

Abstract
3-Phosphoinositide-dependent protein kinase-1 (PDK1) plays a critical role in the development of mammalian brain. Here, we investigated the role of PDK1 in Purkinje cells (PCs) by generating the PDK1-conditional knock-out mice (cKO) through crossing PV-cre or Pcp2-cre mice with Pdk1fl/fl mice. The male mice were used in the behavioral testing, and the other experiments were performed on mice of both sexes. These PDK1-cKO mice displayed decreased cerebellar size and impaired motor balance and coordination. By the electrophysiological recording, we observed the reduced spontaneous firing of PCs from the cerebellar slices of the PDK1-cKO mice. Moreover, the cell body size of PCs in the PDK1-cKO mice was time dependently reduced compared with that in the control mice. And the morphologic complexity of PCs was also decreased after PDK1 deletion. These effects may have contributed to the reduction of the rpS6 (reduced ribosomal protein S6) phosphorylation and the PKCγ expression in PDK1-cKO mice since the upregulation of pS6 by treatment of 3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-1, the agonist of mTOR1, partly rescued the reduction in the cell body size of the PCs, and the delivery of recombinant adeno-associated virus-PKCγ through cerebellar injection rescued the reduced complexity of the dendritic arbor in PDK1-cKO mice. Together, our data suggest that PDK1, by regulating rpS6 phosphorylation and PKCγ expression, controls the cell body maintenance and the dendritic development in PCs and is critical for cerebellar motor coordination.
SIGNIFICANCE STATEMENT Here, we show the role of 3-phosphoinositide-dependent protein kinase-1 (PDK1) in Purkinje cells (PCs). The ablation of PDK1 in PCs resulted in a reduction of cell body size, and dendritic complexity and abnormal spontaneous firing, which attributes to the motor defects in PDK1-conditional knock-out (cKO) mice. Moreover, the ribosomal protein S6 (rpS6) phosphorylation and the expression of PKCγ are downregulated after the ablation of PDK1. Additionally, upregulation of rpS6 phosphorylation by3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-1 partly rescued the reduction in cell body size of PCs, and the overexpression of PKCγ in PDK1-KO PCs rescued the reduction in the dendritic complexity. These findings indicate that PDK1 contributes to the maintenance of the cell body and the dendritic development of PCs by regulating rpS6 phosphorylation and PKCγ expression.
Keywords: cell body, cerebellum, PDK1, PKCγ, pS6, Purkinje cells
Introduction
Purkinje cells (PCs) are the sole output neuron of the cerebellar cortex, serving as an integration center in this region, and are important for motor coordination and motor balance (Lui et al., 2017; Edamakanti et al., 2018; Popa et al., 2019). Although the neurogenesis and neuronal migration of PCs are completed during the embryonic stage, the postnatal development process is critical for PCs and cerebellum-related motor function (Liu et al., 2017; Lui et al., 2017; Edamakanti et al., 2018). During the 8 week postnatal stage, the soma size of PCs gradually increases and the characteristic dendritic trees of PCs are also formed at this stage (Takeo et al., 2015). Previous studies have demonstrated that mTOR (the mammalian target of rapamycin) pathway may be involved in the postnatal development of PCs. For example, in PC-specific deletion of mTORC1 mice, PCs are progressively lost due to apoptosis that is paralleled by the age-dependent motor deficits (Angliker et al., 2015). Moreover, the whole-brain mTORC2 KO mice display motor coordination defects, which correlate with reduced Purkinje cell body size, developmental deficits in climbing fiber elimination, and impaired dendritic self-avoidance. Furthermore, the deletion mTORC2 in cerebellum leads to a striking decrease in the activation and expression of several protein kinase C (PKC) isoforms including the γ isoform of PKC (PKCγ; Thomanetz et al., 2013; Angliker et al., 2015). Previous studies have reported that PKCγ, which is highly and specifically expressed in PCs, plays a critical role in eliminating supernumerary climbing fiber synapses from developing PCs (Kano et al., 1995; Hirai, 2018). Although various molecules and mechanisms have been implicated in the regulation of PC morphogenesis, the mechanisms that regulate postnatal maintenance and development of PCs remain unclear.
The 3-phosphoinositide-dependent protein kinase-1 (PDK1), a serine/threonine kinase of the AGC kinase group, phosphorylates and activates at least 23 other AGC protein kinases such as AKT (protein kinase B), PKC, ribosomal protein S6 kinase B1, and serum/glucocorticoid-regulated kinase (SGK; Castel et al., 2016; Leroux et al., 2018), which contributes to many biological processes, including cell polarization, neuronal migration, and neuronal differentiation (Oishi et al., 2009; Dainichi et al., 2016; Itoh et al., 2016; Wang et al., 2017b). Our previous studies showed that PDK1 is required for the proliferation, differentiation, and migration of neural progenitor cells during dentate gyrus development (Xu et al., 2019). Whole-brain downregulation of PDK1–AKT signaling results in microcephaly, reflected in a reduction in cell size rather than cell number (Lawlor et al., 2002; Cordón-Barris et al., 2016). Additionally, PDK1 is a downstream antagonist of phosphatase and tensin homolog (PTEN), and the hypertrophy of PTEN-deficient neuronal nuclei and soma can be rescued by the PDK1 deletion (Chalhoub et al., 2009; Grego-Bessa et al., 2016). Recent evidence indicates that the loss of PTEN in PCs results in autistic-like traits, including motor-learning deficits, sociability impairment, and repetitive behavior, which may be attributed to the structural abnormalities in neurite and late-onset cell death (Cupolillo et al., 2016). These data indicate that PDK1 is likely involved in the postnatal development of PCs.
In this study, we investigated the role of PDK1 in the postnatal development of PCs by crossing Pdk1fl/fl mice with the PV-cre or Pcp2-cre line. And our data showed that the ablation of PDK1 in PCs decreased cell body size, reduced dendritic complexity, and was associated with substantial abnormal spontaneous electrophysiological properties in PCs, which was paralleled by the motor defects. We further showed that the reduced ribosomal protein S6 (rpS6) phosphorylation and expression of PKCγ was probably contributed to these phenotypes. Together, our results suggested that PDK1 was important for cell body maintenance and dendritic development in postnatal PCs and was correlated with cerebellar motor coordination and balance.
Materials and Methods
Animals and mouse breeding.
Pdk1fl/fl mice have been reported previously (Lawlor et al., 2002). Parvalbumin-cre (PV-cre) mice were provided by Prof. Li-Huei Tsai (Picower Institute for Learning and Memory, MIT, Cambridge, MA). Pcp2-cre mice were provided by Prof. Ying Shen (Zhejiang University, Hangzhou, Zhejiang, People's Republic of China; Barski et al., 2000). ROSA26-LSL-EYFP mice were provided by Prof. Chunjie Zhao (Southeast University, Nanjing, Jiangsu, People's Republic of China; Srinivas et al., 2001). The PV-cre; Pdk1fl/fl mice and the Pcp2-cre; Pdk1fl/fl mice were generated by crossing the Pdk1fl/fl mice with the PV-cre mice and the Pcp2-cre; Pdk1fl/fl mice, respectively. In this study, the PV-cre; Pdk1fl/fl mice and the Pcp2-cre; Pdk1fl/fl mice were named conditional knock-out (cKO)-PV and cKO-Pcp2, respectively, and the Pdk1fl/fl mice were grouped as the control (Ctrl). All of the mice are maintained on a C57BL/6 background and kept in a barrier facility, and all animal experiments were conducted in accordance with the procedures approved by the Animal Ethical and Welfare Committee for Institutional Animal Care and Use Committee (IACUC) of Nanjing Medical University (approval No. IACUC-1907019). The day of birth was considered as postnatal day 0 (P0).
Tissue collection.
Mice (age, P14–P120) were anesthetized by intraperitoneal administration of ketamine/xylazine (ketamine:, 100 mg/kg; xylazine, 10 mg/kg) based on body weight and killed by transcardial perfusion with PBS followed by 4% paraformaldehyde (PFA) in PBS. The cerebellum was removed and postfixed in 4% PFA overnight at 4°C and processed into cryosections by using a freezing microtome (25 μm thick; model CM 1950, Leica).
Genotyping.
Genotype was identified by PCR with genomic DNA obtained from the toes or the tails. The primers sequences used to identify specific genotypes were as follows: Pdk1fl/fl, 5′-CTATGCTGTGTTACTTCTTGGAGCACAG-3′ and 5′-TGTGGACAAACAGCAATGAACATACACGC-3′; Pcp2-cre and PV-cre, 5′-CATACCTGGAAAATGCTTCTGTCC-3′ and 5′-TCCCCAGAAATGCCAGATTACG-3′; and ROSA26-LSL-EYFP, 5′-AAGACCGCGAAGAGTTTGTC-3′ and 5′-AAAGTCGCTCTGAGTTGTTAT-3′. The PCR program used was as follows: 94°C for 5 min, then 35 cycles of 94°C for 30 s for denaturation, 62°C for 45 s for annealing, and 72°C 45 s for elongation.
Behavioral testing.
All behavioral experiments were conducted on male mice between 9:00 A.M. and 4:00 P.M. Each mouse was subjected to each of the following behavioral tests.
Footprint.
Hindpaws and forepaws of mice were painted with nontoxic blue and red watercolor respectively. Mice walked through a tunnel (100 cm long, 9 cm wide, 60 cm high) with white paper lining the floor to a dark box with a hole. Each mouse was trained on 3 consecutive days with three trials per day. Footprint patterns were analyzed for length of stride, stance, and sway and distance between the front and hind footprints on each side. For each measurement, the first and last 20 cm of the prints were excluded. If the mouse stopped in the middle of the tunnel, the trial was repeated.
Rotarod assay.
The rotarod apparatus (ZH-300) was used to measure motor coordination, as previously described (Carter et al., 1999). During the training period, each mouse was placed on the rotarod at a constant speed (20 rpm) for a maximum of 60 s. Mice received six trials per day for 4 consecutive days, by which time a steady baseline level of performance was attained. Mice then received two trials at the following six independent speed levels: 10, 15, 20, 25, 30, and 33 rpm. The mean latency to fall off the rotarod (for the two trials at each speed level) was recorded and used in subsequent analysis.
Elevated beam-walk test assay.
The elevated beam-walk assay for evaluating motor balance was conducted as previously described (Carter et al., 2001). Briefly, the beams consisted of long strips of matte-surface acrylic (130 cm) with a 28- or 12-mm-square cross section and a 30, 16, or 10 mm diameter. The beams were placed horizontally, 40 cm above the platform surface, with one end mounted on a narrow support and the other end attached to an enclosed box (30 cm square) into which the mouse could escape. In the middle part of each beam, an 80-cm-long segment was marked. One angle poise light (40 W) was positioned above and to one side of the start of the beam. During training, mice were placed at the start of the 12-mm-square beam and encouraged to traverse it. Each mouse underwent three consecutive trials and was then returned to its home cage to rest for 30 min, then another three consecutive trials were performed. Each mouse underwent six trials per day over 4 d. On the fifth day, each mouse underwent three consecutive trials on each of the round beams, from the widest to the narrowest beam. The latency to traverse each 80 cm section and the number of times the hindpaws slipped off each beam were recorded for each trial. Mice were allowed up to 60s to cross the beam. If the mouse dropped from the beam, the trial was repeated, and if it could not cross the beam within 60 s after two attempts, the trial was allocated a maximum latency of 60 s for inclusion in the latency analyses. Analysis of each measure was based on the mean scores of the two trials for each beam.
Immunofluorescence.
Immunofluorescence experiments were performed as previously described (Liu et al., 2015). Briefly, brain tissue sectioning was performed using a freezing microtome (25 μm thick; model CM 1950, Leica). For each genotype, at least three histologic sections at middle sagittal levels from three different animals were analyzed for immunostaining, and confocal optical sections were acquired. The primary antibodies and working concentrations were as follows: rabbit anti-calbindin (1 μg/ml; ab49899, Abcam); anti-NeuN (1 μg/ml; ABN78, Millipore); anti-PAX6 (4 μg/ml; catalog #ab5790, Abcam); anti-PDk1 (2 μg/ml; catalog #3377–1, Epitomics); anti-PDk1 (0.36 μg/ml; catalog #ab52893, Abcam); anti-PKC (0.68 μg/ml; phospho-T514; catalog #ab109539, Abcam), anti-phospho-S6 ribosomal protein (3.8 × 10−2 μg/ml; Ser235/236; catalog #2211, Cell Signaling Technology); mouse anti-NeuN (2 μg/ml; catalog #MAB377, Millipore); anti-GFAP (1 μg/ml; catalog #MAB3402, Millipore); anti-GAD67 (glutamic acid decarboxylase 67; 1 μg/ml; catalog #MAB5406, Millipore); anti-mCherry (1 μg/ml; catalog #MB2013, Bioworld Technology); and chicken anti-GFP (2.5 μg/ml; catalog #ab13970, Abcam). The secondary antibodies included DyLight 488 donkey anti-rabbit (1 μg/ml; catalog #SA5-10 038, Thermo Fisher Scientific); Invitrogen Alexa Fluor 488 goat anti-rabbit (4 μg/ml; catalog #A11008, Thermo Fisher Scientific); DyLight 488 donkey anti-mouse (1 μg/ml; catalog #SA5-10 166, Thermo Fisher Scientific); Invitrogen Alexa Fluor 555 donkey anti-rabbit (4 μg/ml; A31572, Thermo Fisher Scientific); DyLight 550 donkey anti-mouse (1 μg/ml; catalog #SA5-10 167, Thermo Fisher Scientific); and Alexa Fluor 488 donkey anti-chicken (0.25 μg/ml; catalog #703–545-155, Jackson ImmunoResearch). Secondary antibodies were diluted in PBS containing 10% fetal bovine serum, 0.1% Triton X-100, and 0.5 μg/ml 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI).
Golgi staining.
Golgi staining was performed using FD Rapid GolgiStain Kit (catalog #PK401, FD NeuroTechnologies) following the manufacturer protocols. Briefly, the cerebellum was dissected from P30 mice and incubated in premixed solutions A and B that were refreshed the next day. The cerebellum was kept in the dark for 2 weeks at room temperature, then incubated in solution C in the dark for 5–7 d at room temperature. The cerebellum was then sectioned into 250-μm-thick slices with a vibratome (catalog #VT1000, Leica Microsystems) and stained according to the manufacturer protocols. Images of PCs and other cerebellar neurons were captured by a Olympus Fluoview FV1200 Confocal Microscope.
The dendritic arbors of each PC were semiautonomous traced by using National Institutes of Health (NIH) ImageJ software, as previously described (Stanko et al., 2015). Dendritic arborization complexity was assessed using a Sholl analysis to examine dendritic intersections per 1 μm concentric radial interval from cell body (Ferreira et al., 2014). The schematic representation of the Sholl analysis method described is seen in Figure 12, G and H. The significance of the differences in complexity was determined using GraphPad Prism software with a two-way ANOVA (genotype and circle radius as factors) followed by the Bonferroni's post hoc test.
Figure 12.
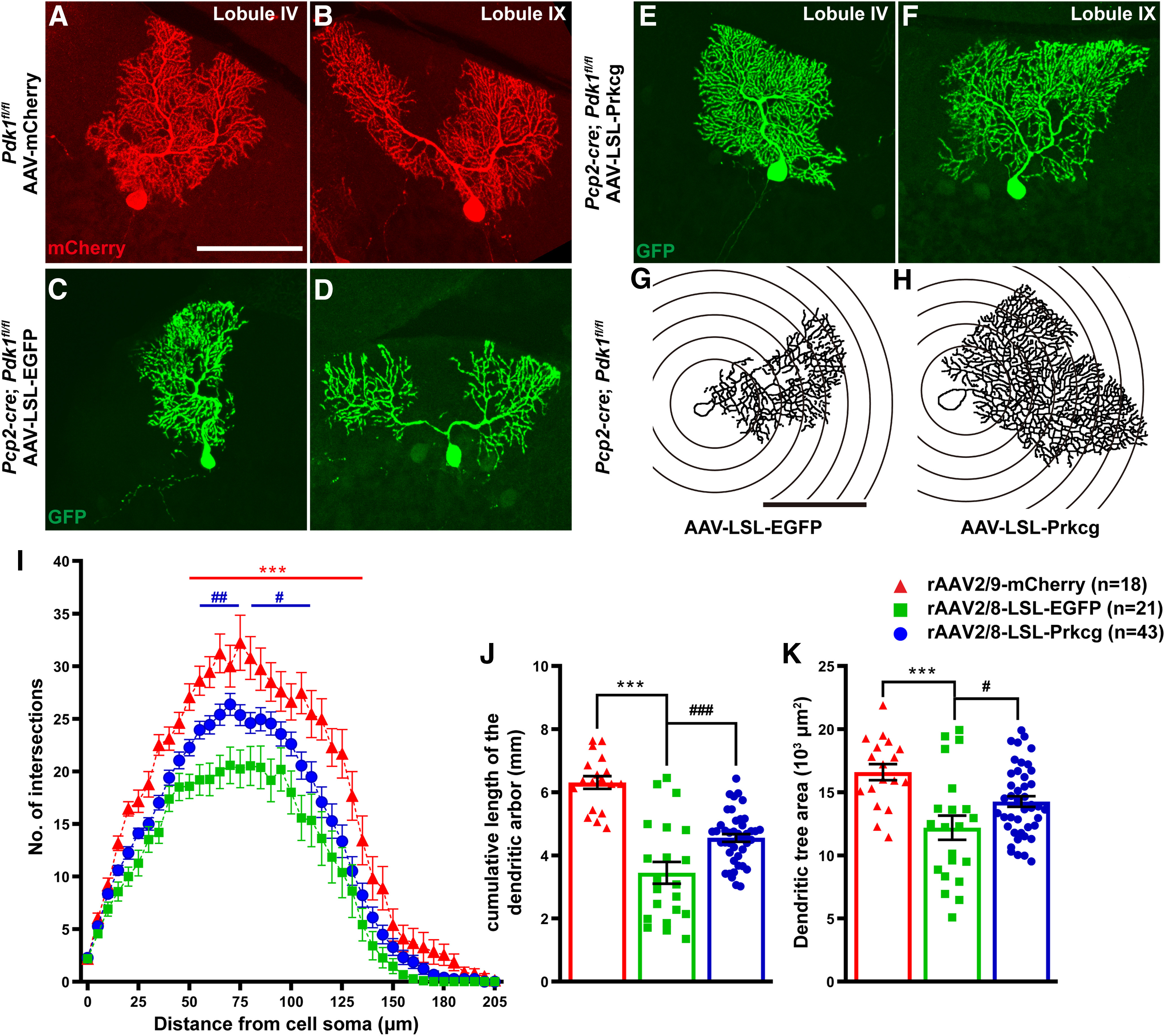
The rescue of deficient dendritic complexity by overexpressing PKCγ in the PDK1-KO PCs from the PDK1 cKO-Pcp2 mice. A, B, Immunostaining for mCherry shows the rAAV-mCherry- infected PCs in lobule IV (A) and lobule IX (B) in Ctrl mice. C–F, Immunostaining for GFP shows the rAAV-EGFP-infected PCs (C, D) and the rAAV-pPKCγ overexpressing PCs (E, F) in lobule IV (C, E), and lobule IX (D, F) in cKO-Pcp2 mice. G, H, The schematic illustration of the Sholl analysis of rAAV-infected PCs. The 20-mm-radius steps are shown in the illustration. AAV2/9-CAG-FLEX-EGFP-infected PCs in cKO-Pcp2 mice (G) and AAV2/9-CAG-FLEX-Prkcg-3xHA-P2A-EGFP-infected PCs in cKO-Pcp2 mice (H). I, Sholl analysis for three groups of PCs in G–L at P30. The red curve represented the rAAV-mCherry-infected PCs in Ctrl mice (n = 18 PCs from 5 mice). The green curve represented the rAAV-EGFP-infected PCs (n = 21 PCs from 3 mice), and the blue curve represented the PCs overexpressing pPKCγ (n = 43 PCs from 3 mice) in cKO-Pcp2 mice. The number of intersections of the dendrite at different distances from the cell body of PCs, and the data are the mean ± SEM. A two-way ANOVA; Bonferroni's post hoc analysis was used: rAAV-mCherry/Ctrl mice versus rAAV-EGFP/cKO-Pcp2 mice, ***p < 0.001 between 50 and 135 mm from the cell body. rAAV-EGFP/cKO-Pcp2 mice versus rAAV-PKCγ/cKO-Pcp2 mice: ##p < 0.01 between 55 and 75 mm from the cell body; and #p < 0.05 between 80 and 110 mm from the cell body. J, The cumulative length of the dendritic arbor of PCs. The data are the mean ± SEM: rAAV-mCherry/Ctrl mice: 6.31 ± 0.20 mm, n = 18 PCs from 5 mice; rAAV-EGFP/cKO-Pcp2 mice: 3.45 ± 0.35 mm, n = 21 PCs from 3 mice (p < 0.0001, compared with that in rAAV-mCherry/Ctrl mice); and rAAV-PKCγ/cKO-Pcp2 mice: 4.55 ± 0.12 mm, n = 43 PCs from 3 mice (p = 0.0007, compared with that in rAAV-EGFP/cKO-Pcp2). K, Dendritic tree area of PCs. The data are the mean ± SEM: rAAV-mCherry/Ctrl mice: 16.59 ± 0.65 × 103 μm2, n = 18 PCs form 5 mice; rAAV-EGFP/cKO-Pcp2 mice: 12.19 ± 0.96 × 103 μm2, n = 21 PCs form 3 mice (p = 0.0001, compared with that in rAAV-mCherry/Ctrl mice); and rAAV-PKCγ/cKO-Pcp2 mice: 14.26 ± 0.42 × 103 μm2, n = 43 PCs form 3 mice (p = 0.0392, compared with that in rAAV-EGFP/cKO-Pcp2 mice). Data in I were analyzed using a two-way ANOVA; Bonferroni's post hoc analysis and a one-way ANOVA; Bonferroni's post hoc analysis was used to analyze data in J and K. ###p < 0.001, ##p < 0.01, #p < 0.05, ***p < 0.001. Scale bars, 100 μm.
Western blotting.
Western blotting experiments were performed as previously described (Xu et al., 2019). Briefly, mouse cerebellums were collected and homogenized, and the lysates from at least four brains per genotype were clarified by centrifugation at 14,000 rpm. The protein concentrations were measured (Thermo Fisher Scientific). Cerebellum lysates (20 μg) were separated by 10% SDS-PAGE and transferred to PVDF membranes. After blocking the membranes with 5% nonfat dry milk in Tris-buffered saline with 0.5% Tween-20, they were incubated at 4°C with primary antibodies, followed by incubation with the HRP-linked anti-rabbit IgG secondary antibody (8.2 × 10−3 μg/ml; catalog #7074, Cell Signaling Technology), and signals were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). The primary antibodies used and the working concentration of these antibodies were as follows: rabbit anti-β-actin (1 μg/ml; catalog #AP0060, Bioworld Technology); anti-GAPDH (8.4 × 10−3 μg/ml; 2118, Cell Signaling Technology); anti-PDk1 (0.5 μg/ml; catalog #3377-1, Epitomics); anti-PDk1 (0.09 μg/ml; catalog #ab52893, Abcam); and anti-PKCγ (0.1 μg/ml; catalog #ab4145, Abcam).
Whole-cell patch-clamp recordings on brain slices.
Whole-cell patch-clamp recordings on brain slices were performed as previously described (Wang et al., 2017c). Briefly, cerebellar slices (300 μm in thickness) were prepared following the standard protocols. During whole-cell patch-clamp recordings, PCs were held at a membrane potential of −70 mV and characterized by the injection of rectangular voltage pulse (5 mV, 50 ms) to monitor the whole-cell membrane capacitance (Cm), membrane resistance, and series resistance. PCs were excluded from the experiments if the series resistance was not stable or exceeded 20 MΩ.
3-Benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-1 treatment.
3-Benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-1 (3BDO; catalog #S8317, Selleck Chemicals) was dissolved in 100% DMSO and stored in a stock solution of 300 mg/ml at −20°C. A working solution was prepared immediately before injection at a final concentration of 6 mg/ml 3BDO, 2% DMSO, and 0.01 m PBS. P14 cKO-PV and Pdk1fl/fl mice were injected intraperitoneally with either 3BDO (80 mg/kg body weight/mouse) or vehicle once a day before harvest or a behavior test.
Virus vector construction.
Total RNA extraction of the cerebellar cortices and cDNA synthesis was performed as previously described (Liu et al., 2015). Full-length PKCγ coding sequence was amplified from cDNA taken from a wild-type C57BL/6 mouse and inserted into a pCAGIG plasmid (provided by Prof. Chunjie Zhao, Southeast University). Recombinant adeno-associated viruses (AAVs) were produced by the Taiting Biological Co, Ltd at the following stock titers [in genome copies (GC)/ml)]: AAV2/9-CAG-FLEX-Prkcg-3xHA-P2A-EGFP (enhanced green fluorescent protein) at 6.7 × 1012, AAV2/9-CAG-FLEX-EGFP at 2.3 × 1013, and AAV2/8-CMV_bGI-mCherry at 2.77 × 1013. Viruses were diluted in 0.01 mol/L sterile PBS, pH 7.4, before injection. The PCs in the Ctrl and cKO-Pcp2 mice were labeled using AAV2/8-CMV_bGI-mCherry constructs that express mCherry under a CMV promoter. Rescue experiments were conducted in Pcp2-cre; Pdk1fl/fl mice using recombinant AAVs (rAAVs) expressing EGFP or full-length PKCγ. rAAVs expressing EGFP under CAG promoters (AAV2/9-CAG-FLEX-EGFP) were used to label PDK1-KO PCs in mice, and rAAVs expressing both EGFP and PKCγ (AAV2/9-CAG-FLEX-Prkcg-3xHA-P2A-EGFP) were used to label PDK1-KO PCs and overexpress PKCγ. The PKCγ-expressing rAAV construct contained a CAG promoter and a floxed stop sequence upstream of an independent EGFP and full-length PKCγ sequence (rAAV2/9-CAG-FLEX-Prkcg-3xHA-P2A-EGFP). Thus, only Cre-positive PCs in the cKO-Pcp2 mice were able to remove the stop sequence and to overexpress EGFP and PKCγ. The rAAV2/9-CAG-FLEX-EGFP was used to express EGFP and to label Cre-positive PDK1-KO PCs in the cKO-Pcp2 mice without overexpression of PKCγ.
Virus injections.
All injections were performed on newborn mouse pups at P0–P1, as previously described (Gibson and Ma, 2011). Briefly, the P0 or P1 mouse pups were divided into the following two groups: one group was injected with a mix of AAV2/8-CMV_bGI-mCherry and AAV2/9-CAG-FLEX-EGFP in a 1:1 ratio, and the other group was injected with a mix of AAV2/8-CMV_bGI-mCherry and AAV2/9-CAG-FLEX-Prkcg-3xHA-P2A-EGFP in a 1:1 ratio. Each pup was injected with 1–1.5 μl of diluted rAAVs, and the concentration is 1 × 1012 GC/ml for each AAV vector, so the final concentration is 1 × 1012 GC/ml. A digital pressure pump and a Hamilton syringe with a beveled 34 ga stainless steel needle were used. Pups were then returned to their mothers and analyzed at P30.
Microscopic analysis.
The images were acquired on a Nikon ECLIPSE Ti microscope or an Olympus FV1200 confocal microscope. All images were processed using Image-Pro Plus 6.0 software (Media Cybernetics), Adobe Photoshop CS6.0 software (Adobe Systems), or the open source FIJI (NIH ImageJ) software (http://fiji.sc/Fiji).
Analysis of PC dendrites.
Confocal z-stacks were collected on an Olympus FV1200 laser-scanning confocal microscope. Confocal stacks of labeled PCs were collected from anatomic positions throughout the cerebellum. GFP-positive or mCherry-positive PC dendrites were manually traced to generate a skeletonized 2D reconstruction of the entire arbor, as previously described (Stanko et al., 2015). The total dendrite area was calculated using the confocal stack images of each PC, and total dendrite lengths were calculated from the 2D reconstruction. Dendritic arbor complexity was assessed using a Sholl analysis method, which was described in the Materials and Methods for Golgi staining. All processes described above were performed using the open source FIJI (NIH ImageJ) software (http://fiji.sc/Fiji).
Morphometric analysis.
The area of cerebellum; the thickness of the molecular layer (ML) of cerebellum; and the total number, cell density, and cell body size of PCs were evaluated in lobules II–III, V–VI, and IX–X of cerebellum of Ctrl, cKO-PV, and cKO-Pcp2 mice at different ages. Immunostaining of cerebellar slices with the specific antibody of calbindin was used to label the cell bodies and proximal primary dendrites of PCs, and a combination of immunostaining for calbindin and DAPI staining was used to label the outlines of sagittal cerebellar slices and to distinguish the three layers of the cerebellar cortex. The area of cerebellum was obtained by drawing the outline of sagittal sections of cerebellum and then calculating the average enclosed area. The size of each Purkinje cell body was obtained by drawing the outline of the PC body, clearly showing a proximal primary dendrite. The total number of PCs was obtained by drawing the outline of Purkinje cell layer (PCL) in the selected lobules of cerebellum and counting the number of PCs in the lobule manually. The length of the PCL was then measured, and the density of PCs was calculated by dividing the total number of PCs by the length of PCL. At least six slices/mouse and three mice (except for vehicle-treated cKO-PV mice) at each age point were analyzed for each condition in parallel experiments. And for the analysis of the cell body size of PCs, at least 32 PCs per mouse were analyzed for each condition in parallel experiments. All processes described above were performed using the open source FIJI (NIH ImageJ) software (http://fiji.sc/Fiji).
Experimental design and statistical analysis.
The male mice were used in the behavioral testing, and in the other experiments, no effects of ewe, fetus, or sex were observed. All the data were statistically analyzed using GraphPad Prism software (version 8; GraphPad Software). The data are expressed as the mean ± SEM. The error bars represent the SEM. For morphometric analysis, Western blotting, electrophysiological analysis and behavior tests, two-tailed unpaired Student's t test, one-way ANOVA followed by the Bonferroni's post hoc test, or two-way ANOVA followed by the Bonferroni's post hoc test were used to analyze statistical significance. The statistical methods used in each experiment are described in the figure legends, and the detailed description for each statistical test, including the degrees of freedom and the sample size, are shown in the figure legends or tables. If the p values are <0.0001 or >0.9999, they were presented as p < 0.0001 or p > 0.9999, respectively. And in the other cases, the exact p values are presented in the figure legends or tables. In the figures, p values are presented as follows: ns, p ≥ 0.05; * or #p < 0.05; ** or ##p < 0.01; and *** or ###p < 0.001.
Results
Cerebellar size was decreased in cKO-PV mice
Previous data demonstrated that Cre expression was evident in PCs no later than postnatal day 8 in PV-cre mice (Buttermore et al., 2012). To confirm the specificity of the PV-cre drivers, we generated PV-cre; ROSA26-LSL-EYFP mice and observed yellow fluorescent protein (YFP)-positive cells located in the ML and PCL of cerebellum (Fig. 1A), suggesting that parvalbumin is expressed in PCs and ML interneurons in the cerebellar cortex. Coimmunostaining for YFP and the PC marker calbindin further confirmed that YFP was expressed in PCs (Fig. 1A–D). Furthermore, our data demonstrated that PDK1 is highly expressed in postnatal PCs (Fig. 1E–J′′). To better understand the role of PDK1 in the postnatal development of PCs and motor dysfunction, we deleted Pdk1 by crossing thePdk1fl/fl mice with the PV-cre line. Immunostaining for PDK1 and GAD67, which is expressed in the cell bodies, dendrites, and axonal projections of PCs, showed a dramatic reduction in PDK1 abundance in PCs at P21, P40, and P60 in cKO-PV mice (Fig. 2A–F′′), and the disruption efficiency was confirmed by Western blot at P60 (Fig. 2G; Nam et al., 2019). Moreover, our data indicated that PDK1-positive cells were detected in the granule cell layer (GCL) and the ML of cerebellum in the Ctrl mice at P60, and in the cKO-PV mice, PDK1-positive cells were detected in the GCL of cerebellum but not in the ML of cerebellum (Fig. 2E–F′′). We found that nearly all cKO-PV mice survived to adulthood. Moreover, the cerebellar size was dramatically reduced, while the body weight, brain weight, and brain architecture of the forebrain or midbrain in adult cKO-PV mice were similar to those in the Ctrl mice (Fig. 3A–D, Table 1). By immunostaining for PC marker calbindin, we observed that the sagittal cross-sectional area of the cerebellar vermis was decreased significantly beginning at P21 after the PDK1 deletion during the postnatal developmental stage, while the density of PCs, and the overall cytoarchitecture and foliation of the cerebellum were not changed in cKO-PV mice compared with those in Ctrl mice at P14, P21, P30, and P60 (Fig. 3E–J, Table 2). Moreover, the total numbers of PCs in cerebellar lobule V and VI from the Ctrl and cKO-PV mice were counted, and no significant differences were observed at P60; however, both the density and the total number of PCs were decreased at P120 in cKO-PV mice (Fig. 3J–L, Table 2). Moreover, to assess whether other types of cells were affected, the immunostaining for NeuN and GFAP, which are expressed in the cerebellar granule neurons and Bergmann glia, respectively, were performed. As shown in Figure 4, the distribution and cytoarchitecture of cerebellar granule neurons and the morphology of Bergmann glial fibers in the cKO-PV mice were similar to those in the Ctrl mice. Together, these data indicated that the deletion of PDK1 contributed to the reduction of cerebellar size in the cKO-PV mice.
Figure 1.

The distribution of PDK1-positive cells in the cerebellum. A–D, YFP-positive cells are detected in the cerebellar PCs and interneurons in the PV-cre; ROSA26-stop-EYFP mice. E–J′′, Immunostaining for PDK1 at P14 (E–H) and P30 (I–J′′). PDK1 is expressed in PCs (F, J, J′, arrows), the cells in the GCL (F, J, J′, open arrowheads), and deep cerebellar nuclei (H, J′′, yellow arrowheads) of the cerebellum. H is the boxed region in G. J, J′ and J″′ are the boxed regions in I, which are labeled 1, 2, and 3, respectively. Scale bars: A–D, F, H, and J–J′′, 100 μm; E, G, and I, 500 μm.
Figure 2.
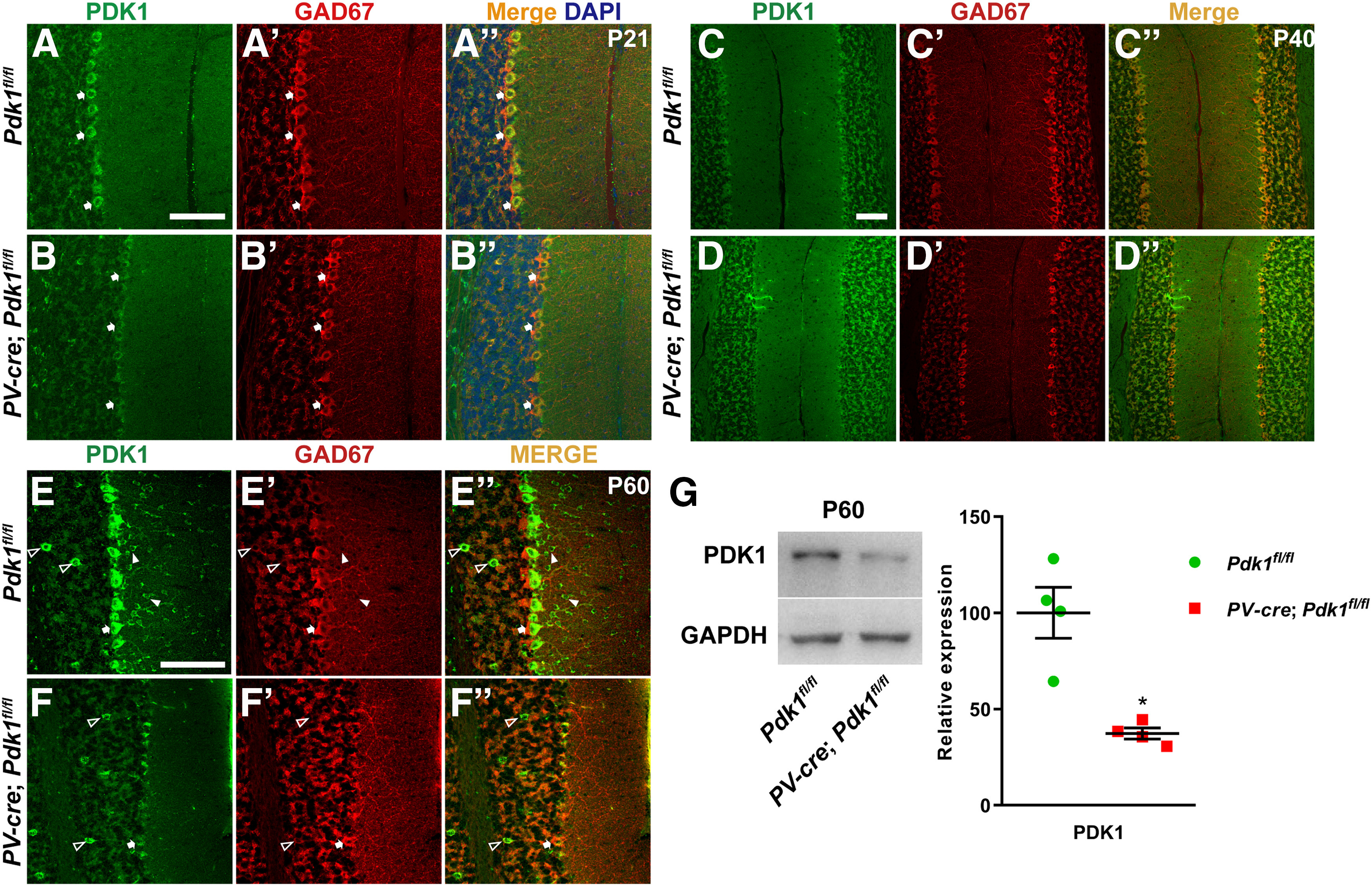
The disruption of PDK1 in PCs and PV-positive cells in the ML of the cerebellum in cKO-PV mice. A–F′′, The expression of PDK1 is disrupted in PCs and PV-positive cells in the ML of cerebellum in cKO-PV mice. Coimmunostaining for PDK1 (A, B, C, D, E, F) and GAD67 (A′, B′, C′, D′, E′, F′) at P21 (A–B′′), P40 (C–D′′), and P60 (E–F′′) is shown. In the Ctrl mice at P60, PDK1 is detected in PCs (arrows) and cells in the GCL (open arrowheads) and the ML (closed arrowheads) of cerebellum (E–E′′), while in cKO-PV mice PDK1 is detected in cells in the GCL but not in PCs and cells in the ML of cerebellum (F–F′′). G, Western blotting for PDK1 from P60 cerebellar cortical lysates confirms the dramatic reduction of PDK1 protein level (left). The levels of PDK1 proteins were shown by the relative density normalized to GAPDH (right) and were analyzed using a two-tailed Student's t test. The data are the mean ± SEM: 100.0 ± 13.23 in the Ctrl mice versus 37.29 ± 2.88 in the cKO-PV mice; n = 4 mice in each group; p = 0.0320 (*p < 0.05). Scale bars, 100 μm.
Figure 3.

Decreased cerebellar size in cKO-PV Mice. A, Gross view of brains from the Ctrl and cKO-PV mice at P60. B, Decreased cerebellar length and width in cKO-PV mice compared with those in Ctrl mice at P60. C, D, Brain (C) and body (D) weights of the 9-week-old mice were not affected by the ablation of PDK1. E–H′, Immunostaining for calbindin at P14 (E, E′), P21 (F, F′), P60 (G, G′), and P120 (H, H′). I, The sagittal cross-sectional area of the cerebellar vermis. J, The number of PCs per 100 μm length. K, L, The total number of PCs in the cerebellar lobule V (K) and lobule VI (L) at P60. The data are the mean ± SEM. Data in B–D were analyzed using a two-tailed Student's t test. A two-way ANOVA with Bonferroni's post hoc analysis was used in I–L. ***p < 0.001; **p < 0.01; *p < 0.05; ns, p ≥ 0.05; ###p = 0.0003. Scale bars: A, 5 mm; E–H′, 500 μm.
Table 1.
The Statistical data of brain morphology in the cKO-PV and Ctrl mice at P60
| Ctrl | cKO-PV | p value | |
|---|---|---|---|
| The cerebellar length (mm) | 3.22 ± 0.08 (n = 4) | 2.75 ± 0.17 (n = 3)* | 0.0425 |
| The cerebellar width (mm) | 6.70 ± 0.20 (n = 4) | 5.87 ± 0.18 (n = 3)* | 0.0324 |
| Brain weight (g) | 0.48 ± 0.01 (n = 7) | 0.47 ± 0.01 (n = 8)ns | 0.8031 |
| Body weight (g) | 29.33 ± 1.76 (n = 3) | 26.17 ± 1.68 (n = 6)ns | 0.2823 |
The data are the mean ± SEM.
*p < 0.05; ns, p ≥ 0.05.
Table 2.
The Statistical data for morphologic analysis of the cerebellum in the cKO-PV and Ctrl mice
| Age | P14 | P21 | P30 | P60 | P120 | |
|---|---|---|---|---|---|---|
| The area of cerebellar middle sagittal sections (mm2) | Ctrl | 6.86 ± 0.07(n = 4) | 8.30 ± 0.09(n = 4) | 8.30 ± 0.16(n = 4) | 8.94 ± 0.18(n = 4) | 8.73 ± 0.39(n = 3) |
| cKO-PV | 6.64 ± 0.13(n = 3)ns | 7.35 ± 0.19(n = 4)* | 6.88 ± 0.15(n = 3)*** | 6.72 ± 0.22(n = 5)*** | 4.90 ± 0.34(n = 4)*** | |
| p value | >0.9999 | 0.0140 | 0.0005 | <0.0001 | <0.0001 | |
| The number of PCs per 100 μm length | Ctrl | 4.20 ± 0.10(n = 3) | 4.05 ± 0.14(n = 4) | 4.04 ± 0.06(n = 4) | 3.89 ± 0.08(n = 4) | 3.05 ± 0.17(n = 3) |
| cKO-PV | 3.96 ± 0.05(n = 3)ns | 4.35 ± 0.33(n = 4)ns | 4.34 ± 0.02(n = 4)ns | 4.53 ± 0.11(n = 5)ns | 1.70 ± 0.27(n = 3)*** | |
| p value | >0.9999 | >0.9999 | >0.9999 | 0.1903 | 0.0005 | |
| Total number of PCs in cerebellar lobule V | Ctrl | 79.74 ± 4.41(n = 4) | 59.75 ± 3.89(n = 3) | |||
| cKO-PV | 79.80 ± 4.51(n = 4)ns | 26.30 ± 4.56(n = 3)** | ||||
| p value | >0.9999 | 0.0012 | ||||
| Total number of PCs in cerebellar lobule VI | Ctrl | 82.61 ± 3.32(n = 4) | 60.83 ± 2.00(n = 3) | |||
| cKO-PV | 79.47 ± 1.55(n = 4)ns | 21.50 ± 4.73(n = 3)*** | ||||
| p value | 0.8988 | <0.0001 | ||||
| Area of PCs cell body (μm2) | Ctrl | 314.21 ± 15.79(n = 4) | 323.85 ± 3.26(n = 4) | 325.91 ± 11.21(n = 4) | 321.06 ± 8.07(n = 4) | |
| cKO-PV | 302.29 ± 15.10(n = 3)ns | 243.71 ± 13.88(n = 4)*** | 214.40 ± 4.47(n = 4)*** | 169.46 ± 5.33(n = 5)*** | ||
| p value | >0.9999 | <0.0001 | <0.0001 | <0.0001 | ||
| The ML thickness of the cerebellar lobule V (μm) | Ctrl | 102.20 ± 9.43(n = 4) | 144.73 ± 6.50(n = 3) | 152.23 ± 1.58(n = 4) | 167.49 ± 4.30(n = 4) | 158.08 ± 5.96(n = 3) |
| cKO-PV | 97.10 ± 3.99(n = 3)ns | 121.94 ± 3.82(n = 4)* | 127.71 ± 4.27(n = 4)** | 110.24 ± 3.66(n = 4)*** | 63.59 ± 2.05(n = 3)*** | |
| p value | >0.9999 | 0.0250 | 0.0072 | <0.0001 | <0.0001 | |
| The ML thickness of the cerebellar lobule VI (μm) | Ctrl | 112.27 ± 11.13(n = 4) | 163.44 ± 6.37(n = 3) | 181.08 ± 2.63(n = 4) | 196.98 ± 5.18(n = 4) | 188.18 ± 7.71(n = 3) |
| cKO-PV | 111.21 ± 6.58(n = 3)ns | 139.97 ± 5.76(n = 4)ns | 148.58 ± 2.36(n = 4)** | 128.78 ± 8.21(n = 4)*** | 72.72 ± 1.51(n = 3)*** | |
| p value | >0.9999 | 0.1030 | 0.0053 | <0.0001 | <0.0001 |
The data are the mean ± SEM.
***p < 0.001;
**p < 0.01;
*p < 0.05; ns, p ≥ 0.05.
Figure 4.

The distribution and cytoarchitecture of cerebellar granule neurons and the morphology of Bergmann glial fibers in cKO-PV mice were comparable to those in Ctrl mice, but the ML thickness in lobule V was decreased after the ablation of PDK1. A–B′′, The distribution and cytoarchitecture of cerebellar granule neurons in cKO-PV mice were comparable to those in Ctrl mice. Immunostaining for NeuN in Ctrl mice (A–A′′) and cKO-PV mice (B–B′′) at P21. C–D′′, The morphology of Bergmann glial fibers in cKO-PV mice was comparable to those in Ctrl mice. Immunostaining for GFAP in Ctrl mice (C–C′′) and cKO-PV mice (D–D′′) at P14. Scale bars: A, B, 1000 μm; A′, A′′, B′, B′′, C–D′′, 100 μm.
The cell body size and the morphologic complexity of PCs were decreased in cKO-PV mice
To further study the morphology of PCs in the absence of PDK1, the cell body size of PCs was measured at P14, P21, P30, and P60. Compared with the Ctrl mice, the Purkinje cell body size was time-dependently reduced, and the significant reduction was detected since P21 in cKO-PV mice (Fig. 5A–F, Table 2). As the cell body size of PCs was decreased in the cKO-PV mice, and the PC dendrites are located in the ML of the cerebellum, we next investigated whether the decreased cell body area after PDK1 ablation affected the cerebellar ML thickness at P14, P21, P30, P60, and P120. We observed that the ML thickness of cerebellar lobules V and VI was significantly increased at P21 in Ctrl mice compared with that at P14, while not in cKO-PV mice. Moreover, the ML thickness of cerebellar lobules V was markedly decreased beginning at P21, and the significant reduction of that in lobule VI was detected beginning at P30 in the cKO-PV mice compared with that in the Ctrl mice (Fig. 5G–J′, Table 2). To confirm the presence of dysmorphic PC dendrites in the absence of PDK1, we used Golgi staining to examine the morphology of PCs from P30 mice and observed a severe decrease in the complexity of the dendritic arbor in cKO-PV mice compared with that in Ctrl mice (Fig. 5K–L). Thus, our data demonstrate that PDK1 is required for postnatal development and maintenance of PC dendrites and suggest that the decreased cerebellum is due to the reduced ML thickness.
Figure 5.
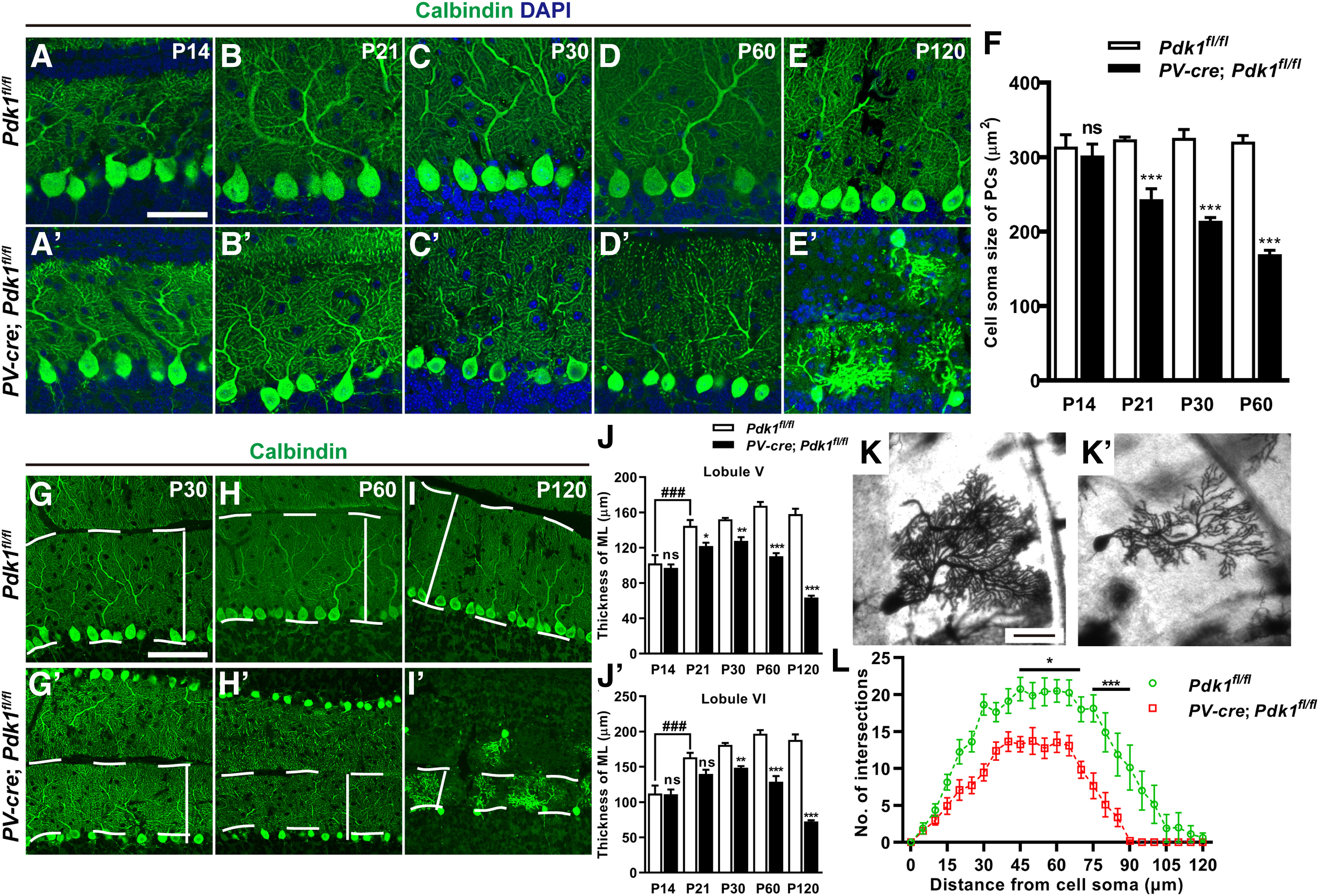
Decrease of PC body size, ML thickness, and morphologic complexity of dendrites in the cKO-PV Mice. A–E′, Immunostaining for calbindin at P14 (A, A′), P21 (B, B′), P30 (C, C′), P60 (D, D′), and P120 (E, E′) indicated the decreased cell body size of PCs in cKO-PV mice. F, Area of PC bodies. G–I′, The ML was immunostained for calbindin at P30 (G, G′), P60 (H, H′), and P120 (I, I′). The straight line between the dotted lines illustrates the thickness of the ML. J, J′, The ML thickness of the cerebellar lobule V (J) and lobule VI (J′). K, K′, Golgi staining of PCs in the Ctrl mice (K) and cKO-PV mice (K′) at P30. L, Sholl analysis: the number of intersections of the dendrite at different distances from the cell body of PCs in the Ctrl and cKO-PV mice. A two-way ANOVA with respect to group effect; intersection number: F(1,2142) = 988.3, p < 0.001; Bonferroni's post hoc comparison: *p < 0.05 between 45 and 65 mm from the cell body; ***p < 0.001 between 70 and 95 mm from the cell body. The data are the mean ± SEM. Statistical analysis used two-way ANOVA with Bonferroni's post hoc analysis. ***p < 0.001; **p < 0.01; *p < 0.05; ns, p ≥ 0.05; ###p < 0.001 (P14 vs P21: p < 0.0001 in lobule V; p = 0.0001 in lobule VI). Scale bars: A–E′, K, K′, 50 μm; G–I′, 100 μm.
The cKO-PV mice displayed motor defects
We observed that the cKO-PV mice staggered and demonstrated ataxia-like behavior, indicating that PDK1 may be attributed to the proper motor control (Movies 1, 2). To quantitatively analyze their gait defects, we performed a footprint assay at 9–10 weeks after the birth of mice (Fig. 6A,A′). Moreover, a statistic analysis was performed by using the parameters illustrated in Figure 6B. Our data demonstrated that the stride distance was similar in between the cKO-PV and the Ctrl mice; however, the stance lengths, the sway distance, and the overlap distance in the cKO-PV mice were significantly greater than those in the Ctrl mice (Fig. 6C–F, Table 3). These data indicated that PDK1 ablation resulted in wider gait and tottering steps in adult mice. We then performed an elevated beam-walk assay to examine whether the motor balance was affected after PDK1 deletion. A schematic diagram of an elevated beam-walk assay is shown in Figure 6G. Most of the cKO-PV mice fell from the square beam (side length, 28 mm) and could not finish the test at the age of 8 weeks (Fig. 6H). Furthermore, the rotarod test was used to investigate the effect the PDK1 ablation on motor coordination. However, at the age of 8 weeks, no cKO-PV mice were able to stand on the rotating rod and to continue the test. These results indicated that the ablation of PDK1 leads to motor deficits at adult stage in cKO-PV mice.
Figure 6.

Motor Defects in cKO-PV mice. A, A′, Gait of 9- to 10-week-old mice tested with a footprint assay was shown in red (forepaws; A) and blue (hindpaws; A′). B, Diagram of parameters measured in footprint analysis. C, The cKO-PV mice show a similar stride length to that of the Ctrl mice. D, The overlap length. E–F, Longer sway lengths (E) and stance lengths (F) of forelimbs and hindlimbs in the cKO-PV mice. G, Schematic illustration of the elevated beam-walk assay for testing motor coordination. H, The latency to cross a 28-mm-wide square beam at 8 weeks after birth. The Ctrl mice at 9.46 ± 2.14 s (n = 8 mice) versus the cKO-PV mice at 49.13 ± 10.87 s (n = 4 mice), p = 0.0005. I, The average time 4-week-old mice took to traverse the round beams. J, The average number of missteps for hindlimbs of 4-week-old mice to traverse the round beams. K, The latency to fall down from the rotarods in the cKO-PV mice (n = 8) was significantly reduced compared with that in the Ctrl mice (n = 9) at 4 weeks after birth. A two-way ANOVA; Bonferroni's post hoc analysis was used: p = 0.0055 for 10 rpm test; p < 0.0001 for 15 rpm test; p < 0.0001 for 20 rpm test; p < 0.0001 for 25 rpm test; p = 0.0154 for 30 rpm test; and p = 0.3137 for 33 rpm test. L–O, Gait of 4-week-old mice tested by footprint assay. Bar graphs show that there were no differences in the lengths of stride (L), overlap (M), stance (N), and sway (O) between the Ctrl and cKO-PV mice at 4 weeks. The data are the mean ± SEM. Data in C–F, H, and L–O were analyzed using a two-tailed Student's t test and a two-way ANOVA; Bonferroni's post hoc analysis was used in I–K. ***p < 0.001; **p < 0.01; *p < 0.05; ns, p ≥ 0.05. Scale bar, 20 mm.
Table 3.
The Statistical data for footprint tests in the cKO-PV and Ctrl mice at 4 and 8–9 weeks after birth
| Age | 4 weeks old | 8∼9 weeks old | |
|---|---|---|---|
| The stride distance | Ctrl | 63.17 ± 2.21(n = 7) | 71.82 ± 1.75(n = 14) |
| cKO-PV | 58.61 ± 4.41 (n = 6)ns | 73.23 ± 1.31 (n = 14)ns | |
| p value | 0.3543 | 0.5239 | |
| The overlap distance (mm) | Ctrl | 7.34 ± 0.90(n = 7) | 7.75 ± 0.45(n = 14) |
| cKO-PV | 9.44 ± 1.360 (n = 6)ns | 10.85 ± 0.45 (n = 14)*** | |
| p value | 0.2126 | <0.0001 | |
| The sway distance offorelimbs (mm) | Ctrl | 12.78 ± 0.58(n = 7) | 14.93 ± 0.60(n = 14) |
| cKO-PV | 14.87 ± 1.66 (n = 5)ns | 18.39 ± 0.82 (n = 14)** | |
| p value | 0.2064 | 0.0020 | |
| The sway distance ofhindlimbs (mm) | Ctrl | 24.88 ± 1.05(n = 7) | 25.49 ± 0.69(n = 14) |
| cKO-PV | 26.98 ± 1.20 (n = 6)ns | 33.13 ± 1.17 (n = 14)*** | |
| p value | 0.2145 | <0.0001 | |
| The stance lengths offorelimbs (mm) | Ctrl | 33.70 ± 1.00(n = 7) | 39.16 ± 0.83(n = 14) |
| cKO-PV | 34.43 ± 2.46 (n = 5)ns | 41.64 ± 0.56 (n = 14)* | |
| p value | 0.7622 | 0.0205 | |
| The stance lengths ofhindlimbs (mm) | Ctrl | 41.55 ± 1.32(n = 7) | 44.38 ± 0.93(n = 14) |
| cKO-PV | 42.74 ± 1.02 (n = 6)ns | 49.85 ± 0.84 (n = 14)*** | |
| p value | 0.5013 | 0.0002 |
The data are the mean ± SEM.
***p < 0.001;
**p < 0.01;
*p < 0.05; ns, p ≥ 0.05.
Pdk1fl/fl mouse at P36.
PV-cre; Pdk1fl/fl mouse at P36.
To test whether the motor deficits observed in the adult cKO-PV mice existed in the developmental stage, the same behavior tests were performed at 4 weeks after birth. The cKO-PV mice took a significantly longer time and showed more hindlimb missteps in crossing beams with diameters of 10 and 16 mm, while not on a test using a 30 mm round beam than did the Ctrl mice (Fig. 6I,J, Table 4). In rotarod assay, the cKO-PV mice fell off the rotarod in less time than the Ctrl mice (Fig. 6K). Furthermore, we analyzed the gait and observed no differences in the stance lengths, the sway distance, the overlap distance, or the stride distance between the cKO-PV mice and the Ctrl mice at 4 weeks after birth (Fig. 6L–O, Table 3). These results indicated that although the 4-week-old cKO-PV mice showed no significant gait abnormality, they displayed the altered motor coordination and motor balance. Together, our results suggest that the ablation of PDK1 in PCs and cerebellar ML interneurons leads to progressive motor deficits.
Table 4.
The statistical data for elevated beam-walk in the 4-week-old cKO-PV and Ctrl mice
| The diameterof the round beam | 30 mm | 16 mm | 10 mm | |
|---|---|---|---|---|
| Time to traversebeam (s) | Ctrl | 4.75 ± 0.65(n = 9) | 4.81 ± 0.92(n = 9) | 8.76 ± 1.97(n = 9) |
| cKO-PV | 6.97 ± 0.74 (n = 8)ns | 9.68 ± 1.59 (n = 8)* | 15.98 ± 1.19 (n = 8)*** | |
| p value | 0.6812 | 0.0293 | 0.0007 | |
| Number offootslips | Ctrl | 0.06 ± 0.06(n = 9) | 0.06 ± 0.06(n = 9) | 0.11 ± 0.11(n = 9) |
| cKO-PV | 0.56 ± 0.32 (n = 8)ns | 2.06 ± 0.50 (n = 8)*** | 1.44 ± 0.50 (n = 8)* | |
| p value | 0.7388 | <0.0001 | 0.0108 |
The data are the mean ± SEM.
***p < 0.001;
*p < 0.05; ns, p ≥ 0.05.
PC-specific ablation of PDK1 resulted in decreased size of PCs and motor defects
Because the PV-cre mice express Cre recombinase in all parvalbumin-positive interneurons in the nervous system, including PCs, basket cells (BCs) in the ML of cerebellum and a part of cerebral cortical GABAergic interneurons, we could not exclude the possibility that the disrupted morphology of PCs and motor dysfunction in the cKO-PV mice were due to a deficient interaction between PCs and cerebellar ML interneurons or to the abnormal inhibitory signal from the cerebral cortical PV-positive GABAergic interneurons. To determine whether the effects of PDK1 on PCs were cell autonomous or non-cell autonomous, Purkinje-cell protein 2-cre (Pcp2-cre) mice were used. Cre recombinase-dependent DNA recombination is evident in the PCs by P6 and fully established by 2–3 weeks in Pcp2-cre mice (Barski et al., 2000). As for the cKO-PV mice, the expression of PDK1 was efficiently disrupted and the sagittal cross-sectional area of the cerebellar vermis was significantly decreased after PC-specific deletion of Pdk1 in cKO-Pcp2 mice at P30 after birth (Figs. 7A–B′′, 8A–E, Table 5). Further analysis showed that the cell body sizes of PCs in lobules II/III, V/VI, and IX of the cerebellum was also decreased in the cKO-Pcp2 mice at P30 (Fig. 8 A′–B′′′,F, Table 5). Although comparable PC density was detected in the Ctrl mice and the cKO-Pcp2 mice, the ML thicknesses were significantly reduced after PDK1 ablation at P30 (Fig. 8G,H, Table 5).
Figure 7.

PDK1 disruption in PCs of cKO-Pcp2 mice at P30. A–B′′, Coimmunostaining for PDK1 (A, B, A′′, B′′) and GAD67 (A′, B′, A′′, B′′) in cKO-Pcp2 mice (B–B′′) and Ctrl mice (A–A′′) at P30.
Figure 8.
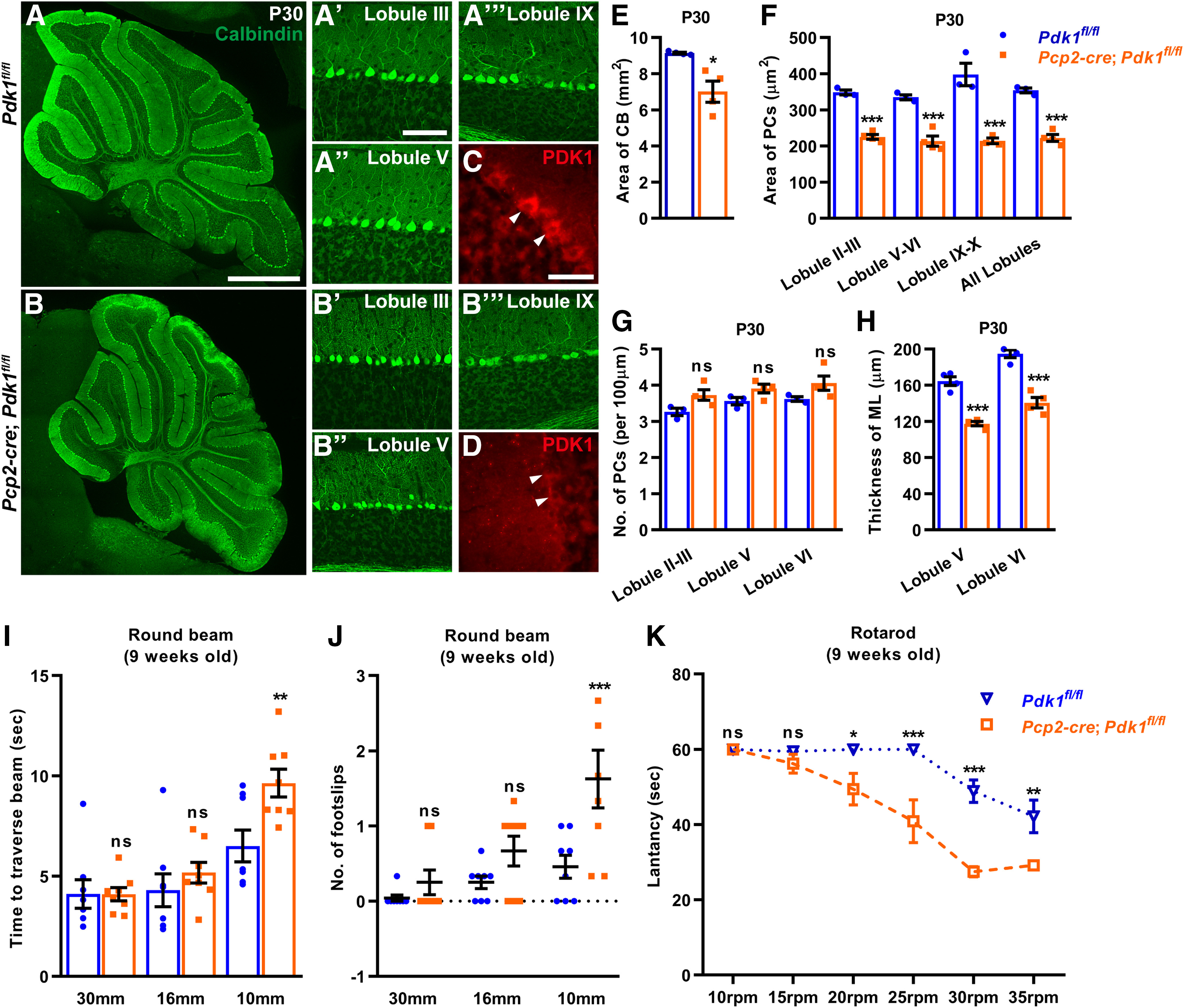
Purkinje cell-specific ablation of PDK1 leads to decreased cell body size, ML thickness, and motor defects in mice. A–B′′′, Immunostaining for calbindin at P30 in Ctrl and cKO-Pcp2 mice. Decreased cell body size of PDK1-deficient PCs in the cerebellar lobule II–III (A′, B′), lobule V–VI (A′′, B′′), and lobule IX–X (A′′′, B′′′). C, D, Immunostaining for PDK1 at P30. E, Decreased cell body area of PDK1-deficient PCs at P30. F, The number of PCs per 100 μm at P30. G, Decreased cerebellar size in cKO-Pcp2 mice at P30. H, The ML thickness of lobules V and VI at P30. I, Latency to traverse the round beams. J, The number of missteps. K, Rotarod assay showed a defect motor coordination in cKO-Pcp2 mice at 9 weeks old. The number of mice used in this experiment are n = 9 mice for the Ctrl and n = 8 for the cKO-Pcp2 mice. A two-way ANOVA; Bonferroni's post hoc analysis was used: p > 0.9999 for 10 rpm test; p > 0.9999 for 15 rpm test; p = 0.0421 for 20 rpm test; p < 0.0001 for 25 and 30 rpm test; and p = 0.0065 for 35 rpm test. The data are the mean ± SEM. Data in G and H were analyzed using a two-tailed Student's t test and a two-way ANOVA; Bonferroni's post hoc analysis was used to analyze data in E, F, and I–K. ***p < 0.001; **p < 0.01; *p < 0.05; ns, p ≥ 0.05. Scale bars: A, B, 1 mm; A′–A′′′ and B′–B′′′, 100 μm; C, D, 50 μm.
Table 5.
The Statistical data for morphologic analysis of the cerebellum in the cKO-Pcp2 and Ctrl mice at P30
| Ctrl | cKO-Pcp2 | p value | |
|---|---|---|---|
| The area of cerebellar middle sagittal sections (mm2) | 9.14 ± 0.05 (n = 4) | 7.01 ± 0.58 (n = 4)* | 0.0104 |
| Area of PCs cell body in the lobules II–III (μm2) | 348.31 ± 6.53 (n = 3) | 225.19 ± 6.74 (n = 4)*** | <0.0001 |
| Area of PCs cell body in the lobules V–VI (μm2) | 334.75 ± 6.96 (n = 3) | 213.67 ± 13.98 (n = 4)*** | <0.0001 |
| Area of PCs cell body in the lobules IX–X (μm2) | 398.02 ± 30.87 (n = 3) | 214.25 ± 7.88 (n = 3)*** | <0.0001 |
| Average area of PCs cell body in the CB (μm2) | 353.87 ± 6.81 (n = 3) | 222.33 ± 9.34 (n = 4)*** | <0.0001 |
| The number of PCs per 100 μm length in the lobules II–III | 3.26 ± 0.10 (n = 3) | 3.73 ± 0.14 (n = 4)ns | 0.1091 |
| The number of PCs per 100 μm length in the lobule V | 3.56 ± 0.10 (n = 3) | 3.91 ± 0.12 (n = 4)ns | 0.3177 |
| The number of PCs per 100 μm length in the lobule VI | 3.62 ± 0.06 (n = 3) | 4.06 ± 0.20 (n = 4)ns | 0.1405 |
| The ML thickness of the cerebellar lobule V (μm) | 164.56 ± 4.86 (n = 4) | 117.45 ± 2.37 (n = 4)*** | <0.0001 |
| The ML thickness of the cerebellar lobule VI (μm) | 194.81 ± 4.16 (n = 4) | 140.65 ± 5.84 (n = 4)*** | <0.0001 |
The data are the mean ± SEM.
***p < 0.001;
*p < 0.05; ns, p ≥ 0.05.
To further study whether specific deletion of PDK1 in PCs resulted in the same motor deficits as that in cKO-PV mice, the elevated beam-walk and rotarod assays were used. Consistent with the above results, the cKO-Pcp2 mice displayed the longer latency to traverse the 10 mm beams, a greater number of missteps, and the shorter latency to fall off the rotarod than the Ctrl mice did at 9 weeks after birth (Fig. 8I–K, Table 6). However, the latency and number of missteps in cKO-Pcp2 mice were similar to those in Ctrl mice, when beams with diameters of 16 or 30 mm were used (Fig. 8I,J, Table 6), suggesting that the motor balance deficits are only detectable in the harder test in cKO-Pcp2 mice at the age of 9 weeks. Together, the results suggest that specific ablation of PDK1 in postnatal PCs leads to the decreased body size, the reduced cerebellar size, and the motor deficits, indicating that the role of PDK1 in postnatal PC development is cell autonomous.
Table 6.
the Statistical data for elevated beam walk in the 9-week-old cKO-Pcp2 and Ctrl mice
| The diameter of the round beam | 30 mm | 16 mm | 10 mm | |
|---|---|---|---|---|
| Time to traverse beam (s) | Ctrl | 4.11 ± 0.71 (n = 8) | 4.29 ± 0.81 (n = 8) | 6.50 ± 0.79 (n = 8) |
| cKO-Pcp2 | 4.10 ± 0.33(n = 8)ns | 5.18 ± 0.51(n = 8)ns | 9.64 ± 0.69(n = 8)** | |
| p value | >0.9999 | >0.9999 | 0.0053 | |
| Number of footslips | Ctrl | 0.04 ± 0.04 (n = 8) | 0.25 ± 0.08 (n = 8) | 0.46 ± 0.15 (n = 8) |
| cKO-Pcp2 | 0.25 ± 0.16(n = 8)ns | 0.67 ± 0.20(n = 8)ns | 1.62 ± 0.3(n = 8)*** | |
| p value | >0.9999 | 0.4624 | 0.0006 |
The data are the mean ± SEM.
***p < 0.001;
**p < 0.01; ns, p ≥ 0.05.
Defects in spontaneous firing activity of PDK1-KO PCs
To test whether the electrophysiological properties of PCs were affected by the absence of PDK1, we performed whole-cell patch-clamp recordings on PCs. To exclude the influence of the cerebellar ML on the PCs, the cerebellar slices were separated from cKO-Pcp2 mice at P21. From recordings of spontaneous PC firings, we observed that both of the PCs from the Ctrl and cKO-Pcp2 mice exhibited regular firing (Fig. 9A). We quantified the PC spontaneous firing patterns by testing the coefficient of variation (CV) of interspike intervals (ISIs) and observed no difference between PDK1-deficient and control PCs; however, the spontaneous firing frequency was remarkably lower in PDK1-KO PCs than that in control PCs (Fig. 9B–D). Furthermore, the membrane capacitance (Cm) of PDK1-KO PCs was decreased compared with that of the control cells (Fig. 9E), which might be due to the decreased cell body size of PCs after PDK1 ablation. Moreover, spontaneous EPSCs (sEPSCs) were recorded and analyzed to test the synaptic transmission to PCs with PDK1 knocked out (Fig. 9F). The amplitude and frequency of sEPSCs recorded in PDK1-KO PCs were similar to those in control cells (Fig. 9G,H). Together, the ablation of PDK1 affected the electrophysiological properties of PCs at P21.
Figure 9.
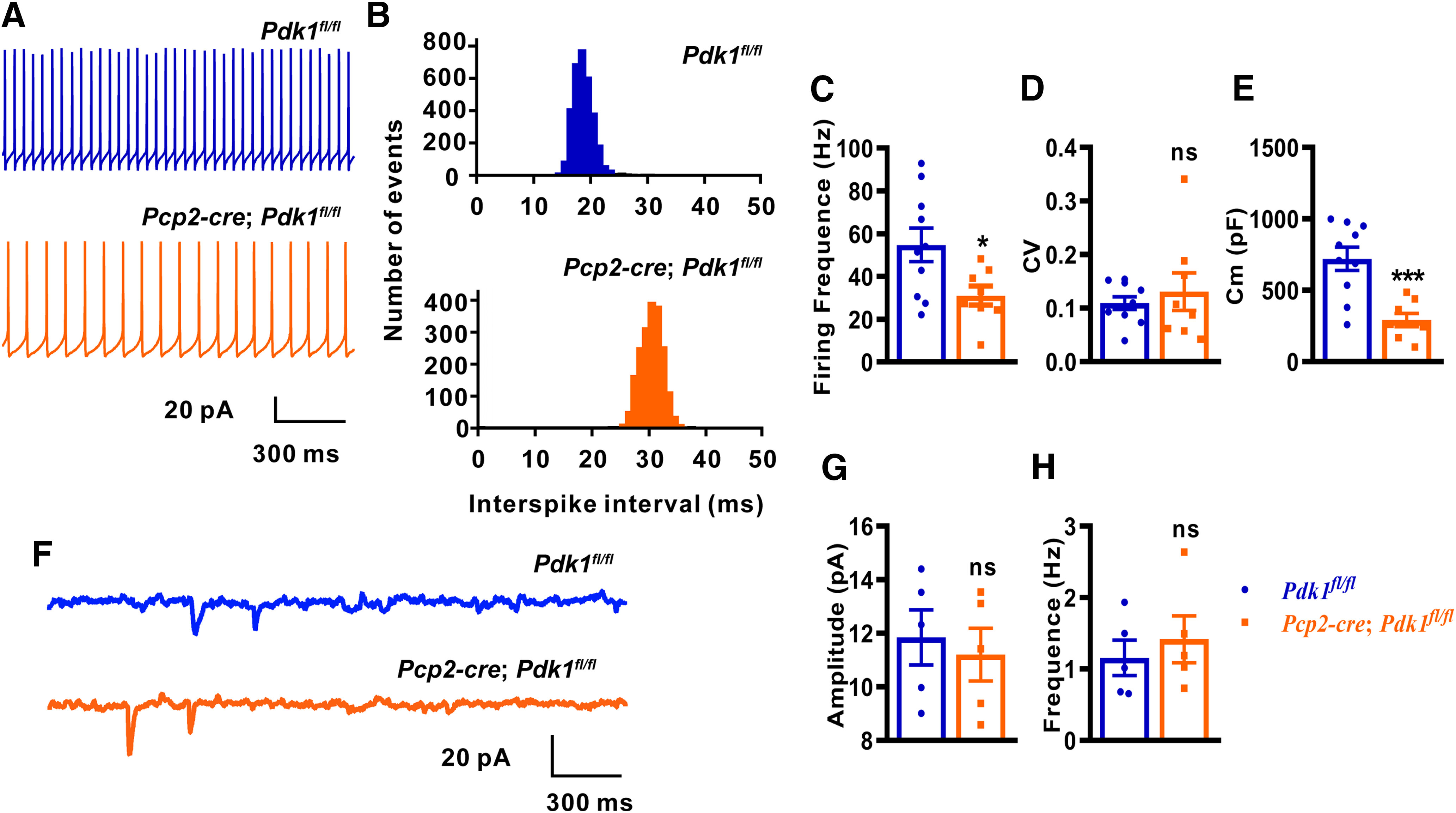
Defects in spontaneous firing activity of PDK1-KO PCs. The electrophysiological characteristic of PCs was evaluated at P21. A, Representative traces of 1.5 s duration from the control PCs (top) and the PDK1-KOs PCs (bottom). B, The ISI histogram of the PCs. C, The spontaneous firing frequencies of PCs. Control PCs, 54.78 ± 7.82 Hz (n = 10 PCs from 6 mice); versus PDK1-KO PCs, 31.07 ± 4.50 Hz (n = 8 PCs from 6 mice), p = 0.0259. D, CVs of spontaneous firing of PCs. Control PCs, 0.11 ± 0.01 (n = 10 PCs from 6 mice); versus PDK1-KO PCs, 0.13 ± 0.04 (n = 8 PCs from 6 mice), p = 0.5343. E, The Cm of PCs. Control PCs, 720.10 ± 81.52 pF (n = 10 PCs from 6 mice); versus PDK1-KO PCs, 291.60 ± 46.35 pF (n = 8 PCs from 6 mice), p = 0.0006. F–H, The spontaneous EPSC of PDK1-KO PCs showed no difference compared with control PCs. F, Representative traces 1.5 s sEPSC duration from the control PCs (top) and the PDK1-KO PCs (bottom). G, The amplitudes of PCs sEPSC. Control PCs, 11.85 ± 1.03 pA (n = 5 PCs from 3 mice); versus PDK1-KO PCs, 11.21 ± 0.98 pA (n = 5 PCs from 3 mice), p = 0.6649. H, The frequencies of sEPSC. Control PCs, 1.16 ± 0.25 Hz (n = 5 PCs from 3 mice); versus PDK1-KO PCs, 1.42 ± 0.33 Hz (n = 5 PCs from 3 mice), p = 0.5434. The data are the mean ± SEM. Data were analyzed using a two-tailed Student's t test. ***p < 0.001; *p < 0.05; ns, p ≥ 0.05.
The reduction of PC body size was partly rescued by treatment of 3BDO in Pdk1 cKO-PV mice
Previous studies have shown that downregulated catalytic activity of PDK1 causes decreased rpS6 phosphorylation (Zurashvili et al., 2013). Immunostaining for pS6 was performed to measure the activation level of mTORC1. We performed coimmunostaining for pS6 and GAD67, which is expressed in the cell bodies, dendrites, and axonal projections of PCs, and observed that rpS6 phosphorylation was significantly lower in PCs at P14 in the cKO-PV mice than that in the Ctrl mice (Fig. 10A–B′′). Furthermore, the staining for pS6 in PCs was completely disrupted at P21 after PDK1 deletion (Fig. 10C–D′′). These results indicated that in PCs, PDK1 ablation decreased rpS6 phosphorylation.
Figure 10.
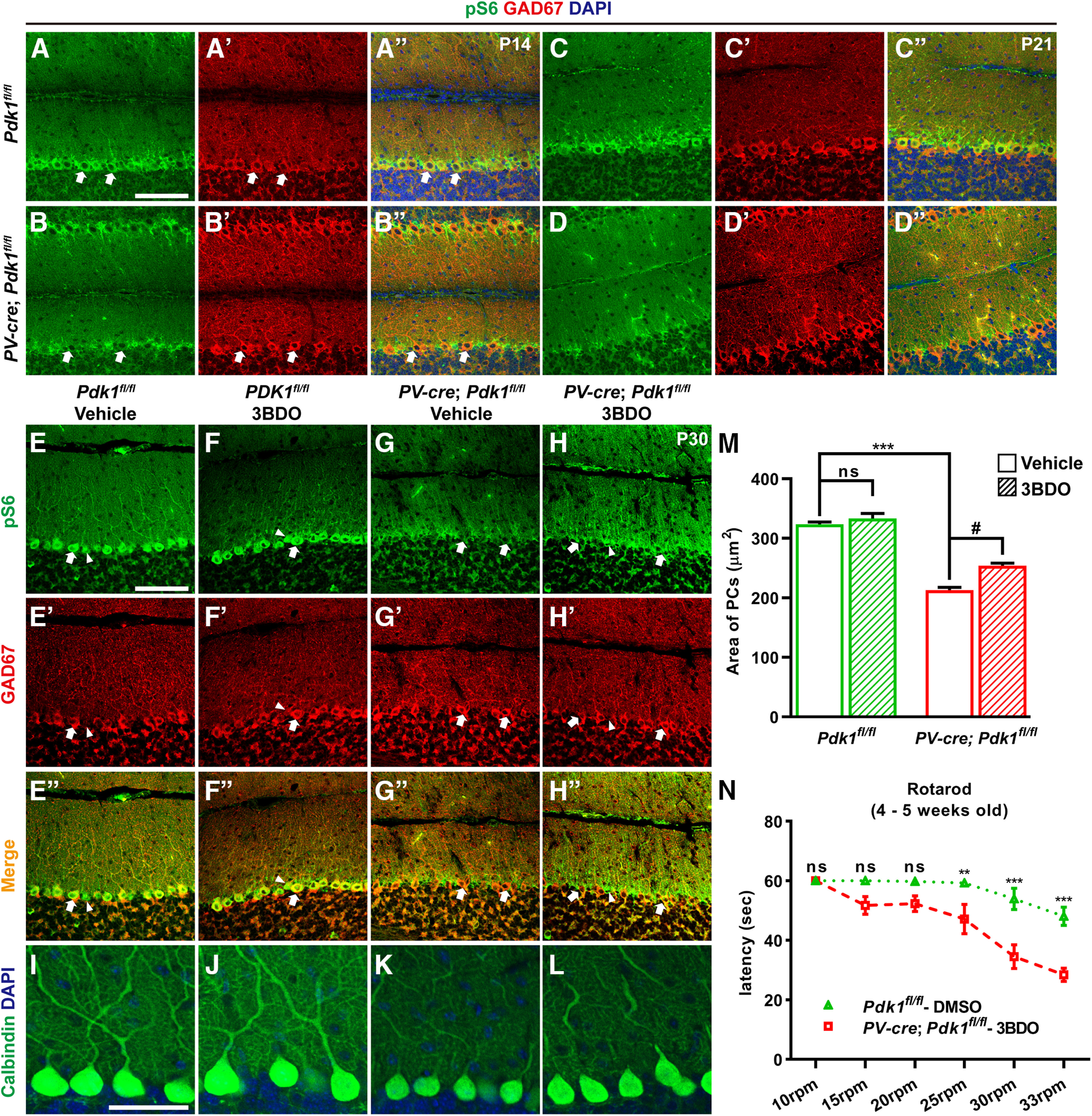
The decreased phosphorylation level of ribosomal protein S6 and the decreased body size of PCs were rescued by 3BDO in the cKO-PV mice. A–D′′, Coimmunostaining for pS6 (A, B, C, D) and GAD67 (A′, B′, C′, D′) at P14 (A–B′′) and P21 (C–D′′). E–H, Coimmunostaining for pS6 (E, F, G, H) and GAD67 (E′, F′, G′, H′) at P30. Arrows indicate the PCs, and arrowheads indicate the Bergmann glia in the cerebellum. I–L, Immunostaining for calbindin at P30. M, Area of PC bodies in mice at P30. The data are the mean ± SEM: 323.70 ± 3.57 μm2 for vehicle-treated Ctrl mice (n = 149 PCs from 3 mice); 333.40 ± 8.24 μm2 for 3BDO-treated Ctrl mice (n = 159 PCs from 3 mice); 213.04 ± 4.68 μm2 for vehicle-treated cKO-PV mice (n = 85 PCs from 2 mice); and 254.00 ± 4.45 μm2 for 3BDO-treated cKO-PV mice (n = 144 PCs from 3 mice). Vehicle/Ctrl mice versus 3BDO/Ctrl mice, no significance; vehicle/Ctrl mice versus vehicle/cKO-PV mice, ***p < 0.0001; vehicle/cKO-PV mice versus 3BDO/cKO-PV mice, #p = 0.0137. N, In the low rotational speed (15 and 20 rpm), 3BDO treatment rescued the motor coordination in the cKO-PV mice. p Values of 3BDO treatment of cKO-PV mice compared with vehicle treatment of Ctrl mice: 10 rpm, p > 0.999; 15 rpm, p = 0.1228; 20 rpm, p = 0.2065; 25 rpm, p = 0.0047; 30 and 33 rpm, p < 0.0001. The number of the mice used in the experiments were n = 12 for the vehicle-treated Ctrl mice group and n = 7 for the 3BDO-treated cKO-PV mice group. The data are the mean ± SEM. A two-way ANOVA; Bonferroni's post hoc analysis was used to analyze data in M and N. ***p < 0.001, **p < 0.01, ns, p ≥ 0.05. Scale bars: A–H, 100 μm; I–L, 50 μm.
To further test whether decreased rpS6 phosphorylation in cKO-PV mice results in decreased cell body size of PCs, 3BDO, an activator of mTORC1, was used (Peng et al., 2014). The cKO-PV and Ctrl mice were treated daily with either 3BDO (80 mg/kg) or vehicle from P14 to P30, and the brains were harvested at P30. As shown in Figure 10E–H′′, the reduced immunostaining intensity of pS6 in the PCs and Bergmann glia of cKO-PV mice was significantly increased by 3BDO treatments at P30. Moreover, the reduced cell body size of PCs in the cKO-PV mice was partly rescued after 3BDO treatment, although the PC body size was not as large as that in the Ctrl mice, and 3BDO treatment did not affect the cell body size of control PCs (Fig. 10I–M). And this 3BDO treatment also partly rescued some behavioral deficits in cKO-PV mice. The latency to fall off the rotarod in the 3BDO-treated cKO-PV mice was similar to those in vehicle-treated Ctrl mice in the lower rotational speed range (≤20 rpm), while not in the high rotational speed range (≥25 rpm; Fig. 10N). These results suggest that when PDK1 is disrupted, decreased cerebellar mTORC1 activity contributes, at least partially, to the reduction of cell body size of PCs and the motor coordination defects.
The decreased dendritic complexity was partly rescued by PKCγ overexpression in Pdk1 cKO-Pcp2 mice
Moreover, PKCγ, a member of the PKC family of proteins that is highly expressed in PCs, has been demonstrated to be involved in the impaired development of dendritic trees in cultured PKCγ-deficient PCs (Schrenk et al., 2002; Takahashi et al., 2017; Hirai, 2018). To investigate whether the expression or the phosphorylation level of PKCγ was affected and contributed to the dendritic deficits after PDK1 ablation in PCs, we performed immunostaining for he phosphorylated form of PKCγ (pPKCγ). Although the immunostaining intensity for pPKCγ in PCs from cKO-PV mice was similar to that from Ctrl mice at P21, it was significantly decreased at P30 and P40 (Fig. 11A–C′). The coimmunostaining for pPKCγ and GAD67 further showed that the staining intensity for pPKCγ was completely disrupted in PCs after PDK1 deletion at P60 (Fig. 11D–E′′). The Western blot with the anti-total PKCγ protein showed that the expression level of total PKCγ protein from cerebellar lysate in cKO-PV mice was lower than that in Ctrl mice at P21 (Fig. 11F,F′). To test whether the reduction of PKCγ protein in PCs is specific, we performed Western blotting with anti-calbindin and found that the expression level of calbindin protein in the cerebellum is comparable in Ctrl and cKO-PV mice (Fig. 11G,G′). Together, our data demonstrated that PDK1 ablation in PCs leads to a reduction in the expression of PKCγ.
Figure 11.
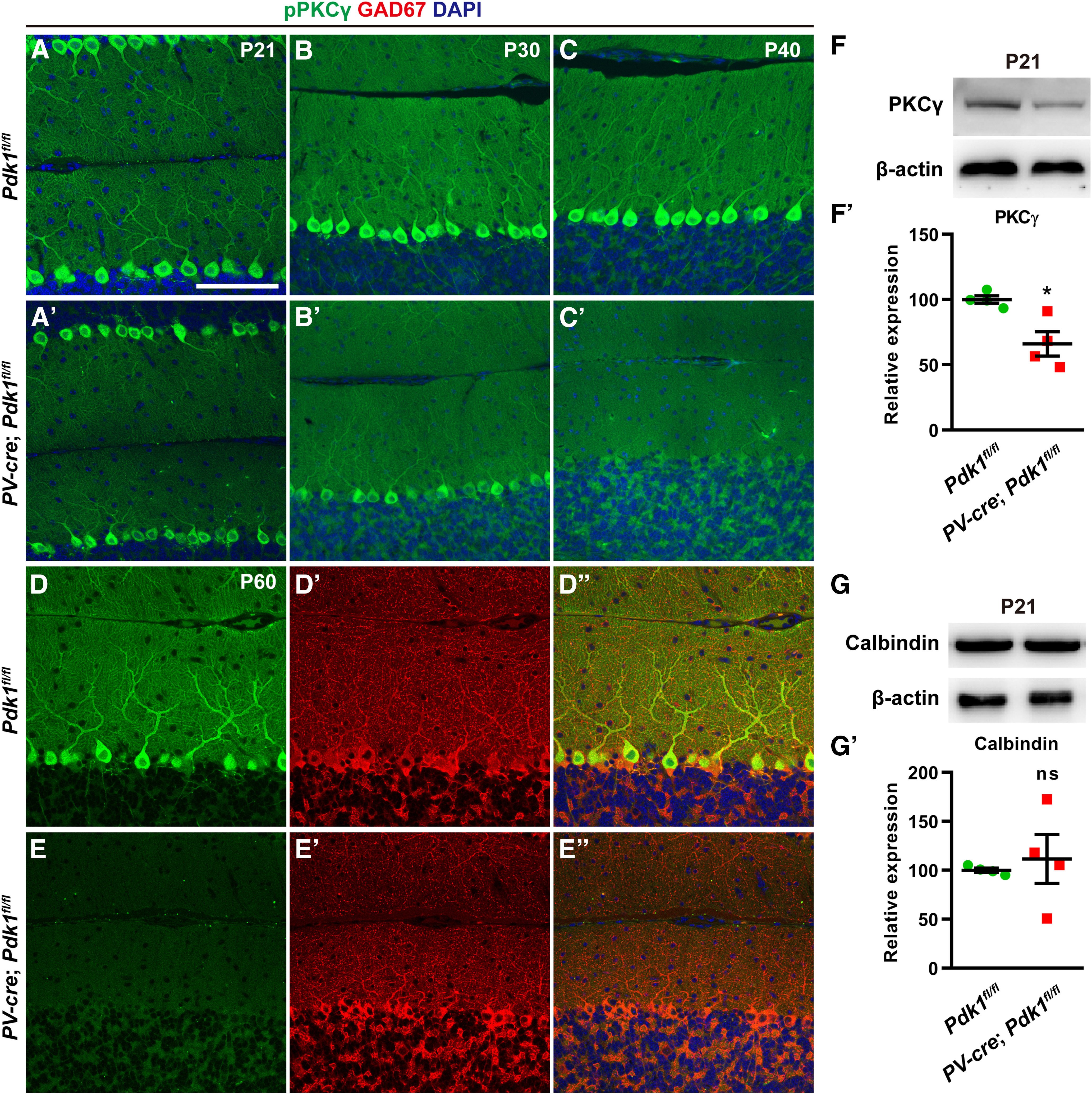
Decreased expression levels of PKCγ in cKO-PV mice and rescue of deficient dendritic complexity by overexpressing PKCγ. A–C′, Immunostaining for pPKCγ at P21 (A, A′), at P30 (B, B′), and at P40 (C, C′) shows progressive loss of staining intensity in cKO-PV mice. D–E′′, pPKCγ was totally disrupted in the PCs after the ablation of PDK1 at P60. pPKCγ, green; GAD67, red. Scale bars, 100 μm. F, Representative blots with the antibody of PKCγ. F′, Quantification of PKCγ levels in the cerebellum lysates from Ctrl and cKO-PV mice at P21. Amount of PKCγ was normalized to β-actin. The value obtained in Ctrl mice was set to 100. The data are the mean ± SEM: Ctrl: 100.0 ± 2.9, n = 4 mice; cKO-PV: 66.0 ± 9.3, n = 4 mice; p = 0.0131. G, Representative blots with the antibody of calbindin. G′, Quantification of calbindin levels. Ctrl: 100.0 ± 2.1, n = 4 mice; cKO-PV: 111.6 ± 25.0, n = 4 mice; p = 0.6609. Data in F′ and G′ were analyzed using a two-tailed Student's t test. *p < 0.05, ns, p ≥ 0.05.
To test the function of PKCγ in the dendritic defects of PDK1-KO PCs, we performed rescue experiments by injecting rAAV into the cerebellum of cKO-Pcp2 mice at P0 or P1 (Gibson and Ma, 2011). In this study, the Cre-positive PDK1-KO PCs in the cKO-Pcp2 mice infected by the rAAV-PKCγ-EGFP constructs were able to overexpress PKCγ and EGFP or express EGFP only. Meanwhile, the PCs in the Ctrl and the cKO-Pcp2 mice infected by mCherry-expressing rAAV construct were labeled by mCherry. Then we analyzed the morphology of the PC dendrites at P30. Our data showed that the complexity of dendritic arbors was significantly decreased in EGFP-positive PDK1-KO PCs compared with that in mCherry-positive control PCs (Fig. 12A–D,I). After the treatment of rAAV-PKCγ-EGFP, the complexity of dendritic arbors of PDK1-KO PCs was partly rescued (Fig. 12C–I). Furthermore, we observed that the PDK1 deletion in PCs decreased the cumulative length of the dendritic arbor and the dendritic tree area of PCs, which was also partly rescued by infection with rAAV-PKCγ-EGFP (Fig. 12A–H,J,K). Our data demonstrated that the abnormal dendritic arbors observed in PCs after PDK1 ablation were partly rescued by PKCγ overexpression, suggesting an important role for PKCγ in PC dendritic development.
Discussion
In this study, we provided novel evidences for the role of PDK1 in the postnatal development of PCs and motor control. The deletion of PDK1 in PCs resulted in a decrease in cell body size, reduced dendritic complexity, abnormal spontaneous firing, and motor defects. PDK1 was shown to regulate the rpS6 phosphorylation and the expression of PKCγ in vivo. Additionally, upregulation of pS6 in the cerebellar cortex partly rescued the reduction in cell body size, and the overexpression of PKCγ in PDK1-KO PCs rescued the reduction in the dendritic complexity observed after PDK1 ablation. These findings reveal an essential role for PDK1 in the maintenance of cell body and postnatal dendritic development of PCs by regulating pS6 phosphorylation and PKCγ expression.
PDK1 regulates cell body size and dendritic development in postnatal PCs
In previous studies, downregulation of PDK1–AKT signaling led to a smaller brain size due to a reduction in neuronal cell size (Lawlor et al., 2002). Moreover, recent work showed that the simultaneous haploinsufficiency of Pdk1 and contiguous genes causes microcephaly, developmental delay, intellectual disability, and epilepsy (Mucha et al., 2019). In this study, we showed that the ablation of PDK1 in PCs and cerebellar ML interneurons using the PV-cre mouse line led to a reduction in cell body size, which is associated with a reduction in dendritic complexity. Meanwhile, we used Pcp2-cre mice to specifically delete Pdk1 in PCs and observed that the dendritic complexity was also decreased compared with those in the Ctrl mice. Because the dendrites of PCs are in the cerebellum ML, the decreased dendritic complexity and the cumulative length of the dendritic arbor are likely to lead to a decrease in cerebellar ML thickness and cerebellar size. Furthermore, in the cKO-Pcp2 mice, we observed deficits in morphology and motor skills similar to those observed in cKO-PV mice, which suggests that the role of PDK1 in regulating the maintenance of PC body size is cell autonomous.
Previous work has shown that downregulation of PDK1–AKT signaling is essential for neuronal survival; however, recent work suggests that PDK1 deficiency in the forebrain causes neuronal apoptosis during cerebral cortical development (Xu et al., 2017, 2019; Wang et al., 2017b). Our data demonstrate that the densities of PCs are not affected by Pdk1 deletion before P60 but are decreased at P120. Together, our data indicate that PDK1 is indispensable for the survival of PCs during postnatal development before adulthood and is likely to have different roles during the aging stage after adulthood.
Specific inactivation of PDK1 in PCs results in deficits in motor balance, coordination, and spontaneous firing
A previous study demonstrated that the observed effects of whole-brain PTEN loss on brain size and neuronal cell size are PDK1 dependent (Chalhoub et al., 2009). Recent work showed that the loss of PTEN in cerebellar PCs results in defects in spontaneous and evoked firing activity of PCs and autistic-like traits, including impaired sociability, repetitive behavior, and deficits in motor learning (Cupolillo et al., 2016). Furthermore, PDK1 was upregulated significantly in patients with Parkinson's disease when compared with normal healthy control subjects, indicating that PDK1 may be associated with the development of movement disorders (Wang et al., 2017a). In this study, decreased cell body size and dendritic complexity of PCs after PDK1 deletion suggest that Pdk1 deletion probably leads to deficits in motor control. Our data show that cKO-PV mice show progressive gait abnormalities with deficient motor balance and coordination compared with Ctrl mice. Similar deficits in motor balance and coordination are observed in adult cKO-Pcp2 mice, in which Pdk1 has been specifically disrupted in PCs. Together, these results demonstrate that PDK1 is required for the postnatal development of PC-related motor function in mice. Moreover, the deficits in motor balance and coordination are more severe in cKO-PV mice than those in cKO-Pcp2 mice. PDK1 is widely expressed in the entire brain, including cerebellum, cerebral cortex, and hippocampus (Yoshida et al., 1999). Previous studies proved that PDK1 is required for the generation of neocortical interneurons, and the neocortical PV-positive interneurons are required for motor coordination (Oishi et al., 2009; Xenos et al., 2018). Additionally, increased connectivity of BCs, PV-positive interneurons in the ML of cerebellum are detected in human patients with spinocerebellar ataxia type 1 (SCA1), which is characterized by progressive loss of motor coordination (Edamakanti et al., 2018). In cKO-PV mice, PDK1 is disrupted not only in PCs but also in the neocortical PV-positive interneurons and the BCs, but in the cKO-Pcp2 mice PDK1 is deleted only in PCs in the cerebellum. Thus, a wider range of PDK1 deletion in the cKO-PV mice than that in the cKO-Pcp2 mice is likely to explain the more severe motor defects in the cKO-PV mice.
The motor defects and decrease in the cell body size of PCs after PDK1 ablation led us to hypothesize that the electrophysiological properties of PCs would also be affected. In PDK1-KO PCs, the spontaneous firing frequency and the Cm are lower than those of control cells, probably due to the decreased cell body size of the PCs (Tolias et al., 2005). Our findings demonstrate a new role for PDK1 in the development of PC-related motor function and, together, greatly improve our overall understanding of the postnatal development of PCs.
PDK1 regulates the postnatal maintenance of PCs by inducing rpS6 phosphorylation and PKCγ expression
Previous studies have shown that the PDK1–SGK1 axis activates mTORC1 by phosphorylating and inhibiting tubercular sclerosis complex 2, and the inactivation of mTORC1 in PCs leads to a reduction in cell body size, dendritic degeneration, axonal swelling, and age-dependent apoptosis (Angliker et al., 2015; Castel et al., 2016). We used the phosphorylation state of rpS6 as a marker for the level of activation of the mTORC1 pathway in PCs. Our data show that the intensity of pS6 in PCs was disrupted beginning at P14, suggesting that mTORC1 activation is disrupted after the ablation of PDK1. Additionally, daily intraperitoneal injection of cKO-PV and Ctrl mice with 3BDO or vehicle was performed, and our data showed that 3BDO treatment partly rescued the reduction in PC body size in cKO-PV mice, indicating that PDK1 regulates the development of PC body size partly through mTORC1 activation. Moreover, our data show that the motor coordination of the 3BDO-treated cKO-PV mice is similar to that of the vehicle-treated Ctrl mice in the low-rotational speed range, but is disrupted in the high-rotational speed range. These data suggest that decreased cerebellar mTORC1 possibly contributes to the motor coordination defects after the ablation of PDK1.
There is fourfold greater expression of PKCγ in the cerebellum than the average expression in the rest of the brain, and PKCγ is expressed solely in PCs in the cerebellum (Takahashi et al., 2017). Previous studies have suggested that PKCγ plays a pivotal role in climbing fiber pruning in developing PCs, and cultured PCs from PKCγ-deficient mice show impaired dendritic trees (Kano et al., 1995; Schrenk et al., 2002; Hirai, 2018). Increased constitutive activity of PKCγ may be one, but not the only, cause of SCA14, a neurologic disease characterized by motor dysfunction and death of PCs (Takahashi et al., 2015; Shimobayashi and Kapfhammer, 2017; Nakazono et al., 2018; Wong et al., 2018; Trzesniewski et al., 2019). In this study, an immunoblotting test with anti-PKCγ antibody demonstrates that the expression level of PKCγ in cKO-PV mice is reduced to 66% compared with that in Ctrl mice at P21. However, immunostaining with the anti-pPKCγ (phospho T514) antibody shows that a significant reduction of phosphorylation level of PKCγ is first detected in cKO-PV mice at P40. One possible explanation is that the remaining PKCγ in cKO-PV mice can be detected by immunostaining of cerebellar frozen sections; further studies are needed to exclude other possibilities. Together, we show a progressive loss of phosphorylation level of PKCγ in cKO-PV mice after P30, which is probably due to the reduction of PKCγ expression. We hypothesize that the deficient dendritic complexity in PCs after PDK1 ablation is a consequence of the downregulation of PKCγ expression. To test this hypothesis, we overexpressed PKCγ in PDK1-KO PCs by injecting an rAAV construct into the cerebellum of cKO-Pcp2 mice at P0. Our data showed that the dendritic size and complexity of the PKCγ-overexpressing PDK1-KO PCs are increased significantly compared with those of the PDK1-KO PCs. Thus, our data indicate that PDK1 regulates the dendritic development of PCs by PKCγ. Recently, the retinoic acid-related orphan receptor-α (RORα) was reported to play multiple roles in PC dendritic genesis, dendrite regression, and maintenance of mature dendrites at specific time windows during development and throughout adulthood (Chen et al., 2013; Takeo et al., 2015). Further studies on the possible role of RORα in PDK1 cKO mice are needed.
In summary, our data show that PDK1 contributes to cell body maintenance and dendritic development in postnatal PCs by rPS6 and PKCγ. Continued work exploring the signaling pathway through which PDK1 regulates rPS6 phosphorylation and PKCγ expression will facilitate an understanding of the postnatal developmental program and cerebellar motor coordination of PCs.
Footnotes
The authors declare no competing financial interests.
This work was financially supported by the Natural Science Foundation of Jiangsu Province, China (Grant BK20171063) and the China Postdoctoral Science Foundation (Grants 2018M632336) to R.L.; and the National Natural Science Foundation of China (Grants 81973308 and 81673416) and the Key R&D Program of Jiangsu Province (Grants BE2016761 and 2017CX010) to J.G.; and the National Natural Science Foundation of China (Grants 81671107) to J.-N.Z. We thank Dr. Dario Alessi for providing the Pdk1fl/fl mice, Prof. Li-Huei Tsai for providing the PV-cre mice, Prof. Chun-Jie Zhao for ROSA26-LSL-EYFP mice and Prof. Ying Shen for providing the Pcp2-cre mice. We also thank Prof. Ying Shen for technical advice and helpful discussions.
References
- Angliker N, Burri M, Zaichuk M, Fritschy JM, Rüegg MA (2015) mTORC1 and mTORC2 have largely distinct functions in Purkinje cells. Eur J Neurosci 42:2595–2612. 10.1111/ejn.13051 [DOI] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, Meyer M (2000) Cre recombinase expression in cerebellar Purkinje cells. Genesis 28:93–98. [DOI] [PubMed] [Google Scholar]
- Buttermore ED, Piochon C, Wallace ML, Philpot BD, Hansel C, Bhat MA (2012) Pinceau organization in the cerebellum requires distinct functions of neurofascin in Purkinje and basket neurons during postnatal development. J Neurosci 32:4724–4742. 10.1523/JNEUROSCI.5602-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Morton J, Dunnett SB (2001) Motor coordination and balance in rodents. Curr Protoc Neurosci Chapter 8:Unit 8.12. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ (1999) Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci 19:3248–3257. 10.1523/JNEUROSCI.19-08-03248.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, Brogi E, Alessi DR, Baselga J, Scaltriti M (2016) PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell 30:229–242. 10.1016/j.ccell.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Zhu G, Zhu X, Baker SJ (2009) Cell type specificity of PI3K signaling in Pdk1- and Pten-deficient brains. Genes Dev 23:1619–1624. 10.1101/gad.1799609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XR, Heck N, Lohof AM, Rochefort C, Morel MP, Wehrlé R, Doulazmi M, Marty S, Cannaya V, Avci HX, Mariani J, Rondi-Reig L, Vodjdani G, Sherrard RM, Sotelo C, Dusart I (2013) Mature Purkinje cells require the retinoic acid-related orphan receptor-α (RORα) to maintain climbing fiber mono-innervation and other adult characteristics. J Neurosci 33:9546–9562. 10.1523/JNEUROSCI.2977-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordón-Barris L, Pascual-Guiral S, Yang S, Giménez-Llort L, Lope-Piedrafita S, Niemeyer C, Claro E, Lizcano JM, Bayascas JR (2016) Mutation of the 3-phosphoinositide-dependent protein kinase 1 (PDK1) substrate-docking site in the developing brain causes microcephaly with abnormal brain morphogenesis independently of Akt, leading to impaired cognition and disruptive behaviors. Mol Cell Biol 36:2967–2982. 10.1128/MCB.00230-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupolillo D, Hoxha E, Faralli A, De Luca A, Rossi F, Tempia F, Carulli D (2016) Autistic-like traits and cerebellar dysfunction in Purkinje cell PTEN knock-out mice. Neuropsychopharmacology 41:1457–1466. 10.1038/npp.2015.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dainichi T, Hayden MS, Park SG, Oh H, Seeley JJ, Grinberg-Bleyer Y, Beck KM, Miyachi Y, Kabashima K, Hashimoto T, Ghosh S (2016) PDK1 is a regulator of epidermal differentiation that activates and organizes asymmetric cell division. Cell Rep 15:1615–1623. 10.1016/j.celrep.2016.04.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edamakanti CR, Do J, Didonna A, Martina M, Opal P (2018) Mutant ataxin1 disrupts cerebellar development in spinocerebellar ataxia type 1. J Clin Invest 128:2252–2265. 10.1172/JCI96765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ, van Meyel DJ (2014) Neuronal morphometry directly from bitmap images. Nat Methods 11:982–984. 10.1038/nmeth.3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DA, Ma L (2011) Mosaic analysis of gene function in postnatal mouse brain development by using virus-based Cre recombination. J Vis Exp. Advance online publication. Retrieved August 1, 2018. doi: 10.3791/2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grego-Bessa J, Bloomekatz J, Castel P, Omelchenko T, Baselga J, Anderson KV (2016) The tumor suppressor PTEN and the PDK1 kinase regulate formation of the columnar neural epithelium. Elife 5:e12034. 10.7554/eLife.12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H. (2018) Protein kinase C in the cerebellum: its significance and remaining conundrums. Cerebellum 17:23–27. 10.1007/s12311-017-0898-x [DOI] [PubMed] [Google Scholar]
- Itoh Y, Higuchi M, Oishi K, Kishi Y, Okazaki T, Sakai H, Miyata T, Nakajima K, Gotoh Y (2016) PDK1-Akt pathway regulates radial neuronal migration and microtubules in the developing mouse neocortex. Proc Natl Acad Sci U S A 113:E2955–E2964. 10.1073/pnas.1516321113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S (1995) Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell 83:1223–1231. 10.1016/0092-8674(95)90147-7 [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR (2002) Essential role of PDK1 in regulating cell size and development in mice. EMBO J 21:3728–3738. 10.1093/emboj/cdf387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux AE, Schulze JO, Biondi RM (2018) AGC kinases, mechanisms of regulation and innovative drug development. Semin Cancer Biol 48:1–17. 10.1016/j.semcancer.2017.05.011 [DOI] [PubMed] [Google Scholar]
- Liu C, Mei M, Li Q, Roboti P, Pang Q, Ying Z, Gao F, Lowe M, Bao S (2017) Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc Natl Acad Sci U S A 114:346–351. 10.1073/pnas.1608576114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Yang Y, Shen J, Chen H, Zhang Q, Ba R, Wei Y, Li KC, Zhang X, Zhao C (2015) Fstl1 is involved in the regulation of radial glial scaffold development. Mol Brain 8:53. 10.1186/s13041-015-0144-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui NC, Tam WY, Gao C, Huang JD, Wang CC, Jiang L, Yung WH, Kwan KM (2017) Lhx1/5 control dendritogenesis and spine morphogenesis of Purkinje cells via regulation of Espin. Nat Commun 8:15079. 10.1038/ncomms15079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha BE, Banka S, Ajeawung NF, Molidperee S, Chen GG, Koenig MK, Adejumo RB, Till M, Harbord M, Perrier R, Lemyre E, Boucher R-M, Skotko BG, Waxler JL, Thomas MA, Hodge JC, Gecz J, Nicholl J, McGregor L, Linden T, et al. (2019) A new microdeletion syndrome involving TBC1D24, ATP6V0C, and PDPK1 causes epilepsy, microcephaly, and developmental delay. Genet Med 21:1058–1064. 10.1038/s41436-018-0290-3 [DOI] [PubMed] [Google Scholar]
- Nakazono A, Adachi N, Takahashi H, Seki T, Hamada D, Ueyama T, Sakai N, Saito N (2018) Pharmacological induction of heat shock proteins ameliorates toxicity of mutant PKCγ in spinocerebellar ataxia type 14. J Biol Chem 293:14758–14774. 10.1074/jbc.RA118.002913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SM, Seo JS, Go TH, Nahm SS, Chang BJ (2019) Ascorbic acid supplementation prevents the detrimental effects of prenatal and postnatal lead exposure on the Purkinje cell and related proteins in the cerebellum of developing rats. Biol Trace Elem Res 190:446–456. 10.1007/s12011-018-1572-y [DOI] [PubMed] [Google Scholar]
- Oishi K, Watatani K, Itoh Y, Okano H, Guillemot F, Nakajima K, Gotoh Y (2009) Selective induction of neocortical GABAergic neurons by the PDK1-Akt pathway through activation of Mash1. Proc Natl Acad Sci U S A 106:13064–13069. 10.1073/pnas.0808400106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J (2014) An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E(-)/(-) mice. Sci Rep 4:5519. 10.1038/srep05519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa LS, Streng ML, Ebner TJ (2019) Purkinje cell representations of behavior: diary of a busy neuron. Neuroscientist 25:241–257. 10.1177/1073858418785628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrenk K, Kapfhammer JP, Metzger F (2002) Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase Cgamma-deficient mice. Neuroscience 110:675–689. 10.1016/s0306-4522(01)00559-0 [DOI] [PubMed] [Google Scholar]
- Shimobayashi E, Kapfhammer JP (2017) Increased biological activity of protein kinase C gamma is not required in spinocerebellar ataxia 14. Mol Brain 10:34. 10.1186/s13041-017-0313-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4. 10.1186/1471-213x-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanko JP, Easterling MR, Fenton SE (2015) Application of Sholl analysis to quantify changes in growth and development in rat mammary gland whole mounts. Reprod Toxicol 54:129–135. 10.1016/j.reprotox.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Adachi N, Shirafuji T, Danno S, Ueyama T, Vendruscolo M, Shuvaev AN, Sugimoto T, Seki T, Hamada D, Irie K, Hirai H, Sakai N, Saito N (2015) Identification and characterization of PKCγ, a kinase associated with SCA14, as an amyloidogenic protein. Hum Mol Genet 24:525–539. 10.1093/hmg/ddu472 [DOI] [PubMed] [Google Scholar]
- Takahashi N, Shuvaev AN, Konno A, Matsuzaki Y, Watanave M, Hirai H (2017) Regulatory connection between the expression level of classical protein kinase C and pruning of climbing fibers from cerebellar Purkinje cells. J Neurochem 143:660–670. 10.1111/jnc.14239 [DOI] [PubMed] [Google Scholar]
- Takeo YH, Kakegawa W, Miura E, Yuzaki M (2015) RORα regulates multiple aspects of dendrite development in cerebellar Purkinje cells in vivo. J Neurosci 35:12518–12534. 10.1523/JNEUROSCI.0075-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomanetz V, Angliker N, Cloëtta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Rüegg MA (2013) Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol 201:293–308. 10.1083/jcb.201205030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME (2005) The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45:525–538. 10.1016/j.neuron.2005.01.024 [DOI] [PubMed] [Google Scholar]
- Trzesniewski J, Altmann S, Jäger L, Kapfhammer JP (2019) Reduced Purkinje cell size is compatible with near normal morphology and function of the cerebellar cortex in a mouse model of spinocerebellar ataxia. Exp Neurol 311:205–212. 10.1016/j.expneurol.2018.10.004 [DOI] [PubMed] [Google Scholar]
- Wang J, Liu Y, Chen T (2017a) Identification of key genes and pathways in Parkinson's disease through integrated analysis. Mol Med Rep 16:3769–3776. 10.3892/mmr.2017.7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Lo CM, Chen L, Ngan ES, Xu A, Poon RY (2017b) CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ 24:38–48. 10.1038/cdd.2016.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen ZP, Zhuang QX, Zhang XY, Li HZ, Wang JJ, Zhu JN (2017c) Role of corticotropin-releasing factor in cerebellar motor control and ataxia. Curr Biol 27:2661–2669.e5. 10.1016/j.cub.2017.07.035 [DOI] [PubMed] [Google Scholar]
- Wong MMK, Hoekstra SD, Vowles J, Watson LM, Fuller G, Németh AH, Cowley SA, Ansorge O, Talbot K, Becker EBE (2018) Neurodegeneration in SCA14 is associated with increased PKCγ kinase activity, mislocalization and aggregation. Acta Neuropathol Commun 6:99. 10.1186/s40478-018-0600-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenos D, Kamceva M, Tomasi S, Cardin JA, Schwartz ML, Vaccarino FM (2018) Loss of TrkB signaling in parvalbumin-expressing basket cells results in network activity disruption and abnormal behavior. Cereb Cortex 28:3399–3413. 10.1093/cercor/bhx173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Yu L, Hou J, Jackson RJ, Wang H, Huang C, Liu T, Wang Q, Zou X, Morris RG, Spires-Jones TL, Yang Z, Yin Z, Xu Y, Chen G (2017) Conditional deletion of PDK1 in the forebrain causes neuron loss and increased apoptosis during cortical development. Front Cell Neurosci 11:330. 10.3389/fncel.2017.00330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Han X, Liu R, Li Y, Qi C, Yang Z, Zhao C, Gao J (2019) PDK1 deficit impairs the development of the dentate gyrus in mice. Cereb Cortex 29:1185–1198. 10.1093/cercor/bhy024 [DOI] [PubMed] [Google Scholar]
- Yoshida S, Sakagami H, Owada Y, Kokubun S, Kondo H (1999) Localization of PDK-1 mRNA in the brain of developing and adult rats. Tohoku J Exp Med 187:249–255. 10.1620/tjem.187.249 [DOI] [PubMed] [Google Scholar]
- Zurashvili T, Cordón-Barris L, Ruiz-Babot G, Zhou X, Lizcano JM, Gómez N, Giménez-Llort L, Bayascas JR (2013) Interaction of PDK1 with phosphoinositides is essential for neuronal differentiation but dispensable for neuronal survival. Mol Cell Biol 33:1027–1040. 10.1128/MCB.01052-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pdk1fl/fl mouse at P36.
PV-cre; Pdk1fl/fl mouse at P36.