

Abstract
Endoplasmic reticulum (ER) stress occurs when ER homeostasis is perturbed with accumulation of unfolded/misfolded protein or calcium depletion. The unfolded protein response (UPR), comprising of inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK) and activating transcription factor 6 (ATF6) signaling pathways, is a protective cellular response activated by ER stress. However, UPR activation can also induce cell death upon persistent ER stress. The liver is susceptible to ER stress given its synthetic and other biological functions. Numerous studies from human liver samples and animal disease models have indicated a crucial role of ER stress and UPR signaling pathways in the pathogenesis of liver diseases, including non-alcoholic fatty liver disease, alcoholic liver disease, alpha-1 antitrypsin deficiency, cholestatic liver disease, drug-induced liver injury, ischemia/reperfusion injury, viral hepatitis and hepatocellular carcinoma. Extensive investigations have demonstrated the potential underlying mechanisms of the induction of ER stress and the contribution of UPR pathways during the development of the diseases. Moreover ER stress and the UPR proteins and genes have become emerging therapeutic targets to treat liver diseases.
Keywords: ER stress, UPR, IRE1α, PERK, ATF6, liver diseases
1. Introduction
Endoplasmic reticulum (ER) is a cellular organelle present in all eukaryote cells that carries out many essential cell functions including protein synthesis and processing, lipid synthesis and calcium storage. ER stress occurs when there is excessive accumulation of unfolded and misfolded protein in the ER or when ER calcium is depleted. The liver is a vital organ with important metabolic, secretory and excretory functions. Hepatocytes are the predominant cell type in the liver and are responsible for producing large amounts of secretory proteins including albumin, alpha-1 antitrypsin and lipoproteins. Given the large requirement for protein synthesis and folding, hepatocytes are enriched in ER and susceptible to ER perturbation and ER stress. Upon ER stress, an adaptive cellular response termed the unfolded protein response (UPR) is activated to restore ER homeostasis and promote cell survival. However, when restoration fails, prolonged ER stress and UPR activation trigger cell death. ER stress and the UPR have been implicated in the pathogenesis of human diseases, including liver diseases.1 This review discusses the contribution of ER stress and three UPR pathways in major liver disorders with a focus on non-alcoholic fatty liver disease, and current therapeutic approaches targeting ER stress.
2. UPR pathways
The UPR is an adaptive response to ER stress.2,3 The UPR is comprised of three transmembrane ER stress sensor proteins, including inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK) and activating transcription factor 6 (ATF6) (Figure 1). The N-terminus of these proteins is positioned in the ER lumen and the C-terminus is in the cytosol, thus connecting the two cellular compartments. When ER homeostasis is perturbed and excess unfolded/misfolded proteins accumulate in the ER, the three ER sensors are activated via dissociation of the ER luminal protein chaperone binding immunoglobulin protein (BIP) and/or by direct association with unfolded/misfolded protein, initiating the downstream UPR signaling cascades.4–6
Figure 1.
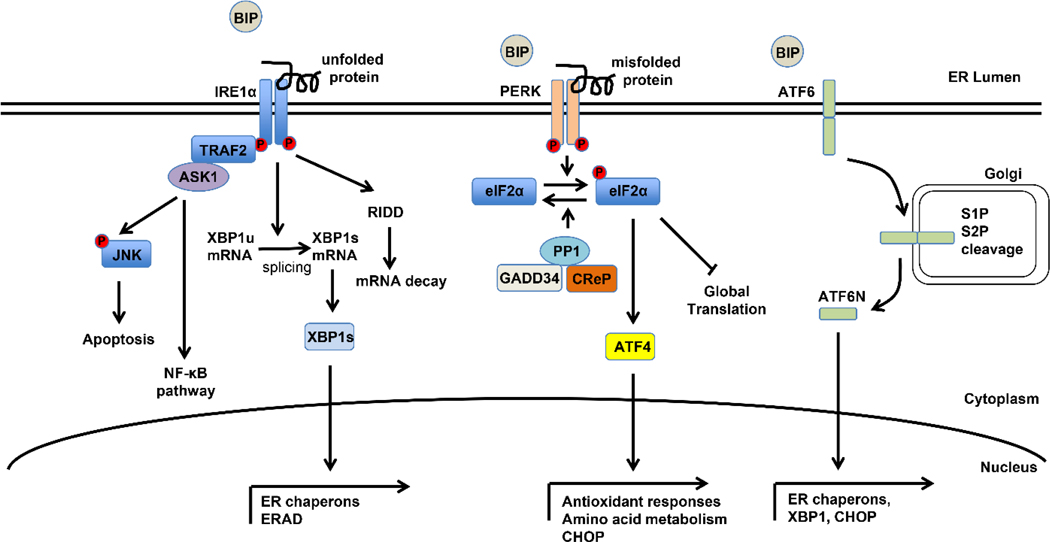
The unfolded protein response pathways.
During ER stress, the three ER sensors are activated via dissociation of the ER protein chaperon BIP and/or direct association with unfolded/misfolded protein. IRE1α dimerizes and auto-phosphorylates to activate its kinase and endoribonuclease activities. Activated IRE1α induces transcriptionally active XBP1s by an atypical splicing mechanism and XBP1s translocates to the nucleus and regulates downstream target genes including ER chaperones and genes involved in ERAD. IRE1α also recruits TRAF2 and ASK1 to mediate the activation of JNK and NF-κB pathways. The ribonuclease activity of IRE1α also negatively regulates gene expression through RIDD-mediated mRNA decay. ER stress causes PERK dimerization and auto-phosphorylation to phosphorylate eIF2α and inhibit global protein translation. The phosphorylation status of eIF2α is also tightly regulated by GADD34 and CReP, which interact and promotes PP1-mediated de-phosphorylation of eIF2α. Phosphorylated-eIF2α also selectively increases translation of a number of mRNAs, such as ATF4. ATF4 promotes adaptation to ER stress by activating UPR target genes encoding proteins necessary for antioxidant responses and amino acid metabolism. ATF4 also transcriptionally activates CHOP, which plays a critical role in ER-stress mediated apoptosis. Dissociation of BIP from ATF6 causes ATF6 translocation from the ER to the Golgi apparatus where it is cleaved by S1P and S2P to generate an active N-terminus cytosolic fragment (ATF6N). ATF6N then translocates into the nucleus to transcriptionally regulate expression of a collection of ER stress target genes. Abbreviations: ER, endoplasmic reticulum; BIP, binding immunoglobulin protein; IRE1α, inositol-requiring enzyme 1α; XBP1s, X-box binding protein 1 spliced; ERAD, ER-associated degradation; TRAF2, TNF receptor associated factor 2; ASK1, apoptosis signal-regulating kinase 1; JNK, c-jun N-terminal kinase; NF-κB, nuclear factor kappa B; RIDD, regulated IRE1α-dependent decay of mRNA; PERK, PKR-like ER kinase; eIF2α, eukaryotic initiation factor 2α; GADD34, growth arrest and DNA damage-inducible protein 34; CReP, constitutive repressor of eIF2α phosphorylation; PP1, protein phosphatase 1; ATF4, activating transcription factor 4; UPR, unfolded protein response; CHOP, CCAAT-enhancer-binding protein homologous protein; ATF6, activating transcription factor 6; S1P, site-1 protease; S2P, site-2 protease.
Among the three ER sensors, IRE1α is a type I ER transmembrane protein and is the evolutionarily most conserved component of the UPR. When ER stress occurs, IRE1α dimerizes and auto-phosphorylates to activate its kinase and endoribonuclease activities. The endoribonuclease property of IRE1α is responsible for the unconventional splicing of X-box binding protein 1 (XBP1) mRNA, removing a 26-nucleotide sequence from unspliced XBP1 (XBP1u) mRNA and causing a translational frameshift to produce the transcriptionally active XBP1 spliced (XBP1s). XBP1s regulates downstream target genes to promote protein folding and ER-associated degradation (ERAD). The cytosolic kinase domain of IRE1α recruits TNF receptor associated factor 2 (TRAF2) and apoptosis signal-regulating kinase 1 (ASK1) to mediate the activation of c-jun N-terminal kinase (JNK) and nuclear factor kappa B (NF-κB) pathways. The ribonuclease activity of IRE1α also negatively regulates gene expression through a process termed regulated IRE1α-dependent decay of mRNA (RIDD). RIDD targets include IRE1α mRNA and mRNAs of several genes involved in lipid metabolism.
PERK is also a type I transmembrane protein and a member of the eukaryotic initiation factor 2α (eIF2α) kinase family. Upon activation by ER stress, PERK phosphorylates eIF2α, which in turn attenuates translation initiation, thus blocking global protein translation to reduce the ER protein folding load. The phosphorylation status of eIF2α is also tightly regulated by growth arrest and DNA damage-inducible protein 34 (GADD34) and constitutive repressor of eIF2α phosphorylation (CReP), which interact with protein phosphatase 1 (PP1) and promote PP1-mediated de-phosphorylation of eIF2α.7 In addition to protein translation inhibition, phosphorylated-eIF2α (p-eIF2α) also selectively increases translation of a number of mRNAs, such as activating transcription factor 4 (ATF4). ATF4 promotes adaptation to ER stress by activating UPR target genes encoding proteins necessary for antioxidant responses and amino acid synthesis and transport. ATF4 also transcriptionally activates CCAAT-enhancer-binding protein homologous protein (CHOP), which plays a critical role in ER-stress mediated apoptosis. Interestingly CHOP is also regulated at the translational level by p-eIF2α. In addition, PERK signaling is part of the integrated stress response (ISR), which will be discussed in the end of this review.
ATF6 is a type II transmembrane protein and a basic Leucine zipper transcription factor. Upon ER stress, ATF6 dissociates with BIP and translocates from the ER to the Golgi apparatus where it is cleaved by site-1 protease (S1P) and site-2 protease (S2P) to generate an active N-terminus cytosolic fragment (ATF6N). ATF6N then translocates into the nucleus to transcriptionally regulate expression of a collection of ER stress target genes, including XBP1, CHOP and protein chaperones such as BIP. ATF6 also forms a heterodimer with XBP1 to activate genes involved in ERAD.8
These three UPR branches orchestrate a response to restore ER homeostasis resulting from various cellular stresses. However unmitigated ER stress and prolonged UPR activation may lead to cell apoptosis. Multiple mechanisms have been proposed for ER stress-induced apoptosis.9–11 CHOP is a major mediator for ER stress-induced apoptosis. CHOP deletion attenuates ER stress-induced apoptosis whereas CHOP overexpression sensitizes cells to apoptosis.12,13 CHOP transcriptionally activates a number of pro-apoptotic genes including death receptor 5 (DR5), Bcl-2-like protein 11 (BIM), GADD34 and tribbles homolog 3 (TRB3) and represses the expression of anti-apoptotic B-cell lymphoma 2 (BCL2), thus inducing apoptosis during ER stress. ER oxidase 1α (ERO1α) is a downstream target of CHOP and has a role in reactive oxygen species (ROS) generation. CHOP induces ERO1α expression which enhances the ROS production and stimulates inositol-1,4,5-trisphosphate receptor (IP3R)-mediated Ca2+ release from the ER, triggering cell death.14 In addition, a recent study shows CHOP promotes cell apoptosis through inhibiting autophagy.15 IRE1α contributes to ER stress-induced apoptosis through JNK activation which mediates multiple stress signaling pathways and promotes inflammation and apoptosis in the liver. ER resident pro-caspase 12 associates with TRAF2 and mediates ER stress-induced apoptosis.16 Mice lacking caspase-12 are resistant to apoptosis induced by pharmacologic ER stress.17 Persistent ER stress also induces apoptosis through RIDD-mediated degradation of pre-microRNAs and mRNAs encoding pro-survival proteins.18,19 Interestingly proapoptotic Bcl-2-associated X protein (BAX) and BCL2 antagonist/killer 1 (BAK) directly interact with IRE1α and modulate the UPR, providing a physical connection between the core apoptotic pathway to the UPR.20
3. ER and Hepatic Steatosis
The ER is the major site of lipid metabolism in hepatocytes and a major function of the UPR is to maintain hepatic lipid homeostasis. The master transcription factors regulating fatty acid synthesis and sterol synthesis, sterol regulatory element-binding protein 1c (SREBP1c) and SREBP2 respectively, are localized in ER membrane and get activated by S1P in a manner similar to ATF6 activation. Triglycerides are synthesized from fatty acid and glycerol mainly in the ER. The ER is also the location where very low-density lipoprotein (VLDL) is assembled before trafficking to the Golgi. Therefore, ER homeostasis is closely related to lipid metabolism. Previous studies have shown that ER stress induces hepatic steatosis in mice.21–27 Multiple mechanisms have been proposed including SREBP activation22–24, reduced VLDL secretion25,26, increased expression of VLDL receptor (VLDLR)27 and decreased fatty acid oxidation28. On the other hand, lipid accumulation triggers ER stress. Palmitate and other saturated fatty acids have been shown to activate the UPR (PERK, XBP1s) in the liver and in cell cultures.29–31 Interestingly the lipid composition of the hepatic ER is altered in obese mice and this shift of lipid composition promotes ER stress through disruption of ER calcium homeostasis.32 The ER stress transducers can also directly sense membrane lipid saturation and activate the UPR.33 This two-way interaction between ER stress and lipid metabolism creates a positive feedback loops to promote hepatic steatosis. UPR activation is protective from transient ER stress-induced hepatic steatosis, unresolved ER stress and defective UPR activation further aggravate ER-stress induced hepatic steatosis.21,34
4. Non-alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is currently the most common cause of abnormal liver chemistry tests in the United States and western world. NAFLD represents a spectrum of diseases associated with the accumulation of triglycerides in hepatocytes, ranging from isolated hepatic steatosis to progressive non-alcoholic steatohepatitis (NASH) that can cause progressive liver injury, fibrosis and cirrhosis.35 With the rapid rise in the prevalence of obesity and the metabolic syndrome, the prevalence of NAFLD is estimated to be approximately 24% globally and 21–24% in the United States.36 Approximately 20% of NAFLD patients will develop NASH, with up to 5% developing cirrhosis. In fact, NASH is expected to be the leading indication for liver transplantation in the United States within the next ten years.37,38 The mechanisms underlying the progression from isolated hepatic steatosis to NASH are poorly understood although proinflammatory cytokines39, oxidative stress, mitochondrial dysfunction40 and genetic factors are believed to contribute to the pathogenesis. ER stress and dysregulation of UPR proteins have been implicated in human NAFLD and NASH.41–43 In fact, NASH is associated with increased p-eIF2α and the inability to adequately express XBP1s41, although other data show increased expression of XBP1s and its target genes in NASH liver biopsies42,44. Studies from animal models of NAFLD also support the important role of the three UPR pathways in the development and progression of NAFLD.45
The hepatic IRE1α/XBP1 pathway is important for lipid metabolism through regulation of hepatic lipid synthesis and VLDL assembly and secretion.46 Hepatic deficiency of IRE1α causes modest elevation of basal liver lipid contents and exacerbates pharmacologic ER stress-induced liver steatosis through increasing lipogenic genes (including CCAAT-enhancer-binding protein β (C/EBPβ), C/EBPδ and peroxisome proliferator-activated receptor gamma (PPARγ)) and reducing VLDL secretion.47 Loss of hepatocyte IRE1α aggravates high-fat diet-induced steatosis, in part by up-regulating miR-200 and miR-34 and down-regulating their targets (PPARα and sirtuin 1 (SIRT1)).48 However, mice with hyperactive IRE1α RNase activity induced by deleting the negative IRE1α regulator bax inhibitor-1 (BI-1) are more vulnerable to pharmacologic ER stress activator tunicamycin- or high fat diet-induced hepatosteatosis, as well as hepatocellular injury with enhanced inflammasome signaling and cell death.49 In addition, overexpressing BI-1 protects from obesity-associated insulin resistance by down-regulating genes involved in lipid metabolism (C/EBPα, SREBP1 and peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC1α)).50 On the other hand, XBP1s transcriptionally regulates fibroblast growth factor 21 (FGF21) to protect from ER stress-induced hepatic steatosis.51 XBP1 overexpression using adenovirus leads to reduced hepatic triglyceride and diacylglycerol in diet-induced and genetically obese mice, which is associated with decreased hepatic fatty acid synthesis rate and enhanced macrolipophagy.52 Loss of hepatic XBP1 results in lower serum triglyceride and cholesterol level due to reduced hepatic de novo lipogenesis.53 XBP1 directly regulates a subset of lipid metabolism genes (such as Fdps, Hsd17b7).54,55 However multiple key lipogenic genes (Dgat2, Angptl3, and etc) have also been shown to be primarily regulated by RIDD. The activation of RIDD pathway in XBP1 deficient mice and subsequent degradation of the mRNA of these key lipogenic genes contributes to the hypolipidemic phenotype in these mice.55 Mice with hepatic XBP1 deficiency have decreased hepatic steatosis when fed lipogenic diets, however, these mice can be protected or predisposed to diet-induced liver injury under different diet compositions and strain backgrounds.31,55 Given that XBP1 deletion causes a feedback activation of IRE1α, whether the observed modulation of liver injury is a direct effect of XBP1 deficiency and/or IRE1α activation is yet to be fully determined.
The PERK/p-eIF2α/ATF4 arm of the UPR also regulates lipid homeostasis. The PERK/ATF4 pathway is essential for tunicamycin-induced VLDLR expression, which mediates pharmacologic ER stress-induced hepatic steatosis.27 Genetic ablation of eIF2α in the liver exacerbates tunicamycin-induced lipid accumulation in the liver.21 GADD34 transgenic mice, which have defective p-eIF2α-mediated UPR signaling, have lower expression of C/EBPα. C/EBPβ, their downstream target PPARγ and improved glucose tolerance in response to a high fat diet, and are protected from high fat diet-induced hepatic steatosis.56 ATF4 null mice are protected from high fructose or high-carbohydrate diet-induced liver steatosis.57,58 ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis in zebrafish.59
ATF6 null mice demonstrate aggravated hepatic steatosis in response to tunicamycin, which is associated with sustained expression of CHOP, inhibition of C/EBPα, decreased fatty acid β oxidation, reduced VLDL formation and enhanced lipid droplet formation.21,60 Hepatocyte ATF6 enhances fatty acid oxidation through interacting with PPARα and increasing PPARα transcriptional activity. Overexpression of dominant negative ATF6 exacerbates diet-induced hepatic steatosis and insulin resistance, whereas overexpression of active ATF6 protects against diet-induced hepatic steatosis.61 ATF6 inhibits hepatic gluconeogenesis by disrupting the cAMP response element binding protein (CREB) and CREB regulated transcription coactivator 2 (CRTC2) interaction and thereby improving glucose metabolism.62 Active ATF6α also interacts and suppresses SREBP2-mediated transcription of lipogenic genes in cultured hepatocyte.63
5. Alcoholic Liver Disease
Alcoholic liver disease (ALD) is a major cause of chronic liver disease worldwide. ALD can cause simple steatosis or can progress to steatohepatitis, fibrosis, cirrhosis and hepatocellular carcinoma.64 Multiple factors associated with alcohol consumption can trigger ER stress, including increased hepatic cytochrome P450 2E1 (CYP2E1) expression, alcohol-induced accumulation of ROS, reduced liver S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH) ratio, excessive serum homocysteine level and epigenetic regulation.65–67 Indeed, ER stress and UPR pathway activation are observed both in livers of patients with chronic ALD68 and in many experimental ALD models67. Intragastric alcoholic feeding in mice up-regulates UPR genes, such as BIP and CHOP.69 In ALD, ER stress contributes to steatosis by activating lipogenic pathways in hepatocytes through SREBP induction.22,69 Alcohol-induced ER stress also plays a role in inflammation through NF-κB and JNK pathway activation. In addition, enhanced ER stress also induces apoptosis via CHOP and interferon regulatory factor 3 (IRF3) pathway. Loss of hepatic BIP causes ER damage and aggravates alcohol-induced liver injury.70 CHOP-null mice are protected from alcohol-induced hepatocellular apoptosis, but not fatty liver, ER stress or hyperhomocysteinemia.71 ATF6 expression is significantly increased in zebrafish model of ALD and blocking ATF6 prevents alcohol-induced hepatic steatosis whereas overexpressing ATF6N induces steatosis in a SREBP-independent/fatty acid synthetase (FASN)-dependent manner.72 Recent studies have shown that ATF4 expression is upregulated by alcohol and liver-specific ATF4 deficient mice are protected from ethanol-induced hepatosteatosis through activating AMP-activated protein kinase (AMPK) signaling.73 The precise role of the IRE1α branch of the UPR in ALD is yet to be determined, though it is anticipated that the IRE1α/XBP1 pathway may modulate ALD-induced hepatic steatosis.
6. Alpha-1 Antitrypsin Deficiency
Alpha-1 antitrypsin (AAT) deficiency (AATD) is a genetic disorder caused by mutations in the AAT-encoding SERPINA1 gene, which results in the retention of the mutant AAT protein in the liver and decreased serum AAT levels. AATD can cause lung disease due to the lack of the protease inhibitor activity in the lung and can be treated by replacement therapy. In contrast, replacement therapy is ineffective for the liver disease, since the liver injury occurs due to the accumulation of the misfolded protein in the liver. The most frequent liver disease-associated “Z” allele mutation leads to conformational change and polymerization of the Z-AAT protein. While most of this mutated protein is degraded through ERAD and autophagy, a large amount of this protein accumulates in the ER of hepatocytes and other AAT-producing cells, stimulating ER stress. Therefore AATD is considered a prototypic protein misfolding and ER stress disease. The retention of mutant Z-AAT protein in hepatocytes leads to hepatocyte damage and predisposes to cirrhosis and hepatocellular carcinoma.74 Studies from liver biopsy samples of AATD patients demonstrate dilated and abnormal ER structures, supporting the existence of ER stress.75 However, the activation of UPR in AATD is inconsistent. Studies using cell line expressing mutant AAT or transgenic mice with liver-specific inducible expression of mutant AAT show no evidence of UPR activation, but specific activation of caspase-12 and NF-κB pathway.76 Whereas other data have shown that BIP promoter and protein expression is increased in cells expressing AAT mutant protein and treated with pharmacologic ER stress inducer thapsigargin.77 UPR activation is observed with accumulation of nonpolymerogenic null Hong Kong (NHK) AAT mutant, which cannot fold properly.76,78 Recently mutant AAT proteins have been shown to interact with IRE1α and induce ATF6 binding responsive reporter activity.79 These suggest that the activation of UPR is context- and cell type-specific. Indeed, the UPR activation is observed in monocytes from individuals with AATD, providing a connection between ER stress and inflammatory response in AATD.80 Nonetheless, one of the current therapeutic approaches is to enhance ERAD and autophagy to decrease ER load of mutant Z-AAT. Activation of the ATF6 pathway is shown to attenuate Z-AAT accumulation and mitochondrial damage in liver cells through promoting ERAD.81 Recently nor-ursodeoxycholic acid (nor-UDCA), a synthetic bile acid under investigation for treatment of cholangiophathies, is shown to be protective in a mouse model of AATD.82
7. Cholestatic Liver Diseases
Cholestatic liver diseases including primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), genetic liver diseases and biliary obstruction are highly prevalent causes of progressive liver diseases that are characterized by the impairment of bile formation and bile flow. ER stress markers BIP and protein disulfide isomerase (PDI) expression increase in small bile ducts in PBC patients, indicating a possible involvement of ER stress in PBC.83 Excess accumulation of toxic bile acids in the liver has been shown to induce ER stress in vivo and in vitro. Feeding mice bile acid increases hepatic expression of UPR genes (Xbp1s, Bip and Chop).84,85 In a genetic model of intrahepatic cholestasis, liver-specific Foxa2 ablation results in accumulated bile acids with resultant ER stress, dilated ER and increased BIP expression.85 In α-naphthyl isothiocyanate (ANIT) model of intrahepatic cholestasis, expression of UPR genes (Bip, Perk, eIF2α, Ire1α and Atf6) and proteins (BIP, p-IRE1α) is up-regulated.86 Wild-type mice subjected to bile duct ligation, a model of obstructive cholestasis, demonstrate robust and transient ER stress with increased hepatic expression of IRE1α, XBP1s, p-PERK and CHOP.84,87,88 CHOP deficiency attenuates obstructive cholestatic liver injury and fibrosis and CHOP-deficient hepatocytes display decreased cell death in response to the bile acid glycochenodeoxycholic acid (GCDCA).89 The hepatoprotective role of global CHOP knockout mice subjected to bile duct ligation may also be due, in part, to suppression of intestinal stem cell proliferation and intestinal epithelial cell regeneration. CHOP can disrupt intestinal barrier tight junction, increase intestinal bacteria translocation, kupffer cell activation and inflammatory responses, supporting the liver-gut axis in the pathogenesis of cholestatic liver injury.88 Treating HepG2 cells with the bile acid sodium deoxycholate induces promoter activity of BIP and CHOP.90 Hepatocytes treated with various bile acids also demonstrate increased expression of BIP, XBP1s and CHOP, ER-related calcium release and mitochondria oxidative stress.84,91–94 Interestingly ER stress and the UPR have been shown to regulate bile acid homeostasis, which may serve as a feedback mechanism to modulate the cellular response to cholestasis.95,96
8. Viral Hepatitis
Hepatitis B virus (HBV) and hepatitis C virus (HCV) cause chronic infection that can progress to fibrosis, cirrhosis and eventually hepatocellular carcinoma. ER stress occurs during viral infection as the ER in the host cells is utilized to synthesize a large amount of viral proteins over a short period, which leads to perturbation of the ER homeostasis. The activation of UPR by HBV and HCV infection has been shown in human liver tissues.97,98 The role of ER stress and UPR activation by HBV and HCV infection is also well described in vivo and in vitro.99–101 The multifunctional regulatory protein of HBV, hepatitis B x protein (HBx) activates IRE1α/XBP1 and ATF6 pathways of the UPR to promote HBV replication and expression in liver cells, contributing to liver disease pathogenesis.102 HBx also activates the PERK/ATF4 pathway to induce cyclooxygenase-2 (COX-2) expression which mediates inflammation.103 However other data have shown that HBx represses PERK/ATF4 pathway to inhibit cell apoptosis to promote HBV-associated carcinogenesis.104 In addition, pre-S mutant proteins have been shown to accumulate in the ER and can trigger ER stress to induce oxidative DNA damage and genomic instability.105,106 Recently the middle S protein has been shown to initiate ER stress and activate the p38/NF-kB pathway to produce IL-6.107 HCV infection in Huh7 cells induces an acute and sustained activation of all three UPR pathways.108 However, in a liver-specific Huh7 line stably expressing HCV replicons, XBP1s transcriptional activity and its downstream ERAD pathway are repressed (even though XBP1s expression is increased), allowing the viral proteins to escape degradation and accumulate in the cells.109 A number of HCV proteins triggers ER stress. Expression of HCV envelope proteins in Hela cells induces PERK/ATF4-depedent CHOP expression and XBP1 splicing.110 HCV core protein is processed in the ER and expressing HCV core protein elicits ER stress and calcium depletion leading to apoptosis.111 Nonstructural HCV protein NS4B modulates the UPR by inducing ATF6 cleavage and XBP1 splicing, though the expression of XBP1s downstream ERAD target gene EDEM is repressed to favor HCV replication.112,113 Viral infection-induced ER stress and adaptive UPR pathway are essential for viral productive life cycles; however chronic ER stress induced by hepatic virus contribute to hepatic inflammation and the progression of liver diseases. Pharmacologic inhibition of ER stress by administration of 4-phenylbutyric acid (4-PBA) or the specific IRE1α inhibitor 4μ8C impaired the HBV production induced by cisplatin treatment.114
9. Ischemia/Reperfusion Injury
Liver ischemia/reperfusion (I/R) injury can occur during systemic hypotension, vascular occlusion, and surgery including liver transplantation. I/R injury in the donor liver is a major determinant for clinical outcomes. I/R injury causes liver damage through oxidative stress, inflammation, calcium release from the ER and apoptosis.115,116 ER stress and activation of the UPR have been shown to play a role in I/R injury. In ischemic and reperfused human livers, UPR activation, as assessed by expression of BIP, CHOP, GADD34, XBP1s, p-PERK and p-eIF2α, occurs in a biphasic manner.117 Similar UPR activation is also observed in cold preserved liver grafts118 and mouse models of I/R injury. Hepatic XBP1 splicing and increased BIP expression occur in a traumatic-hemorrhagic shock model119 and cleaved ATF6 expression increases in ischemic mice livers and promotes ER stress-medicated inflammatory responses.120,121 I/R induces cytoprotective protein BI-1 expression and loss of BI-1 enhances IRE1α and ATF6 activation and increases hepatic liver damage.122 The involvement of ER stress in IR injury is further supported by the protective effect of chemical chaperone 4-PBA and tauroursodeoxycholic acid (TUDCA) against I/R injury.123,124
10. Drug-Induced Liver Injury
The majority of drugs and xenobiotics are metabolized in the liver and drug-induced liver injury (DILI) is often a dose-limiting side effect for many medications. The pathogenesis of DILI is complex, including hepatocyte apoptosis, inflammatory response activation, mitochondrial dysfunction and bile duct injury.125 The smooth ER of the hepatocyte is the primary site of drug metabolism and ER stress has recently been shown to have an important role in DILI126,127, and has been linked to the adverse events of many drugs128. Acetaminophen (APAP) overdose is the most common cause of acute liver failure, and APAP causes hepatocellular injury by depleting glutathione level and altering the redox balance in the ER. APAP toxicity results in increased expression of p-eIF2α and CHOP and mice with CHOP deficiency are protected from APAP-induced liver damage, with decreased liver necrosis and increased survival.129 A sublethal dose of APAP also induces ATF6 and transient caspase12 activation in mouse liver.130 Liver-specific XBP1 deficiency protects the mice from APAP-induced hepatotoxicity through IRE1α/RIDD-mediated down-regulation of Cyp1a2 and Cyp2e1.131 The role of ER stress in APAP-induced liver injury is further supported by the evidence that treating mice with 4-PBA decreases APAP-induced hepatocyte apoptosis/necrosis.132 In addition to APAP, HIV protease inhibitors (PI) increase ER stress and UPR activation in hepatocytes, intestinal epithelial cells, macrophages and adipocytes contributing to PI-induced hepatocyte injury and metabolic syndrome.133–136
11. Hepatocellular Carcinoma
ER stress occurs when the microenvironment changes in cancer cells and has been implicated in many forms of cancer including hepatocellular carcinoma (HCC). In the United States, the incidence and mortality from HCC is rising and HCC occurs predominantly in the presence of pre-existing cirrhosis. HBV can cause HCC in the absence of cirrhosis, and is a common cause of liver cancer death worldwide. In addition, recent data indicate that HCC may occur in non-alcoholic steatohepatitis prior to the development of cirrhosis.137 Activated gene expression of the ATF6, XBP1s and BIP has been reported in human HCC138 and UPR pathways are activated at different stage of tumorigenesis in an orthotopic mouse model of HCC139. IRE1α signaling may be crucial during HCC initiation. Liver-specific IRE1α deficient mice have decreased HCC incidence in diethylnitrosamine (DEN)-treated mice irrespective of their adiposity status. This is associated with STAT3 activation and decreased hepatocyte proliferation, in spite of increased hepatic apoptosis, and reduced production of tumor necrosis factor α (TNFα) and interleukin 6 (IL-6).140 CHOP also has a role in ER stress-induced HCC cell apoptosis through inhibiting autophagy.15 The activation of the UPR in response to tumorigenesis-induced ER stress is a protective mechanism for cancer cells survival, adaptation to adverse environmental conditions, and resistance to conventional chemotherapy. Therefore, the UPR may serves as a therapeutic target for cancer treatment. Ongoing clinical studies are investigating the role of XBP1 inhibitors in multiple myeloma and other malignancies.
12. Modulators of ER Stress and UPR in Liver Diseases
ER stress and UPR activation are implicated in the etiology of many liver diseases; therefore modulators of ER stress and the UPR are of interest for treatment of liver diseases.141,142 Several compounds have been developed either targeting a single UPR pathway or acting as protein chaperones in order to modulate ER stress. Ursodeoxycholic acid (UDCA) and 4-PBA are chemical chaperones that promote protein folding and assembly and are FDA-approved to treat primary biliary cholangitis and urea-cycle disorder, respectively. TUDCA and 4-PBA have been shown to be beneficial in several murine models of fatty liver diseases.143,144 4-PBA has also been shown to increase secretion of the mutant AAT protein145, while TUDCA inhibits apoptosis induced by mutant AAT protein146 and reduces hepatocarcinogenesis in a diethylnitrosamine (DEN) model of HCC147. Berberine, a natural plant alkaloid, has been shown to prevent the progression from steatosis and steatohepatitis by reducing ER stress.148 The IRE1α inhibitor 4μ8C suppresses carbon tetrachloride (CCl4)-induced liver injury and fibrosis.149 Similarly PERK pathway modulator salubrinal prevents eIF2α dephosphorylation and improves HepG2 cell viability in response to tunicamycin.139 Other small inhibitors of UPR pathways are currently being developed for several liver and other benign and malignant diseases.
13. Integrated Stress Response
The integrated stress response (ISR) is an adaptive response to cellular stress, including ER stress and UPR pathways are important in the ISR. In addition to PERK, the ISR comprises three additional eIF2α kinases, general control nonrepressed 2 kinase (GCN2), dsRNA activated protein kinase (PKR) and heme-regulated eIF2α kinase (HRI), that phosphorylate eIF2α under different stress conditions.150 Similar to the UPR, transient activation of the ISR is considered pro-survival, whereas prolonged ISR activation can lead to induction of cell death. The activation of ISR promotes ATF6 activation during ER stress.151 The ISR is important in cardiovascular disease152, lung disease153, inherited retinal degeneration154 and central nervous system injury155. Recently ISR has been connected to metabolic disease through FGF21 induction in diet-induced obesity and insulin resistance.156 The activation of the ISR has been implicated in the pathogenesis of liver steatosis.56–58 The role of ISR in host of liver diseases remains unclear, providing avenues for future research in liver diseases.
14. Conclusion
ER stress and subsequent adaptive UPR activation are important in the pathogenesis of many liver disorders. The three UPR pathways are not only essential to restore ER homeostasis and promote cell survival, but also have important roles in regulating metabolic functions including glucose and lipid metabolism. ER stress often occurs initially as a result of the pathological conditions, and then subsequent defective UPR activation or prolonged ER stress can predispose the system to further injury. ER stress also occurs concomitantly with oxidative stress, mitochondrial dysfunction and autophagy. The complex interplay between these hepatic signaling pathways may collectively determine the overall response to the injurious stimuli. The causative or resultant role of ER stress in liver diseases and the precise contribution of each individual UPR pathways are incompletely understood. Further investigation on the mechanisms of ER stress and UPR activation during hepatic diseases may allow for the identification of novel, effective therapeutic targets to treat liver diseases.
Table 1.
Human studies of the involvement of hepatic ER stress and UPR activation in liver diseases.
| Liver disease | Findings | Reference(s) |
|---|---|---|
| Non-alcoholic fatty liver disease/ | ↓XBP1s | 41 |
| Non-alcoholic steatohepatitis | ↑p-eIF2α | 41 |
| ↑XBP1s and targets | 42–44 | |
| ↓BIP, PDI | 43 | |
| Alcoholic liver disease | ↑BIP | 68 |
| ↑IRE1α | 68 | |
| ↑PERK, p-PERK, ATF4, CHOP | 68 | |
| Alpha-1 antitrypsin deficiency | Dilated and abnormal ER | 75 |
| Primary biliary cholangitis | ↑BIP, PDI | 83 |
| Hepatitis B | ↑BIP, XBP1s | 97 |
| Hepatitis C | Abnormal ER | 98 |
| ↑XBP1s, ATF6 and PERK, ATF4 pathway | 98 | |
| ↑BIP | 97 | |
| Ischemia/reperfusion injury | ↑BIP, CHOP, GADD34, XBP1s, p-PERK and p-eIF2α | 117 |
| Hepatocellular carcinoma | ↑ATF6, XBP1s and BIP | 138 |
Table 2.
UPR pathways in experimental models of liver diseases.
| Liver Disease | UPR component | Functions | Ref |
|---|---|---|---|
| Non-alcoholic fatty liver disease/ | IRE1α | Regulates hepatic lipogenesis through RIDD | 55 |
| Non-alcoholic steatohepatitis | IRE1α | Prevents tunicamycin-induced hepatic steatosis | 47 |
| IRE1α | Protects from diet-induced steatosis | 48 | |
| IRE1α | Promotes the progression of NAFLD to NASH | 49 | |
| XBP1 | Protects from steatosis | 51,52 | |
| XBP1 | Protects from diet-induced liver injury | 31 | |
| XBP1 | Regulates hepatic lipogenesis | 53–55 | |
| XBP1 | Contributes to diet-induced liver injury | 55 | |
| PERK/ATF4 | Essential for tunicamycin-induced hepatic steatosis | 27 | |
| ATF4 | Contributes to diet-induced hepatic steatosis | 57,58 | |
| ATF4 | Induces early onset of steatosis in zebrafish | 59 | |
| eIF2α | Protects from tunicamycin-induced fatty liver | 21 | |
| GADD34 | Protects from diet-induced steatosis | 56 | |
| ATF6 | Protects from tunicamycin-induced fatty liver | 21,60 | |
| ATF6 | Protects from diet-induced steatosis | 61 | |
| Alcoholic liver disease | BIP | Protects from alcohol-induced liver injury | 70 |
| ATF4 | Responsible for alcohol-induced hepatic steatosis | 73 | |
| CHOP | Promotes alcohol-induced liver injury | 71 | |
| ATF6 | Induces alcohol-induced hepatic steatosis | 72 | |
| Alpha-1 antitrypsin deficiency | IRE1α | Interacts with mutant AAT to induce ATF6 activity | 79 |
| ATF6 | Attenuates liver mitochondrial damage through promoting ERAD | 81 | |
| Cholestasis | IRE1α/XBP1 | Increases with bile acids treatment or bile duct ligation | 84–87 |
| IRE1α/XBP1 | Regulates bile acid metabolism | 95 | |
| PERK | Phosphorylation increases with bile duct ligation | 87 | |
| CHOP | Increases with bile duct ligation | 87,88 | |
| CHOP | Promotes cholestatic cell death and liver injury | 88,89 | |
| Hepatitis B | IRE1α/XBP1 | Activated by HBx to promote HBV replication | 102 |
| PERK/ATF4 | Activated by HBx to induce inflammation mediator COX-2 | 103 | |
| PERK/ATF4 | Repressed by HBx to inhibit apoptosis | 104 | |
| ATF6 | Activated by HBx to promote HBV replication | 102 | |
| Hepatitis C | IRE1α/XBP1 | Activated by HCV protein | 108, 110 |
| IRE1α/XBP1 | Transcriptional activity repressed by HCV | 109, 112, 113 | |
| PERK/ATF4 | Activated by HCV protein | 110 | |
| ATF6 | Activated by HCV protein | 112, 113 | |
| Ischemia/reperfusion injury | XBP1s | Increases in mouse model of Ischemia/reperfusion injury | 119 |
| ATF6 | Increases in ischemic mouse livers | 120 | |
| ATF6 | Promotes ER stress-medicated inflammatory responses | 120 | |
| Acetaminophen overdose | IRE1α | Protects from acetaminophen toxicity through RIDD | 131 |
| p-eIF2α | Increased with acetaminophen toxicity | 129 | |
| CHOP | Increased with acetaminophen toxicity | 129 | |
| CHOP | Mediates acetaminophen toxicity-induced liver injury | 129 | |
| ATF6 | Activated by a sublethal dose of Acetaminophen | 130 | |
| Hepatocellular carcinoma | IRE1α/XBP1s | Increases in HCC model, promotes HCC development | 139,140 |
| PERK/ATF4 | Increases in HCC model | 139 | |
| CHOP | Increases in HCC model, promotes HCC cell apoptosis | 15 | |
| ATF6 | Downstream targets increase in HCC model | 139 | |
Acknowledgements:
This work is supported by NIDDK R01 DK093807.
Abbreviations:
- 4-PBA
4-phenylbutyric acid
- AAT
alpha-1 antitrypsin
- AATD
alpha-1 antitrypsin deficiency
- ALD
alcoholic liver disease
- AMPK
AMP-activated protein kinase
- ANIT
α-naphthyl isothiocyanate
- APAP
acetaminophen
- ASK1
apoptosis signal-regulating kinase 1
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- BAK
Bcl2 antagonist/killer 1
- BAX
Bcl2-associated X protein
- BCL2
B-cell lymphoma 2
- BI-1
bax inhibitor-1
- BIM
Bcl-2-like protein 11
- BIP
binding immunoglobulin protein
- CCl4
carbon tetrachloride
- C/EBPα
CCAAT-enhancer-binding protein α
- C/EBPβ
CCAAT-enhancer-binding protein β
- C/EBPδ
CCAAT-enhancer-binding protein δ
- CHOP
CCAAT-enhancer-binding protein homologous protein
- COX-2
Cyclooxygenase-2
- CREB
cAMP response element binding protein
- CReP
constitutive repressor of eIF2α phosphorylation
- CRTC2
CREB regulated transcription coactivator 2
- CYP2E1
cytochrome P450 2E1
- DEN
diethylnitrosamine
- DILI
drug-induced liver injury
- DR5
death receptor 5
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERO1α
ER oxidase 1α
- FASN
fatty acid synthetase
- FGF21
fibroblast growth factor 21
- GADD34
growth arrest and DNA damage-inducible protein 34
- GCDCA
glycochenodeoxycholic acid
- GCN2
general control nonrepressed 2 kinase
- HBV
hepatitis B virus
- HBx
hepatitis B x protein
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HRI
heme-regulated eIF2α kinase
- IL-6
interleukin 6
- I/R
ischemia/reperfusion
- IP3R
inositol-1,4,5-trisphosphate receptor
- IRE1α
inositol-requiring enzyme 1α
- IRF3
interferon regulatory factor 3
- ISR
integrated stress response
- JNK
c-jun N-terminal kinase
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NF-κB
nuclear factor kappa B
- NHK
null Hong Kong
- nor-UDCA
nor-ursodeoxycholic acid
- p-eIF2α
phosphorylated-eIF2α
- PBC
primary biliary cholangitis
- PDI
protein disulfide isomerase
- PERK
PKR-like ER kinase
- PGC1α
peroxisome proliferator-activated receptor-gamma coactivator 1α
- PI
protease inhibitor
- PKR
dsRNA activated protein kinase
- PP1
protein phosphatase 1
- PPARα
peroxisome proliferator-activated receptor α
- PPARγ
peroxisome proliferator-activated receptor γ
- PSC
primary sclerosing cholangitis
- RIDD
regulated IRE1α-dependent decay of mRNA
- ROS
reactive oxygen species
- S1P
site-1 protease
- S2P
site-2 protease
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SIRT1
sirtuin 1
- SREBP1c
sterol regulatory element-binding protein 1c
- SREBP2
sterol regulatory element-binding protein 2
- TNFα
tumor necrosis factor α
- TRAF2
TNF receptor associated factor 2
- TRB3
tribbles homolog 3
- TUDCA
tauroursodeoxycholic acid
- UPR
unfolded protein response
- VLDL
very low-density lipoprotein
- VLDLR
very low-density lipoprotein receptor
- XBP1
X-box binding protein 1
- XBP1s
XBP1 spliced
- XBP1u
XBP1 unspliced
Reference:
- 1.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. [DOI] [PubMed] [Google Scholar]
- 3.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. [DOI] [PubMed] [Google Scholar]
- 5.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P, Li J, Tao J, Sha B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J Biol Chem. 2018;293:4110–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci U S A. 2009;106:1832–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto K, Sato T, Matsui T and et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. [DOI] [PubMed] [Google Scholar]
- 9.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. [DOI] [PubMed] [Google Scholar]
- 10.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marciniak SJ, Yun CY, Oyadomari S and et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li G, Mongillo M, Chin KT and et al. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol. 2009;186:783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lei Y, Wang S, Ren B and et al. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS One. 2017;12:e0183680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoneda T, Imaizumi K, Oono K and et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa T, Zhu H, Morishima N and et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. [DOI] [PubMed] [Google Scholar]
- 18.Upton JP, Wang L, Han D and et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han D, Lerner AG, Vande Walle L and et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hetz C, Bernasconi P, Fisher J and et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. [DOI] [PubMed] [Google Scholar]
- 21.Rutkowski DT, Wu J, Back SH and et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Werstuck GH, Lentz SR, Dayal S and et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JN, Ye J. Proteolytic activation of sterol regulatory element-binding protein induced by cellular stress through depletion of Insig-1. J Biol Chem. 2004;279:45257–45265. [DOI] [PubMed] [Google Scholar]
- 24.Kammoun HL, Chabanon H, Hainault I and et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caviglia JM, Gayet C, Ota T and et al. Different fatty acids inhibit apoB100 secretion by different pathways: unique roles for ER stress, ceramide, and autophagy. J Lipid Res. 2011;52:1636–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo H, Choe SS, Shin KC and et al. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology. 2013;57:1366–1377. [DOI] [PubMed] [Google Scholar]
- 28.DeZwaan-McCabe D, Sheldon RD, Gorecki MC and et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017;19:1794–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–281. [DOI] [PubMed] [Google Scholar]
- 30.Cao J, Dai DL, Yao L and et al. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell Biochem. 2012;364:115–129. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Henkel AS, LeCuyer BE, Schipma MJ, Anderson KA, Green RM. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am J Physiol Gastrointest Liver Physiol. 2015;309:G965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu S, Yang L, Li P and et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A. 2013;110:4628–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olivares S, Henkel AS. Hepatic Xbp1 Gene Deletion Promotes Endoplasmic Reticulum Stress-induced Liver Injury and Apoptosis. J Biol Chem. 2015;290:30142–30151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angulo P Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. [DOI] [PubMed] [Google Scholar]
- 36.Younossi Z, Anstee QM, Marietti M and et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- 37.Adams LA, Lymp JF, St Sauver J and et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. [DOI] [PubMed] [Google Scholar]
- 38.Lazo M, Hernaez R, Eberhardt MS and et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol. 2013;178:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diehl AM, Li ZP, Lin HZ, Yang SQ. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut. 2005;54:303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. [DOI] [PubMed] [Google Scholar]
- 41.Puri P, Mirshahi F, Cheung O and et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. [DOI] [PubMed] [Google Scholar]
- 42.Lake AD, Novak P, Hardwick RN and et al. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci. 2014;137:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S, Kim S, Hwang S, Cherrington NJ, Ryu DY. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget. 2017;8:63370–63381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim RS, Hasegawa D, Goossens N and et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci Rep. 2016;6:39342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henkel A, Green RM. The unfolded protein response in fatty liver disease. Semin Liver Dis. 2013;33:321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S, Chen Z, Lam V and et al. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012;16:473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K, Wang S, Malhotra J and et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30:1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang JM, Qiu Y, Yang Z and et al. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci Signal. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lebeaupin C, Vallee D, Rousseau D and et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology. 2018;68:515–532. [DOI] [PubMed] [Google Scholar]
- 50.Bailly-Maitre B, Belgardt BF, Jordan SD and et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem. 2010;285:6198–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang S, Yan C, Fang QC and et al. Fibroblast growth factor 21 is regulated by the IRE1alpha-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J Biol Chem. 2014;289:29751–29765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrema H, Zhou Y, Zhang D and et al. XBP1s Is an Anti-lipogenic Protein. J Biol Chem. 2016;291:17394–17404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.So JS, Hur KY, Tarrio M and et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16:487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao G, Zhang T, Yu S and et al. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J Biol Chem. 2013;288:25350–25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H, Meng Q, Xiao F and et al. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem J. 2011;438:283–289. [DOI] [PubMed] [Google Scholar]
- 59.Yeh KY, Lai CY, Lin CY, Hsu CC, Lo CP, Her GM. ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis and enhances adipogenesis in zebrafish. Sci Rep. 2017;7:16362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamamoto K, Takahara K, Oyadomari S and et al. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21:2975–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X, Zhang F, Gong Q and et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor alpha. Diabetes. 2016;65:1904–1915. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng L, Lu M, Mori K and et al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28 Suppl 1:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–242. [DOI] [PubMed] [Google Scholar]
- 66.Ji C Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J Gastroenterol Hepatol. 2008;23 Suppl 1:S16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ji C New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int J Hepatol. 2014;2014:513787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Longato L, Ripp K, Setshedi M and et al. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Longev. 2012;2012:479348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. [DOI] [PubMed] [Google Scholar]
- 70.Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54:229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Howarth DL, Lindtner C, Vacaru AM and et al. Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish. PLoS Genet. 2014;10:e1004335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li K, Xiao Y, Yu J and et al. Liver-specific Gene Inactivation of the Transcription Factor ATF4 Alleviates Alcoholic Liver Steatosis in Mice. J Biol Chem. 2016;291:18536–18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004;286:G851–862. [DOI] [PubMed] [Google Scholar]
- 75.Hultcrantz R, Mengarelli S. Ultrastructural liver pathology in patients with minimal liver disease and alpha 1-antitrypsin deficiency: a comparison between heterozygous and homozygous patients. Hepatology. 1984;4:937–945. [DOI] [PubMed] [Google Scholar]
- 76.Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and −12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280:39002–39015. [DOI] [PubMed] [Google Scholar]
- 77.Lawless MW, Greene CM, Mulgrew A, Taggart CC, O’Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol. 2004;172:5722–5726. [DOI] [PubMed] [Google Scholar]
- 78.Ordonez A, Snapp EL, Tan L, Miranda E, Marciniak SJ, Lomas DA. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology. 2013;57:2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sundaram A, Appathurai S, Plumb R, Mariappan M. Dynamic changes in complexes of IRE1alpha, PERK, and ATF6alpha during endoplasmic reticulum stress. Mol Biol Cell. 2018;29:1376–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carroll TP, Greene CM, O’Connor CA, Nolan AM, O’Neill SJ, McElvaney NG. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J Immunol. 2010;184:4538–4546. [DOI] [PubMed] [Google Scholar]
- 81.Smith SE, Granell S, Salcedo-Sicilia L and et al. Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem. 2011;286:41563–41577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang Y, Fickert P, Trauner M, Marcus N, Blomenkamp K, Teckman J. Autophagy induced by exogenous bile acids is therapeutic in a model of alpha-1-AT deficiency liver disease. Am J Physiol Gastrointest Liver Physiol. 2016;311:G156–165. [DOI] [PubMed] [Google Scholar]
- 83.Sasaki M, Yoshimura-Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J Gastroenterol. 2015;50:984–995. [DOI] [PubMed] [Google Scholar]
- 84.Liu X, Guo GL, Kong B and et al. Farnesoid X receptor signaling activates the hepatic X-box binding protein 1 pathway in vitro and in mice. Hepatology. 2018;68:304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bochkis IM, Rubins NE, White P, Furth EE, Friedman JR, Kaestner KH. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat Med. 2008;14:828–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yao X, Li Y, Cheng X, Li H. ER stress contributes to alpha-naphthyl isothiocyanate-induced liver injury with cholestasis in mice. Pathol Res Pract. 2016;212:560–567. [DOI] [PubMed] [Google Scholar]
- 87.Huang YH, Yang YL, Huang FC and et al. MicroRNA-29a mitigation of endoplasmic reticulum and autophagy aberrance counteracts in obstructive jaundice-induced fibrosis in mice. Exp Biol Med (Maywood). 2018;243:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu R, Li X, Huang Z and et al. C/EBP homologous protein-induced loss of intestinal epithelial stemness contributes to bile duct ligation-induced cholestatic liver injury in mice. Hepatology. 2018;67:1441–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tamaki N, Hatano E, Taura K and et al. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498–505. [DOI] [PubMed] [Google Scholar]
- 90.Bernstein H, Payne CM, Bernstein C, Schneider J, Beard SE, Crowley CL. Activation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholate. Toxicol Lett. 1999;108:37–46. [DOI] [PubMed] [Google Scholar]
- 91.Tsuchiya S, Tsuji M, Morio Y, Oguchi K. Involvement of endoplasmic reticulum in glycochenodeoxycholic acid-induced apoptosis in rat hepatocytes. Toxicol Lett. 2006;166:140–149. [DOI] [PubMed] [Google Scholar]
- 92.Iizaka T, Tsuji M, Oyamada H, Morio Y, Oguchi K. Interaction between caspase-8 activation and endoplasmic reticulum stress in glycochenodeoxycholic acid-induced apoptotic HepG2 cells. Toxicology. 2007;241:146–156. [DOI] [PubMed] [Google Scholar]
- 93.Adachi T, Kaminaga T, Yasuda H, Kamiya T, Hara H. The involvement of endoplasmic reticulum stress in bile acid-induced hepatocellular injury. J Clin Biochem Nutr. 2014;54:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cai SY, Ouyang X, Chen Y and et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight. 2017;2:e90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu X, Henkel AS, LeCuyer BE and et al. Hepatic deletion of X-box binding protein 1 impairs bile acid metabolism in mice. J Lipid Res. 2017;58:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Henkel AS, LeCuyer B, Olivares S, Green RM. Endoplasmic Reticulum Stress Regulates Hepatic Bile Acid Metabolism in Mice. Cell Mol Gastroenterol Hepatol. 2017;3:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yeganeh B, Rezaei Moghadam A, Alizadeh J and et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J Gastroenterol. 2015;21:13225–13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Asselah T, Bieche I, Mansouri A and et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J Pathol. 2010;221:264–274. [DOI] [PubMed] [Google Scholar]
- 99.Lazar C, Uta M, Branza-Nichita N. Modulation of the unfolded protein response by the human hepatitis B virus. Front Microbiol. 2014;5:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim SY, Kyaw YY, Cheong J. Functional interaction of endoplasmic reticulum stress and hepatitis B virus in the pathogenesis of liver diseases. World J Gastroenterol. 2017;23:7657–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chan SW. Unfolded protein response in hepatitis C virus infection. Front Microbiol. 2014;5:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li B, Gao B, Ye L and et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007;124:44–49. [DOI] [PubMed] [Google Scholar]
- 103.Cho HK, Cheong KJ, Kim HY, Cheong J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem J. 2011;435:431–439. [DOI] [PubMed] [Google Scholar]
- 104.Li J, He J, Fu Y and et al. Hepatitis B virus X protein inhibits apoptosis by modulating endoplasmic reticulum stress response. Oncotarget. 2017;8:96027–96034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang HC, Wu HC, Chen CF, Fausto N, Lei HY, Su IJ. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am J Pathol. 2003;163:2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang HC, Huang W, Lai MD, Su IJ. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006;97:683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li YX, Ren YL, Fu HJ, Zou L, Yang Y, Chen Z. Hepatitis B Virus Middle Protein Enhances IL-6 Production via p38 MAPK/NF-kappaB Pathways in an ER Stress-Dependent Manner. PLoS One. 2016;11:e0159089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Merquiol E, Uzi D, Mueller T and et al. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One. 2011;6:e24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J Biol Chem. 2004;279:17158–17164. [DOI] [PubMed] [Google Scholar]
- 110.Chan SW, Egan PA. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005;19:1510–1512. [DOI] [PubMed] [Google Scholar]
- 111.Benali-Furet NL, Chami M, Houel L and et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. [DOI] [PubMed] [Google Scholar]
- 112.Zheng Y, Gao B, Ye L and et al. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J Microbiol. 2005;43:529–536. [PubMed] [Google Scholar]
- 113.Li S, Ye L, Yu X and et al. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology. 2009;391:257–264. [DOI] [PubMed] [Google Scholar]
- 114.Li X, Pan E, Zhu J and et al. Cisplatin Enhances Hepatitis B Virus Replication and PGC-1alpha Expression through Endoplasmic Reticulum Stress. Sci Rep. 2018;8:3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cannistra M, Ruggiero M, Zullo A and et al. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int J Surg. 2016;33 Suppl 1:S57–70. [DOI] [PubMed] [Google Scholar]
- 117.Emadali A, Nguyen DT, Rochon C, Tzimas GN, Metrakos PP, Chevet E. Distinct endoplasmic reticulum stress responses are triggered during human liver transplantation. J Pathol. 2005;207:111–118. [DOI] [PubMed] [Google Scholar]
- 118.Folch-Puy E, Panisello A, Oliva J and et al. Relevance of Endoplasmic Reticulum Stress Cell Signaling in Liver Cold Ischemia Reperfusion Injury. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Duvigneau JC, Kozlov AV, Zifko C and et al. Reperfusion does not induce oxidative stress but sustained endoplasmic reticulum stress in livers of rats subjected to traumatic-hemorrhagic shock. Shock. 2010;33:289–298. [DOI] [PubMed] [Google Scholar]
- 120.Zhou H, Zhu J, Yue S and et al. The Dichotomy of Endoplasmic Reticulum Stress Response in Liver Ischemia-Reperfusion Injury. Transplantation. 2016;100:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rao J, Yue S, Fu Y and et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transplant. 2014;14:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bailly-Maitre B, Fondevila C, Kaldas F and et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:2809–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vilatoba M, Eckstein C, Bilbao G and et al. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138:342–351. [DOI] [PubMed] [Google Scholar]
- 124.Ben Mosbah I, Alfany-Fernandez I, Martel C and et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee SJ, Lee YJ, Park KK. The pathogenesis of drug-induced liver injury. Expert Rev Gastroenterol Hepatol. 2016:1–11. [DOI] [PubMed] [Google Scholar]
- 126.Chen S, Melchior WB Jr., Guo L. Endoplasmic reticulum stress in drug- and environmental toxicant-induced liver toxicity. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2014;32:83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hinton MLY, Kwong E., Zhou H. in Cellular Injury in Liver Diseases. (ed Yin XM. Ding WX.) 37–53 (Springer, Cham, 2017). [Google Scholar]
- 128.Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect. 2016;4:e00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Uzi D, Barda L, Scaiewicz V and et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol. 2013;59:495–503. [DOI] [PubMed] [Google Scholar]
- 130.Nagy G, Kardon T, Wunderlich L and et al. Acetaminophen induces ER dependent signaling in mouse liver. Arch Biochem Biophys. 2007;459:273–279. [DOI] [PubMed] [Google Scholar]
- 131.Hur KY, So JS, Ruda V and et al. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kusama H, Kon K, Ikejima K and et al. Sodium 4-phenylbutyric acid prevents murine acetaminophen hepatotoxicity by minimizing endoplasmic reticulum stress. J Gastroenterol. 2017;52:611–622. [DOI] [PubMed] [Google Scholar]
- 133.Zhou H, Gurley EC, Jarujaron S and et al. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1071–1080. [DOI] [PubMed] [Google Scholar]
- 134.Zhou H HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Methods Enzymol. 2011;490:107–119. [DOI] [PubMed] [Google Scholar]
- 135.Zhou H, Pandak WM Jr., Lyall V, Natarajan R, Hylemon PB. HIV protease inhibitors activate the unfolded protein response in macrophages: implication for atherosclerosis and cardiovascular disease. Mol Pharmacol. 2005;68:690–700. [DOI] [PubMed] [Google Scholar]
- 136.Zha BS, Wan X, Zhang X and et al. HIV protease inhibitors disrupt lipid metabolism by activating endoplasmic reticulum stress and inhibiting autophagy activity in adipocytes. PLoS One. 2013;8:e59514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nakagawa H, Umemura A, Taniguchi K and et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shuda M, Kondoh N, Imazeki N and et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605–614. [DOI] [PubMed] [Google Scholar]
- 139.Vandewynckel YP, Laukens D, Bogaerts E and et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: a PERK for hepatocellular carcinoma therapy. Hepatol Int. 2015;9:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wu Y, Shan B, Dai J and et al. Dual role for inositol-requiring enzyme 1alpha in promoting the development of hepatocellular carcinoma during diet-induced obesity in mice. Hepatology. 2018. [DOI] [PubMed] [Google Scholar]
- 141.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–597. [DOI] [PubMed] [Google Scholar]
- 142.Maly DJ, Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014;10:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ozcan U, Yilmaz E, Ozcan L and et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cho EJ, Yoon JH, Kwak MS and et al. Tauroursodeoxycholic acid attenuates progression of steatohepatitis in mice fed a methionine-choline-deficient diet. Dig Dis Sci. 2014;59:1461–1474. [DOI] [PubMed] [Google Scholar]
- 145.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97:1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Miller SD, Greene CM, McLean C and et al. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of Bad. Hepatology. 2007;46:496–503. [DOI] [PubMed] [Google Scholar]
- 147.Vandewynckel YP, Laukens D, Devisscher L and et al. Tauroursodeoxycholic acid dampens oncogenic apoptosis induced by endoplasmic reticulum stress during hepatocarcinogen exposure. Oncotarget. 2015;6:28011–28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang Z, Li B, Meng X and et al. Berberine prevents progression from hepatic steatosis to steatohepatitis and fibrosis by reducing endoplasmic reticulum stress. Sci Rep. 2016;6:20848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wu T, Zhao F, Gao B and et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Teske BF, Wek SA, Bunpo P and et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22:4390–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Santos-Ribeiro D, Godinas L, Pilette C, Perros F. The integrated stress response system in cardiovascular disease. Drug Discov Today. 2018;23:920–929. [DOI] [PubMed] [Google Scholar]
- 153.van ‘t Wout EF, Hiemstra PS, Marciniak SJ. The integrated stress response in lung disease. Am J Respir Cell Mol Biol. 2014;50:1005–1009. [DOI] [PubMed] [Google Scholar]
- 154.Starr CR, Pitale PM, Gorbatyuk M. Translational attenuation and retinal degeneration in mice with an active integrated stress response. Cell Death Dis. 2018;9:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morris A Neuroendocrinology: Integrated stress response linked to TBI. Nat Rev Endocrinol. 2017;13:501. [DOI] [PubMed] [Google Scholar]
- 156.Xu X, Krumm C, So JS and et al. Preemptive activation of the integrated stress response protects mice from diet-induced obesity and insulin resistance via fibroblast growth factor 21 induction. Hepatology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]