

Abstract
Objective:
Cardiac troponin I (cTnI) is an essential physiological and pathological regulator of cardiac relaxation. Significant to this regulation, the post-translational modification of cTnI through phosphorylation functions as a key mechanism to accelerate myofibril relaxation. Similar to phosphorylation, post-translational modification by acetylation alters amino acid charge and protein function. Recent studies have demonstrated that the acetylation of cardiac myofibril proteins accelerates relaxation and that cTnI is acetylated in the heart. These findings highlight the potential significance of myofilament acetylation; however, it is not known if site-specific acetylation of cTnI can lead to changes in myofilament, myofibril, and/or cellular mechanics. The objective of this study was to determine the effects of mimicking acetylation at a single site of cTnI (lysine-132; K132) on myofilament, myofibril, and cellular mechanics and elucidate its influence on molecular function.
Methods:
To determine if pseudo-acetylation of cTnI at 132 modulates thin filament regulation of the acto-myosin interaction, we reconstituted thin filaments containing WT or K132Q (to mimic acetylation) cTnI and assessed in vitro motility. To test if mimicking acetylation at K132 alters cellular relaxation, adult rat ventricular cardiomyocytes were infected with adenoviral constructs expressing either cTnI K132Q or K132 replaced with arginine (K132R; to prevent acetylation) and cell shortening and isolated myofibril mechanics were measured. Finally, to confirm that changes in cell shortening and myofibril mechanics were directly due to pseudo-acetylation of cTnI at K132, we exchanged troponin containing WT or K132Q cTnI into isolated myofibrils and measured myofibril mechanical properties.
Results:
Reconstituted thin filaments containing K132Q cTnI exhibited decreased calcium sensitivity compared to thin filaments reconstituted with WT cTnI. Cardiomyocytes expressing K132Q cTnI had faster relengthening and myofibrils isolated from these cells had faster relaxation along with decreased calcium sensitivity compared to cardiomyocytes expressing WT or K132R cTnI. Myofibrils exchanged with K132Q cTnI ex vivo demonstrated faster relaxation and decreased calcium sensitivity.
Conclusions:
Our results indicate for the first time that mimicking acetylation of a specific cTnI lysine accelerates myofilament, myofibril, and myocyte relaxation. This work underscores the importance of understanding how acetylation of specific sarcomeric proteins affects cardiac homeostasis and disease and suggests that modulation of myofilament lysine acetylation may represent a novel therapeutic target to alter cardiac relaxation.
Keywords: Troponin I, Acetylation, Relaxation, Calcium sensitivity
1. Introduction
The troponin (Tn) regulatory complex is a requisite component in establishing calcium-dependent myofilament activationand thus, myocardial acto-myosin force production, as well as enhancing the inhibition of myofilament protein interactions that mediates relaxation. The Tn complex is made up of a calcium-binding subunit, troponin C (TnC), an actin-binding inhibitory subunit, troponin I (TnI), and a tropomyosin (Tm)-binding subunit, troponin T (TnT). In the absence of calcium, myosin’s interaction with actin is sterically inhibited by Tm and TnI’s interaction with actin. Upon calcium binding, the Tn complex orchestrates the release of TnI from actin that results in azimuthal movement of Tm over the actin surface to expose the binding sites that allow actin-myosin cross-bridge formation, which in turn, leads to contraction [1,2]. The process of relaxation is initiated when calcium dissociates from Tn and the Tn complex mediates TnI binding to actin and the movement of Tm to a position on actin that together sterically prevent myosin binding. This intricate process is essential for cardiac function. Since these proteins are so integral to cardiac function, it is not surprising that mutations in each of the Tn subunits have been linked to hypertrophic, restrictive, and dilated cardiomyopathies [3–5].
In addition to mutations, many studies have demonstrated that post-translational modifications (PTMs) to Tn are altered both physiologically (e.g. TnI phosphorylation), and in cardiomyopathies [4–6]. These changes influence both the calcium sensitivity of myofilament steadystate isometric force production as well relaxation kinetics [1,7]. While phosphorylation has been the most studied PTM of sarcomeric proteins, it has become evident that many different modifications occur normally and in disease, including acetylation, glycation, oxidation, citrullination, and succinylation [8–12]. These PTMs have the potential to impact sarcomeric protein interactions and thereby alter the properties of contraction and relaxation.
Acetylation, where lysine residues can be reversibly modified by addition of an acetyl group to the epsilon-amino group, has been shown to occur on multiple sarcomeric proteins [11,13]. There are 2 families of enzymes that regulate acetylation: lysine acetyl transferases (KATs, also known as histone acetyltransferases) reversibly acetylate lysine, and lysine deacetylases (KDACs, also known as histone deacetylases) remove acetyl groups from lysines. While the identity of the sarcomeric KATs and KDACs, as well as their target-site specificity, have yet to be determined, Jeong et al. (2018) recently demonstrated that myofibrils treated, ex vivo, with p300 (a promiscuous acetyltransferase), increased sarcomeric acetylation and accelerated relaxation [14]. Thus, sarcomeric acetylation is sufficient to modulate contractile dynamics, though the precise sarcomeric acetylation sites that can impact myofilament contractility have not been determined.
The goal of the current study was to evaluate the role of site-specific acetyl-mimetic modification of TnI on myofilament, myofibril, and myocyte function. Specifically, we designed acetyl-mimetic (K ➔ Q) and acetyl-null (K ➔ R) substitutions on cTnI K132 and used cell-based and ex vivo myofibril mechanics in addition to molecular-based in vitro motility assays to determine the impact of the PTM on contractile force and kinetics.
2. Materials and methods
2.1. Immunoprecipitation of acetylated proteins
All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Colorado Denver. Male Dahl Salt Sensitive (DSS) rats (Envigo) were fed a normal salt diet (Teklad; 2020×; 0.4% NaCl) or a high salt diet (Teklad; 2020×; 4% NaCl) for 10 weeks and myofibrils were isolated from the left ventricles. Two micrograms of myofibril protein was incubated with antibodies directed against acetylated-lysine epitopes (5.0 μL, PTM Biolabs) overnight at 4 °C. A 100 μL aliquot of protein G agarose (Sigma) was then applied to the sample and allowed to rotate overnight at 4°C. Supernatants were removed and the beads were washed 5 times with TBS-T. The sample was boiled for 5 min in SDS loading buffer and then applied to 10% SDS-PAGE gels, where the sample was run into the gel approximately 3 cm. The gel was then coomassie stained for 10 min and destained overnight. Protein bands were excised from the gel and digested with trypsin, as previously described [15]. Additionally, acetyl-peptide enrichment was utilized following the manufacturers protocol (PTM Biolabs).
2.2. LC-MS/MS identification of myofibril acetylated proteins
Protein digests were analyzed using nanoliquid chromatography (EASY-nLC, Proxeon) at a flow rate of 300 nL/min with a gradient of 5 to 40% ACN (0.2% formic acid) over 40 min on C-18 Proxeon EASY-Column trapping (20 × 0.1 mm) and analytical columns (100 × 0.075 mm). The nLC was coupled to a nano-ESI source and amaZon speed ion trap mass spectrometer using data-dependent CID MS/MS (Bruker Daltonics, Inc., Billerica, MA) [16]. Data analysis was performed using Mascot (version 2.2.04, www.matrixscience.com). Search parameters included trypsin as the digestion enzyme with 3 missed cleavages. Peptide and MS/MS tolerance were 1.2 and 0.6 Da, respectively. Variable modifications of cysteine carbamidomethyl, methionine oxidation, and lysine acetylation were also included. Mascot generic files were then imported into Scaffold (version 3.3.3, Proteome Software, Inc., Portland, OR) to analyze, interpret, and organize the MS/MS data. Reported MS/MS spectra were manually validated for confirmation.
2.3. cDNA constructs
cDNA encoding the rat cardiac TnI acetyl-mimetic, K132 mutated to Q (cTnI K132Q), was synthesized, cloned into pET17b and sequence verified by Genewiz. The generation of wild type rat cardiac TnI, N-terminal myc tagged (MMEQKLISEEDL) rat cardiac TnT, and rat cardiac troponin C plasmids have been previously described [17]. Further, it has been demonstrated that TnT containing an N-terminal myc tag does not affect myofibril function [18]. Genes encoding the individual recombinant human cardiac Tn subunits were expressed by transformation into Escherichia coli and purified to homogeneity similar to that previously described [19,20]. Tn complexes were prepared by mixing equimolar amounts of cardiac Tn subunits, reconstituted by sequential dialysis to remove urea and decrease salt followed by purification using Mono-Q chromatography as previously described [21].
2.4. Ex vivo exchange of exogenous recombinant human cardiac troponin I into rat myofibrils
Myofibrils were isolated from the hearts of Sprague Dawley rats as described [14]. Briefly, a small section of left ventricle was cut into thin slices and bathed in 0.05% Triton X-100 in rigor solution with protease inhibitors (50 mM Tris, 100 mM KCl, 2 mM MgCl2, 1 mM EGTA, pH 7.0) overnight at 4°C overnight. Skinned tissue was washed three times in rigor solution and resuspended in bath solution with protease inhibitors and homogenized for 10 s three times. The final myofibril suspension was then centrifuged at 1500 ×g for 5 min at 4 °C, resuspended in 15 μM cold recombinant troponin complex in recombination buffer (200 mM KCl, 5 mM MgCl2, 5 mM EGTA, 0.5 mM DTT, 20 mM MOPS, pH 6.5), and incubated overnight at 4 °C. Myofibrils were centrifuged at 1500 ×g for 5 min at 4 °C and washed twice in 1 mL cold relaxing solution (pCa 9.0; 100 mM Na2EGTA; 1 M potassium propionate; 100 mM Na2SO4; 1 M MOPS; 1 M MgCl2; 6.7 mM ATP; and 1 mM creatine phosphate; pH 7.0) with protease inhibitors (10 μM leupeptin, 5 μM pepstatin, 200 μM phenyl-methylsuphonylfluoride, 10 μM E64, 500 μM NaN3, 2 mM dithioerythritol). Finally myofibrils were resuspended in 1 mL relaxing solution and myofibril mechanics measurements were completed. A sample was collected from every exchange to determine TnI exchange percentage by Western blot.
2.5. Calcium-regulated in vitro motility assay
Calcium regulated in vitro motility parameters of reconstituted thin filaments (RTFs) were determined via established procedural modifications [22]. Freshly purified myosin from white New Zealand rabbit psoas muscles [23,24] was introduced into a flow cell and allowed two minutes to bind a nitrocellulose-coated cover slip. The surface was blocked with 2 mg/mL BSA, and enzymatically active myosin was bound to ~10 nM Alexa568-phalloidin-labeled rabbit skeletal F-actin (Cytoskeleton Cat. # AKL95). A mixture containing 300 nM each of tissue-purified, bovine cardiac tropomyosin (provided by Dr. Larry Tobacman, UIC) and E. coli-expressed, rat cardiac Tn, that included either WT or K132Q TnI, was introduced and allowed 5 min to decorate myosin-bound, F-actin. Calcium-dependent motility of RTFs at 30 °C was assessed using motility buffer containing 25 mM KCl, 25 mM imidazole, 25 mM MOPS (pH 7.4), 2 mM MgCl2, 10 mM DTT, 1 mM ATP, 150 nM Tpm and corresponding Tn, oxygen scavengers (17 units/mL glucose oxidase and 125 units/mL catalase), 0.5% methyl cellulose, and varying calcium (10−9 to 10−4 M). Imaging was performed on an Olympus IX73 microscope, and Alexa568-phalloidin was excited using an X-CITE 120 LED lamp and 531/40 filter. Emitted light was captured at 593/40, detected on a Hamamatsu Flash 4LT EMCCD camera at 1–10 fps controlled by HCI imaging software, and converted to multipage TIF’s. Average velocities were determined via manual tracking [25] of ~20–25 filaments at each calcium concentration tested. Flow chambers were partitioned in half and RTFs containing Tn complexes composed of WT or K132Q TnI were assessed in parallel. The experiment was repeated four times (n = 4); therefore, ~100 RTFs were analyzed per RTF type at each calcium concentration (exceptions being at pCa ≥ 6.25 when motility was substantially reduced). A weighted calcium sensitivity curve was generated as described previously [26]. Briefly, weighted average velocities (VAvg.weight) at each calcium concentration were calculated as shown below:
where VAvg is average filament velocity measured in n experiment, and xn is the number of filaments analyzed. VAvg.weight values were plotted as a function of calcium along with weighted standard errors (S.E.weight) determined from weighted standard deviations, S.D.weight:
Weighted mean calcium sensitivity plots for RTFs containing WT or K132Q TnI were fit by the least squares method (GraphPad Prism) to a 3-parameter Hill equation of the form:
where Vavg is the predicted velocity at a given calcium concentration x, and the fit parameters are Vmax, velocity at maximum calcium activation, Kd, concentration of calcium required to achieve 0.5* Vmax, and h, the Hill coefficient. Significant differences in fit parameters (Vmax, pCa50, and cooperativity) were determined via extra sum of squares F-test, with p < .05 deemed significant. Weighted averages of raw velocity measures at each pCa were then normalized to corresponding Vmax’s determined from the fits and re-plotted.
2.6. Myofibril mechanics
Mechanical measurements were completed on isolated myofibrils using the fast solution switching method as described [27,28]. Briefly, a small bundle of myofibrils (average diameter of 4.43 μm and average length of 7.96 μm) were mounted between two microtools at 15 °C in relaxing solution (pCa 9.0). One microtool was attached to a motor that produces rapid length changes (Mad City Labs) and the second micro-tool was a calibrated cantilevered force probe (7.42 μm/μN; frequency response 2–5 KHz). Myofibril length was set at ~2.2 μm. Mounted myofibrils were activated and relaxed by rapidly translating the interface between two flowing streams of solutions of different pCa (6 concentrations; pCa range 9–4.5) [28,29]. Data were collected and analyzed using customized LabView software. Measured mechanical and kinetic parameters were defined as follows: resting tension (mN/mm2) – myofibril basal tension in fully relaxing condition; maximal tension (mN/mm2) – maximal tension generated at full calcium activation (pCa 4.5); the rate constant of tension development following maximal calcium activation (kACT); the rate constant of tension redevelopment following a release-restretch applied to the activated myofibril (kTR) [30]; duration of early slow force decline - measured from onset of solution change to the beginning of the exponential force decay (tREL, LINEAR), the rate constant of the first, linear phase of relaxation – normalized linear slope of the linear relaxation (kREL,LINEAR); the rate constant of the final exponential phase of force decline (kREL,LINEAR). Calcium sensitivity was determined by measuring maximal force development over 6 concentrations of pCa (4.5, 5.2, 5.4, 5.6, 5.8, 7, 9) and normalizing submaximal tension (P) to tension generated at pCa 4.5 (P0). Each myofibril was activated at all calcium concentrations. Data were fitted to the Hill equation: P/P0 = 1/(1 + 10(−nh(pCa50-pCa))).
2.7. Adenoviral production
Adenoviral vectors were produced as described [31]. cDNA was obtained from the pET-24 vector containing a full-length rat cTnI sequence generously provided by Dr. Michael Regnier (University of Washington) [32]. cTnI was cloned into pcDNA3.1 with an in-frame N-terminal cMyc epitope tag. To create the cTnI mutants (K132Q and K132R), the PfuTurbo DNA Polymerase System (Aglient Technologies, 600,250) was used with primer sequences shown in Table 1. WT and mutant cDNAs were excised and subcloned into Gateway pENTR2B Selection Vector (Thermo Fisher Scientific). LR-recombination was performed to transfer cDNA into pAd-CMV-V5 DEST (ThermoFisher Scientific). Adenoviral vectors were transfected into 293A cells using polyethyleneimine (PEI). Viruses were amplified and recovered from 293A cells, and tittered using the SeaPlaque agarose method (Lonza).
Table 1.
Primers used to generate site-directed mutagenesis.
| cTnI K132Q Forward Primer | ATCTGACCCAGCAAATCTATGACCTGCGTG |
| cTnI K132Q Reverse Primer | CACGCAGGTCATAGATTTGCTGGGTCAGAT |
| cTnI K132R Forward Primer | ATCTGACCCAGAGAATCTATGACCTGCGTG |
| cTnI K132R Reverse Primer | CACGCAGGTCATAGATTCTCTGGGTCAGAT |
2.8. Cardiomyocyte culture
Adult rat left ventricular myocytes (ARVMs) were cultured from female Sprague Dawley rats (250–300 g) as described [33]. Briefly, the heart was removed and retrograde perfused (120.5 mM NaCl, 14.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 4.6 mM NaHCO3, 10 mM Na-HEPES, 30 mM Taurine, 10 mM 2,3-butanedione monoxime, 5.5 mM Glucose, pH 7.2) for 10 min at 37 °C, then digested with Liberase DH (Roche, 0.33 mg/mL) for 8 min. The heart was then minced and the slurry filtered through sterile 150-nm mesh. Myocytes were separated by centrifugation at 400 g for 4 min and the myocyte suspension was layered over 60 μg/mL of BSA and allowed to settle for 15 min. Myocytes were plated on 100-mm laminin-coated plastic culture dishes at a density of 100 to 150 cells/mm2. ARVM culture was maintained in serum-free DMEM supplemented with albumin (2 mg/mL), carnitine (2 mmol/L), creatine (5 mmol/L), taurine (5 mmol/L), BDM (1 mg/mL), and penicillin-streptomycin (100 μg/mL). After overnight culture, ARVMs were infected with adenoviruses at a multiplicity of infection (MOI) of 400 for 4 days unless otherwise specified.
2.9. Measurement of cellular contraction and relaxation
Cellular shortening and relengthening of ARVMs were measured using the IonOptix system. ARVMs were stimulated at 1 Hz in Tyrode’s solution (40 mM NaCl, 6 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 2.5 mM pyruvate, 5 mM HEPES, pH 7.4, 37 °C) and cell shortening and relengthening were measured by detecting edge movement of adherent, paced cells. Each plate of cardiomyocytes was stimulated for no > 20 min and 8–10 independent traces per plate were measured. IonWizard software (IonOptix) was used to analyze data and data were normalized to WT for each experiment. Peak height (H) as a percentage of baseline (BL) was calculated and is the percentage of myocytes shortened in relation to baseline. This measurement is indicative of peak ventricular contractility. Single exponential tau measures the exponential decay time constant of relaxation and represents the rate of relaxation.
2.10. Myofibrils from cardiomyocyte culture
Myofibrils were isolated from ARVMs as described [34]. ARVMs were washed in room temperature phosphate buffered saline (PBS) twice and then collected in 1 mL of cold 20% sucrose in relaxing solution (pCa 9.0) with protease inhibitors. The demembraned ARVM slurry was vortexed (30 s) then settled on ice for 10 min. The cell suspension was centrifuged (300G for 15 min in 4 °C) and the pellet was resuspended in fresh relaxing solution with protease inhibitor cocktail to remove the sucrose. The resuspension/wash cycle was repeated twice. The final demembraned ARVMs were resuspended in relaxing solution with protease inhibitor cocktail and homogenized at medium speed (Tissue Tearer) for 15–20 s to make the final myofibril suspension.
2.11. Indirect immunofluorescence
ARVMs were fixed with 4% paraformadelhyde at room temperature for 30 min. Cells were permeablized with PBST (PBS containing 0.3% Triton-X-100) for 30 min and then blocked with 10% fetal bovine serum (Gemini Bio-Products) in PBST for 30 min at room temperature. Following blocking, cells were incubated in primary antibodies overnight (mouse anti-cMyc (1:200; Santa Cruz Biotechnology, sc-40)). Secondary antibodies (anti-mouse (1:1000; Alex Fluor 488; A11029)) were applied for 3 h at room temperature, followed by phalloidin staining (100 nM; Cytoskeleton; PHDH1-A) for 30 min. Coverslips were mounted on glass slides using mounting medium with DAPI (Vector Laboratories, H-1200). Confocal images were obtained using an Olympus FLUOVIEW FV1000 confocal laser scanning microscope (UC Denver, Advanced Light Microscopy Core).
2.12. SDS-PAGE and Western blots
In order to confirm cTnI expression and exchange efficiency as well as verify that phosphorylation of cTnI was not altered, proteins were separated by SDS-PAGE and Western blotted or stained with ProQ Diamond PhosphoProtein Gel Stain (Invitrogen) (as described [17,18,34]). ARVMs were washed in PBS twice then collected in isoelectric focusing buffer (IEF; 8 M urea, 2.5 M thiourea, 4% CHAPS, 2 mM EDTA, 1 mM DTT, 1% TBP, phosphatase and protease inhibitors (Sigma Aldrich)). Ten micrograms of protein was separated by SDS-PAGE on a 12% gel. For phosphorylation analysis gels were stained with ProQ Diamond, visualized on a Typhoon 9410 Gel Imager (GE Healthcare). The gels were washed in methanol and restained with Coomassie Blue (BioRad) to visualize total protein. To determine exchange efficiency, cTnI expression, or phosphorylation of cTnI at Ser23/24, gels were transferred to PVDF membrane. The membranes were blocked with 5% milk in TBS for an hour then incubated in primary antibodies overnight at 4 °C (cTnI: 1:1000, Fitzgerald, 10R-T123k; cTnI phosphoserine 23/24: 1:1000, Phosphosolutions p2010–2324; cTnI phosphoserine 150: 1:1000, Phosphosolutions 9094, GAPDH: 1:10,000, Santa Cruz Biotechnology sc-20,357). Membranes were washed 3 times in TBS with 1% tween and then incubated in secondary antibody (goat-anti-mouse or rabbit, 1:2000, SouthernBiotech). Membranes were incubated with Pico ECL reagent (ThermoFisher Scientific, Pittsburg, PA) and exposed using the FluorChem HD scanner (Alpha Innotech). Protein was quantified using ImageJ and normalized to GAPDH. To quantify phosphorylation differences, phospho-specific signal was normalized to total cTnI for each sample.
Recombinant rat cardiac Tn in the present experiments included TnT with an N-terminal myc tag to allow for Tn exchange quantification. Following exchange experiments, the remaining myofibrils were solubilized in denaturing buffer (8 M Urea, 2 M thiourea, 3% SDS, 0.05% bromophenol blue, 0.75 mM DTT, 50 mM tris-HCl, pH 6.8), heated for five min at 80 °C and clarified by centrifugation for five min. Exogenous Tn exchange was determined by Western blot with slight modification from that previously described [35]. Briefly, proteins were separated on 12% (200:1) Laemmli gel and transferred to PVDF. Following blocking with 1% BSA in TBS resultant membranes were incubated with an anti-cardiac TnT antibody (Iowa Hybridoma; clone CT3). Following washes, membranes were incubated with a fluorescently labeled secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) and visualized on a Typhoon 9410 imager (GE Healthcare, Piscataway, NJ). The differential migration of endogenous rat cardiac TnT vs. exogenous exchanged rat cardiac myc-TnT that results from the addition of the myc tag allowed for quantification of both TnT bands by densitometry and the percent exchange calculated as the densitometry of the exogenous myc-TnT divided by the endogenous TnT + the exogenous myc-TnT.
2.13. Data analysis and statistics
Statistical analyses were performed using Prism Version 8 (GraphPad Software, San Diego, CA). Data were checked for normality using a Shapiro Wilks test and if not normally distributed, they were log transformed before statistical testing. Comparison of 2 groups was completed using the Student’s t-test with Welch’s correction. Comparison of 3 or more groups was completed using One-way ANOVA and Sidak’s multiple comparison tests. Data that were not normally distributed were tested for statistical differences using Kruskal-Wallis nonparametric tests with Dunn’s multiple comparison test.
3. Results
3.1. Identification and selection of acetylated lysine residue K132 within cTnI
We used tandem mass spectrometry to determine acetylated residues in a pooled sample of myofibrils isolated from DSS rats fed a normal salt diet or DSS rats fed a high salt diet because DSS rats fed high salt diets develop diastolic dysfunction after 10 week [14]. An initial database search identified MS/MS 3 spectra matching the acetyl-peptide NITEIADLNQkIFDLR (Fig. 1A), where k designates the position of acetylated lysine. We were able to discern not only the presence of the lysine-acetylated peptide (+42 m/z), but two other peptide species containing the acetylated-lysine and a single deamidation event (+43 m/z) or a double deamidation event (+44 m/z). Moreover, the MS/MS spectrum for NITEIADLNQkIFDLR closely mirrors previously identified matching spectra identified in the guinea pig acetyl-lysine proteome [13]. TnI K132 is one of 4 lysine acetylation sites found in both guinea pigs and rats as well as the rat model of diastolic dysfunction that we assessed [11,13]. Of the lysine residues found to be acetylated, K132 is of particular interest because it is located adjacent to the regulatory cTnI inhibitory peptide (amino acids 148–164) (Fig. 1B) [36]. The inhibitory peptide of cTnI is critical to force production as this region of cTnI directly binds actin in the absence of calcium binding to TnC to inhibit the acto-myosin interaction.
Fig. 1.
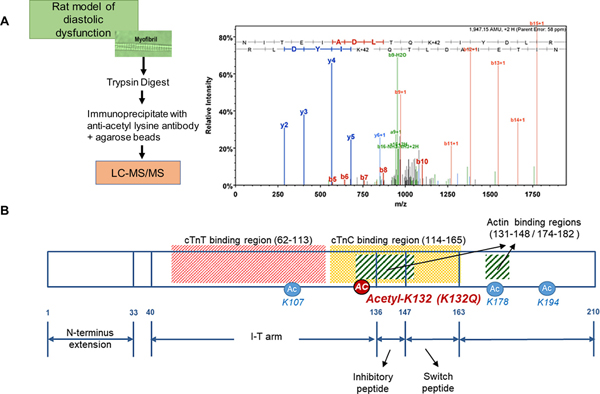
Identification of acetyl-K132 TnI. A. Schematic illustrating the experimental approach used to determine that K132 of cTnI is acetylated in rat left ventricle tissue obtained from a model of diastolic dysfunction. The detailed fragmentation spectrum showing b and y ion identification confirms the site-specific acetylation of K132 on TnI. B. Schematic illustrating troponin I binding regions demonstrating the location of the common lysines (4: K107, K132, K178, K194) found to be acetylated in cTnI by proteomic screens in rat and guinea pig [10,12]. K132 is within a region that binds cTnC and in the actin binding region.
3.2. Thin filaments reconstituted with acetyl-mimetic, K132Q cTnI exhibited decreased calcium sensitivity in the in vitro motility assay
In order to determine if pseudo-acetylation of cTnI at K132 modulates thin filament regulation of the acto-myosin interaction, calcium-regulated in vitro motility was performed on RTFs containing either WT cTnI or acetyl-mimetic K132Q cTnI. Vmax-normalized plots of velocity vs. pCa (Fig. 2) revealed no difference in Hill coefficient between WT-(2.14 ± 0.39) and K132Q-containing (1.75 ± 0.26) RTFs, however, K132Q filament pCa50 (5.85 ± 0.04) was significantly decreased compared to WT (5.98 ± 0.04), indicating a loss in calcium sensitivity in the presence of the cTnI K132Q acetyl-mimetic.
Fig. 2.
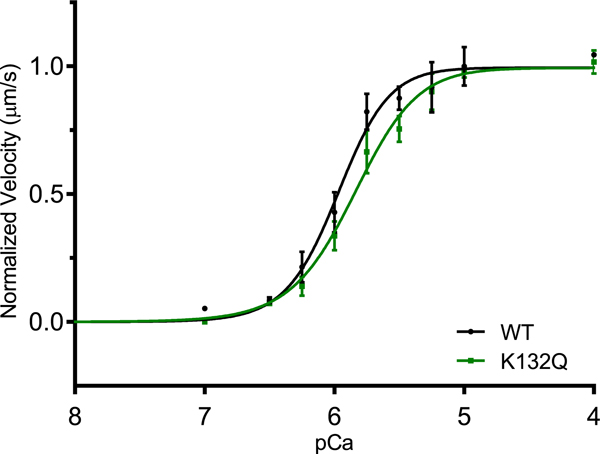
Decreased calcium sensitivity of thin filaments reconstituted with acetyl-mimetic K132Q cTnI. Vmax of RTFs containing WT TnI (5.36 ± 0.21 μm/s) and K132Q TnI (5.17 ± 0.20 μm/s) was indistinguishable (data not shown). Normalized velocities revealed no difference in cooperativity between WT TnI- and K132Q TnI-containing RTFs (2.14 ± 0.39 and 1.75 ± 0.26 respectively), however, pCa50 for WT (5.98 ± 0.04) vs. K132Q (5.85 ± 0.04) was significantly greater, indicating decreased calcium sensitivity of K132Q-containing RTFs (p < .05; n = 4 experiments).
3.3. ARVMs expressing cTnI K132Q had faster cell relengthening
To test if acetyl-mimetic cTnI K132Q alters relaxation, adult rat ventricular myocytes (ARVMs) were infected with adenoviruses expressing cMyc-tagged WT, K132Q, or K132R cTnI (non-acetylatable) for 96 h (% expression: WT = 70.8 ± 2.7%; K132Q = 73.9 ± 3.1%; K132R = 72.7 ± 2.9%; Fig. 3A and B). Immunostaining demonstrated that each of the cMyc-tagged cTnI proteins localized within the sarcomere (Fig. 3C). Cell shortening was measured using an IonOptix system and the total reduction of cell length (baseline to peak H) was decreased in cells expressing cTnI K132Q (WT = 100 ± 4.5%; K132Q = 66.4 ± 3.4%; K132R = 86.7 ± 3.8%; WT v K132Q p = .0003; K132Q v K132R p = .03; Fig. 3E). The velocity of shortening was not significantly different between cells expressing WT, K132Q or K132R cTnI. However, the rate of relengthening (Tau; specifically related to the duration of relaxation) was faster in cells expressing K132Q cTnI when compared to WT or K132R cTnI (WT = 1.00 ± 0.01 ms; K132Q = 0.88 ± 0.03 ms; K132R = 0.96 ± 0.06 ms; WT v K132Q p = .03; Fig. 3E).
Fig. 3.
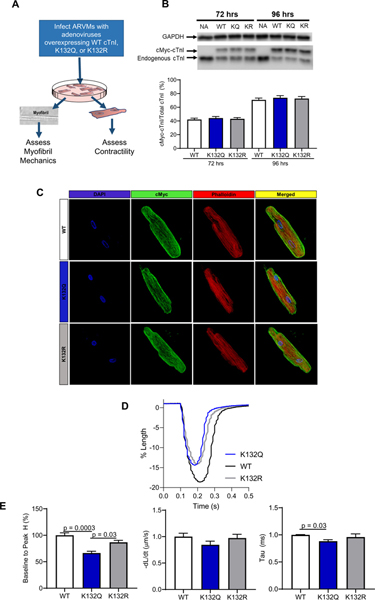
ARVMs infected with K132Q cTnI demonstrate shorter duration of relengthening and decreased shortening. A. Schematic illustrating the experimental design. B. Representative blot and quantification of expression of cTnI constructs in ARVMs. Exogenous WT, K132Q, and K132R cTnI expression was 75% greater than endogenous cTnI expression 96 h after infection [n = 4 ARVM experiments]. C. ARVMs stained with anti-cMyc and fluorescently labeled-phalloidin demonstrate incorporation of cMyc tagged constructs into the ARVM sarcomere. [n = 4 ARVM experiments]. D. Representative trace of cell shortening of ARVMs expressing WT, K132Q, or K132R. Baseline of each trace was normalized to 1. E. ARVMs expressing K132Q cTnI have decreased peak height of contraction compared to ARVMs expressing WT cTnI (p = .0003) and ARVMs expressing K132R cTnI (p = .03). There is no significant change in rate of contraction of ARVMs expressing WT, K132Q, or K132R. ARVMs expressing K132Q cTnI have a faster exponential decay time constant (representing the speed of relaxation) than ARVMs expressing WT cTnI (p = .03). Statistical analyses of normal data were assessed by one way ANOVA with Sidak’s multiple comparison test. [3 ARVM experiments; WT n = 1–2 plates per experiment, 8 cardiomyocytes per plate; K132Q n = 2 plates per experiment, 8 cardiomyocytes per plate; K132R n = 1 plate per experiment, 8 cardiomyocytes per plate].
3.4. Myofibrils isolated from ARVMs expressing cTnI K132Q relaxed faster and were less sensitive to calcium
Expression of cTnI K132Q or cTnI K132R differentially impacted mechanical parameters and calcium sensitivity in myofibrils isolated from ARVMs (Table 2; Fig. 4). Myofibrils isolated from cells expressing WT, K132Q, and K132R cTnI did not have differences in resting tension. Additionally, there was no difference in the rate of force generation between any of the groups at maximum calcium. Previous studies have demonstrated that cardiac myofibrils relax in a biphasic manner [37,38]. The first phase of relaxation in response to calcium withdrawal is isometric, slow, and linear (duration = tREL, LINEAR; rate = kREL, LINEAR), attributable to crossbridges transitioning from strongly-bound to weakly-bound states and the re-engagement between actin and troponin [39,40]. Myofibrils isolated from cells expressing K132Q cTnI had a shorter duration for the linear phase of relaxation compared to myofibrils expressing WT or K132R cTnI (WT = 51.1 ± 1.7 ms; K132Q = 42.0 ± 1.4 ms; K132R = 47.6 ± 1.5 ms; WT v K132Q p = .002; K132Q v K132R p = .05; Fig. 4B). At the same time the rate of the linear phase of relaxation in myofibrils from cells expressing K132Q cTnI was faster than the rate of linear relaxation from myofibrils with WT cTnI and trending faster than the rate of linear relaxation from myofibrils containing K132R cTnI (WT = 1.07 ± 0.1 s−1; K132Q = 2.03 ± 0.2 s−1; K132R = 1.52 ± 0.2 s−1; WT v K132Q p = .01; K132Q v K132R p = .09). In contrast, the second phase of relaxation, which represents sequential sarcomere lengthening that stems from rapid crossbridge detachment (kREL, EXP), was not altered in myofibrils with cTnI K132Q compared to WT or K132R. Myofibril mechanical parameters were also measured at submaximal calcium to better understand interactions at calcium concentrations closer to physiologic conditions. While maximal force did not differ between myofibrils isolated from each group, K132Q cTnI myofibrils demonstrated significantly decreased relative forces at pCa 5.4, 5.6, 5.8, and 6 compared with WT or K132R cTnI myofibrils (Relative force: pCa50 WT = 5.75 ± 0.01; K132Q = 5.57 ± 0.01; K132R = 5.70 ± 0.01; p < .0001; Fig. 4C). In line with this, the rates of activation and re-activation also demonstrated this decreased calcium sensitivity (kACT pCa50 WT = 5.86 ± 0.02; K132Q = 5.75 ± 0.04; K132R = 5.83 ± 0.03; p < .05; kTR pCa50 WT = 5.92 ± 0.02; K132Q = 5.77 ± 0.03; K132R = 5.92 ± 0.04; p < .002; Supplemental Fig. 1). At submaximal calcium concentrations, the relaxation parameters also demonstrate the same changes observed at maximal calcium concentrations (Supplemental Fig. 2).
Table 2.
Mechanical parameters at pCa 4.5 from myofibrils isolated from ARVMs expressing WT, K132Q, or K132R cTnI.
| RT (mN/mm2) | Po (mN/mm2) | kACT (s−1) | kTR (s−1) | t REL, LINEAR (ms) | k REL, LINEAR (s−1) | kREL, EXP (s−1) | SL (μm) | |
|---|---|---|---|---|---|---|---|---|
| cTnI WT | 3.80 ± 0.4 (n = 27) | 69.1 ± 5.6 (n = 27) | 3.38 ± 0.2 (n = 27) | 3.36 ± 0.2 (n = 27) | 51.1 ± 1.7 (n = 27) | 1.07 ± 0.1 (n = 27) | 55.6 ± 5.2 (n = 27) | 2.21 ± 0.02 (n = 27) |
| cTnI K132Q | 4.29 ± 0.4 (n = 25) | 53.6 ± 4.5 (n = 25) | 3.32 ± 0.2 (n = 25) | 3.28 ± 0.2 (n = 25) | 42.0 ± 1.4** (n = 25) | 2.03 ± 0.2**(n = 24) | 43.5 ± 3.2 (n = 25) | 2.19 ± 0.02 (n = 25) |
| cTnI K132R | 4.44 ± 0.4 (n = 24) | 68.9 ± 4.8 (n = 24) | 3.12 ± 0.2 (n = 24) | 3.36 ± 0.2 (n = 24) | 47.6 ± 1.5*(n = 24) | 1.40 ± 0.1 (n = 20) | 49.5 ± 3.5 (n = 24) | 2.21 ± 0.02 (n = 24) |
Data is expressed as mean ± standard error. N = 4 per experiment, n = total number myofibrils measured between 4 different experiments.
p < .05 K132Q v K132R.
p < .01 WT v K132Q by One-Way ANOVA with Sidak’s multiple comparison test.
Fig. 4.
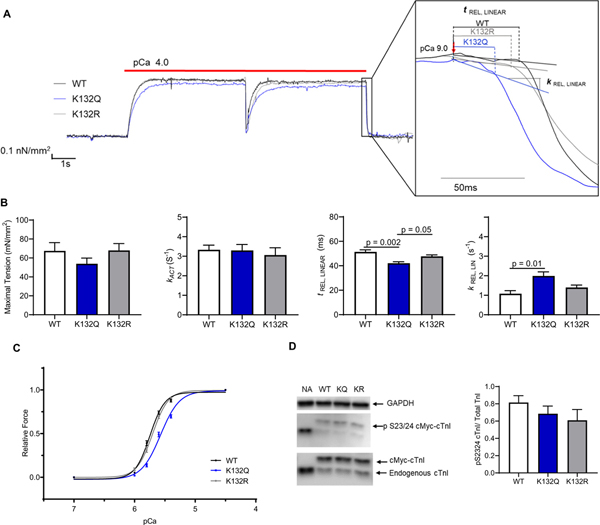
Faster relaxation and decreased calcium sensitivity of myofibrils isolated from ARVMs expressing acetyl-mimetic K132Q cTnI. A. Representative traces of myofibrils isolated from ARVMs expressing WT, K132Q, or K132R cTnI. Subset expands the linear phase relaxation. Force from each trace was normalized to WT force. B. Mechanical parameters in myofibrils isolated from ARVMs expressing K132Q cTnI compared with myofibrils isolated from ARVMs expressing WT cTnI or K132R cTnI. Maximal tension and rate of activation (kACT) are not significantly different in myofibrils exchanged with WT, K132Q, or K132R cTnI. Myofibrils isolated from ARVMs expressing K132Q cTnI have significantly faster relaxation as demonstrated by shorter duration (tREL,LINEAR) and steeper rate of linear relaxation (kREL,LINEAR) compared to myofibrils isolated from ARVMs expressing WT cTnI (p = .002; p = .01). Duration of linear phase relaxation was faster in myofibrils expressing K132Q cTnI than in myofibrils isolated from ARVMs expressing K132R cTnI (p = .05). The rate of linear relaxation trended faster in myofibrils isolated from ARVMs expressing K132Q cTnI compared to myofibrils expressing K132R cTnI (p = .096). Statistical analyses of normal data were assessed by one way ANOVA with Sidak’s multiple comparison test. [4 ARVM experiments; myofibrils from each experiment were averaged; WT n = 27 total myofibrils; K132Q n = 25 total myofibrils; K132R n = 24 total myofibrils]. C. Relative force is less in myofibrils isolated from ARVMs expressing K132Q cTnI (blue) for given calcium concentration compared to myofibrils isolated from ARVMs expressing WT (black) and K132R (gray). Data were assessed by log (agonist) v response variable slope parameters. WT pCa50 = 5.75; K132Q pCa50 = 5.57; K132R pCa50 = 5.70. p < .0001. [3 ARVM experiments; WT n = 17 myofibrils; K132Q n = 20 myofibrils; K132R n = 17 myofibrils]. D. ARVMs expressing WT, K132Q, or K132R do not have differences in phosphorylation of cTnI at serine 23/24. Representative image and quantification demonstrating phosphorylation of cTnI at serine 23/24 is not significantly different between ARVMs expressing WT, K132Q, or K132R cTnI [n = 4 ARVM experiments]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To determine if altering K132 through mimicking acetylation or by blocking acetylation would alter phosphorylation, protein from ARVMs was assessed for total phosphorylation and cTnI phosphorylation at serine 23/24. Phosphorylation measured by ProQ Diamond PhosphoProtein stain show no changes between ARVMs expressing WT, K132Q, or K132R cTnI (Supplemental Fig. 2). Since phosphorylation of cTnI at serine 23/24 has been shown to affect relaxation [32], phosphorylation of cTnI at serine 23/24 was specifically assessed and was not significantly different between cells expressing different constructs (cTnI pS23/24: WT = 0.82 ± 0.08; K132Q = 0.69 ± 0.09; K132R = 0.61 ± 0.13; Supplemental Fig. 2 and Fig. 4D).
3.5. Ex vivo incorporation of K132Q cTnI into myofibrils quickened relaxation and decreased calcium sensitivity
To verify that the observed changes in ARVMs expressing K132Q are directly due to myofibril function and not other non-myofilament effects of adenovirus expression, we exchanged endogenous Tn with exogenous Tn containing either WT cTnI or K132Q cTnI into isolated rat myofibrils (Fig. 5A). There was no difference in exchange of Tn containing WT or K132Q cTnI (percent exogenous Tn exchange: WT = 75.4 ± 4.3%; K132Q = 74.8 ± 7.5%; Fig. 5B). Average data from myofibril mechanics at maximum calcium concentration are shown in Table 3. Exchanging endogenous cTnI with WT cTnI or K132Q cTnI did not alter resting tension of the myofibrils. Similarly, WT or K132Q cTnI exchange did not alter tension at maximal calcium concentration (pCa 4.5) (WT = 34.8 ± 3.7 mN/mm2; K132Q = 26.1 ± 4.2 mN/mm2; p = .09) or the rate of activation (kACT) (WT = 2.52 ± 0.2 s−1; K132Q = 2.25 ± 0.2 s−1). Notably, myofibrils exchanged with cTnI K132Q demonstrated a shorter duration of linear relaxation than myofibrils exchanged with WT cTnI (WT = 57.9 ± 2.2 ms; K132Q = 48.4 ± 1.7 ms; p = .002; Fig. 5D). Additionally, the rate of linear relaxation was faster in myofibrils exchanged with K132Q cTnI compared to myofibrils exchanged with WT cTnI (WT = 1.34 ± 0.2 s−1; K132Q = 2.9 ± 0.2 s−1; p = .0005; Fig. 5D). Furthermore, myofibrils with cTnI K132Q had significantly decreased calcium sensitivity with relative forces lower in myofibrils exchanged with K132Q cTnI (Relative force: pCa50 WT = 5.68 ± 0.01; K132Q = 5.548 ± 0.13; p < .0001; Fig. 5E). Myofibrils exchanged with K132Q cTnI also had slower rates of activation and reactivation at submaximal calcium (kACT pCa50 WT = 6.00 ± 0.08; K132Q = 5.79 ± 0.04; p < .05; kTR pCa50 WT = 5.96 ± 0.03; K132Q = 5.78 ± 0.03; p = .002; Supplement Fig. 3). Finally, at submaximal calcium concentrations, rate and duration of linear phase relaxation was significantly faster in myofibrils exchanged with K132Q cTnI (Supplement Fig. 3).
Fig. 5.
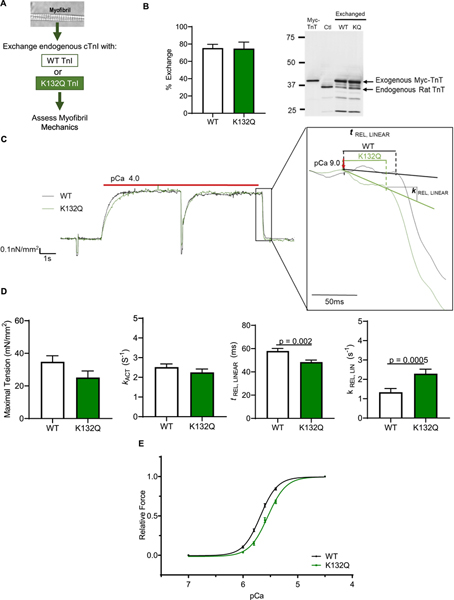
Ex vivo incorporation of K132Q cTnI into myofibrils quickened relaxation and decreased calcium sensitivity. A. Schematic illustrating the experimental design. B. Representative image and quantification of Western blot analysis to measure the exchange of cTnI constructs. Since recombinant Tn with myc tagged TnT and either WT or K132Q cTnI, we measured exchange by blotting for myc-TnT. Exchange of recombinant Tn with WT cTnI is 75.4% and recombinant Tn with K132Q cTnI is 74.8%. [WT n = 3 exchange experiments; K132Q n = 3 exchange experiments]. C. Representative traces of myofibrils exchanged with WT or K132Q cTnI. Subset expands linear phase relaxation. Force for each trace was normalized to WT force. D. Mechanical parameters are different in myofibrils with K132Q cTnI compared to myofibrils exchanged with WT cTnI. Maximal tension and rate of activation (kACT) are not significantly different but myofibrils exchanged with K132Q cTnI have faster linear phase relaxation as demonstrated by shorter duration of linear phase (tREL, LINEAR; p = .005) and faster rate of linear relaxation (kREL, LINEAR; p = .001). Statistical analysis of normal data were assessed by Student’s t-test with Welch’s correction. [3 exchange experiments; WT n = 21 total myofibrils; K132Q = 17 total myofibrils]. E. Relative force is less in myofibrils exchanged with cTnI K132Q for given calcium concentration. Data assessed by log (agonist) v response variable slope parameters. Relative force: WT pCa50 = 5.68; K132Q pCa50 = 5.55. p < .0001. [3 exchange experiments; WT n = 21 myofibrils; K132Q = 17 myofibrils].
Table 3.
Mechanical parameters at pCa 4.5 from myofibrils with cTnI exchanged with WT or K132Q.
| RT (mN/mm2) | Po (mN/mm2) | kACT (s−1) | kTR (s−1) | t REL, LINEAR (ms) | k REL, LINEAR (s−1) | kREL, EXP (s−1) | SL (μm) | |
|---|---|---|---|---|---|---|---|---|
| cTnI WT | 8.98 ± 1.0(n = 19) | 34.8 ± 3.7 (n = 21) | 2.52 ± 0.2 (n = 21) | 3.20 ± 0.2 (n = 21) | 57.9 ± 2.2 (n = 21) | 1.34 ± 0.2 (n = 21) | 40.3 ± 3.8 (n = 21) | 2.15 ± 0.02 (n = 21) |
| cTnI K132Q | 8.05 ± 0.9 (n = 17) | 26.1 ± 4.2 (n = 17) | 2.25 ± 0.2 (n = 17) | 3.41 ± 0.3 (n = 17) | 48.4 ± 1.7* (n = 17) | 2.9 ± 0.2*(n = 17) | 48.0 ± 5.8 (n = 16) | 2.12 ± 0.02 (n = 17) |
Data is expressed as mean ± standard error. N = 3 experiments, n = total number of myofibrils measured between 3 experiments.
p = 0.05 by Student’s t-test with Welch’s correction.
4. Discussion
This is the first study to demonstrate that site-specific pseudo-acetylation of cTnI alters calcium sensitivity of contraction and myofibril mechanics. Our findings indicate that targeted acetylation of particular amino acid residues in sarcomeric proteins results in functional changes.
Acetylation of myofibril proteins is of particular interest because a recent study demonstrated that treatment with a KDAC inhibitor ITF2357 prevents development of diastolic dysfunction in animal models [14]. Diastolic dysfunction is a hallmark of heart failure with preserved ejection fraction (HFpEF) which currently does not have any effective therapies. Dahl Salt Sensitive (DSS) rats fed 4% salt diet for 10 weeks developed diastolic dysfunction, which was prevented via treatment with the KDAC inhibitor. Isolated myofibrils from rat hearts characterized with diastolic dysfunction relaxed more slowly than control myofibrils, and treatment with ITF2357 induced faster relaxation. Identical findings were observed in a model of aging-induced diastolic dysfunction. Further, acetylation of myofibril proteins using p300 (a lysine acetyltransferase) was sufficient to accelerate myofibril relaxation, illustrating that acetylation of myofilament proteins is sufficient to alter relaxation; however, the question remained as to which myofilament proteins are acetylated and if specific residues affect mechanical function. Furthermore, it is important to consider site-specific stoichiometry when assessing physiologically relevant lysine acetylation and functional changes in myofibril mechanics. Our assessment of protein acetylation from myofibrils isolated from the hearts of DSS rats determined that a specific lysine residue on cTnI is acetylated: K132.
cTnI mutations have been implicated in many different diseases including, hypertrophic, dilated, and restrictive cardiomyopathies. There are 24 lysines in cTnI. Lundby et al. (2012) showed that 10 lysines are acetylated physiologically in Sprague Dawley rats and Foster et al. (2013) found that 8 lysines are acetylated physiologically in guinea pigs [11,13]. There are 4 sites reported by both groups, K107, K132, K178, and K194 (Fig. 1B). Importantly, Foster et al. (2013) points out that the acetylation sites identified in cTnI are at specific loci where proteins interact and are close to known mutation sites, indicating that modification by acetylation (through physiology or disease) has the potential to induce functional changes. K132 of cTnI is adjacent to the regulatory inhibitor peptide (amino acids 149–164) [36]. Further, this residue has been reported as a locus for a familial mutation associated with HCM [4].
The mechanisms by which acetylation impacts cardiomyocyte contractility and relaxation have only begun to be addressed. Interestingly, treating primary cultured cardiomyocytes with the KDAC inhibitor ITF2357 increases cellular levels of acetylation and causes changes in relaxation similar to those noted here [14]. The cardiac acetylome clearly shows that acetylation targets include both calcium-handling proteins (e.g. RYR2, SERCA2, phospholamban) as well as myofilament proteins (e.g. actin, TnI, TnT, and myosin, among others). Studies have recently shown SERCA2 acetylation also leads to increased ATPase activity and faster cellular relaxation and actin acetyl-mimetics can engender excessive force production, in vivo [26,41]. Therefore, acetylation may impact multiple targets along the excitation-contraction coupling axis, whose relative influence on contractility have yet to be determined. Further, it is likely that different KATs and KDACs may target various proteins. Investigating site-specific changes and mechanical consequences in correlation with pan-changes in acetylation has the potential to illuminate how acetylation changes in the sarcomere impact function and mechanisms by which targeting specific acetylation sites improve cardiac performance in disease.
We found that reconstituting thin filaments with K132Q cTnI decreased calcium sensitivity in the in vitro motility assay using skeletal muscle myosin. It should be noted, however, that use of skeletal myosin may not fully recapitulate the cardiac-specific effects of K132Q cTnI. While absolute RTF velocity and myosin-mediated force are expected to be different in the presence of cardiac RTFs and cardiac myosin, previous studies have determined that myosin isoforms do not affect calcium sensitivity, which was shown to be altered by the K132Q cTnI mutation [42]. Since the main objective was to ascertain the direct impact(s) of the K132Q acetyl-mimetic TnI mutation on molecular function, in vitro motility data, generated with skeletal myosin, still provide important information regarding those effects. Following the observed decrease in calcium sensitivity of thin filaments reconstituted with K132Q cTnI, we wanted to understand how acetylation of K132 cTnI affected whole cell function. This is particularly important as mechanics and crossbridge kinetics, are highly dependent on experimental factors, including concentrations of calcium, ADP and inorganic phosphate. Specifically, intact cultured adult rat ventricular cardio-myocytes were infected with adenoviral constructs bearing either WT cTnI, K132Q cTnI, or K132R cTnI. Cardiomyocytes expressing K132Q cTnI, had faster relengthening and isometric force production was decreased, likely due to altered calcium sensitivity. At physiologic calcium concentrations, cardiomyocytes expressing K132Q cTnI had a decreased percentage of contraction and faster return to baseline length suggesting faster relaxation. In addition, myofibrils isolated from ARVMs expressing K132Q cTnI relaxed faster and have decreased calcium sensitivity compared to myofibrils from ARVMs expressing WT or K132R cTnI. It is important to note that maximal tension of the myofibrils expressing K132Q cTnI was not significantly different from maximal tension from myofibrils expressing either WT or K132R cTnI, suggesting that the decreased cell shortening observed at the whole cell level was indeed due to decreased calcium sensitivity.
While mass spectrometry studies have identified that K132 is acetylated physiologically, expression of K132R cTnI myofibril mechanics, calcium sensitivity, and whole cell mechanics were similar to WT parameters. First, this suggests that preservation of charge at K132 is important for function. In addition, the similarity between WT cTnI and K132R cTnI indicates the stoichiometry of endogenous TnI acetylation is low under these conditions. It is possible that culturing cells may remove physiologic acetylation on K132 which would make WT cTnI calcium sensitivity and myofibril mechanical parameters resemble K132R. In fact, several other studies have demonstrated differences in acetylation based on cell morphology or cultured cells [43,44]. Regardless of this difference, our findings demonstrate how changes that mimic or block acetylation at specific sites affect mechanical parameters and suggest that exploration of these differences in disease and physiologically will highlight important drivers of cellular function and important targets for modification.
In order to verify that the cellular effects observed in ARVMs expressing K132Q cTnI are due to direct myofibril effects and not due to mechanisms outside the myofibril, we exchanged WT or K132Q cTnI into rat myofibrils. Myofibrils exchanged with K132Q cTnI relaxed faster than those exchanged with WT cTnI at maximum and submaximal calcium concentrations. Further, myofibrils exchanged with K132Q cTnI have decreased calcium sensitivity, consistent with the RTF and ARVM studies. These findings suggest that mimicking acetylation at a specific site of cTnI can modulate calcium sensitivity and relaxation. In particular, the acetyl-mimetic mutation at K132 increases the rate of linear phase relaxation which is thought to represent the rate of detachment of cycling cross-bridges (reviewed in [45,46]). Taken together, these findings suggest that mimicking acetylation at this particular lysine residue may impact both calcium binding as well as the interaction between the thin filament and myosin. Interestingly, Rao et al. explored the effect of phosphorylation of cTnI at Ser23/24 and demonstrated that phosphorylation at these sites leads to decreased calcium sensitivity and changes in the duration of relaxation [32]. Their study suggests that phosphorylation of cTnI leads to faster relaxation by reducing calcium-TnC affinity, accelerating the calcium dissociation rate, coincidentally reducing affinity of TnI for TnC, particularly at physiological calcium concentrations. Similarly, we found that incorporation of the acetyl-mimetic mutant at K132 of cTnI also decreased calcium sensitivity for all parameters of contraction and accelerated relaxation. However, other studies have demonstrated that calcium sensitivity is an inherent property of the troponin complex and interactions of the thin filament proteins, but that relaxation mechanics are more closely related to actin and myosin binding and release [47]. In fact, when cTnI is exchanged into skeletal muscle myofibrils, the myofibrils have decreased calcium sensitivity but no changes in relaxation parameters [47]. Further, treatment with bepridil (which alters calcium binding to troponin C) does not change relaxation kinetics. In context with our findings, it is possible that site-specific acetyl-mimetic modification at K132 may impact both troponin C (affecting calcium sensitivity) and actin binding (affecting cross-bridge cycling). In fact, both the rate of reactivation at submaximal calcium and the rate of linear phase relaxation suggest that cross-bridge detachment is impacted in the myofibrils with the K132Q mutation. Further investigation is required to verify this hypothesis.
Our results with K132Q do not fully phenocopy myofibrillar mechanical changes detailed in DSS rats treated systemically with ITF2357. K132Q cTnI recapitulates acceleration of the slow relaxation phase seen in the rat model with ITF2357 treatment, however, the prior study did not find any calcium-desensitization [14]. This difference likely stems from an offsetting effect of acetylation, or some other PTM, at another site where additional PTMs may alter sarcomeric protein interactions in concert with acetylation at K132. Another possibility is that acetylation of other residues of cTnI could also have different effects on the protein’s function. These findings demonstrate the importance of understanding the regulation of acetylation and how acetylation at different myofilament sites impacts overall function.
With these issues in mind, we examined the impact of K132Q on the overall total phosphorylation of myofibrillar proteins as well as specific phosphorylation of cTnI at Ser23/24. Phosphorylation status of cTnI has been largely attributed to phosphorylation within its N-terminal domain by PKA [6,32]. We show that acetyl-mimetic mutation of cTnI does not alter the phosphorylation of cTnI in cells. Thus our K132Q results are not confounded by functional sequelae of altered TnI phosphorylation. Nevertheless, the fact that both myofilament phosphorylation and acetylation are perturbed in heart failure, will make it important to unravel the relative contribution of each to pathogenesis and pathophysiology.
5. Conclusions
This study demonstrates that a site-specific change in cTnI to mimic acetylation leads to functional changes in the sarcomeric mechanical parameters indicating that acetylation of a single amino acid residue of cTnI is sufficient to induce functional differences. This highlights the need to better understand regulation of acetylation of sarcomeric proteins. Further, while this study focused on a single amino acid change to mimic acetylation, this is unlikely to occur in isolation and further studies are necessary to determine the interactions of these PTMs. In addition, it is clear that acetylation is a key post-translational modification mediating sarcomeric function and further studies are necessary to determine specificity of the various acetyltransferases and deacetylases regulating these changes. Detailed knowledge of the mechanisms controlling site-specific sarcomeric protein acetylation should guide efforts to manipulate this post-translational modification via deacetylase or acetyltransferase inhibition or gain-of-function as a potential therapeutic strategy for heart muscle disease.
Supplementary Material
Acknowledgements
This study was supported by National Institute of Health (NIH) by grants HL 116848, HL 147558, DK 119594, HL 127240 (TAM), HL 124091 (AC), HL 114940 (BJB), AA 0022146 (KSF), and 2K12HD057022–11 (KCW). This study was also funded by the American Heart Association by grants 16POST30960017 (YHL), 17POST33630159 (WS) and 16SFRN31400013 (TAM). We thank Tianjing Hu for advice on adenovirus production.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2020.01.007.
Declaration of Competing Interest
None.
References
- [1].Biesiadecki BJ, Davis JP, Ziolo MT, Janssen PML, Tri-modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross-bridge cycling kinetics, Biophys. Rev. 6 (3–4) (2014) 273–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kobayashi T, Solaro RJ, Calcium, thin filaments, and the integrative biology of cardiac contractility, Annu. Rev. Physiol. 67 (2005) 39–67. [DOI] [PubMed] [Google Scholar]
- [3].Lu QW, Wu XY, Morimoto S, Inherited cardiomyopathies caused by troponin mutations, J. Geriatr. Cardiol. 10 (1) (2013) 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mogensen J, Hey T, Lambrecht S, A systematic review of phenotypic features associated with cardiac troponin I mutations in hereditary cardiomyopathies, Can. J. Cardiol. 31 (11) (2015) 1377–1385. [DOI] [PubMed] [Google Scholar]
- [5].Vikhorev PG, Vikhoreva NN, Cardiomyopathies and related changes in contractility of human heart muscle, Int. J. Mol. Sci. 19 (8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ, Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle, Circ. Res. 88 (10) (2001) 1059–1065. [DOI] [PubMed] [Google Scholar]
- [7].Biesiadecki BJ, Westfall MV, Troponin I modulation of cardiac performance: plasticity in the survival switch, Arch. Biochem. Biophys. 664 (2019) 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cuello F, Wittig I, Lorenz K, Eaton P, Oxidation of cardiac myofilament proteins: priming for dysfunction? Mol. Asp. Med. 63 (2018) 47–58. [DOI] [PubMed] [Google Scholar]
- [9].Fert-Bober J, Giles JT, Holewinski RJ, Kirk JA, Uhrigshardt H, Crowgey EL, Andrade F, Bingham CO 3rd, Park JK, Halushka MK, Kass DA, Bathon JM, Van Eyk JE, Citrullination of myofilament proteins in heart failure, Cardiovasc. Res. 108 (2) (2015) 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Janssens JV, Ma B, Brimble MA, Van Eyk JE, Delbridge LMD, Mellor KM, Cardiac troponins may be irreversibly modified by glycation: novel potential mechanisms of cardiac performance modulation, Sci. Rep. 8 (1) (2018) 16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV, Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns, Cell Rep. 2 (2) (2012) 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rattanasopa C, Kirk JA, Bupha-Intr T, Papadaki M, de Tombe PP, Wattanapermpool J, Estrogen but not testosterone preserves myofilament function from doxorubicin-induced cardiotoxicity by reducing oxidative modifications, Am. J. Physiol. Heart Circ. Physiol. 316 (2) (2019) H360–H370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Foster DB, Liu T, Rucker J, O’Meally RN, Devine LR, Cole RN, O’Rourke B, The cardiac acetyl-lysine proteome, PLoS One 8 (7) (2013) e67513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, Ambardekar AV, Granzier HL, Dinarello CA, McKinsey TA, Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism, Sci. Transl. Med. 10 (427) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fritz KS, Green MF, Petersen DR, Hirschey MD, Ethanol metabolism modifies hepatic protein acylation in mice, PLoS One 8 (9) (2013) e75868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fritz KS, Kellersberger KA, Gomez JD, Petersen DR, 4-HNE adduct stability characterized by collision-induced dissociation and electron transfer dissociation mass spectrometry, Chem. Res. Toxicol. 25 (4) (2012) 965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hanft LM, Biesiadecki BJ, McDonald KS, Length dependence of striated muscle force generation is controlled by phosphorylation of cTnI at serines 23/24, J. Physiol. 591 (18) (2013) 4535–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chandra M, Rundell VL, Tardiff JC, Leinwand LA, De Tombe PP, Solaro RJ, Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T, Am. J. Physiol. Heart Circ. Physiol. 280 (2) (2001) H705–H713. [DOI] [PubMed] [Google Scholar]
- [19].Engel PL, Kobayashi T, Biesiadecki B, Davis J, Tikunova S, Wu S, Solaro RJ, Identification of a region of troponin I important in signaling cross-bridge-dependent activation of cardiac myofilaments, J. Biol. Chem. 282 (1) (2007) 183–193. [DOI] [PubMed] [Google Scholar]
- [20].Salhi HE, Hassel NC, Siddiqui JK, Brundage EA, Ziolo MT, Janssen PM, Davis JP, Biesiadecki BJ, Myofilament calcium sensitivity: mechanistic insight into TnI Ser-23/24 and Ser-150 phosphorylation integration, Front. Physiol. 7 (2016) 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PM, Davis JP, Biesiadecki BJ, Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation, J. Mol. Cell. Cardiol. 76 (2014) 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liang B, Chen Y, Wang CK, Luo Z, Regnier M, Gordon AM, Chase PB, Ca2+ regulation of rabbit skeletal muscle thin filament sliding: role of cross-bridge number, Biophys. J. 85 (3) (2003) 1775–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Margossian SS, Lowey S, Preparation of myosin and its subfragments from rabbit skeletal muscle, Methods Enzymol. 85 (Pt B) (1982) 55–71. [DOI] [PubMed] [Google Scholar]
- [24].Pollard TD, Myosin purification and characterization, Methods Cell Biol. 24 (1982) 333–371. [DOI] [PubMed] [Google Scholar]
- [25].Meijering E, Dzyubachyk O, Smal I, Methods for cell and particle tracking, Methods Enzymol. 504 (2012) 183–200. [DOI] [PubMed] [Google Scholar]
- [26].Viswanathan MC, Blice-Baum AC, Schmidt W, Foster DB, Cammarato A, Pseudo-acetylation of K326 and K328 of actin disrupts Drosophila melanogaster indirect flight muscle structure and performance, Front. Physiol. 6 (2015) 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Colomo F, Nencini S, Piroddi N, Poggesi C, Tesi C, Calcium dependence of the apparent rate of force generation in single striated muscle myofibrils activated by rapid solution changes, Adv. Exp. Med. Biol. 453 (1998) 373–381 (discussion 381–2). [DOI] [PubMed] [Google Scholar]
- [28].Tesi C, Colomo F, Nencini S, Piroddi N, Poggesi C, The effect of inorganic phosphate on force generation in single myofibrils from rabbit skeletal muscle, Biophys. J. 78 (6) (2000) 3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tesi C, Colomo F, Nencini S, Piroddi N, Poggesi C, Modulation by substrate concentration of maximal shortening velocity and isometric force in single myofibrils from frog and rabbit fast skeletal muscle, J. Physiol. 516 (Pt 3) (1999) 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brenner B, Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction, Proc. Natl. Acad. Sci. U.S. A. 85 (9) (1988) 3265–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bagchi RA, Ferguson BS, Stratton MS, Hu T, Cavasin MA, Sun L, Lin YH, Liu D, Londono P, Song K, Pino MF, Sparks LM, Smith SR, Scherer PE, Collins S, Seto E, McKinsey TA, HDAC11 suppresses the thermogenic program of adipose tissue via BRD2, JCI Insight 3 (15) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rao V, Cheng Y, Lindert S, Wang D, Oxenford L, McCulloch AD, McCammon JA, Regnier M, PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils, Biophys. J. 107 (5) (2014) 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jeong MY, Walker JS, Brown RD, Moore RL, Vinson CS, Colucci WS, Long CS, AFos inhibits phenylephrine-mediated contractile dysfunction by altering phospholamban phosphorylation, Am. J. Physiol. Heart Circ. Physiol. 298 (6) (2010) H1719–H1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Woulfe KC, Ferrara C, Pioner JM, Mahaffey JH, Coppini R, Scellini B, Ferrantini C, Piroddi N, Tesi C, Poggesi C, Jeong M, A novel method of isolating myofibrils from primary cardiomyocyte culture suitable for myofibril mechanical study, Front. Cardiovasc. Med. 6 (2019) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP, Biesiadecki BJ, Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment, J. Mol. Cell. Cardiol. 72 (2014) 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li MX, Spyracopoulos L, Sykes BD, Binding of cardiac troponin-I147–163 induces a structural opening in human cardiac troponin-C, Biochemistry 38 (26) (1999) 8289–8298. [DOI] [PubMed] [Google Scholar]
- [37].Belus A, Piroddi N, Scellini B, Tesi C, D’Amati G, Girolami F, Yacoub M, Cecchi F, Olivotto I, Poggesi C, The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils, J. Physiol. 586 (15) (2008) 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Piroddi N, Belus A, Scellini B, Tesi C, Giunti G, Cerbai E, Mugelli A, Poggesi C, Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium, Pflugers Arch. 454 (1) (2007) 63–73. [DOI] [PubMed] [Google Scholar]
- [39].Tesi C, Piroddi N, Colomo F, Poggesi C, Relaxation kinetics following sudden Ca (2+) reduction in single myofibrils from skeletal muscle, Biophys. J. 83 (4) (2002) 2142–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stehle R, Solzin J, Iorga B, Gomez D, Blaudeck N, Pfitzer G, Mechanical properties of sarcomeres during cardiac myofibrillar relaxation: stretch-induced cross-bridge detachment contributes to early diastolic filling, J. Muscle Res. Cell Motil. 27 (5–7) (2006) 423–434. [DOI] [PubMed] [Google Scholar]
- [41].Meraviglia V, Bocchi L, Sacchetto R, Florio MC, Motta BM, Corti C, Weichenberger CX, Savi M, D’Elia Y, Rosato-Siri MD, Suffredini S, Piubelli C, Pompilio G, Pramstaller PP, Domingues FS, Stilli D, Rossini A, HDAC inhibition improves the sarcoendoplasmic reticulum Ca(2+)-ATPase activity in cardiac myocytes, Int. J. Mol. Sci. 19 (2) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schoffstall B, Brunet NM, Williams S, Miller VF, Barnes AT, Wang F, Compton LA, McFadden LA, Taylor DW, Seavy M, Dhanarajan R, Chase PB, Ca2+ sensitivity of regulated cardiac thin filament sliding does not depend on myosin isoform, J. Physiol. 577 (Pt 3) (2006) 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Le Beyec J, Xu R, Lee SY, Nelson CM, Rizki A, Alcaraz J, Bissell MJ, Cell shape regulates global histone acetylation in human mammary epithelial cells, Exp. Cell Res. 313 (14) (2007) 3066–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wisniewski JR, Zougman A, Kruger S, Mann M, Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue, Mol. Cell. Proteomics 6 (1) (2007) 72–87. [DOI] [PubMed] [Google Scholar]
- [45].Poggesi C, Tesi C, Stehle R, Sarcomeric determinants of striated muscle relaxation kinetics, Pflugers Arch. 449 (6) (2005) 505–517. [DOI] [PubMed] [Google Scholar]
- [46].Stehle R, Solzin J, Iorga B, Poggesi C, Insights into the kinetics of Ca2+ −regulated contraction and relaxation from myofibril studies, Pflugers Arch. 458 (2) (2009) 337–357. [DOI] [PubMed] [Google Scholar]
- [47].deTombe PP, Belus A, Piroddi N, Scellini B, Walker JS, Martin AF, Tesi C, Poggesi C, Myofilament calcium sensitivity does not affect cross-bridge activation-relaxation kinetics, Am. J. Phys. Regul. Integr. Comp. Phys. 292 (3) (2007) R1129–R1136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.