections/mouse were counted and are indicated as the number (left) or area (right) of plaques per area (left) or section area (right). Error bars, S.E. (unpaired t test; *, p < 0.05; 4 mice for APP23, 3 mice for APP23/Alcα-deficient background).
Alcβ deficiency does not affect APP metabolism
Alcα has closely related family members alcadein β/calsyntenin 3 (Alcβ) and alcadein γ/calsyntenin 2 (Alcγ). Alcβ is also highly expressed in the brain (Fig. S2D). It is therefore plausible that Alcβ may also be involved in amyloidogenic processing of APP. To address this question, we generated Alcβ-deficient mice by replacing exons 1–3 with the PGKNeo cassette (Fig. S2A) and verified their APP metabolism in vivo (6 months old) (Fig. 4). As demonstrated in Fig. 2, Alcα deficiency significantly augmented amyloidogenic processing of endogenous APP and subsequent generation of Aβ-peptides, without affecting BACE1 levels (Fig. 4, A and B). However, Alcβ-deficient mice did not show any significant differences in APP metabolism or Aβ peptide generation (Fig. 4, A and B). Alcβ deficiency did not augment amyloidogenic processing in an Alcα-deficient background, even in older (12-month-old) mice (Fig. S3), suggesting that Alcβ likely plays a different role in Alzheimer's disease pathobiology in vivo. Therefore, we focused our subsequent analyses on Alcα.
Alcα deficiency attenuates APP-X11L association to enhance APP β-site cleavage in endosomes
Our previous reports indicated that X11L deficiency increases β-site cleavage of APP in the brain due to the lack of association between APP and X11L (16, 17). Alcα is also identified as an X11L-binding protein and forms a complex with APP, mediated by X11L. Furthermore, the interaction between APP and X11L is enhanced by the association of Alcα with X11L in vitro (20). Alcα and X11L were co-immunoprecipitated with anti-APP antibody from mouse brain lysate and were found to be co-localized in the hippocampus (20). To further confirm the co-expression of APP, Alcα, and X11L in neurons, we performed triple labeling of single primary cultured cortical neurons with mouse anti-X11L, rabbit anti-APP, and guinea pig anti-Alcα antibodies (20, 35, 36). Co-localization was readily observed for these proteins, especially around the perinuclear structures in neurons (Fig. S4). We therefore postulated that the association of APP with X11L would be attenuated by Alcα deficiency in the brain. To examine this possibility, we performed a co-immunoprecipitation assay of APP with X11L in the brain (hippocampus and cerebral cortex) lysates of WT and Alcα-deficient mice (Fig. 5). Anti-X11L antibody recovered APP along with X11L from solubilized brain membranes of WT (+/+) and Alcα-deficient (−/−) mice. A smaller amount of APP was recovered from the brains of Alcα-deficient mice compared with the WT, indicating that the association of APP with X11L was attenuated in the brains of Alcα-deficient mice. These results supported the idea that the failure to form a tripartite complex comprising of APP, X11L, and Alcα in the brains of Alcα-deficient mice increased the β-site cleavage of APP by BACE1.
Figure 5.
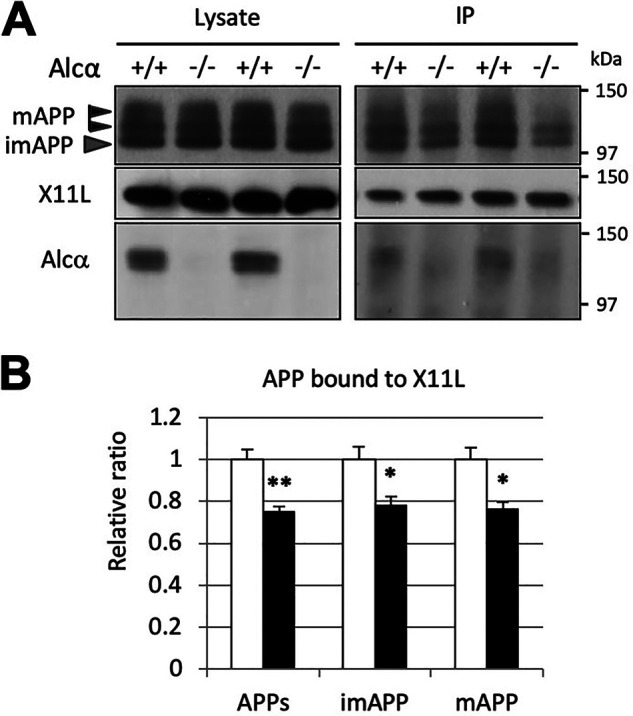
Attenuated association of APP with X11L in the brains of Alcα-deficient mice. A, co-immunoprecipitation of APP with X11L in the presence or absence of Alcα. Crude membrane fractions (500 µg of protein) of the hippocampus and cerebral cortex of WT (+/+) and Alcα-deficient (−/−) mice were subject to immunoprecipitation with anti-X11L antibody, and the immunoprecipitates were analyzed by immunoblotting with anti-APP, anti-X11L, and anti-Alcα antibodies. B, the band densities of APP in A were quantified and standardized against X11L in the immunocomplex. APPs, mature APP plus immature APP; imAPP, immature APP; mAPP, mature APP. WT was assigned a reference value of 1.0. Statistical significance was analyzed using three independent experiments (n = 3 mice/group; unpaired t test; *, p < 0.05; **, p < 0.01). Data represent means ± S.E. (error bars).
BACE1 is active in the acidic compartment of the endosomal pathway, and CTFβ and CTFβ′ are largely generated in endosomes (27, 37). Therefore, we examined whether the β-site cleavage of APP was facilitated in endosome-enriched fractions from the brains of Alcα-deficient mice (Fig. 6). Endosome-enriched fractions of the brain were prepared by ultracentrifugation of the post-nuclear supernatant of the brain homogenate with a discontinuous sucrose gradient solution (Fig. 6A). A typical fractionation profile of a WT mouse brain is shown in Fig. 6B. ∼10% of post-nuclear proteins were recovered in the endosome-enriched fraction (fraction 5), and others containing cytoplasmic proteins were recovered in fractions 9 and 10 (Fig. S5). EEA1, a cytoplasmic protein associating with early endosomes through phosphatidylinositol 3-phosphate, and BACE1 were readily detected in the endosome-enriched fraction. The majority of APP CTFs were recovered in the endosome-enriched fractions of WT (+/+) and Alcα-deficient (−/−) mouse brains (Fig. 6C) and were examined and compared with the endosome-subtracted fractions. Greater amounts of CTFβ/C99 and CTFβ'/C89 were recovered from the endosome-enriched fractions of Alcα-deficient mouse brains than from the WT mouse brains. The amounts of APP CTFs of other protein fractions were not significantly different between the WT and Alcα-KO mouse brains (Fig. 6, C and D). These results suggest that APP cleavage at the β-sites in endosomes was facilitated in the brains of Alcα-KO mice, thus supporting the notion that the presence of Alcα attenuates APP cleavage by BACE1.
Figure 6.

Increased β-site cleavages of APP in the endosome-enriched fraction of Alcα-deficient mouse brains. A, preparation of endosome-enriched membrane fraction. Post-nuclear supernatant (PNS) prepared from brain homogenate was adjusted to 42.5% sucrose and set at the bottom. Buffer with 35% sucrose was overlaid, and the same buffer with 5% sucrose was subsequently applied as shown in the figure. Very light membrane (VLM) largely composed of late endosomes entered the 5% sucrose layer, and heavy membrane (HM)-containing plasma membrane and rough endoplasmic reticulum membrane with cytosol proteins resided in the 42.5% sucrose layer. Light membrane (LM)-containing early endosomes with Golgi and other membranes accumulated underneath the interface between 5 and 35% sucrose layers after ultracentrifugation. See Fig. S5 for briefly illustrated preparation scheme. B, typical isolation of endosome-enriched fraction from WT mouse hippocampus and cerebral cortex. The endosome-enriched fraction (fr. 5) and other protein fractions (fr. 9) are shown in boldface type. PNS and the respective fractions were analyzed by immunoblotting with antibodies to detect indicated proteins. Some EEA1 and BACE1 resided in the endosome-enriched fraction 5. ∼10% of the protein was separated in fraction 5, and ∼80% of protein resided in fractions 9 and 10 (Fig. S5). TfR, transferrin receptor. C, immunoblot analysis of the endosome-enriched (fr. 5) and other protein (fr. 9) fractions of WT and Alcα-deficient mouse brains. Samples of WT (+/+) and Alcα-deficient (−/−) mice were analyzed with antibodies to detect the indicated proteins. D, the band densities of APP and APP CTFs in C were quantified and standardized against transferrin receptor. The value of WT was assigned a reference value of 1.0. Statistical significance was analyzed using three independent experiments (n = 3 mice/group; unpaired t test; *, p < 0.05). Data represent means ± S.E. (error bars). APP, mature APP plus immature APP; C99, CTFβ; C89, CTFβ′; C83, CTFβ of APP CTFs.
Discussion
In this study, we explored the role of Alcα in the amyloidogenic metabolism of APP, especially in the regulation of β-site cleavage of APP, in brains in vivo using Alcα gene KO mice. The generation and neurotoxic oligomer formation of Aβ in the brain causes neurodegeneration and promotes the onset of Alzheimer's disease, the most common neurodegenerative disease of aged subjects (38–40). Therefore, precise elucidation of the mechanisms regulating Aβ generation in the brain will be crucial to understand AD pathogenesis and inform therapeutic development. We demonstrated that Alcα is involved in the regulation of amyloidogenic processing of endogenous APP, and Alcα deficiency sufficiently augments Aβ generation to enhance amyloid plaque formation in human APP-Tg mice, with a reduced association of X11L with APP.
The neuronal adaptor protein X11L is a regulator of β-site cleavage of APP, and X11L forms a complex with APP and Alcα. The presence of the tripartite complex comprising APP, X11L, and Alcα has been demonstrated in mouse brains by co-immunoprecipitation (20). Our observations strongly support the idea that the tripartite complex formation is a physiologically relevant step that affects APP metabolism.
The present study using Alcα-deficient mice clearly demonstrated that in Alcα-deficient brains, (i) β-site cleavages of APP to generate CTFβ/C99 and CTFβ′/C89 were significantly enhanced, (ii) Aβ generation was significantly increased, (iii) AD pathology progressed in human APP-Tg mice, and (iv) association of APP with X11L was also attenuated. Although the role of Alcα in the amyloidogenic cleavage of APP is an indirect effect mediated by X11L, these results indicate that Alcα plays an important role in the regulation of APP amyloidogenic metabolism in the brain. Our previous observations suggested that one function of X11L is to prevent APP from being transported into membrane microdomains in which active BACE1 resides (17). Our present analyses suggest that Alcα enhances the functions of X11L by forming a tripartite complex comprising APP, X11L, and Alcα; the absence of Alcα may facilitate localization of APP in these membrane microdomains, including in endosomes. The mechanism underlying the formation of the tripartite complex that prevents APP subcellular localization and enhances its amyloidogenic processing is yet to be elucidated. Sortilin and its related family members affect APP subcellular localization through their direct interaction with APP to modulate amyloidogenic processing of APP (reviewed in Ref. 41). The tripartite complex formation could affect their accessibility to APP in a manner attenuating its amyloidogenic processing. It would also be plausible that the tripartite complex formation may prevent APP from being sorted to endocytic pathway in Golgi apparatus. APP and Alcα have similar functions as the cargo receptor molecule of kinesin-1 in neurons (35, 42, 43). Transport membrane vesicles, including APP, bind to kinesin-1 via the cytoplasmic region of APP; this binding is mediated by c-Jun N-terminal kinase–interacting protein 1 (JIP1) interaction between APP and kinesin light chain of kinesin-1, by which APP vesicles are anterogradely transported with high efficiency in axons (43–45). Alcα also associates with kinesin light chain of kinesin-1 directly, using its cytoplasmic WD motifs, by which transport membrane vesicles, including Alcα, transport cargos toward nerve terminals, as does APP (35, 42). Transport cargos of Alcα are largely independent of APP cargos in functions with different transport velocity (35, 43). When the functions of either APP or Alcα cargos are impaired, they are likely to compensate for each other's roles to maintain neuronal function (46). Therefore, functional deficiency in Alcα may also influence neuronal function and proteolytic metabolism of APP possibly by affecting its intracellular transport and association with other factors, such as X11L. Indeed, altered function of Alcα disturbs APP axonal transport and increases the production of Aβ. Previous observations suggest that APP and Alcα may function cooperatively around Golgi exit sites (47, 48), and release of APP from X11L may be regulated by phosphorylation of X11L (49). These observations lead to the hypothesis of a possible mechanism by which Alcα and X11L collectively suppress premature dispatching of APP into the endocytic membrane traffic system. It was recently reported that activity-dependent APP endocytosis to endosome is required and enhanced by the X11 family proteins to augment Aβ production in cultured neurons (50). Given that a considerable amount of Alcα resides in the endosome-enriched fraction (Fig. 6C, and Fig. S5), it would also be plausible that X11 family proteins, free from Alcα, make APP become more efficiently internalized into the endosome in Alcα-deficient neurons. Further investigations are required to verify this. Recent analysis with integrative genomics reports that the APBA2 gene, which encodes X11L, is a modulator of late-onset AD (51). Although this report was retracted later (52), the integrative genomics analysis itself was not fabricated (53), and our recent observation suggested that X11L significantly affects gene expression profile in the human APOE ε4 knock-in mouse brain.6 These analyses may collectively support the idea that Alcα plays an important role in the regulation of X11L function in the AD onset. It should be noted that X11L is a family member of the X11 family proteins: X11, X11L, and X11L2. Previous studies showed that X11L is widely expressed in the brain and appears to be responsible for attenuating amyloidogenic processing of endogenous APP (16, 17, 54). However, other X11 family members are known to share the same properties in the regulation of APP metabolism (20, 50), and X11s could possess an equivalently important function in the brain, especially in the neurons predominantly expressing other family members.
Alcα is also a closely related family member of Alcs: Alcα, Alcβ, and Alcγ. Contrary to the aforementioned expectation of X11 family members, Alcβ deficiency did not augment amyloidogenic processing of APP, suggesting that Alcβ has different functions. Our recent observations suggested that Alcβ may be differently involved in AD pathogenesis in terms of p3-Alcβ peptide generation (55), which would further support this notion. Alcβ is also reported to have synaptogenic activity through association with α-neurexin; however, Alcα does not exhibit such activity (56). Alcadein family members likely have different physiological roles despite their similar metabolisms and primary structures (20, 26).
Overall, our study provided evidence to strengthen the notion that the neuronal membrane protein Alcα is closely related to APP function and AD pathogenesis through intracellular interactions between APP and Alcα. Alcα deficiency leads to augmented amyloidogenic processing of endogenous APP in vivo. Further analysis of the roles of Alcα in APP metabolism and neurodegeneration will deepen our understanding of AD pathogenesis and contribute to novel therapeutic development for AD.
Experimental procedures
Generation of Alcα-KO, Alcβ-KO, and human APP-Tg/Alcα-KO mice
All experimental protocols were approved by the animal care and use committees of Hokkaido University, RIKEN Kobe Branch, and Kagawa University. All experiments were conducted in compliance with the ARRIVE guidelines. Mouse genomic DNA containing the first exon of the Alcα gene (CLSTN1) was obtained from a C57BL/6 BAC clone (Invitrogen) and used to construct a targeting vector. The coding sequence was replaced by a LacZ-pA-PGK-Neo-pA cassette from DT-A/LacZ/Neo plasmid to construct the targeting vector, which was electroporated into TT2 embryonic stem (ES) cells (57). The successful recombinants were identified by PCR using the primer sets 5′-ACCGCTTCCTCGTGCTTTACGGTATC-3′ and 5′-TAAGAACCTATTTAACAGGGGCTAGC-3′ and further confirmed by Southern blotting analysis. The recombinant ES cells were injected into ICR eight-cell stage embryos to generate chimeric founders, which were crossed to C57BL/6 females to obtain mice carrying the disrupted allele. The resultant mice (accession no. CDB0472K; RIKEN Center for Biosystems Research) were backcrossed to C57BL/6 mice for more than 10 generations. The PGK-Neo region of the cassette was removed by crossing to transgenic C57BL/6 mice ubiquitously expressing flippase (58). The presence of the WT allele and floxed LacZ-pA-pA allele was verified by PCR using the following primer sets: 5′-CGGGGTCTGGGCCGCGCGAGGTAA-3′ and 5′-CCACCTCCTTGCACCCGGTTACTAT-3′ for WT (416 bp); 5′-CGGGGTCTGGGCCGCGCGAGGTAA-3′ and 5′-GCTGGCTGCCATGAACAAAGGTTGG-3′ for LacZ-pA-pA (1,224 bp) (see Fig. 1).
For generating Alcβ-KO mice, the genomic DNA fragment of 129 Sv mice containing Alcβ exons 1–5 was isolated, and its EcoRV-EcoRI region containing exons 1–3 was replaced with the pGKneo (59) cassette. The resultant targeting vector was electroporated into ES-D3 cells, and successful recombinants were identified by Southern blotting. The recombinant ES cells were injected into ICR eight-cell stage embryos to generate chimeric founders, which were crossed to C57BL/6 females to obtain mice carrying the disrupted allele. The resultant mice were backcrossed to C57BL/6 mice for more than 10 generations. The presence of the WT allele and pGKneo allele was verified by PCR using the following primer sets: 5′-GGTACCCTTCGAGGCTGTGATC-3′ and 5′-GAGACTTCTTGGTATTGGTGCCATC-3′ for WT (158 bp); 5′-ATACCGTAAAGCAGGAAGCGGTC-3′ and 5′-CATTCGACCACCAAGCGAAACATCGC-3′ for pGKneo (343 bp) (see Fig. S1).
The human APP751swe-tg APP23 mouse was kindly supplied from Novartis Pharma Inc. (60). APP23/Alcα-KO mice were generated by mating APP23 mice with Alcα-KO mice. Heterozygous human APPswe transgenic (Tg+/−, Alcα−/−) and (Tg+/−, Alcα +/+) mutant mice were used for experimentation.
Immunohistochemistry
Frozen mouse brain sections (20-μm-thick) were prepared as described previously (61). Sections were incubated with 0.1% (v/v) Triton X-100 in PBS and blocked with PBS including 5% (v/v) heat-inactivated goat serum, incubated with rabbit polyclonal anti-Alcα 958 antibody (serum 1:1,000 dilution) (48), followed by anti-rabbit IgG Alexa Fluor 488 (Invitrogen). After washing the sections with PBS, the sections were mounted on glass slides with Shandon Immu-Mount (catalog no. 9990402, Thermo Fisher Scientific) and observed by fluorescence microscopy with a ×10 objective and ×10 eyepiece lens followed by merging respective images (BZ-9000, Keyence (Osaka, Japan)). Mouse primary cultured cortical neurons were prepared as described previously (17). Fixed neurons were incubated with 0.1% (v/v) Triton X-100 in PBS, blocked with PBS including 5% (v/v) heat-inactivated goat serum, and then incubated with guinea pig polyclonal anti-Alcα antibody (col90, 1:200 dilution) (36), mouse monoclonal anti-X11L (MINT2, 1:250 dilution, BD Bioscience), and rabbit anti-APP (G369, 1:100 dilution) (17) followed by a secondary incubation with anti-rabbit IgG Alexa Fluor 488, anti-mouse IgG Alexa Fluor 633 (Invitrogen), and anti-guinea pig IgG Cy3 (Jackson ImmunoResearch Laboratories). After washing the sections with PBS, the slides were mounted using Shandon Immu-Mount (catalog no. 9990402, Thermo Fisher Scientific), observed by fluorescence microscopy with a ×63 objective and a ×10 eyepiece lens, and then merged into respective images (BZ-9000, Keyence). Specificities of antibodies were verified using corresponding deficient mice whenever possible (see Fig. 1E and Fig. S2D).
Measurement of Aβ plaque load
APP23 mice brains were fixed and sliced to prepare 35-µm-thick sections (−2.8 to +0.7 mm to bregma), and 10 slices per brain with a 315-µm interval were further processed. The slices were incubated in PBS containing 0.3% (v/v) hydrogen peroxide, washed in PBS three times, and incubated in PBS containing 70% (w/v) formic acid for 1 min prior to blocking. The sections were incubated with mouse monoclonal anti-human Aβ 82E1 antibody (1 µg/ml; IBL, Fujioka, Japan) and washed three times for 10 min. The sections were then incubated for 1 h at room temperature with horse anti-mouse IgG conjugated with biotin (Vector Laboratories, Burlingame, CA, USA), followed by VECTASTATIN ABC kit (Vector Laboratories). Peroxidase activity was revealed using diaminobenzidine as a chromogen. The sections were viewed using a BZ-9000 microscope with a ×10 objective and ×10 eyepiece lens followed by merging of respective images (Keyence). Numbers of plaques in these 10 slices per brain were counted manually, and the resultant number was divided by the area of the slices. For measurement of the plaque area, the same images were turned to black-and-white images with ImageJ software by thresholding nonplaque signals after manually removing artifacts derived from edges or folds. The sum of the numbers of still positively stained pixels in each image was counted with ImageJ software, and the resultant number was divided by the pixel numbers of the area occupied by the slices.
Immunoblot analysis of APP and Alcα in mouse brains
For preparing whole-brain lysates, mouse brains were homogenized in a 10-fold volume of radioimmunoprecipitation assay lysis buffer (50 mm Tris-HCl, pH 8.0, containing 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1% (v/v) Nonidet P-40, and 150 mm NaCl) containing a protease inhibitor mixture (5 µg/ml cymostatin, 5 µg/ml leupeptin, and 5 µg/ml pepstatin) and centrifuged at 14,200 × g for 20 min at 4 °C. The supernatant (crude lysate) was used for immunoblot analysis. For preparing brain membrane fractions, mouse cerebral cortex and hippocampus were homogenized (five strokes of a Dounce homogenizer) in an 8-fold volume of buffer H (20 mm HEPES (pH 7.4), 150 mm NaCl, 10% (v/v) glycerol, 5 mm EDTA) containing a protease inhibitor mix. The homogenate was centrifuged at 1,000 × g for 10 min at 4 °C. The supernatant was subject to ultracentrifugation at 100,000 × g (TLA-55 rotor; Beckman Coulter) for 1 h at 4 °C, and the resultant precipitates (P100 fraction) were used for immunoblot analysis. The indicated amounts of proteins were separated by SDS-PAGE (8% (w/v) polyacrylamide Tris-glycine gels for APP, Alcα, Alcβ, BACE1, and X11L; 12% (w/v) polyacrylamide Tris-glycine gels for BACE1 (Fig. 4A); or 15% (w/v) polyacrylamide Tris-Tricine gels for APP CTF and Alcα CTF, which are carboxyl-terminal fragments of APP and Alcα cleaved by α- or β-secretase, respectively). To identify APP CTFβ/C99, CTFβ′/C89, and CTFα/C83 precisely, samples were treated with λ protein phosphatase (400 units; Sigma–Aldrich) for 2 h as phosphorylation of APP CTFs at Thr-668 (amino acid number for APP695 isoform) causes the complex protein pattern on immunoblotting (62, 63) (reviewed in Ref. 34) (see Fig. S1). The separated proteins were transferred onto a nitrocellulose membrane and probed with primary antibodies. Immunoreactive proteins were detected using Clarity Western ECL substrate (catalog no. 170-5061, Bio-Rad) and quantitated on LAS-4000 (FUJIFILM, Tokyo, Japan). Rabbit polyclonal anti-Alcα UT83 (1:500) (20), anti-Alcβ UT99 (1:500) (11), anti-APP 369 (1:4,000) (64), and anti-BACE1 Ab-2 (1:1,000) (Millipore, Burlington, MA, USA) antibodies; rabbit monoclonal anti-BACE1 D10E5 (for Fig. 4 (A and B), 1:1,000; Cell Signaling Technology, Danvers, MA, USA); and mouse monoclonal anti-actin (1 µg/ml; Chemicon International, Temecula, CA, USA), anti-X11L Mint2 (1:1,000, BD Transduction Laboratories/BD Bioscience), anti-α-tubulin (1:10,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-flotillin-1 (1:1,000; BD Transduction Laboratories), anti-EEA1 (1:10,000; Santa Cruz Biotechnology), and anti-transferrin receptor (1:1,000, BD Transduction Laboratories) were as described or purchased. Results were derived from multiple independent experiments. The numbers of experiments (n) are indicated in the figure legends. Specificities of antibodies were verified using corresponding deficient mice whenever possible (see Figs. 1D and 4A).
Aβ levels in mouse brains
Mouse endogenous Aβ40 and Aβ42 were measured as described previously (16). Hippocampus and cortex from mice were homogenized in a 6-fold volume of TBS (50 mm Tris-HCl, pH 7.6, 150 mm NaCl) containing a protease inhibitor mixture. The homogenates were centrifuged at 200,000 × g for 20 min, and the pellet was washed in TBS at 200,000 × g for 5 min (TLA 100.4 rotor; Beckman Coulter, Brea, CA, USA), and 9.1 µl of 6 m guanidine chloride in TBS was added to the washed pellet. The pellet was sonicated for 30 s, allowed to stand for 60 min at room temperature, and subjected to centrifugation at 200,000 × g for 20 min The supernatant was diluted with a 12-fold volume of ELISA buffer (PBS containing 0.05% (v/v) Tween 20 and 1% (w/v) BSA) and centrifuged at 14,000 × g for 5 min. The resulting supernatant was used for quantification of mouse Aβ with an ELISA kit (IBL (Fujioka, Japan), catalog no. 27720 for Aβ40 and catalog no. 27721 for Aβ42).
Co-immunoprecipitation of APP and X11L from solubilized brain membrane fraction
Mouse cerebral cortex and hippocampus were homogenized (five strokes of a Dounce homogenizer) in an 8-fold volume of buffer H (20 mm HEPES (pH 7.4), 150 mm NaCl, 10% (v/v) glycerol, 5 mm EDTA) containing a protease inhibitor mix. The homogenate was centrifuged at 1,000 × g for 10 min at 4 °C. The supernatant was subjected to ultracentrifugation at 100,000 × g (TLA-55 rotor; Beckman Coulter) for 1 h at 4 °C, and the resultant precipitates (P100 fraction) were suspended in PBS-CHAPS buffer (10 mm PBS (pH 7.4), 150 mm NaCl, 10 mm CHAPS) containing 1 mm NaF, 1 mm Na3VO4, and a protease inhibitor mix under rotation for 30 min at 4 °C. The suspension was centrifuged at 10,000 × g for 5 min, and the supernatant was used for immunoprecipitation samples. Samples (1 mg of protein) were subject to preclearing with Protein G–Sepharose (Thermo Fisher Scientific). Anti-X11L antibody (2.5 µg each; Mint2, BD Transduction Laboratories) was added to samples and incubated for 8 h at 4 °C, and the immunocomplex was recovered by the addition of Protein G–Sepharose. The immunoprecipitates were analyzed by immunoblotting with the indicated antibodies following SDS-PAGE (Tris-glycine buffered 12% polyacrylamide gel). The band densities were quantified and standardized against X11L in the immunocomplex.
Preparation of endosome-enriched fraction
The cerebral cortex and hippocampus of mouse brains were homogenized (30 strokes of Dounce homogenizer) in an 8-fold volume of solution (0.25 m sucrose, 3 mm imidazole, pH 7.5) and subjected to centrifugation at 1,000 × g for 10 min at 4 °C. The resultant supernatant (1 ml) was suspended in 2 ml of buffer (final 42.5% sucrose, 3 mm imidazole) in a centrifugation tube (Beckman Coulter PA13.2) and mixed. Solutions of 35 and 8% sucrose containing 3 mm imidazole, respectively, were layered in the tube, and the sample was subjected to centrifugation at 35,000 rpm for 3 h at 4 °C (SW41 Ti rotor, Beckman Coulter). Samples were collected with a 1-ml fraction from top to bottom. Fraction 5 (Fig. 6, fr. 5), including the interface between 35 and 8% sucrose density solution, was recovered as the endosome-enriched fraction along with fraction 9 (fr. 9), containing other membranes with cytoplasmic proteins (65). An equal volume of solution (0.25 m sucrose/3 mm imidazole) was added to both fractions and centrifuged at 100,000 × g for 1 h at 4 °C (TLA-55 rotor, Beckman Coulter). Resultant precipitates were analyzed for immunoblotting.
Statistical analysis
Data are expressed as means ± S.E. Statistical differences were assessed using unpaired two-tailed Student's t tests for two comparisons and two-way nonrepeated measures ANOVA with Tukey's post hoc test for multiple comparisons. A p value of <0.05 was considered statistically significant. No sample size calculation, tests for normal distribution, or tests for outliers were performed. The study was not preregistered.
Data availability
All data described are contained in the article and the supporting information.
Supplementary Material
This article contains supporting information.
Author contributions—N. G., Y. S., S. H., H. S., D. O., C. M., T. S., and T. Y. formal analysis; N. G., Y. S., T. S., and T. Y. investigation; Y. S., T. S., and T. Y. writing-review and editing; S. H., T. A., D. K., F. M., T. S., and T. Y. supervision; M. S., T. A., D. K., F. M., and T. Y. resources; T. S. and T. Y. conceptualization; S. H., T. S., and T. Y. funding acquisition; T. S. and T. Y. writing-original draft; T. S. and T. Y. project administration; T. S. and T. Y. methodology.
Funding and additional information—This work was supported in part by KAKENHI, Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) JP18K07384 (to S. H.); JP26293010 and JP18H02566 (to T. S.); and JP25460059, JP16K08237, and JP19K07065 (to T. Y.) and by Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development Grant JP19dm0107142h0004.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
T. Nakaya and T. Suzuki, unpublished observation.
- AD
- Alzheimer's disease
- APP
- amyloid β protein precursor
- BACE1
- β-site APP-cleaving enzyme 1
- Aβ
- amyloid β protein
- CTF
- C-terminal fragment
- Alc
- alcadein
- SYP
- synaptophysin
- TBS
- Tris-buffered saline
- X11L
- X11-like
- KO
- knockout
- ES
- embryonic stem
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- ANOVA
- analysis of variance
- Tg
- transgenic.
References
- 1. Takami M., and Funamoto S. (2012) γ-Secretase-dependent proteolysis of transmembrane domain of amyloid precursor protein: successive tri- and tetrapeptide release in amyloid β-protein production. Int. J. Alzheimers Dis. 2012, 591392 10.1155/2012/591392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kakuda N., Shoji M., Arai H., Furukawa K., Ikeuchi T., Akazawa K., Takami M., Hatsuta H., Murayama S., Hashimoto Y., Miyajima M., Arai H., Nagashima Y., Yamaguchi H., Kuwano R., et al. (2012) Altered γ-secretase activity in mild cognitive impairment and Alzheimer's disease. EMBO Mol. Med. 4, 344–352 10.1002/emmm.201200214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forman M. S., Cook D. G., Leight S., Doms R. W., and Lee M.-Y. V. (1997) Differential effects of the Swedish mutant amyloid precursor protein on β-amyloid accumulation and secretion in neurons and nonneuronal cells. J. Biol. Chem. 272, 32247–32253 10.1074/jbc.272.51.32247 [DOI] [PubMed] [Google Scholar]
- 4. Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., and Lannfelt L. (1992) A pathogenic mutation for probable Alzheimer's disease in the APP gene at N-terminus of β-amyloid. Nat. Genet. 1, 345–347 10.1038/ng0892-345 [DOI] [PubMed] [Google Scholar]
- 5. Goate A., Chartier-Harlin M.-C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L., Mant R., Newton P., Rooke K., Roques P., Talbot C., et al. (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- 6. Hashimoto T., Adams K. W., Fan Z., McLean P. J., and Hyman B. T. (2011) Characterization of oligomer formation of amyloid-β peptide using a split-luciferase complementation assay. J. Biol. Chem. 286, 27081–27091 10.1074/jbc.M111.257378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nilsberth C., Westlind-Danielsson A., Eckman C. B., Condron M. M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D. B., Younkin S. G., Näslund J., and Lannfelt L. (2001) The “Arctic” APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat. Neurosci. 4, 887–893 10.1038/nn0901-887 [DOI] [PubMed] [Google Scholar]
- 8. Tcw J., and Goate A. M. (2017) Genetics of β-amyloid precursor protein in Alzheimer's disease. Cold Spring Harb. Perspect. Med. 7, a024539 10.1101/cshperspect.a024539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., and Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med 3, 89ra57 10.1126/scitranslmed.3002156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iwata N., Tsubuki T., Takaki Y., Shirotani K., Lu B., Gerard N. P., Gerard C., Hama E., Lee H. J., and Saido T. C. (2001) Metabolic regulation of brain Aβ by neprilysin. Science 292, 1550–1552 10.1126/science.1059946 [DOI] [PubMed] [Google Scholar]
- 11. Vekrellis K., Ye Z., Qiu W.-Q., Walsh D., Hartley D., Chesneau V., Rosner M. R., and Selkoe D. J. (2000) Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665 10.1523/JNEUROSCI.20-05-01657.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kakuda N., Yamaguchi H., Akazawa K., Hata S., Suzuki T., Hatsuta H., Murayama S., Funamoto S., and Ihara Y. (2020) γ-Secretase activity is associated with Braak senile plaque stages. Am. J. Pathol. 190, 1323–1331 10.1016/j.ajpath.2020.02.009 [DOI] [PubMed] [Google Scholar]
- 13. Hata S., Hu A., Piao Y., Nakaya T., Taru H., Morishima-Kawashima M., Murayama S., Nishimura M., and Suzuki T. (2020) Enhanced amyloid-β generation by γ-secretase complex in detergent-resistant membrane microdomain accompanied by the reduced cholesterol level. Human. Mol. Genet. 29, 382–393 10.1093/hmg/ddz297 [DOI] [PubMed] [Google Scholar]
- 14. Tomita S., Ozaki T., Taru H., Oguchi S., Takeda S., Yagi Y., Sakiyama S., Kirino Y., and Suzuki T. (1999) Interaction of a neuron-specific protein containing PDZ domains with Alzheimer's amyloid precursor protein. J. Biol. Chem. 274, 2243–2254 10.1074/jbc.274.4.2243 [DOI] [PubMed] [Google Scholar]
- 15. McLoughlin D. M., Irving N. G., Brownlees J., Brion J. P., Leroy K., and Miller C. C. (1999) Mint2/X11-like colocalizes with the Alzheimer's disease amyloid precursor protein and is associated with neuritic plaques in Alzheimer's disease. Eur. J. Neurosci. 11, 1988–1994 10.1046/j.1460-9568.1999.00610.x [DOI] [PubMed] [Google Scholar]
- 16. Sano Y., Syuzo-Takabatake A., Nakaya T., Saito Y., Tomita S., Itohara S., and Suzuki T. (2006) Enhanced amyloidogenic metabolism of the amyloid β-protein precursor in the X11L-deficient mouse brain. J. Biol. Chem. 281, 37853–37860 10.1074/jbc.M609312200 [DOI] [PubMed] [Google Scholar]
- 17. Saito Y., Sano Y., Vassar R., Gandy S., Nakaya T., Yamamoto T., and Suzuki T. (2008) X11 proteins regulate the translocation of Amyloid β-protein precursor (APP) into detergent-resistant membrane and suppress the amyloidogenic cleavage of APP by β-site-cleaving enzyme in brain. J. Biol. Chem. 283, 35763–35771 10.1074/jbc.M801353200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kondo M., Shiono M., Itoh G., Takei N., Matsushima T., Maeda M., Taru H., Hata S., Yamamoto T., Saito Y., and Suzuki T. (2010) Increased amyloidogenic processing of transgenic human APP in X11-like deficient mouse brain. Mol. Neurodegener. 5, 35 10.1186/1750-1326-5-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J.-H., Lau K.-F., Perkinton M. S., Standen C. L., Rogelj B., Falinska A., McLoughlin D. M., and Miller C. C. (2004) The neuronal adaptor protein X11β reduces amyloid β-protein levels and amyloid plaque formation in the brain of transgenic mice. J. Biol. Chem. 279, 49099–49104 10.1074/jbc.M405602200 [DOI] [PubMed] [Google Scholar]
- 20. Araki Y., Tomita S., Yamaguchi H., Miyagi N., Sumioka A., Kirino Y., and Suzuki T. (2003) Novel cadherin-related membrane proteins, Alcadeins, enhance the X11-like protein-mediated stabilization of amyloid β-protein precursor metabolism. J. Biol. Chem. 278, 49448–49458 10.1074/jbc.M306024200 [DOI] [PubMed] [Google Scholar]
- 21. Vagnoni A., Perkinton M. S., Gray E. H., Francis P. T., Noble W., and Miller C. C. (2012) Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum. Mol. Genet. 21, 2845–2854 10.1093/hmg/dds109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vogt L., Schrimpf S. P., Meskenaite V., Frischknech R., Kinter J., Leone D. P., Ziegler U., and Sonderegger P. (2001) Calsyntenin-1, a proteolytically processed postsynaptic membrane protein with a cytoplasmic calcium-binding domain. Mol. Cell. Neurosci. 17, 151–166 10.1006/mcne.2000.0937 [DOI] [PubMed] [Google Scholar]
- 23. Araki Y., Miyagi N., Kato N., Yoshida T., Wada S., Nishimura M., Komano H., Yamamoto T., De Strooper B., Yamamoto K., and Suzuki T. (2004) Coordinated metabolism of Alcadein and amyloid β-protein precursor regulates FE65-dependent gene transactivation. J. Biol. Chem. 279, 24343–24354 10.1074/jbc.M401925200 [DOI] [PubMed] [Google Scholar]
- 24. Cam M., Durieu E., Bodin M., Manousopoulou A., Koslowski S., Vasylieva N., Barnych B., Hammock B. D., Bohl B., Koch P., Omori C., Yamamoto K., Hata S., Suzuki T., Karg F., et al. (2018) Induction of amyloid-β42 production by fipronil and other pyrazole insecticides. J. Alzheimers Dis. 62, 1663–1681 10.3233/JAD-170875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Portelius E., Durieu E., Bodin M., Cam M., Pannee J., Leuxe C., Mabondzo A., Oumata N., Galons H., Lee J. Y., Chang Y.-T., Stüber K., Koch P., Fontaine G., Potier M.-C., et al. (2016) Specific triazine herbicides induce amyloid-β42 production. J. Alzheimers Dis. 54, 1593–1605, 10.3233/JAD-160310 [DOI] [PubMed] [Google Scholar]
- 26. Hata S., Fujishige S., Araki Y., Kato N., Araseki M., Nishimura M., Hartmann D., Saftig P., Fahrenholz F., Taniguchi M., Urakami K., Akatsu H., Martins R. N., Yamamoto K., Maeda M., et al. (2009) Alcadein cleavages by APP α- and γ-secretases generate small peptides p3-Alcs indicating Alzheimer disease-related γ-secretase dysfunction. J. Biol. Chem. 284, 36024–36033 10.1074/jbc.M109.057497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cole S. L., and Vassar R. (2008) The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer's disease pathophysiology. J. Biol. Chem. 283, 29621–29625 10.1074/jbc.R800015200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hata S., Fujishige S., Araki Y., Taniguchi M., Urakami K., Peskind E., Akatsu H., Araseki M., Yamamoto K., Martins N. R., Maeda M., Nishimura M., Levey A., Chung K. A., Montine T., et al. (2011) Alternative γ-secretase processing of γ-secretase substrates in common forms of mild cognitive impairment and Alzheimer disease: evidence for γ-secretase dysfunction. Ann. Neurol. 69, 1026–1031 10.1002/ana.22343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konno T., Hata S., Hamada Y., Horikoshi-Sakuraba Y., Nakaya T., Saito Y., Yamamoto T., Yamamoto T., Maeda M., Ikeuchi T., Gandy S., Akatsu H., and Suzuki T., and Japanese Alzheimer's Disease Neuroimaging Initiative (2011) Coordinated increase of γ-secretase reaction products in the plasma of some female Japanese sporadic Alzheimer's disease patients: quantitative analysis with a new ELISA system. Mol. Neurodegener. 6, 76 10.1186/1750-1326-6-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kamogawa K., Kohara K., Tabara Y., Takita R., Miki T., Konno T., Hata S., and Suzuki T. (2012) Utility of plasma levels of soluble p3-Alcadein α as a biomarker for sporadic Alzheimer's disease. J. Alzheimers Dis. 31, 421–428 10.3233/JAD-2012-120601 [DOI] [PubMed] [Google Scholar]
- 31. Hata S., Taniguchi M., Piao Y., Ikeuchi T., Fagan A. M., Holtzman D. M., Bateman R., Sohrabi H. R., Martins R. N., Gandy S., Urakami K., and Suzuki T., and Japanese Alzheimer's Disease Neuroimaging Initiative (2012) Multiple γ-secretase product peptides are coordinately increased in concentration in the CSF of a subpopulation of sporadic Alzheimer's disease subjects. Mol. Neurodegener. 7, 16 10.1186/1750-1326-7-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Omori C., Kaneko M., Nakajima E., Akatsu H., Waragai M., Maeda M., Morishima-Kawashima M., Saito Y., Nakaya T., Taru H., Yamamoto T., Asada T., Hata S., Suzuki T., and Japanese Alzheimer's Disease Neuroimaging Initiative (2014) Increased levels of plasma p3-Alcα35, a major fragment of Alcadeinα by β-secretase cleavage, in Alzheimer's disease. J. Alzheimers Dis. 39, 861–870 10.3233/JAD-131610 [DOI] [PubMed] [Google Scholar]
- 33. Kimura A., Hata S., and Suzuki T. (2016) Alternative selection of β-site APP cleaving enzyme 1 (BACE1) cleavage sites in amyloid β-protein precursor (APP) harboring protective and pathogenic mutations within the Aβ sequence. J. Biol. Chem. 291, 24041–24053 10.1074/jbc.M116.744722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suzuki T., and Nakaya T. (2008) Regulation of amyloid β-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 283, 29633–29637 10.1074/jbc.R800003200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Araki Y., Kawano T., Taru H., Saito Y., Wada S., Miyamoto K., Kobayashi H., Ishikawa H. O., Ohsugi Y., Yamamoto T., Matsuno K., Kinjo M., and Suzuki T. (2007) The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 26, 1475–1486 10.1038/sj.emboj.7601609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maruta C., Saito Y., Hata S., Gotoh N., Suzuki T., and Yamamoto T. (2012) Constitutive cleavage of the single-pass transmembrane protein Alcadeinα prevents aberrant peripheral retention of kinesin-1. PLoS ONE 7, e43058 10.1371/journal.pone.0043058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huse J. T., Kangning L., Pijak D. S., Carlin D., Lee V. M.-Y., and Doms R. W. (2002) Beta-secretase processing in the trans-Golgi network preferentially generated truncated amyloid specific that accumulate in Alzheimer's disease brain. J. Biol. Chem. 277, 16278–16284 10.1074/jbc.M111141200 [DOI] [PubMed] [Google Scholar]
- 38. Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., and Rogers J. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–886 10.1016/S0002-9440(10)65184-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., and Masters C. L. (1999) Soluble pool of Aβ amyloid as determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 40. Lesne S. E., Sherman M. A., Grant M., Kuskowski M., Schneider J. A., Bennett D. A., and Ashe K. H. (2013) Brain amyloid-β oligomers in aging and Alzheimer's disease. Brain 136, 1383–1398 10.1093/brain/awt062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eggert S., Thomas C., Kins S., and Hermey G. (2018) Trafficking in Alzheimer's disease: modulation of APP transport and processing by the transmembrane proteins LRP1, SorLA, SorCS1c, Sortilin, and Calsyntenin. Mol. Neurobiol. 55, 5809–5829 10.1007/s12035-017-0806-x [DOI] [PubMed] [Google Scholar]
- 42. Kawano T., Araseki M., Araki Y., Kinjo M., Yamamoto T., and Suzuki T. (2012) A small peptide sequence is sufficient for initiating kinesin-1 activation through part of TPR region of KLC1. Traffic 13, 834–848 10.1111/j.1600-0854.2012.01350.x [DOI] [PubMed] [Google Scholar]
- 43. Chiba K., Araseki M., Nozawa K., Furukori K., Araki Y., Matsushima T., Nakaya T., Hata S., Saito Y., Uchida S., Okada Y., Nairn A. C., Davis R. J., Yamamoto T., Kinjo M., et al. (2014) Quantitative analysis of APP axonal transport in neurons—role of JIP1 in APP anterograde transport. Mol. Biol. Cell 25, 3569–3580 10.1091/mbc.E14-06-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chiba K., Chien K., Sobu Y., Hata S., Kato S., Nakaya T., Okada Y., Nairn A. C., Kinjo M., Taru H., Wang R., and Suzuki T. (2017) Phosphorylation of KLC1 modifies interaction with JIP1 and abolishes the enhanced fast velocity of APP transport by kinesin-1. Mol. Biol. Cell 28, 3857–3869 10.1091/mbc.E17-05-0303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsukamoto M., Chiba K., Sobu Y., Shiraki Y., Okumura Y., Hata S., Kitamura A., Nakaya T., Uchida S., Kinjo M., Taru H., and Suzuki T. (2018) The cytoplasmic region of the amyloid β-protein precursor (APP) is necessary and sufficient for the enhanced fast velocity of APP transport by kinesin-1. FEBS Lett. 592, 2716–2724 10.1002/1873-3468.13204 [DOI] [PubMed] [Google Scholar]
- 46. Sobu Y., Furukori K., Chiba K., Nairn A. C., Kinjo M., Hata S., and Suzuki T. (2017) Phosphorylation of multiple sites within an acidic region of Alcadein α is required for kinesin-1 association and Golgi exit of Alcadein α cargo. Mol. Biol. Cell 28, 3844–3856 10.1091/mbc.E17-05-0301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ludwig A., Blume J., Diep T. M., Yuan J., Mateos J. M., Leuthäuser K., Steuble M., Streit P., and Sonderegger P. (2009) Calsyntenins mediate TGN exit of APP in a kinesin-1-dependent manner. Traffic 10, 572–589 10.1111/j.1600-0854.2009.00886.x [DOI] [PubMed] [Google Scholar]
- 48. Takei N., Sobu Y., Kimura A., Urano S., Piao Y., Araki Y., Taru H., Yamamoto T., Hata S., Nakaya T., and Suzuki T. (2015) Cytoplasmic fragment of Alcadein α generated by regulated intramembrane proteolysis enhances amyloid β-protein precursor (APP) transport into the late secretory pathway and facilitates APP cleavage. J. Biol. Chem. 290, 987–995 10.1074/jbc.M114.599852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakuma M., Tanaka E., Taru H., Tomita S., Gandy S., Nairn A. C., Nakaya T., Yamamoto T., and Suzuki T. (2009) Phosphorylation of the amino-terminal region of X11L regulates its interaction with APP. J. Neurochem. 109, 466–475 10.1111/j.1471-4159.2009.05988.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sullivan S. E., Dillon G. M., Sullivan J. M., and Ho A. (2014) Mint proteins are required for synaptic activity-dependent amyloid precursor protein (APP) trafficking and amyloid β generation. J. Biol. Chem. 289, 15374–15383 10.1074/jbc.M113.541003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rhinn H., Fujita R., Qiang L., Cheng R., Lee J. H., and Abeliovich A. (2013) Integrative genomics identifies APOE ε4 effectors in Alzheimer's disease. Nature 500, 45–50 10.1038/nature12415 [DOI] [PubMed] [Google Scholar]
- 52. Rhinn H., Fujita R., Qiang L., Chen R., Lee J. H., and Abeliovich A. (2015) Retraction: Integrative genomics identifies APOE ε4 effectors in Alzheimer's disease. Nature 523, 626 10.1038/nature14591 [DOI] [PubMed] [Google Scholar]
- 53. National Institutes of Health (2015) Findings of research misconduct, NIH Guide for Grants and Contracts (Online), NOT-OD-15-092 [Google Scholar]
- 54. Motodate R., Saito Y., Hata S., and Suzuki T. (2016) Expression and localization of X11 family proteins in neurons. Brain Res. 1646, 227–234 10.1016/j.brainres.2016.05.054 [DOI] [PubMed] [Google Scholar]
- 55. Hata S., Omori C., Kimura A., Saito H., Kimura N., Gupta V., Pedrini S., Hone E., Chatterjee P., Taddei K., Kasuga K., Ikeuchi T., Waragai M., Nishimura M., Hu A., et al. (2019) Decrease in p3-Alcβ37 and p3-Alcβ40, products of Alcadeinβ generated by γ-secretase cleavages, in aged monkeys and patients with Alzheimer's disease. Alzheimers Dement. (NY) 5, 740–750 10.1016/j.trci.2019.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pettem K. L., Yokomaku D., Luo L., Linhoff M. W., Prasad T., Connor S. A., Siddiqui T. J., Kawabe H., Chen F., Zhang L., Rudenko G., Wang Y. T., Brose N., and Craig A. M. (2013) The specific α-neurexin interactor calsyntenin-3 promotes excitatory and inhibitory synapse development. Neuron 80, 113–128 10.1016/j.neuron.2013.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yagi T., Tokunaga T., Furuta Y., Nada S., Yoshida M., Tsukada T., Saga Y., Takeda N., Ikawa Y., and Aizawa S. (1993) A novel ES cell line, TT2, with high germline-differentiating potency. Anal. Biochem. 214, 70–76 10.1006/abio.1993.1458 [DOI] [PubMed] [Google Scholar]
- 58. Tian E., Kimura C., Takeda N., Aizawa S., and Matsuo I. (2002) Otx2 is required to respond to signals from anterior neural ridge for forebrain specification. Dev. Biol. 242, 204–223 10.1006/dbio.2001.0531 [DOI] [PubMed] [Google Scholar]
- 59. Soriano P., Friedrich G., and Lawinger P. (1991) Promoter interactions in retrovirus vectors introduced into fibroblasts and embryonic stem cells. J. Virol. 65, 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sturchler-Pierrat C., Abramowski D., Duke M., Wiederhold K. H., Mistl C., Rothacher S., Ledermann B., Bürki K., Frey P., Paganetti P. A., Waridel C., Calhoun M. E., Jucker M., Probst A., Staufenbiel M., et al. (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292 10.1073/pnas.94.24.13287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ishikawa T., Gotoh N., Murayama C., Abe T., Iwashita M., Matsuzaki F., Suzuki T., and Yamamoto T. (2011) IgSF molecule MDGA1 is involved in radial migration and positioning of a subset of cortical upper-layer neurons. Dev. Dyn. 240, 96–107 10.1002/dvdy.22496 [DOI] [PubMed] [Google Scholar]
- 62. Iijima K., Ando K., Takeda S., Satoh Y., Seki T., Itohara S., Greengard P., Kirino Y., Nairn A. C., and Suzuki T. (2000) Neuron-specific phosphorylation of Alzheimer's β-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 75, 1085–1091 10.1046/j.1471-4159.2000.0751085.x [DOI] [PubMed] [Google Scholar]
- 63. Nakaya T., and Suzuki T. (2006) Role of APP phosphorylation in FE65-dependent gene transactivation mediated by AICD. Genes Cells 11, 633–645 10.1111/j.1365-2443.2006.00968.x [DOI] [PubMed] [Google Scholar]
- 64. Oishi M., Nairn A. C., Czernik A. J., Lim G. S., Isohara T., Gandy S. E., Greengard P., and Suzuki T. (1997) The cytoplasmic domain of Alzheimer's amyloid precursor protein is phosphorylated at Thr654, Ser655, and Thr668 in adult rat brain and cultured cell. Mol. Med. 3, 111–123 10.1007/BF03401803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pasquali C., Fialka I., and Huber L. A. (1999) Subcellular fractionation, electromigration analysis and mapping of organelles. J. Chromatogr. B Biomed. Sci. Appl. 722, 89–102 10.1016/S0378-4347(98)00314-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described are contained in the article and the supporting information.