

Abstract
Although there are only two bispecific antibody (bsAb) drugs in the market, around 100 bsAb drug candidates are in clinical development. bsAbs have gained fast growing investment and attractions from the biopharmaceutical industry and academia in recent years. Antibody Engineering and Therapeutics 2019 (AET 2019) was held in San Diego, USA, from 9 to 13 December 2019. This year’s AET certainly reflected the trend. In this report, we selected 11 presentations from AET 2019 to highlight bsAbs’ design and their potentials in cancer therapy. These presentations have discussed emerging strategies to improve bispecific antibody drugs in efficacy, safety and production. As compared to CAR-Ts, some T cell-redirecting bsAbs may potentially achieve comparable efficacies with less side effects and toxicities, as evidenced with both preclinical and clinical data reviewed at the conference. Several approaches to reduce T cell engagers’ toxicities including conditionally active bsAbs and IgM-based bsAbs were also presented and discussed at the conference. For the first time, The Antibody Society and the Chinese Antibody Society jointly held a special session at the AET.
Keywords: bispecific antibodies, AET, bispecific T cell engager, antibody engineering, trispecific antibodies, synthetic immunity, cancer immunotherapy, cytotoxicity
Statement of Significance: Antibody Engineering and Therapeutics 2019 (AET 2019) was held in San Diego, USA, from 9 to 13 December 2019. This meeting report highlights 11 presentations with a focus on bispecific antibodies and discusses emerging strategies to improve bispecific antibody drugs in efficacy, safety and production.
INTRODUCTION
Antibody Engineering and Therapeutics (AET) is the annual meeting of The Antibody Society. It is also the flagship conference for reviewing and discussing the advances and trends in antibody drug discovery and development. AET 2019 was held during December 9–13 in San Diego, participated by over 1000 attendees from industry and academia, surpassing all of the previous annual meetings.
For the first time, The Antibody Society and the Chinese Antibody Society jointly held a special session, debuting on the afternoon of December 13. The joint session was co-chaired by Dr Kerry Chester of the University College London, UK, a board member of The Antibody Society and an advisor of the Chinese Antibody Society, and Dr Mitchell Ho of the National Institutes of Health, a board member of both The Antibody Society and the Chinese Antibody Society, and the editor in chief of “Antibody Therapeutics.” The six talks featured in this special session not only covered the progress of global antibody drug research and development but also showcased some technologies and products developed by Chinese companies including WuXi Biologics and Innovent Biologics.
Overall, AET 2019 offered about 27 sessions and 137 talks, covering topics including antibody engineering, tissue-specific delivery of antibodies, antibody drugs for cancer and neurodegenerative diseases and many more. One recurring theme of this conference is bispecific antibody (bsAb) therapeutics, reflecting one of the most important trends in drug development. Here in this report, 11 talks were selected in chronological order from the conference to highlight bsAbs’ potentials and flexibilities.
DAY 1 (9 DECEMBER 2019): PRECONFERENCE WORKSHOP A—BISPECIFIC ANTIBODIES, NEW STRATEGIES AND CASE STUDIES
Obligate mechanisms
Bispecific antibodies hold greater promises than monospecific antibodies for certain therapeutic applications. Dr Aran Labrijn from Genmab kick-started a preconference workshop by reviewing the new strategies and case studies for bispecific antibodies. An attractive promise of bispecific antibodies is their potential to display obligate activity which is a new functionality that cannot be obtained by combining separate antibodies with the same specificities [1,2]. Obligate effects can be generated either spatially or temporally. In the former situation, a bsAb has to bind to its two targets simultaneously and position them near each other to further induce downstream actions. Most T cell or NK cell redirection strategies using bispecific antibodies are examples of the spatial obligate mechanisms. Emicizumab developed by Chugai is another good example of spatial obligates. It mimics the function of FVIIIa by targeting FIXa and FX simultaneously. In temporal obligates, binding to the first target facilitates or enables the consequential binding to the second target, which may not be accessible to the bsAb molecules without the first binding event. MEDI3902 (anti PsI × PcrV) developed by AstraZeneca is an example of temporal obligates. By targeting sequential steps of Pseudomonas infection, it leads to increased recognition, phagocytosis and killing of bacteria by neutrophils [3].
High-throughput screening of obligate bsAbs
A major challenge for bispecific antibody discovery is to generate a large, diverse bispecific antibody library at first place before subjecting it to high-throughput functional screening. Dr Helene Finney presented the bispecific antibody discovery platform at UCB Biopharma. One of the highlights is their proprietary Fab-KD-Fab format [4] suitable for quickly constructing a flexible, assay-ready, large bispecific library. Briefly, they fused scFv against peptide Y to Fab antibodies of interest (Fab-X against antigen B) and peptide Y to Fabs (Fab-Y against antigen A). A simple mixing of Fab-X and Fab-Y will generate bispecific anti-A × anti-B Fab antibodies in a monovalent format in vitro. In addition to its simplicity, the Fab-KD-Fab format provides multiple controls to detect potential obligate effects. Most importantly, compared with traditional method, this technology dramatically reduces the time, cost, and workload to build a bispecific library. For example, to screen bispecifics targeting any combination of two antigens from a space of 10 different antigens, assuming that for each antigen 4 different Fabs need to be generated to cover different epitopes and affinities, 820 purified proteins in total—780 different bispecific combinations and 40 bivalent monospecific controls—are required to build a comprehensive testing pool. With the Fab-KD-Fab screening format, only 80 purified proteins (40 Fab-Ys and 40 Fab-Xs) are needed, reducing the protein preparation workload by 10-fold.
The UCB team have generated purified Fab-X and Fab-Y library containing single-arm antibodies against over 140 different targets, with which they identified potential therapeutic combinations for different indications. Dr Finney also showed that their quick “mix and go” format is amenable/translatable to the normal bispecific IgG format.
Eliminating mispairing by design
Manufacturing bispecific antibody containing two different light chains can result in a light chain mispairing problem. In order to deal with this issue, different platforms have been invented, such as the CrossMab technology from Roche [5] and engineered CH1:CL interfaces by Merck KGaA [6]. Dr Jonathan Davis from Invenra introduced their B-body™ platform. In a B-body construct, one arm of the bispecific IgG is a wildtype Fab, and the other Fab arm contains CH1/CL domain substitution derived from another human antibody domain (Fig. 1). According to their experience, light chain mispairing issue is not present in high-throughput screening, and the yield after Protein A purification for monovalent bispecific reaches 300 mg/L. By comparison, the purification yield using CrossMab is in the range of 10–40 mg/L [5], and the yield using engineered CH1:CL interfaces is usually around 200 mg/L [6]. Using B-body™ method, the Invenra team developed a series of multi-specific and multivalent anti-OX40 agonist antibodies, each of which targets two different epitopes on OX40. INV531 is their lead molecule which induces clustering of OX40 on cells and reduces IL10 level independent of Fc function.
Figure 1.
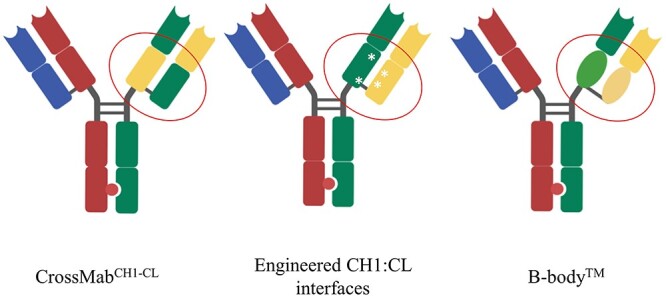
Technologies to eliminate light chain mispairing problem. CrossMab technology is based on the domain crossover in the Fab region of one arm of the bsAb. The left panel shows the CrossMabCH1-CL format, in which CH1 domain and CL domain are exchanged on the right arm of bsAb. In the engineered CH1:CL interface technology (middle panel), several mutations were introduced to the CH1 and CL domains on the right arm, making them repulsive towards a wildtype antibody chain but suitable for pairing with each other. In a B-body construct, one arm of the bispecific IgG is a wildtype Fab, and the other Fab arm has its CH1/CL domains substituted with pairing domains derived from other human antibody domains.
T cell-redirecting bsAbs vs. CAR-T: preclinical studies
In terms of clinical or preclinical progress of bispecific antibody, several speakers presented exciting updates. Dr David DiLillo from Regeneron compared effects of CD3-engaging bispecific antibodies with that of chimeric antigen receptor T (CAR-T) cell therapies. REGN5458, a BCMA x CD20 bispecific antibody, demonstrated similar multiple myeloma (MM) cell killing effect to an anti-BCMA CAR-T in vitro. The two agents generated similar therapeutic responses in a xenogenic mouse model in vivo. The comparisons of bispecific CD20 × CD3 and CD20-CAR-T also gave very similar results. Interestingly, comparing with CAR-T therapy, the bispecific antibody works much faster upon administration in vivo: treatment with REGN5458 led to a rapid clearance of tumors within 4 days, whereas treatment with BCMA CAR-T cells resulted in a delayed clearance of tumors, allowing tumors to growing for 10–14 days following CAR-T injection.
DAY 2 (10 DECEMBER 2019): KEYNOTE PRESENTATIONS
Enhancing efficacy of T cell engagers: trispecific antibodies
A bispecific T cell engager usually binds to CD3, part of the T cell receptor (TCR) complex on the surface of T cells, and a tumor-specific cell surface antigen, redirecting T cells’ killing capabilities towards the cancer cells. However, engaging CD3 alone often leads to T cell anergy or exhaustion instead of the activation desired.
In a Keynote presentation, Dr Gary Nabel from Sanofi introduced a new format of T cell engager designed to overcome such problem: a trispecific antibody targeting CD38, CD3 and CD28. The anti-CD38 domain directs T cells to myeloma cells, whereas engaging both CD3 and CD28 on T cells induces efficient, durable T cell stimulation. CD28 is a co-stimulatory receptor, and the extra agonistic engagement of CD28 can help to achieve the sustained T cell proliferation which is required for an effective immune response. In nature, when antigen-presenting cells (APCs) present antigens to T cells, T cell proliferation can be induced only after two signals are received: MHC-antigen-TCR interaction and B7-CD28 engagement. This strategy of co-activating CD28 signal has also been employed in another type of synthetic immunity—CAR-T. In the second generation of CAR-T therapy, CAR was designed to include the intracellular activation domain of CD28, enhancing and prolonging the killing effects of CAR-T cells.
Based on in vitro studies, the trispecific antibody has been shown to inhibit apoptosis of CD4+ or CD8+ T cells, stimulate human CD4 and CD8 T cell proliferation in the central and effector memory pool and display superior cytolytic activity against human myeloma cell lines than daratumumab (DARZALEX, the anti-CD38 monoclonal antibody (mAb) approved by the FDA for patients with multiple myeloma). Another benefit of the trispecific antibody is its enhanced induction of T cell killing against CD28-expressing multiple myeloma cells. The CD38/CD3 x CD28 trispecific also demonstrated significant protection against disseminated human MM cell tumor growth in a humanized mouse model.
To reduce the drug candidate’s associated risk of cytokine release syndrome (CRS) is a key goal for the drug designers. When designing the molecule, a medium affinity anti-CD3ε (KD = ~20 nM, vs. KD = 2 nM for anti-CD28, and KD = 4 nM for anti-CD38) was used, a distal-CD28 × proximal CD3 format was selected, and the Fc region made immune silent by eliminating all Fc receptor binding sites to reduce the risk of CRS [7]. In a nonhuman primate (NHP) study, CRS was observed when the trispecific antibody was administered by intravenous injection. However, the toxicity was much reduced when the drug was delivered subcutaneously, likely due to a more gradual antibody exposure.
Using a similar format, the Sanofi team constructed another trispecific antibody targeting Her2, CD3 and CD28. It demonstrated a superior immune killing of breast cancer in mouse models compared to the CD3 × CD28 bispecific antibody or anti-Her2 monoclonal antibody. In addition, Sanofi has also developed trispecific antibodies for HIV prevention and treatment.
Although the trispecific antibodies are still evaluated in preclinical studies, the encouraging data have shown their flexibility and promise. As T cell engagers, they can be tailored to optimize lymphocyte activation, T cell survival and tumor targeting to make cancer immunotherapy more precise and potent. The approach also has the potential to broaden the application of immunotherapy to many types of difficult-to-treat cancers.
DAY 3 (11 DECEMBER 2019): REVERSE TRANSLATION—ANTIBODY ENGINEERING, CLINICAL DATA AND LESSONS FROM CANCER IMMUNOTHERAPY
T cell-redirecting bsAbs vs. CAR-T: clinical studies
CAR-T therapy has been proven effective against some blood cancer, but its limitations (such as high cost and tendency to cause CRS) have hampered its wider application in clinical settings. Can bispecific antibodies, specifically T cell engagers, offer a more practical alternative treatment approach? After all, like CAR-T, T cell engagers are a type of synthetic immunity and are capable of unleashing the killing power of T cells (both CD4+ and CD8+ types) on tumor cells that express specific antigens on their cell surface. Compared with CAR-T, bispecific antibodies can be used off-the-shelf and with repetitive dosing, incur lower costs to patients/payers and do not require lymphodepletion prior to treatment [8]. The key question is whether bispecific T cell engagers can achieve efficacy comparable to, if not better than, CAR-T’s, and at the same time incur less severe side effects and toxicities. The short answer is maybe, at least for several full-length, IgG-bearing anti-CD20 x CD3 bispecific antibodies in development, according to Dr Elizabeth Budde from City of Hope.
Dr Budde reviewed recent clinical data of bispecific antibody drugs for B cell lymphoma. Although there have been no clinical studies that provide head-to-head comparisons between CAR-T and bispecific antibodies, their respective clinical data for the same indications may give us some clues.
Blinatumomab is a scFv-based bispecific T cell engager (anti-CD19 x CD3). Because of its short in vivo half-life, blinatumomab requires 28 days of continuous intravenous infusion for each treatment cycle. As shown in the Phase III clinical trial (TOWER study) for acute lymphocytic leukemia (ALL), blinatumomab’s efficacy is impressive but lower than CAR-T’s from historical clinical data: ~45% of blinatumomab-treated patients attained complete remission (CR), vs. 81–93% of CR for CAR-T treatment. Blinatumomab has also shown promise in relapsed/refractory non-Hodgkin’s lymphoma (NHL) and diffuse large B cell lymphoma (DLBCL) in Phase II studies. However, the toxicities pose significant concerns. For example, in the Phase II study for DLBCL, neurotoxicity was observed in almost 70% of the patients, with 21.7% displaying severe neurotoxicity (≥Grade 3). 32% of the patients discontinued the treatment after the first cycle due to either adverse events (AE) (20%) or physician decision (12%).
Although the first generation of T cell-redirecting bsAbs, as represented by Blinatumomab, are considered to be less potent and efficacious than CAR-Ts, displaying similar safety profiles, the new generation of IgG-like bsAbs in development may change that picture.
Several full-length bispecific antibody drug candidates, including mosunetuzumab, REGN1979, CD20-TCB, and GEN3013 offered more promise in beating CAR-T therapy in treating B cell lymphoma. All of these bispecific antibodies target CD3 and CD20. Since they are of full-length antibody format, they have much better PK profiles and longer half-lives in circulation.
In an open-label, multicenter Phase I/Ib study in relapsed or refractory (r/r) B cell NHL patients (NCT02500407), mosunetuzumab (from Genentech/Roche) achieved 37.1% in ORR and 19.4% in CR in patients with aggressive NHL (N = 124), 25 of which had prior CAR-T therapy and 62.7% in ORR and 43.3% in CR in patients with indolent NHL (N = 67). Moreover, mosunetuzumab displayed a good safety profile: most adverse events (AEs) are mild and transient, and only 2.6% (7 out of 270) of patients discontinued the treatment due to AEs.
In an open-label Phase I study in r/r B-NHL, for the follicular lymphoma (FL) patients with higher doses (≥5 mg, 12 weekly doses followed by 12 biweekly doses, N = 14), REGN1979 (from Regeneron) achieved 93% in ORR and 71% in CR. For the DLBCL patients with higher doses (≥80 mg, N = 19), it achieved 58% and 42% in ORR and CR, respectively.
Unlike mosunetuzumab and REGN1979, CD20-TCB (RG6026, from Roche) is a 1:2 CD3/CD20 bispecific antibody with higher binding avidity for CD20 on B cells. In an open-label Phase I study in r/r B-NHL, CD20-TCB has an ORR rate of 55–60% and a CR rate of 30–40% across different dose groups in aggressive B-NHL (N = 80 in total). Its main safety concern is CRS—55% of patients experienced CRS, though still less common than CAR-T therapies (74–94%). The percentage of CRS incidence is highly correlated with the drug dose.
Overall, compared with CAR-T treatments (tisagenlecleucel and axicabtagene ciloleucel), anti- CD20 × CD3 bispecific antibodies have shown better or comparable efficacies and more favorable safety profiles in patients with aggressive B-NHL (Tables 1 and 2).
Table 1.
Anti-CD20 bispecific antibodies have encouraging activities. ORR, objective response rate; CR, complete remission
| Study | Indolent NHL | Aggressive B-NHL | |||
|---|---|---|---|---|---|
| ORR | CR | ORR | CR | ||
| bsAbs | Mosunetuzumab Group B | 63% N = 67 (2.8–40 mg) | 43% | 38% N = 98 (2.8–40 mg) | 20% |
| Regeneron1979 | 93% N = 14 | 71% | 57% N = 7 (80–160 mg) | 57% | |
| CD20-TCB ≥600 μg | 53% N = 70 | 36% | 100% N = 8 | 100% | |
| CAR-T | JULIET (tisagenlecleucel) | n/a | n/a | 50% N = 68 | 32% |
| ZUMA-1 (axicabtagene ciloleucel) | n/a | n/a | 73% N = 101 | 52% | |
Source: adapted from Dr Budde’s presentation with her permission.
Table 2.
Anti-CD20 bispecific antibodies have a more favorable safety profile compared with CAR-Ts. CRS, cytokine release syndrome
| Study | CRS | Neurotoxicity | |||
|---|---|---|---|---|---|
| All grades | Grade 3/4 | All grades | Grade 3/4 | ||
| bsAbs | Mosunetuzumab Group B | 25.3% | Gr3: 1.1% Gr4: 0% | 45.1% | 2.7% |
| Regeneron1979 | 56.8% | Gr3: 7.4% Gr4: 0% | 49.5% | 1.1% | |
| CD20-TCB ≥600 μg | 55% | Gr3: 5% Gr4: 1.3% | 16% | 1% | |
| CAR-T | JULIET (tisagenlecleucel) | 74% | 21% Penn Scale | 58% | 11% |
| ZUMA-1 (axicabtagene ciloleucel) | 94% | 13% Lee Criteria | 87% | 31% | |
Source: adapted from Dr Budde’s presentation with her permission.
IgM bispecifics—a unique approach to reduce T cell toxicities
Is it possible to separate synthetic immunity’s ability to induce T cells’ tumor cell killing power (cytotoxicity) from its tendency to cause unwanted toxicities, particularly CRS? The answer is yes according to Dr Daniel Chen from IGM Biosciences.
Both cytotoxicity and CRS depend on antigenic stimulus, but they require different intensity levels of CD3 engagement. There are two different activation thresholds in cytotoxic T lymphocytes (CTLs), with lytic threshold being more sensitive to CD3 engagement, whereas cytokine release threshold much higher, requiring at least a 2-log higher CD3 occupancy. Therefore, a safer bispecific T cell engager can be designed by either attenuating its avidity for T cell engagement or increasing its CD20/CD3 binding moiety ratio (such as CD20-TCB).
IGM-2323 is such a molecule engineered to hit the sweet spot between the two thresholds using a completely different format. It consists of an IgM pentamer, capable of binding to 10 CD20 molecules, and an anti-CD3 fused to J-chain of the pentamer (Fig. 2). This IgM-based CD20 × CD3 (10:1) T cell engager has high avidity for CD20 and may be advantageous in treating low CD20-expressing cancers.
Figure 2.
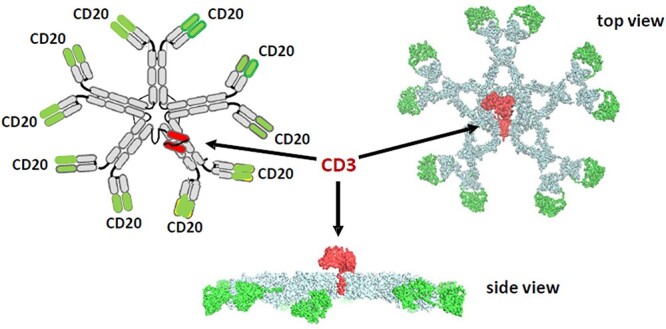
IGM’s anti-CD20 x anti-CD3 antibody. Its high CD20 avidity (10:1) plus anti-CD3 fused to J-chain leads to a better safety profile and an enhanced function via more effective T cell-directed cell-mediated cytotoxicity (TDCC) and complement-dependent cytotoxicity (CDC). Source: Dr Budde’s presentation with her permission.
As shown in in vitro data, IGM-2323 is significantly more potent than an IgG-based CD20 × CD3 antibody with the same CD20 and CD3 binding units against rituximab-resistant Ramos lymphoma cells expressing low levels of CD20. It has also been shown to have a lower cytokine release profile in vitro compared to the IgG CD20 × CD3 antibody, inducing a much more muted elevation, if any elevation at all, of IL-6, IL-2, IFNγ and TNFα. Such dissociation of cytotoxicity and cytokine release has been replicated in NHPs studies.
A Phase I study to evaluate IGM-2323 in r/r NHL was initiated in the third quarter of 2019 and is still ongoing.
DAY 4 (12 DECEMBER 2019): TISSUE-SPECIFIC DELIVERY OF ANTIBODIES
Bispecific antibody therapy in retinal disease
Dr Jörg Moelleken presented Roche/Genentech’s bispecific antibody faricimab, which was designed to target vascular endothelial growth factor A (VEGF-A) and angiopoietin-2 (ANG-2) simultaneously in treating retinal diseases. Currently, anti-VEGF treatments with Lucentis (Eylea) are the standard care for neovascular age-related macular degeneration (nAMD) and diabetic macular edema (DME), but faricimab aims for unmet medical needs with both improved efficacy and less frequent dosing. Advantages of faricimab over current treatments were discussed. Firstly, inhibiting ANG-2 leads to activation of the tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains 2 (Tie2), which is critical for angiogenesis as well as for vascular stability. ANG-2 levels are elevated in retinal vascular diseases, including nAMD, diabetic retinopathy (DR) and retinal vein occlusion (RVO). Secondly, faricimab was designed using CrossMab technology to ensure correct light chain assembly while preventing unwanted side products during manufacturing [9]. Thirdly, to optimize faricimab for ophthalmological use, its Fc region was engineered to abolish binding interactions with all FcɣRs and FcRn. Lacking FcɣRs interaction eradicates antibody effector functions including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cell phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC), which would also reduce drug provoked inflammation. While eliminating the FcRn binding site usually reduces an antibody’s systemic half-life, faricimab’s half-life has not been affected in the vitreous humor after intravitreal administration as shown in primate models. Several Phase II clinical trials in patients with DME or nAMD have concluded and established the superior efficacy and durability of faricimab over standard care. Currently, four ongoing Phase III clinical trials to evaluate faricimab for the treatment of nAMD and DME have completed patient recruitment.
DAY 4 (12 DECEMBER 2019): EFFECTOR FUNCTIONS OF THERAPEUTIC ANTIBODIES
The effect of IgG subclasses on T cell-redirecting bsAbs’ activities
Dr Mark Chiu from Janssen BioTherapeutics shed lights on how different IgG subclasses regulate bispecific T cell engager’s efficiency by examining experimental data involving IgG1, IgG2 and IgG4. T cell killing requires the formation of immunological synapses (ISs). IS is an intercellular structure connecting T cell receptor to antigen complex presented on the target cell. This structure allows T cells to properly release cytotoxic molecules and execute T cell killing. The intercellular space resulted from ISs varies from 10 to 15 nm, which is roughly the same dimension of the inter-Fab arm span of an IgG1 molecule. The significant progress of T cell engager-based bispecifics in current immune-oncology (I-O) therapy suggests that such bsAbs successfully mimic this structure in redirecting T cell cytotoxicity. Different IgG subclasses vary in hinge region length, sequence and disulfide bond structure, which naturally leads to different Fab spatial span and flexibility. Electron microscopy evidence suggests that IgG1 has the most flexible hinge while IgG2 is most rigid. To assess the impact of the IgG subclass on the T cell-redirected cytotoxicity, a series of CD19 × CD3 model molecules with IgG1, IgG2 or IgG4 subclasses were studied. Only CD19 × CD3 IgG2 bispecific did not bind to both antigens simultaneously. However, IgG2 bispecific activity was fully restored when chimeric IgG2s were engineered by grafting IgG1- or IgG4-F(ab)2 to IgG2 Fc [10]. This work clearly demonstrated that the spatial dimensions of IS and IgG subclass are critical elements when designing bispecifics involving T cell redirection.
DAY 5 (13 DECEMBER 2019): TUMOR-CONDITIONAL IMMUNOTHERAPY
Conditionally active bsAbs targeting solid tumors
Dr Bob DuBridge from Maverick Therapeutics introduced a bispecific antibody strategy (named COBRA™) to tackle two significant challenges in current T cell-redirecting therapy. One challenge is the T cell engagers’ significant off-tumor, on-target toxicity problem, and the other one is the poor response rate of solid tumors, which accounts for about 90% of cancer types, to current bispecific approaches. COBRA was developed to resolve these two challenges by delivering an inactive bispecific antibody which can only be activated by enzyme digestion in solid tumor microenvironment (TME). Dr DuBridge elucidated this mechanism using Maverick Therapeutics’ MVC-101, a molecule targeting the EGFR antigen on several types of solid tumors. This drug candidate is designed to carry the bispecific domains, one targeting EGFR (αEGFR sdAb) and the other recruiting T cells (αCD3 VH/VL scFv), which is linked to inhibitory domains (VHi/VLi) via a protease cleavable linker (Fig. 3). The inhibitory domains fold with the bispecific domains to form the inactive prodrug. A human serum albumin-binding domain (αHSA sdAb) is fused at the C-terminus to allow this prodrug’s sufficient exposure to TME. According to Dr DuBridge, a protease enriched within TME would digest and detach the inhibitory and albumin-binding domains, and then two molecules of digested bispecific molecules will interact to make an active drug dimer. This activated drug has T cell-recruiting and tetravalent EGFR-binding capabilities. In this way, MVC-101 minimizes unwanted toxicity from typical T cell engager bispecifics and greatly enhanced the tumor antigen recognition by tetravalent EGFR binding, with KD decreasing over 10-fold. In preclinical animal models, MVC-101 also demonstrated efficacious, dose-dependent tumor killing. Together with an improved tolerance, MVC-101 predicted a well-expanded therapeutic window. Multiple solid tumor clinical trials employing this strategy including EGFR targeting are being planned by Maverick Therapeutics.
Figure 3.
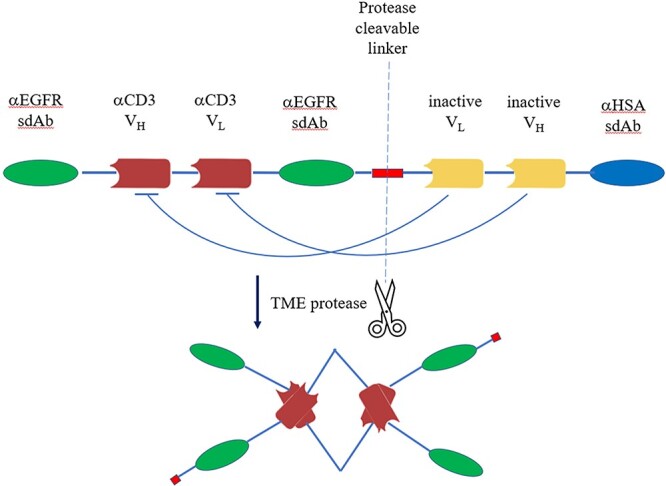
A scheme diagram of COBRA™ format using MVC-101 as an example. MVC-101 is designed to carry the bispecific domains, one targeting EGFR (αEGFR sdAb) and the other recruiting T cells (αCD3VH/VL scFv), which is linked to inhibitory domains (VHi/VLi) via a protease cleavable linker. The inhibitory domains fold with the bispecific domains to form the inactive prodrug. A human serum albumin-binding domain (αHSA sdAb) is fused at the C-terminus to extend its half-life. Specific protease activity within TME would digest and detach the inhibitory and albumin-binding domains, and then two molecules of digested bispecific molecules will interact to make an active drug dimer, which is tetravalent for EGFR binding and bivalent for CD3 binding. TME, tumor microenvironment.
DAY 5 (13 DECEMBER 2019): LOOKING AT TARGETS DIFFERENTLY
NK cell engagers
Dr Michael Tesar from Affimed presented a unique bispecific platform, Redirected Optimized Cell Killing (ROCK®). ROCK is aimed to activate the body’s innate immune system or NK cells, to destroy tumor cells. The advantage of this ROCK platform, as described by Dr Tesar, is that it would avoid the treatment-related toxicities associated with T cell engaging approaches and lead to better tolerance. The ROCK platform achieves this by employing a high-affinity anti-CD16A (CD16A is also known as FcγRIIIa) arm, which will recognize a different epitope from that of Fc binding. This can also minimize the competitive binding against circulating plasma Fc. The anti-CD16 arm is responsible for recruiting NK cells or macrophages, taking the similar role of the anti-CD3 arm in a T cell-redirecting bsAb but activating the innate immunity instead of the adaptive immunity. Secondly, Affimed has created four families and over 50 types of ROCK structures to demonstrate its high versatility and potential. Largely, an underlying mechanism of action (MOA) of ROCK allows Affimed to exploit current antibody formats with the possibility of creating novel intellectual property. The serum half-lives of different structures vary over 20 folds, and the area under the curves (AUC), which define the maximal amount or exposure of a molecule in the organism, vary over 100-fold. Affimed has multiple programs based on the ROCK platform moving into clinical trials, including BCMA and CD30 tumor antigen-targeting drugs [11]. In summary, Dr Tesar demonstrated that ROCK is a highly differentiated and fit-for-purpose bispecific platform allowing the engineering of NK cell engagers to overcome some of the limitations from the current T cell redirection approaches.
DISCUSSION AND PERSPECTIVE
Although there are only two bsAbs in the market, around 100 bsAb drug candidates are in clinical development [2]. bsAbs have gained fast growing investment and attractions from biopharmaceutical industry and academia in the past several years. This conference certainly reflected this trend. Here we highlighted 11 talks in AET 2019 that are related to bsAbs.
The main advantage of bsAbs over simple combination of parent antibodies is their obligate mechanisms, as summarized in Dr Labrijn’s talk. The obligate effects can be shown through either spatial obligate mechanisms (e.g. T cell-redirecting bsAbs) or temporal obligate mechanisms (e.g. MEDI3902). Other advantages of bsAbs include unified PK profiles (and hence single dosing schedules) and simpler regulatory pathways.
The most common formats of bsAbs are fragment-based molecules and full-length Fc-bearing molecules. Because of the FcRn binding sites on Fc, Fc-bearing bsAbs have longer half-lives and better PK profiles than fragment-based bsAbs. Fragment-based bsAbs’ PK can be improved by adding an HSA binding domain, as shown in Dr DuBridge’s talk, a CH3 domain or an Fc region [12,13]. For full-length bsAbs, the Fc domain is usually engineered to be immune silent to reduce unwanted toxicities and side effects.
When manufacturing full-length bsAbs, light chain-heavy chain and heavy chain-heavy chain mispairing will reduce the yield and efficiency of drug production. The mispairing issue can be eliminated with proper molecular designs. The industry has developed and practiced many technologies to solve the mispairing problem, including knobs into holes, common light chain, common heavy chain, CrossMab (e.g. faricimab from Dr Moelleken’s talk), engineered CH1:CL interfaces, WuXiBody [14] and B-body, the last of which was thoroughly discussed in Dr Davis’ talk. It is noteworthy that Dr Jing Li from WuXi Biologics introduced the WuXiBody technology in the special joint session hosted by The Antibody Society and Chinese Antibody Society. For more details of this technology, please refer to our previous summary of a similar talk in our 2019 PEGS report [14].
Currently, over half of the bsAbs in clinical development are T cell-redirecting bsAbs [2]. T cell engagers, as one type of synthetic immunity, may help treat difficult tumors, especially those of immune-excluded or “immune-desert” phenotypes [15]. Compared to CAR-Ts, bispecific T cell engagers may be able to achieve better or comparable efficacies and incur less severe side effects and toxicities as shown in preclinical studies (Dr DiLillo’s talk) and clinical data (Dr Budde’s talk). In addition, T cell-redirecting bsAbs are certainly easier and cheaper to manufacture than CAR-Ts.
T cell engagers’ efficacy and safety profile can be further improved, in some occasions by going beyond the bispecific format. In terms of efficacy, as demonstrated in Dr Nabel’s talk, a CD38/CD3 × CD28 trispecific antibody induces more robust and durable T cell killing of tumor cells. In terms of safety profile, the IgM-based CD20 × CD3 T cell engager has been shown to be more potent than and have a lower cytokine release profile than the IgG-based CD20 x CD3 bispecifics in cell and animal studies (Dr Chen’s talk).
Other ways to reduce T cell engagers’ unwanted toxicities include using conditionally active bsAbs, such as COBRA technology described by Dr DuBridge, and attenuating their CD3 binding affinity. Instead of recruiting T cells, Affimed’s ROCK format-based bsAbs engage NK cells or macrophages, avoiding the toxicities associated with T cell engagers all together (Dr Tesar’s talk).
In conclusion, bsAbs have emerged as one of the most exciting classes of drugs to treat cancers and other diseases. The presentations related to bsAbs presented at AET 2019 collectively demonstrate that there are numerous ways to improve the bispecific antibody drugs in efficacy, safety and manufacture. Within the next decade, more and more bispecific antibodies will come into the market in different forms. These bsAbs can not only provide better safety and efficacy profiles over current treatments but also expand the indications of antibody therapeutics to many diseases that are currently hard to tackle.
ACKNOWLEDGEMENTS
Dr Mitchell Ho’s work is supported by the Intramural Research Program of the National Institutes of Health, NCI. The authors thank Dr Elizabeth Budde for allowing us to include in this report the two tables and one figure adapted from her presentation at 2019 AET conference. We also thank Dr Finney Helene for revising the section of high-throughput screening of obligate bsAbs. Finally, we thank Dr Yaping Sun and Jessica Hong from the NCI for the editorial assistance. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose.
ABBREVIATIONS
ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cell phagocytosis; AE, adverse events; AET, antibody engineering and therapeutics; ALL, acute lymphocytic leukemia; ANG-2, angiopoietin-2; APCs, antigen-presenting cells; AUC, area under the curve; bsAb, bispecific antibody; CAR-T, chimeric antigen receptor T cells; CDC, complement-dependent cytotoxicity; CR, complete remission; CRS, cytokine release syndrome; CTL, cytotoxic T lymphocytes; DLBCL, diffuse large B cell lymphoma; DME, diabetic macular edema; DR, diabetic retinopathy; Fab, antigen-binding fragment; HSA, human serum albumin; I-O, immune-oncology; IS, immunological synapse; mAb, monoclonal antibody; MHC, major histocompatibility complex; MM, multiple myeloma; MOA, mechanism of action; nAMD, neovascular age-related macular degeneration; NHL, non-Hodgkin’s lymphoma; NHPs, nonhuman primates; ORR, objective response rate; PD, pharmacokinetics; ROCK, redirected optimized cell killing; RVO, retinal vein occlusion; rr, relapsed and or refractory; scFv, single chain variable fragment; sdAb, single domain antibody; TCR, T cell receptor; TDCC, T cell-directed cell-mediated cytotoxicity; TME, tumor microenvironment; VEGF, vascular endothelial growth factor.
REFERENCES
- 1. Spiess, C, Zhai, Q, Carter, PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67: 95–106. [DOI] [PubMed] [Google Scholar]
- 2. Labrijn, AF, Janmaat, ML, Reichert, JMet al. . Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev 2019; 18: 585–608. [DOI] [PubMed] [Google Scholar]
- 3. DiGiandomenico, A, Keller, AE, Gao, Cet al. . A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med 2014; 6: 262ra155. [DOI] [PubMed] [Google Scholar]
- 4. Finney, H, Rapecki, S, Wright, M. New Bispecific Format Suitable for Use in High-through-put Screening. UCB Biopharma, Patent Application WO2015181282A1, WIPO/PCT, 2015. [Google Scholar]
- 5. Schaefer, W, Regula, JT, Bahner, Met al. . Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Scie U S A 2011; 108: 11187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonisch, M, Sellmann, C, Maresch, Det al. . Novel CH1:CL interfaces that enhance correct light chain pairing in heterodimeric bispecific antibodies. Protein Eng Des Sel 2017; 30: 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu, L, Seung, E, Xu, Let al. . Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nature Cancer 2020; 1: 86–98. [DOI] [PubMed] [Google Scholar]
- 8. Strohl, WR, Naso, M. Bispecific T-cell redirection versus chimeric antigen receptor (CAR)-T cells as approaches to kill cancer cells. Antibodies (Basel) 2019; 8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Regula, JT, Lundh von Leithner, P, Foxton, Ret al. . Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med 2016; 8: 1265–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kapelski, S, Cleiren, E, Attar, RMet al. . Influence of the bispecific antibody IgG subclass on T cell redirection. MAbs 2019; 11: 1012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellwanger, K, Reusch, U, Fucek, Iet al. . Redirected optimized cell killing (ROCK(R)): a highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. MAbs 2019; 11: 899–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu, H, Saxena, A, Sidhu, SSet al. . Fc engineering for developing therapeutic Bispecific antibodies and novel scaffolds. Front Immunol 2017; 8: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kontermann, RE. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs 2009; 23: 93–109. [DOI] [PubMed] [Google Scholar]
- 14. Li, H, Li, Y, Wang, Cet al. . Highlights of 2019 protein engineering summit (PEGS) in Boston, USA: advancing antibody-based cancer therapies to the clinic. Antibody Therapeutics 2019; 2: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hegde, PS, Chen, DS. Top 10 challenges in cancer immunotherapy. Immunity 2020; 52: 17–35. [DOI] [PubMed] [Google Scholar]