

Gafson et al. review the neurobiology of neurofilament proteins, including their role in synapses and their contributions to disease pathogenesis, before evaluating the evidence supporting the use of neurofilament concentration measures as biomarkers of neurodegeneration.
Keywords: neurodegeneration, biomarkers, neurofilaments, neuroinflammation, traumatic brain injury
Abstract
Interest in neurofilaments has risen sharply in recent years with recognition of their potential as biomarkers of brain injury or neurodegeneration in CSF and blood. This is in the context of a growing appreciation for the complexity of the neurobiology of neurofilaments, new recognition of specialized roles for neurofilaments in synapses and a developing understanding of mechanisms responsible for their turnover. Here we will review the neurobiology of neurofilament proteins, describing current understanding of their structure and function, including recently discovered evidence for their roles in synapses. We will explore emerging understanding of the mechanisms of neurofilament degradation and clearance and review new methods for future elucidation of the kinetics of their turnover in humans. Primary roles of neurofilaments in the pathogenesis of human diseases will be described. With this background, we then will review critically evidence supporting use of neurofilament concentration measures as biomarkers of neuronal injury or degeneration. Finally, we will reflect on major challenges for studies of the neurobiology of intermediate filaments with specific attention to identifying what needs to be learned for more precise use and confident interpretation of neurofilament measures as biomarkers of neurodegeneration.
Introduction
Neurofilaments are assembled from a family of five intermediate filaments (Julien and Mushynski, 1983) that are distinguishable based on their relative apparent molecular masses on SDS-polyacrylamide gels. The largest of these is neurofilament heavy chain (NfH), followed (in order of descending molecular weight) by the medium chain (NfM), the light chain (NfL), α-internexin and peripherin (Fig. 1). Neurofilaments contribute to growth and stability of axons in both central and peripheral nerves as well as to maintaining mitochondrial stability (Gentil et al., 2015) and microtubule content (Bocquet et al., 2009). Roles for distinct neurofilament isoforms in maintaining the structure and function of dendritic spines and in regulating glutamatergic and dopaminergic neurotransmission synapses have also been discovered (Schwartz et al., 1994, 1995).
Figure 1.

Schematic representation of the structure of neuronal intermediate filament proteins. All intermediate filament proteins have a highly conserved central domain of 310 amino acid residues that is responsible for the formation of coiled-coil structures. Flanking this central rod domain are the amino- and carboxyl-terminal domains. These latter domains confer functional specificity to the different types of intermediate filaments proteins. For example, the NfM and NfH carboxyl-terminal domains contain multiple repeats of phosphorylation sites KSP (Lys–Ser–Pro) that account for the unusual high content of phosphoserine residues for these proteins. The N- and C-terminal regions contain multiple O-linked glycosylation sites. Neurofilament proteins NfL, NfM and NfH are obligate heteropolymers. Although α-internexin or peripherin can form homopolymers in vitro, these intermediate filaments proteins usually co-polymerize with the neurofilament triplet proteins in vivo.
The fundamental importance of neurofilaments to neurons has been highlighted by molecular characterization of diseases of the brain and peripheral nerves associated with abnormal neurofilament structure and function. Mutations in the NEFL gene, which encodes NfL, lead to peripheral neurodegeneration in Charcot-Marie-Tooth (CMT) disease types 2E (Mersiyanova et al., 2000) and type 1F (Jordanova et al., 2003) and G (Zuchner et al., 2004). While polymorphisms in NEFH, encoding NfH, are associated with amyotrophic lateral sclerosis (ALS) (Figlewicz et al., 1994), mutations in this gene also are a cause of CMT type 2 (Rebelo et al., 2016). Neurofilament dysfunction or aggregation also may play roles in the neuropathology of Alzheimer’s disease, Parkinson’s disease and other neurodegenerative disorders (Khalil et al., 2018).
Neurofilaments' turnover in healthy neurons is slow. Their expression is regulated by neuronal activity acting through developmentally regulated promoter regions (Yaworsky et al., 1997). Post-transcriptional regulation of neurofilament mRNA stability also may contribute to determining levels of expression of neurofilament protein (Schwartz et al., 1994, 1995). Additional insights into mechanisms for turnover of neurofilament have come through studies of rare diseases arising from gigaxonin E3 ligase mutations causing giant axonal neuropathy (GAN) (Bomont et al., 2000) and mutations in TRIM2 (another E3 ligase) and sacsin (which includes both ubiquitin-like and chaperone domains) that are responsible, respectively for a form of CMT (Ylikallio et al., 2013) and for the cerebellar degeneration occurring in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) (Engert et al., 2000). Study of these diseases has elucidated major pathways responsible for the degradation of neurofilament protein, which is a consequence of combined activities of proteasomal and, possibly, autophagocytic mechanisms (Bomont, 2016). Neurofilament or its fragments can be released from neurons secondary to axonal damage or neurodegeneration, although the predominant peptide species released and the mechanisms responsible for the release have not been clearly characterized. Release may occur actively (e.g. by means of exosomes; Faure et al., 2006; Lachenal et al., 2011) or passively with loss of neuronal membrane integrity. Neurofilaments in different supramolecular structures or with different isoforms may show differences in degradation rates (Nixon and Logvinenko, 1986; Millecamps et al., 2007). Studies of pathways for trafficking of other proteins (Szentistvanyi et al., 1984) suggest that degraded neurofilament proteins may enter the peripheral circulation via perivascular drainage along basement membranes of arteries (Carare et al., 2008) to drain into cervical or lumbar lymph nodes and then into the blood.
Reliable, sensitive assays for measuring concentrations of neurofilaments in CSF have been available for many years (Norgren et al., 2002). These provided an early foundation for exploration of the associations of increased CSF neurofilaments with neurological diseases (Rosengren et al., 1996; Lycke et al., 1998). Ultra-sensitive assays of neurofilaments in blood are now available routinely for clinical applications in many centres (Kuhle et al., 2016a). This has enabled several studies assessing the potential utility of neurofilament (particularly NfL) peptide concentrations in the CSF or peripheral blood as clinical biomarkers of neuronal injury or neurodegeneration (Kuhle et al., 2016a; Disanto et al., 2017; Khalil et al., 2018). For example, CSF and peripheral blood neurofilament protein concentrations are increased after stroke or traumatic brain injury (Khalil et al., 2018) and are associated with ageing (Disanto et al., 2017) and primary neurodegenerative diseases including Alzheimer’s disease (Mattsson et al., 2016; Weston et al., 2017). Neurofilament concentrations in both CSF and peripheral blood are increased in some individuals with multiple sclerosis (Amor et al., 2014; Kuhle et al., 2016b; Disanto et al., 2017; Novakova et al., 2017; Barro et al., 2018; Hakansson et al., 2018; Piehl et al., 2018) and have potential roles as clinical biomarkers of disease activity, treatment responses or prediction of future disease progression and disability. Both peripheral blood and CSF concentrations are correlated with radiological (Kuhle et al., 2016b; Disanto et al., 2017; Novakova et al., 2017; Siller et al., 2018) and clinical measures of disease activity (Disanto et al., 2017; Novakova et al., 2017; Barro et al., 2018; Hakansson et al., 2018; Piehl et al., 2018; Siller et al., 2018). Evidence for treatment responsiveness further supports a causal link between disease activity and increased neurofilament concentrations in CSF and peripheral blood (Gunnarsson et al., 2011; Disanto et al., 2017; Piehl et al., 2018; Gafson et al., 2019; Kuhle et al., 2019). NfL levels have a potential role in assessing prognosis in multiple sclerosis. For example, the predictive association between increased NfL and longer-term brain and spinal cord atrophy (Barro et al., 2018) likely arises from the sensitivity of NfL to the neuroinflammatory neuronal injury and degeneration that provides a substrate for future disease progression (Matthews, 2019).
While the concentration in CSF is ∼20–50-fold greater than in peripheral blood (Bergman et al., 2016), moderate to high correlation between concentrations measured in the two compartments have been reported (Gaiottino et al., 2013; Gisslen et al., 2016). Nevertheless, given that neurofilaments can also be released from peripheral nerves, depending on the pathological context, peripheral blood and CSF measures should not necessarily be correlated (Bergman et al., 2016). Longitudinal measures of CSF and peripheral blood concentration changes after brain injury (Shahim et al., 2016) suggest that turnover times in the blood and CSF compartments are similar. This is consistent with models positing that central and peripheral turnover are linked functionally. However, important questions regarding the neurobiology, mechanisms of turnover and kinetics in the brain and blood compartments remain. These will be highlighted as the current understanding of neurofilament neurobiology in health and disease is reviewed in more detail below (Box 1).
Box 1.
Summary points
Neurofilaments are a family of neuronal intermediate filaments involved in both the growth and stability of axons, and, through incorporation into different supramolecular assemblies, also in synaptic organization and function in the CNS.
The fundamental importance of neurofilaments to axonal structure and function was first appreciated with serial discoveries of causal neurofilament gene mutations for rare forms of CMT disease and ALS.
Evidence that their normal intracellular assembly and turnover involves the ubiquitin-proteasomal pathway (and possibly also the autophagy pathway) came from studies of rare genetic diseases characterized by prominent accumulations of neurofilaments: CMT type 2R, GAN and ARSACS.
Increased concentrations of extra-neuronal neurofilament peptides in CSF and blood now are measured routinely using ultra-sensitive immunoassays after peripheral nerve or brain injury or in association with clinical progression of several chronic neurodegenerative disorders and neuropathies including multiple sclerosis, Alzheimer’s disease, ALS, Huntington’s disease and CMT.
However, as the mechanisms and kinetics of neurofilament protein release from neurons and trafficking between brain and blood compartments are ill-defined, interpretations of increased CSF or blood neurofilament concentrations in terms of the specific nature or extent of any associated neuronal dysfunction or injury or the rate of neurodegeneration should be made with caution.
Given the growing interest in using soluble neurofilament proteins as biomarkers for clinical decision making, elucidating the identities of peptides detected by current assays and the mechanisms by which these are released from neurons are particularly urgent questions to be addressed.
Structure and assembly of neurofilaments
Neurofilaments are major cytoskeletal components in mature neurons. They are found in the cytoplasm of neurons within the peripheral nervous system (PNS) and CNS, most abundantly in axons, but also in cell bodies, dendrites and synapses (Yuan et al., 2015b). They are expressed more highly in large myelinated axons, where they are organized in parallel structures maintained by side arms projecting outwards from a filament core (Yuan et al., 2017). However, the relative abundance of neurofilament proteins can widely differ along the course of even a single axon, e.g. amounts of neurofilaments are 3-fold greater in myelinated regions of axons than at the nodes of Ranvier (Hsieh et al., 1994; Nixon et al., 1994).
Neurofilaments are hetero-polymers composed of core neurofilament proteins (NfL, α-internexin or peripherin) (Yuan et al., 2006, 2012b) co-assembling with NfM and NfH (Fig. 2). α-Internexin interacts with NfL to form a backbone to which NfM and NfH attach. Multiple types of post-translational modifications to neurofilament occur (e.g. phosphorylation, ubiquitination, nitration and addition of O-linked N-acetylglucosamine) (Nixon and Sihag, 1991; Yuan et al., 2017). NfM and NfH can be phosphorylated extensively (Fig. 1) (Beck et al., 2012). The relative proportions of the neurofilament protein components and their post-translational modifications change with development and vary between different types of neurons and neuronal functional states. NfL is uniquely important among the neurofilaments as it is required for neurofilament protein assembly in some neuronal subtypes. NfL knockout mice exhibit severe atrophy of peripheral myelinated axons (Zhu et al., 1997). The latter observation provides evidence that neurofilaments are necessary for the radial growth of large myelinated axons and associated fast nerve conduction (Kriz et al., 2000).
Figure 2.
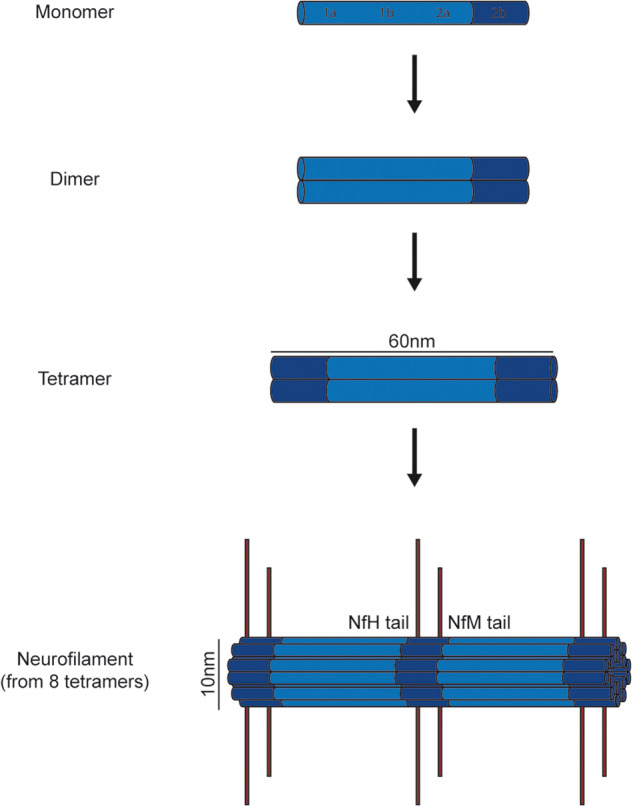
Intermediate filaments are formed by the assembly of intermediate filament protein dimers. Two polypeptide chains form a coiled-coil dimer and two coiled-coil dimers form a 3-nm protofilament. These protofilaments associate in a staggered manner to form filaments of 10-nm in diameter (32 chains). The carboxy-terminal domains of NfM and NfH form side-arm projections at the filament periphery.
The complexity of mechanisms by which neurofilament proteins play their roles in maintaining axonal organization have begun to be defined. Their phosphorylation appears to be a fundamental element. The carboxyl terminals of NfH and NfM proteins form side-arm projections at the periphery of neurofilament structures that contain multiple Lys–Ser–Pro (KSP) repeats, which can be phosphorylated by proline-directed kinases, Erk1/2, Cdk5/p35, and JNK3 (Lee et al., 2014). Because phosphorylation of KSP repeats, especially in NfH (Julien and Mushynski, 1982, 1983), increases the negative charge of these projections (and, by inference, the spacing between neurofilament in the axon), it was believed initially that NfH must play a major role in determining axonal calibre. However, surprisingly, targeted disruption of the NEFH gene had little effect on the radial growth of myelinated axons (Rao et al., 1998; Zhu et al., 1998). In contrast, deletion of NEFM gene (Jacomy et al., 1999) or deletion of the NfM carboxy-terminal tail domain substantially reduced the calibres of large myelinated axons (Rao et al., 2003). The exact NfM domain that modulates axon calibre remains to be elucidated. A mouse knock-in substitution of KSP repeats by KAP (NF-MS→A) repeats (which cannot be phosphorylated) in NfM demonstrated that phosphorylation of NfM KSP repeats does not determine axonal calibre (Garcia et al., 2009). The current model for their structural organization proposes that the tail domains of NfM and NfH form side arms that interconnect neurofilaments and link them to other cytoskeletal elements and organelles such as mitochondria and microtubules (Yuan et al., 2017). Phosphorylation of neurofilament stabilizes this structure by inhibiting neurofilament proteolysis and increasing the half-life of the whole supramolecular assembly (Rao et al., 2012).
Most neurofilament proteins are synthesized and assembled in the neuronal perikaryon and must be transported along axons for functional integration into the axonal cytoskeleton (Yuan et al., 2012a). The newly synthesized neurofilament population enters axons in the form of short filaments and possibly also smaller polymeric/oligomeric assemblies (Pachter and Liem, 1984). It was proposed initially that, after their assembly in the perikaryon, neurofilaments are transported unidirectionally by slow transport mechanisms (0.1–1 mm/day) distally along the axon, where they ultimately are proteolytically degraded (Hoffman and Lasek, 1975). This classical model of neurofilament transport and turnover in the axon was based on pulse chase radiolabelling techniques with low time resolution (Hoffman and Lasek, 1975). However, subsequent time-lapse microscopy of fluorescently tagged neurofilament proteins in growing axons of neurons in vitro revealed fast transport of tagged-neurofilament at rates up to 2 μm/s that is interrupted by long pauses resulting in an average rate approximating the slow neurofilament transport estimates (Roy et al., 2000; Wang et al., 2000; Li et al., 2012). Live imaging of immature or regenerating axons in vitro has also identified a pool of labelled neurofilaments that, after entering the axon, remains there for very long periods (Trivedi et al., 2007; Yuan et al., 2009). This pool reflects the initial stage of construction of a stable stationary neurofilament network that maintains calibre sizes of mature large PNS and CNS fibres by integration of neurofilaments with other cytoskeletal elements (Nixon and Logvinenko, 1986; Yuan et al., 2017). These and more recent observations have contributed to a revised model positing that neurofilaments undergoing transport can move bi-directionally along microtubules in the axon via motors such as kinesin and dynein (Shea and Flanagan, 2001). Earlier observations, such as the directional reversibility and alternations between rapid movements and long pauses leading to a net slow movement of the neurofilament population undergoing axonal transport, are well explained by this model. The precise kinetics can also be related to local axonal structure; pulse-escape fluorescence photoactivation recently demonstrated slowing of neurofilament transport at nodes of Ranvier, where there is constriction of the axon (Walker et al., 2019).
Neurofilament protein stoichiometry is essential for appropriate neurofilament assembly and axonal integrity (Julien et al., 1987). Abnormal neurofilament accumulation in the perikaryon of motor neurons can be induced by overexpressing any neurofilament protein alone. Overexpression of NfH in mice led to formation of large perikaryal neurofilament aggregates in spinal neurons and reduction of neurofilament transported into axons (Cote et al., 1993). Maintaining a higher ratio of NfL to NfH and NfM is critical for the normal growth of axons and dendrites (Kong et al., 1998); NfL plays an essential and distinct role in neurofilament assembly from those of NfM and NfH. Additional observations further emphasize the importance of maintaining proper intermediate filament protein stoichiometry in an axon. In the mouse, large perikaryal accumulations of neurofilaments due to human NEFH transgene overexpression were associated with severe atrophy of peripheral axons, but did not cause substantial neuronal death (Cote et al., 1993). However, even with normal stoichiometry of the major proteins, neurofilament disorganization alone appears sufficient to lead to neuronal death; overexpression of peripherin (Beaulieu et al., 1999) or of a mutated NfL protein (Lee et al., 1994) in transgenic mice induced the formation of ALS-like neurofilament aggregates and selective degeneration of spinal motor neurons.
Neurofilament pathologies are commonly expressed as aggregates, but their functional significance appears to depend on their localization within the neuron. Sequestration of peripherin in the perikaryon of motor neurons by NfH overexpression rescued the peripherin-mediated death of motor neurons in transgenic mice (Beaulieu and Julien, 2003), suggesting that axonal neurofilament aggregates (or spheroids) are more toxic than perikaryal neurofilament aggregates, perhaps because of interference with axonal transport of organelles by the former.
Genetic abnormalities in neurofilaments and disease
Historically, the fundamental importance of neurofilament to neuronal structure and function became apparent through the discovery of associations between neurofilament gene mutations and disease (Table 1). Abnormal accumulation of neurofilament and of the related intermediate filament, peripherin, is an early pathological hallmark of ALS (Corbo and Hays, 1992). Several factors could be responsible for the abnormal neurofilament accumulations observed, such as dysregulation of neurofilament gene expression, neurofilament mutations, defective axonal transport, abnormal post-translational modifications, and proteolysis. Degenerating neurons in ALS show a 70% decrease in levels of NfL (NEFL), α-internexin (INA) and peripherin (PRPH) mRNA post-mortem (Campos-Melo et al., 2013). Modifications in the stability of the associated neurofilament mRNA contribute to this; the TAR DNA-binding protein 43 (TDP-43), which forms cytoplasmic aggregates in ALS, was found to bind and destabilize or sequester NEFL mRNA (Strong et al., 2007; Volkening et al., 2009), a phenomenon that also could contribute to alterations of neurofilament protein synthesis, consequent stoichiometry changes and aggregation of neurofilament (Rosengren et al., 1996).
Table 1.
Diseases associated with mutations in neurofilament genes or genes involved in proteins for neurofilament assembly, turnover and degradation
| Neurofilament pathology | Proteins affected | Associated diseases |
|---|---|---|
| Primary neurofilament gene mutations | NfH, peripherin | ALS |
| NfL, NfH | CMT | |
| Mutations in genes involved in NF assembly, turnover and degradation | Sacsin | ARSACS |
| TRIM2 | CMT type 2R | |
| Gigaxonin | GAN |
Evidence for a potentially causal pathogenic role of neurofilament abnormalities in ALS (Table 1) came from the discovery of codon deletions or insertions in the KSP repeat motifs of NfH in a small number of patients with sporadic ALS (Figlewicz et al., 1994; Tomkins et al., 1998). A frameshift deletion and an amino acid substitution in the peripherin (PRPH) gene also have been discovered in two sporadic ALS cases (Gros-Louis et al., 2004; Leung et al., 2004). However, other studies have failed to identify common polymorphisms or rare genetic variants of neurofilament genes in association with familial and sporadic ALS (Rooke et al., 1996; Vechio et al., 1996), suggesting either that neurofilament gene mutations define a rare subtype of ALS or that they make only a minor contribution to general ALS susceptibility.
Pathogenic roles for neurofilaments in ALS may extend beyond associations with rare genetic coding variants: neurofilament abnormalities in ALS also occur as a result of post-translational protein modifications. Phosphorylation changes can alter the axonal transport of neurofilaments, leading to their accumulation in cell bodies and axons. Treatment of neurons with glutamate activates mitogen-activated protein kinase (MAPK), resulting in phosphorylation of neurofilaments and slowing of their axonal transport, hence potentially defining another mechanism by which glutamate might affect its excitotoxicity in ALS (Ackerley et al., 2000; Veyrat-Durebex et al., 2014). Additional observations link glutamate excitotoxicity to neurofilament phosphorylation. Glutamate induces caspase cleavage and activation of protein kinase N1 (PKN1), a kinase targeting the neurofilament head rod domain to disrupt neurofilament organization and axonal transport (Manser et al., 2008). The peptidyl-prolyl isomerase PIN1, which selectively binds to phosphorylated proline-directed serine/threonine residues in NfH, also may play a significant regulatory role in glutamate stress-induced neurofilament phosphorylation. In ALS, PIN1 is co-localized with spinal cord neuronal inclusion bodies. Glutamate-stressed neurons exhibit increased phosphorylated NfH in perikaryal accumulations, which co-localize with PIN1 (Kesavapany et al., 2007). In addition, downregulation of PIN1 by small interfering RNA reduces glutamate-induced NfH phosphorylation and neuronal apotosis (Kesavapany et al., 2007). However, over-expression of human NfL alone also is associated with the potentiation of N-methyl-d-aspartate (NMDA)-dependent calcium entry and apoptosis (Sanelli et al., 2007).
Indirect evidence suggests that additional post-translational modifications of neurofilaments may contribute to ALS. Advanced glycation end-products have been detected in neurofilament aggregates of motor neurons in familial and sporadic ALS (Chou et al., 1998). This observation is of clinical interest given associations discovered between diabetes and ALS, although there is still uncertainty about the clinical significance of the abnormal glycosylation (Kioumourtzoglou et al., 2015; Mariosa et al., 2015).
Mutations in NEFH have been associated with the type 2CC axonal form of CMT (Jacquier et al., 2017) (Fig. 3). Causal mutations in the NEFL gene have been linked to several other (also less common) forms of CMT (Horga et al., 2017). Some of these mutations cause the axonal CMT type 2E (Mersiyanova et al., 2000), but other NEFL mutations are associated with slow nerve conduction velocities and clinical presentations resembling demyelinating type CMT (type 1F) (Jordanova et al., 2003) or the type G intermediate form (Zuchner et al., 2004). While the majority of NEFL mutations resulting in CMT are dominantly inherited, autosomal recessive NEFL mutations that lead to truncated NfL proteins and a severe early onset axonal form of CMT have also been reported (Abe et al., 2009; Yum et al., 2009; Sainio et al., 2018).
Figure 3.

Mutations in the NEFL gene encoding NfL account for a small percentage of CMT disease. It is noteworthy that mutations have been detected in various regions of NfL. Some mutations have been shown to disrupt self-assembly of NfL into a filamentous network. The mutations highlighted here are not exhaustive (Horga et al., 2017) and do not include recently identified recessive mutations.
The P22S mutation in NfL was first discovered in a Slovenian patient (Georgiou et al., 2002) with an early onset form of type 2E CMT (axonal type) associated with axonal deformation and swelling. The P22S mutation abolishes the Thr–Pro Proline-directed protein kinase (PDPK) consensus phosphorylation sequence within the head domain and perturbs normal regulation of neurofilament assembly through phosphorylation (Sasaki et al., 2006); the mutated NfL proteins do not self-assemble with NfM and NfH and cause neurofilament aggregation in cultured cells (Perez-Olle et al., 2002, 2004). Similar neurofilament aggregates caused by CMT-associated mutations in NfL (e.g. P22S, as well as P8R, Q333P) trap motor proteins and organelles in the cytoplasm, contributing to axonal transport defects (Zhai et al., 2007). Interestingly, mutations in the NEFH gene also have been recently identified in an axonal form of CMT (Rebelo et al., 2016; Jacquier et al., 2017).
Transgenic mouse models based on mutations in CMT provide powerful tools for study of the disease pathology. An NfL mutant model that recapitulates the cellular neuropathology found in human axonal CMT was generated by a knock-in strategy replacing one mouse NfL allele with the N98S mutation in the rod region of NfL (Adebola et al., 2015), a mutation that has been described in sporadic cases of CMT with early age of onset (<2 years). Consistent with the clinical presentation, mutant NFLN98S/+ mice were symptomatic at an early age. Tremor was observed at 1 month of age. The N98S mutation caused a severe reduction of neurofilament in myelinated axons of the PNS and CNS, axonal hypotrophy and distal axonal loss in the PNS. Cellular immunopathology revealed abnormal neurofilament aggregates in neuronal cell bodies and axons of the cerebellum and spinal cord from an early age. The mice exhibited hind limb clasping, a likely behavioural expression of the axonopathy. The NFLN98S mice thus provide a model with face validity for testing potential therapeutic strategies directed towards preventing or reversing neuropathic symptoms in humans. In contrast, knock-in of a different point mutation (P8R) that causes symptoms in humans with variable ages of onset was associated with weak phenotypes without neurofilament accumulation. Together, these results suggest the hypothesis that phenotypic severity in mouse models of CMT caused by NfL mutations may be related to both the extent of neurofilament aggregation found in neurons and clinical severity in corresponding human diseases.
A critical proof of principle that selective suppression of mutant neurofilament expression alone could not only slow the progression of, but also reverse disease-related pathology came from a transgenic mouse study with conditional mutant NfLP22S expression. The model was generated using a tetracycline-responsive (tet-off) gene system that allowed suppression of mutant NFLP22S expression in mature neurons after administration of doxycycline (Dequen et al., 2010). The NFLP22S mice recapitulated the key features of CMT type 2E neuropathy: progressive development of an abnormal hind limb posture with motor deficits, hypertrophy of muscle fibres and muscle denervation. Suppression of mutant NFLP22S production after clinical disease onset reversed these pathological features. This important observation suggests that therapies able to abolish mutant NfL expression also may be able to reverse pathology and disability in the clinic. However, whether such dramatic results would be seen in patients still is uncertain given the species differences and also that symptoms in the mouse model occurred very late, expression of mutant NfL was low and neurofilament aggregates were not seen in motor neurons. Additional work is needed to determine whether neuronal function is restored if pathological neurofilament aggregates are present and whether any functional recovery can be related directly to clearing of the aggregates.
Neurofilaments in synapses
Although neurofilaments have been viewed traditionally as structural components primarily of axons and dendrites, recent evidence has shown that distinctive assemblies of neurofilament subunits also are integral components of synapses. For decades, synaptic terminals were associated only with degradation of neurofilaments transported distally along the axon. However, early observations supporting this concept (Roots, 1983), have not been confirmed. For example, recent proteomic analyses show that many synaptosomal proteins have half-lives of weeks to months (Heo et al., 2018), which is longer in some cases than the half-lives of neurofilaments in axons. Other observations of neurons in the intact, mature brain also proved difficult to reconcile with models of neurofilament transport and distribution that mostly relied on in vitro observations made on embryonic neuronal axons, which have few neurofilaments and reflect an early developmental or regenerative state (Nixon, 1998). Most notable are in vivo studies of mature brains showing that only a small pool of newly synthesized neurofilament subunit precursors needs to be transported to maintain the large stationary neurofilament network in myelinated axons because of the exceptionally slow turnover of this network (Yuan et al., 2017). In this model, the small amount of neurofilament protein reaching terminals accords with evidence for the long half-lives of synaptic proteins (Heo et al., 2018) implying low synaptic proteolytic activity and similar rates of local turnover of a predominantly stationary neurofilament network uniformly along axons (Nixon and Logvinenko, 1986). The role of neurofilaments as a critical determinant of axon calibre—their accepted principal role in peripheral nerves—appears much less important for axons of CNS neurons. Despite the presence of abundant neurofilament proteins in the CNS, intrinsic axons of the brain, even the larger corpus callosum axons, have a relatively low neurofilament density and exhibit minimal volume reductions when their neurofilament expression is suppressed genetically (Dyakin et al., 2010).
Direct evidence has now established unequivocally that synapses contain a unique pool of neurofilament that has distinctive functional roles (Yuan et al., 2015a, b). Neurofilament assemblies isolated from brain synaptosomes are distinguishable both morphologically and biochemically from those in other parts of the neuron; neurofilament subunits in synapses exist in unconventional assemblies and even likely in small hetero-oligomeric forms (Yuan et al., 2003). The latter are capable of axonal transport (Yuan et al., 2003). Electron microscopy combined with immunogold labelling has identified short, irregularly oriented and bent 10-nm filaments that often are associated with the postsynaptic density (PSD) or with vesicular organelles (Fig. 4). Synaptic neurofilament proteins are distinctive in both stoichiometry and states of phosphorylation and respond differently to genetic subunit perturbations than the larger neurofilament protein pool in brain white matter (Yuan et al., 2015b). Changes in synaptic NfL phosphorylation associated with calcium/calmodulin-dependent protein kinase II activation during modulation of long-term potentiation (LTP) suggest a role for synaptic neurofilament proteins to enable the latter (Hashimoto et al., 2000) and hint at a broader functional significance of the complex regulation of neurofilament subunits by phosphorylation (Nixon and Sihag, 1991; Sihag et al., 2007).
Figure 4.

Functions of neurofilament subunit assemblies in synapses. Left: Immunogold labelled antibodies against the NfM subunit decorating mouse brain synaptic structures in a linear pattern (immunogold particles outlined in blue) suggest the presence of short neurofilaments and protofilament/protofibrils. In the top inset, a filament within a postsynaptic bouton is decorated by immunogold antibodies to both NfL (large gold dots) and NfH (small gold dots). Graphic inset: Morphometric analyses indicate a higher density of immunogold labelling in postsynaptic boutons than in preterminal dendrites or presynaptic terminals. Middle: Ultrastructural image of a human brain synapse illustrates membranous vesicles [tentatively identified as endosomes (ENDO)], most associated with short 10-nm filaments in the postsynaptic region. Right: Evidence (Yuan et al., 2015a) supports a biological mechanism whereby D1Rs internalized on endosomes from the postsynaptic surface dock on synaptic neurofilament subunit assemblies (outlined in blue) where they remain available to recycle from endosomes to the synaptic surface in response to ligand stimulation. In the absence of NfM, retention of D1R on the plasma membrane surface induces hypersensitivity to D1R agonists, as observed in vivo. Selective NfL deletion in mice induces an NMDAR hypofunction phenotype by lowering membrane surface levels of the GluN1 subunit. Evidence (Yuan et al., 2018a) supports a mechanism in which NfL binds GluN1 associated with NMDAR on postsynaptic terminals and stabilizes the receptor on the membrane by directly anchoring GluN1 and preventing access of the ubiquitin ligase that ubiquitinates GluN1 and targets it for degradation by the proteasome (UPS) leading to reduced NMDAR function. The key below the figure identifies the depicted cellular elements that are depicted. NF = neurofilament; POST = postsynaptic; PRE = presynaptic; SV = synaptic vesicles; UPS = ubiquitin proteasome system.
Synaptic neurofilament proteins have been found to be more abundant in the postsynaptic compartment than in adjacent dendritic areas or presynaptic terminals using quantitative immunogold analysis with electron microscopy (Yuan et al., 2015b). Immunocytochemical studies (Bragina and Conti, 2018) have confirmed neurofilament subunit immunoreactivity (NFIR) in pre-and post-synaptic compartments and greater NFIR in GABAergic than in glutamatergic synapses (Bragina and Conti, 2018).
Recent findings that individual subunits serve unique roles in neurotransmission provide indirect, but compelling evidence for the functional importance of synaptic neurofilament (Yuan et al., 2015b). NMDA receptors (Li and Tsien, 2009) are highly concentrated in postsynaptic membranes of glutamatergic synapses (Huntley et al., 1994). NfL has long been known to interact directly with the cytoplasmic C-terminal domain of GluN1 through its rod domain (Ehlers et al., 1998; Ratnam and Teichberg, 2005). NfL co-expression with GluN1 and GRIN2B subunits in HEK293 cells increases the surface abundance of GluN1 (Ehlers et al., 1998) and blocks its ubiquitination (Ratnam and Teichberg, 2005). Both of these actions of NfL are expected to stabilize NMDA receptors within the neuronal plasma membrane. Consistent with this hypothesis, the abundance of synaptic GluN1 subunits is reduced and ubiquitin-dependent GluN1 subunit turnover is greater in NEFL−/− than in wild-type mice (Yuan et al., 2018a). Binding of antibody that only recognizes poly-ubiquitin chains formed with the Lys48 (K48) residue is greater in GluN1-rich postsynaptic membranes of the hippocampus; GluN1 interactions with NfL may inhibit their accessibility to the ubiquitin ligases known to initiate GluN1 degradation (Kato et al., 2005; Ratnam and Teichberg, 2005; Yuan et al., 2018b). Additionally, NfL binding to protein phosphatase-1(PP1), a protein/serine/threonine phosphatase in the PSD (Terry-Lorenzo et al., 2000), suggests possible regulation of the phosphorylation states of neurofilament subunits and NMDA GluN1 receptors in ways that may influence the cellular distribution of the receptor (Ehlers et al., 1998). These observations may be relevant for understanding the role for regulating the phosphorylation state of NfL with LTP and long-term depression (LTD) (Hashimoto et al., 2000).
Loss of surface GluN1 receptors and NMDAR hypofunction associated with NfL deletion could contribute to clinical presentations of psychiatric and neurodegenerative disorders including Alzheimer’s disease (Lin et al., 2014). NEFL gene deletion in mice, which depresses GluN1 protein levels, both reduces dendritic spine number and length and leads to increased hippocampal glutamate levels as an adaptive response (van Elst et al., 2005; Homayoun and Moghaddam, 2007; de la Fuente-Sandoval et al., 2011; Merritt et al., 2016; Yuan et al., 2018b). Responses to NMDAR antagonists are also lost with NEFL deletion, although effects on NMDA-independent motor activity are minimal. Multiple NMDAR-related behaviours such as pup retrieval, spatial and social memory, prepulse inhibition and night-time activity are also impaired. Importantly, similar NMDAR-related synaptic and behavioural deficits (albeit in milder forms than in NfL-null mice) are seen in NEFL+/− mice that have 40–50% lower brain NfL levels than in the wild-type mice; a relative reduction within the range of NfL loss seen in some brain regions with schizophrenia (Kristiansen et al., 2006). Interestingly, neurofilament genes map to chromosomal regions implicated in schizophrenia (Badner and Gershon, 2002; Lewis et al., 2003) and concentrations of NfL are reduced consistently in this disease (Kristiansen et al., 2006; Pennington et al., 2008).
Different neurofilament subunits likely have distinct roles in synaptic function. NfM co-localizes with the G-protein-coupled dopamine D1 receptor (D1R) in synaptic boutons (Girault and Greengard, 2004) and the deletion of NfM but not any other neurofilament subunit causes postsynaptic D1Rs to redistribute from a reserve pool on endosomes to the synaptic plasma membrane, which significantly increases D1R-stimulated hippocampal LTP and greatly amplifies dopamine D1R-mediated motor responses to cocaine (Yuan et al., 2015b). Furthermore, deletion of the NEFM gene in mice enhances D1R-mediated motor responses to cocaine (Yuan et al., 2015b). The lack of NfM leads to a redistribution of postsynaptic D1Rs from endosomes to plasma membrane, implying that NfM is playing a role in the recycling of the D1R. NEFM deletion also inhibits the desensitization response to cocaine and amphetamine, while enhancing and prolonging ERK activation and ERK mediated NfM phosphorylation. Notably, basal neurotransmission and induction of LTP are normal in NfM-null mice, distinguishing them from mice lacking NfH, which exhibit deficient LTP maintenance and NfL-null mice that display both deficient basal neurotransmission and LTP (Yuan et al., 2015b).
Furthermore, NfL is known to interact with the GluN1 subunit of the NMDA receptor. NfL deletion in mice reduces GluN1 protein levels and dendritic spines and elevates ubiquitin-dependent turnover of GluN1 (Yuan et al., 2018a). Interactions between D1R and NMDA receptors are facilitated through neurofilament subunit assemblies (Yuan et al., 2015b, 2018b). The motor stimulant effect of the NMDA antagonist phencyclidine is blocked by D1R antagonists and deletions of NfL and NfM, which regulate NMDAR and D1R, respectively and have opposing effects on D1R-dependent motor activity induced by NMDA inhibition (Yuan et al., 2018b). Thus, the known functional interdependence of these two distinct receptor complexes appears to depend on a synaptic scaffold containing assemblies of multiple neurofilament subunits.
Degradation and turnover of neurofilaments
Mechanisms responsible for the turnover of neurofilaments are poorly understood. Neurofilaments appear to undergo degradation all along axons by mechanisms regulated by their density and phosphorylation status (Nixon and Logvinenko, 1986). To investigate the turnover and axonal transport of neurofilaments quantitatively, Millecamps et al. (2007) generated mice with the human NEFL transgene under doxycycline control in the presence or absence of endogenous mouse NfL proteins. In these mouse models, although the human NEFL mRNA expression was turned off 1 week after administration of doxycycline, the human NfL proteins persisted with a half-life of ∼3 weeks. The half-life was extended to months when an intermediate filament scaffold was present. These findings are broadly consistent with the half-lives of neurofilament proteins estimated from the decay of 3H-proline radiolabelling proteins in mouse retinal ganglion cell neurons (Nixon and Logvinenko, 1986; Rao et al., 2012).
Studies with conditional NEFL transgene suppression revealed that the turnover of neurofilament proteins is slower in large-calibre axons of the PNS having a high content of neurofilaments: human NfL protein levels expressed in transgenic mouse sciatic nerves were unchanged even after 3 months of suppression of the human NEFL transgene transcription by doxycycline treatment (Millecamps et al., 2007). Neurofilament proteins might last several months or even years in large axons with dense neurofilament networks. In conjunction with the observation that the rate of human NfL transport is enhanced by an order of magnitude (10 mm/day) in peripheral axons lacking a neurofilament network, these results support the notion that a stationary neurofilament network in axons (Nixon and Logvinenko, 1986) contributes to slowing both the turnover of neurofilament and its net transport. The local neurofilament density thus is a key determinant of the half-life of neurofilament proteins in axons.
Phosphorylation of neurofilaments, which causes their dissociation from the kinesin motor for incorporation into stable cytoskeletal networks in axons (Yabe et al., 2000), is one molecular mechanism by which this resistance to degradation is conferred (Yuan et al., 2017); proteolysis of neurofilaments is increased in NF-(H/M)tailΔ mice, in which the heavily phosphorylated carboxyl-terminal tail domains of NfH and NfL are deleted (Rao et al., 2012). Extrapolation from these observations suggests that the degradation of neurofilaments may be regulated differentially in different types of neurons, during different developmental stages and depending on whether an axon is myelinated.
Various proteases can contribute to neurofilament proteolysis (Perrot et al., 2008). The calcium-activated proteases have a high degree of substrate specificity for intermediate filaments. Calpain is capable of a limited proteolysis of neurofilaments. One provocative study, still to be replicated to our knowledge, proposed that neurofilament is cleared after transport to the synaptic terminal, at least in part through activities of calcium-activated proteases such as calpain (Roots, 1983). Calpain proteolysis is one of the key molecular processes in Wallerian degeneration (Wang et al., 2012), as well as in growth cone formation. Other non-specific proteases can also trigger neurofilament turnover and generate neurofilament peptides (Perrot et al., 2008). These include cathepsin D (Nixon and Marotta, 1984) and caspases 6 and 8 (Shabanzadeh et al., 2015).
Important insights into degradation pathways more specific to neurofilaments have come from the study of rare, inherited genetic diseases characterized by prominent abnormal neurofilament accumulation in axons (Table 1): ARSACS (Engert et al., 2000), the early onset CMT type 2R (Ylikallio et al., 2013) and GAN (Bomont et al., 2000).
ARSACS is an early onset autosomal recessive CNS disorder caused by mutations in the gene encoding sacsin (SACS), a protein with both putative ubiquitin and chaperone functions. ARSACS is found world-wide and is the second most common inherited cause of ataxia (Engert et al., 2000). Accumulation and abnormal bundling of neurofilaments is the most prominent neuropathological finding in affected neurons, which include Purkinje cells (Lariviere et al., 2015). Fibroblasts derived from ARSACS patients or SACS knockout fibroblasts show pathological intermediate filament structures, with vimentin filaments collapsed around or beside the nucleus, rather than radiating outwards to the plasma membrane (Duncan et al., 2017). There is also altered distribution of organelles (including autophagosomes and lysosomes), displacement of the nucleus (Duncan et al., 2017) and mitochondrial pathology (Girard et al., 2012; Lariviere et al., 2015; Bradshaw et al., 2016).
Sacsin has several functionally distinct domains: a C-terminal ubiquitin-like domain (Ubl), three SIRPT domains (SIRPT1 bearing homology with the ATP-binding domain of HSP90), a J-domain, and an N-terminal HEPN domain (Engert et al., 2000). The presence of the Ubl and domains with chaperone homology suggests that sacsin is involved in neurofilament assembly and/or turnover. The Ubl domain has been shown to interact with a proteasomal component (Parfitt et al., 2009). In cell models, expression of the Ubl and J-domain peptides both inhibited normal assembly of neurofilament, whereas SIRPT1 and HEPN domain peptides promoted neurofilament protein assembly. In cultured SACS−/− motor neurons modelling the pathology in ARSACS, selective expression of both the SIRPT1 and J-domain peptides led to the clearance of neurofilament bundles in a similar way to that seen with overexpression of heat shock proteins (Gentil et al., 2019). These data highlight multifunctional roles of sacsin as a key player in organizing neurofilament proteins and in regulating subunit levels, assembly, maturation of their supramolecular structures and turnover (Engert et al., 2000).
Mutations in TRIM2 (tripartite motif containing 2) cause a rare early-onset, recessive form of CMT type 2R (Ylikallio et al., 2013). TRIM2 is an E3 ubiquitin ligase that binds and ubiquitinates NfL (Balastik et al., 2008). The pathology shows swollen axons with abnormal aggregation of neurofilaments in myelinated fibres. CNS neurodegeneration with tremor, ataxia and seizures are seen in a Trim2 gene trap mouse line (Balastik et al., 2008).
A central role for E3-ligase activity in the turnover of neurofilaments was discovered through studies of GAN (Bomont et al., 2000), a fatal autosomal recessive neuropathy (Cavalier et al., 2000) in which giant axons (up to 50 μm in diameter) filled with densely packed and disorganized neurofilaments are found throughout the PNS and CNS (Asbury et al., 1972). Begining early in infancy, the disease is associated with progressive loss of motor and sensory function (Kuhlenbaumer et al., 1993; Johnson-Kerner et al., 2014). CNS symptoms arise later from cerebellar dysfunction and cognitive impairments. In the most severe cases, GAN is fatal in young adulthood, usually before the third decade of life. The pathological aggregates of GAN, found both in neuronal and in non-neuronal tissues, include multiple subtypes of intermediate filaments (Prineas et al., 1976). The neurofilament accumulation in nerves within the so-called ‘giant axons’ identified in nerve biopsies of patients are most characteristic (Asbury et al., 1972). However, with the broad range of abnormal intermediate filaments aggregates seen in patients (e.g. extending from desmin in muscles to GFAP in astrocytes, keratin in hair and vimentin in numerous cell types), GAN is considered as a unique disease of the intermediate filaments network.
With its N-terminal BTB domain and C-terminal Kelch domain (Bomont et al., 2000), gigaxonin belongs to the large family of BTB-Kelch proteins. It is presumed to act in the ubiquitin proteasome system (UPS) through interactions between its BTB domain and the Cul3 subunit of E3 ubiquitin ligase complexes (Furukawa et al., 2003). Through interaction with the Kelch domain, gigaxonin is predicted to target its partners for ubiquitin-mediated degradation. Among putative partners identified by mass spectrometry approaches or double screening in yeast (Johnson-Kerner et al., 2015b), intermediate filaments are so far the major biological targets for gigaxonin, as confirmed in cellular and animal models of the pathology. Indeed, numerous types of intermediate filaments are abnormally aggregated in disease, e.g. vimentin in primary fibroblasts from patients (Bomont and Koenig, 2003), peripherin and NfL in motor neuron-like cells derived from induced pluripotent stem cells (Johnson-Kerner et al., 2015a), and vimentin, NfL, NfM, NfH and α-internexin in two different GAN mouse models (Dequen et al., 2008; Ganay et al., 2011). While the neurological phenotypes of these mice are mild in comparison to the human pathology, both GAN mouse models exhibit pronounced alterations of the abundance and spatial distribution of neuronal intermediate filaments throughout the PNS and CNS. The putative role of gigaxonin in regulating the steady state intermediate filament levels also has been demonstrated compellingly in vitro: lentiviral over-expression of gigaxonin was sufficient to drive the clearance of multiple wild-type intermediate filaments (vimentin, peripherin and NfL) and intermediate filaments bundles (NfL, NfM, NfH, peripherin and α-internexin) in GAN cells (Mahammad et al., 2013; Israeli et al., 2016). This effect is mediated by the interaction of gigaxonin with the central rod domain common to all intermediate filament types (Mahammad et al., 2013), supporting the clinical and mouse model data suggesting a key role of gigaxonin in controlling the degradation of the whole intermediate filament family (Bomont, 2016).
Gigaxonin appears to be the only E3 ligase able to target neurofilaments (and intermediate filament proteins more generally) for degradation. However, several questions remain to be answered. What forms of intermediate filament (e.g. short intermediate filaments, single unit length filaments, mature filaments or multimeric forms) are targeted by gigaxonin? What specific ubiquitination chain type and degradative route is involved? Surprisingly, while a role for gigaxonin in controlling NfL, NfM and NfH abundance has been demonstrated in cells in vitro and in two distinct GAN mouse models (Dequen et al., 2008; Ganay et al., 2011), direct evidence for neurofilament ubiquitination by gigaxonin remains to be discovered. Experimental challenges to addressing this are the multi-subunit nature of the gigaxonin-E3 ligase complex and the insolubility of gigaxonin when ectopically expressed; ubiquitin laddering of intermediate filaments upon gigaxonin expression has been extremely challenging, although reported once for peripherin in GAN dorsal root ganglion (Israeli et al., 2016).
The proteasome has been partially implicated in vimentin degradation (Mahammad et al., 2013) and, as the observations above suggest, this may contribute to neurofilament turnover mediated by proteins such as TRIM2 or gigaxonin. However, recent findings also demonstrate a central role for the gigaxonin-E3 ligase in controlling the autophagy pathway through the ATG16L1 protein (Scrivo et al., 2019), presenting the additional possibility that the autophagy pathway may also play a role in neurofilament turnover; activation of autophagy is accompanied by reduced neurofilament levels (Chen et al., 2013).
Release and clearance of neurofilament peptides and proteins
While the nature of the neurofilament species detected with current immunoassays has not been determined because of the technical challenges posed by their low concentrations, it seems likely that most or all of the neurofilament detected in the CSF or peripheral blood compartments are peptides generated from partial degradation of neurofilament in the neuron. With injury to peripheral nerves, these might be expected to arise from axons, but, major contributions from synaptic neurofilaments are possible in the brain and spinal cord because of the relative abundance in the synaptic compartment within the CNS.
The mechanisms for release of these neurofilament peptides from neurons are not yet defined (Khalil et al., 2018) and we can do little more than speculate at this time. However, testable hypotheses regarding mechanisms of release can be made based on what is known about pathways for release of other neuronal peptides and proteins. Intracellular endosomal organelles known as multivesicular bodies may play important roles in the release of peptides (Von Bartheld and Altick, 2011). This may occur through ‘back-fusion’ events and budding from the plasma membrane to generate microvesicles (100–2000 nm diameter) (Kleijmeer et al., 2001; Murk et al., 2002) or through release of smaller endosomally-derived exosomes (30–140 nm) (Faure et al., 2006; Lachenal et al., 2011). By contrast, neuronal multivesicular bodies have been shown to contain protein aggregates that accumulate in Parkinson’s and Alzheimer’s disease, for example (Nixon et al., 2005). They are more abundant with neurodegenerative diseases and in ageing (Nakadate et al., 2006), where they are associated with enhanced autophagy (Truant et al., 2008). Microvesicle production can also increase with higher intracellular [Ca2+] (as with excitotoxic injury), cell stress or with inflammation (Sproviero et al., 2018). Upregulation of molecular chaperones rescues the neurofilament phenotype in SACS knockout neurons (Gentil et al., 2019) and in motor neurons expressing mutant NfL associated with CMT (Tradewell et al., 2009). Chaperones have the potential to increase a more mobile pool of neurofilament proteins accessible to secretory mechanisms such multivesicular bodies (Manek et al., 2018).
Pathways for degradation [e.g. proteasomes, autophagy (Nixon, 2006) or release into the extracellular space (Wang et al., 2006) for degradation by extracellular proteases] could be differentially important in the context of healthy, injured or chronically damaged neurons. The varicosities or large spheroids which occur with neurodegeneration may modify this (Coleman, 2005; Beirowski et al., 2010).
We speculate that the different mechanisms of release may have different kinetics and that they could lead to variable relative levels of different types of neurofilament peptide fragments in blood or CSF. Quantitative interpretations of the relative concentrations of neurofilament or their peptide fragments in either compartment (or between compartments) demands a better understanding of the mechanisms of these release pathways, as well as how peptides are transported from the parenchyma into the fluid compartments and between the CSF and blood.
The mechanisms by which neurofilament traffic between parenchymal, CSF and blood compartments also are unknown. However, the apparently general pathways by which large molecules such as amyloid-β pass from the interstitial fluid (ISF) of the brain into the CSF and blood suggest a tentative model for how neurofilament species could be transported between compartments.
Soluble metabolites or peptides released from cells in most organs are absorbed directly into the blood or drain via lymphatic vessels to regional lymph nodes (Engelhardt et al., 2017). Lymphatic drainage may contribute to neurofilament peptide distribution with peripheral nerve injury. However, the brain constitutes a specialized compartment both because of the selective permeability of the blood–brain barrier and because there are no conventional lymphatic vessels in the CNS. Soluble tracers such as radioionodated serum albumin (RISA) injected into the ISF of the brain drain to cervical lymph nodes along the walls of cerebral arteries (Szentistvanyi et al., 1984). This drainage occurs initially along basement membranes that surround capillaries and then along the basement membranes between smooth muscle cells in the tunica media of intracerebral and leptomeningeal arteries (Carare et al., 2008). Together, this constitutes an intramural peri-arterial drainage (IPAD) pathway (Albargothy et al., 2018) (Fig. 5).
Figure 5.

Fluid balance in the brain and the IPAD pathway. Entry and drainage of fluid into and from the brain is along basement membranes associated with the walls of arteries: (1) CSF enters the surface of the brain along pial-glial basement membranes on the outer aspects of cortical arteries; (2) CSF mixes with ISF; to then, (3) leave the brain along IPAD pathways. (B) A length of cerebral artery in a mouse brain showing fluorescent amyloid-β protein co-localizing (magenta) with collagen IV in basement membranes between smooth muscle cells in the tunica media of the artery wall; this is part of the IPAD pathway (indicated by arrows) along which amyloid-β is draining out of the brain. The IPAD pathway for amyloid-β and, by inference, perhaps that for neurofilament peptides, forms a spiral pattern along the artery wall (smooth muscle cells in the section of artery wall illustrated are stained green). This figure is modified from an original figure reproduced as Fig. 1d of Albargothy et al. (2018).
IPAD might provide a route for the drainage of soluble peptides and proteins from the extracellular spaces in the brain to cervical lymph nodes (Szentistvanyi et al., 1984; Carare et al., 2008; Albargothy et al., 2018). With impaired IPAD, tracer labelled protein injected intracerebrally also accumulates around veins draining from the brain (Hawkes et al., 2011), the walls of which appear to provide a downstream drainage pathway (Iliff et al., 2012), although the specific route for transport of molecules in the paravenous compartment to lymph nodes along veins has not been defined. Modelling studies suggest that the motive force for IPAD could be derived from waves of contraction of smooth muscle cells (vasomotion) in the walls of cerebral arteries and arterioles (Aldea et al., 2019). Any additional motive force along veins, if it is needed, has not been characterized.
Clearance through this mechanism may change with ageing or pathology. For example, age-related changes in artery walls (Hawkes et al., 2011) impair IPAD and appear to be a factor limiting elimination of amyloid-β from the ageing brain in the genesis of Alzheimer’s disease (e.g. reflected by the accumulation of amyloid-β within the IPAD pathways in cerebral amyloid angiopathy) (Weller et al., 2015; Keable et al., 2016).
Levels of proteins or peptides in the CSF cannot be assumed to reflect levels in the ISF directly. Although some reports have suggested that ISF and solutes from the brain drain directly into CSF, this conclusion is confounded in most cases by uncertainty because direct leakage of tracer from intracerebral injections into the CSF cannot be excluded. In better controlled studies, only 10–15% of tracer injected into cerebral hemispheres passes into the CSF (Szentistvanyi et al., 1984; McIntee et al., 2016); 85% of the ISF passes to cervical lymph nodes via IPAD (Szentistvanyi et al., 1984). As yet, there are no direct measurements of the proportion of neurofilament released from the brain that reaches the CSF. It also is not known whether there is any regional neuroanatomical selectivity for this.
CSF drainage from the subarachnoid space into the lymphatic system occurs through mechanisms distinct from those of the IPAD pathway for the drainage of ISF. In experimental animals and in humans, drainage of CSF into lymphatic vessels of the nasal mucosa via the cribriform plate appears to be a major lymphatic drainage pathway (Kida et al., 1993; de Leon et al., 2017), although other routes, including dural lymphatics, have been described (Kida et al., 1993; Aspelund et al., 2015; Louveau et al., 2015). The proportion of CSF that drains directly into the blood through the arachnoid granulations is uncertain.
Together, these observations raise cautions for inferences regarding the extent or severity of CNS neuronal pathology based on neurofilament concentration measures in CSF or peripheral blood. The relationships between neurofilament concentrations in CSF and peripheral blood may be influenced by the rate of release of neurofilament species from the injured or degenerating neuron, where it is occurring in the CNS and variation in the kinetics of clearance related to ageing or direct effects of pathology on the clearance mechanisms themselves.
Changes either in rates of synthesis of neurofilament proteins or differences in mechanisms and rates of peptide release could lead to differences in measured levels of neurofilament or its peptide fragments in peripheral blood or CSF. As far as we are aware, there are no data describing how turnover in any compartment might change with disease in individuals. Nor, as is described above, is anything specific known about mechanisms of release of neurofilaments or their peptides from injured neurons. This knowledge gap substantively limits quantitative interpretations of neurofilament peptide levels in peripheral blood or CSF.
Data defining the dynamics of neurofilament turnover in healthy individuals, with ageing and in those with diseases associated with increased neurofilament peptide concentrations in peripheral blood or CSF are needed. Although not yet applied to these problems, a promising approach for obtaining precise estimates of the kinetics of synthesis and elimination of neurofilament in blood and CSF in healthy humans and those with disease is the Stable Isotope Labelling Kinetic (SILK) method (Bateman et al., 2006; Paterson et al., 2019). SILK can measure protein turnover rate and half-life minimally invasively in humans. In the past decade, SILK has been used to characterize the kinetics of turnover of pathological protein in a range of neurological disorders, e.g. amyloid-β, APOE and tau in Alzheimer’s disease and SOD1 in motor neuron disease (Mawuenyega et al., 2010; Basak et al., 2012; Crisp et al., 2015). The method relies on serial analyses of the body fluid or tissue of interest after a single period of infusion of stable isotope [e.g. 2H (deuterium) or 13C]-labelled amino acids. These then are incorporated into proteins. The subsequent enrichment and decay of enrichment in target proteins after this ‘pulse-labelling’ is measured in the fluid compartment of interest over time by mass spectrometry (Bateman et al., 2006). Studies using SILK have demonstrated that structural proteins such as tau have a significantly longer half-life (∼20 days) than do membrane proteins such as amyloid-β (∼10 h) (Bateman et al., 2006; Sato et al., 2018).
Based on this observation and rodent data demonstrating very slow turnover of neurofilaments incorporated into filamentous structures in the axon (Nixon and Logvinenko, 1986), SILK neurofilament studies may require the use of a SILK protocol specialized for use with very long half-life proteins. This does not promise to be a straightforward task. Protocols for very long-lived peptides face three major technical challenges: (i) sampling of biofluids from participants may need to be performed over periods of several months; (ii) the dilution of the incorporated tracer with non-labelled proteins synthesized over the study period leads to low tracer incorporation rate; and (iii) a mass spectrometry assay that is sufficiently sensitive to detect the low fraction of tracer incorporated into neurofilament present in biofluid has to be developed.
The last point is likely a major technical hurdle for the analysis of neurofilament peptides because of their low concentrations in CSF (1–10 ng/ml range) and peripheral blood (10–100 pg/ml range) (Petzold et al., 2006; Miyazawa et al., 2007; Zetterberg et al., 2016; Mattsson et al., 2017). Mass spectrometric (as opposed to the immunoglobulin capture assays described below) assays of neurofilaments in CSF or peripheral blood have not yet been reported; neurofilament SILK in CSF will require a limit of detection below 10 pg/ml. However, this sensitivity could be achieved theoretically using immuno-purification (IP) combined with the latest generation of mass spectrometers operating in targeted mass spectrometry mode (Gallien et al., 2012; Peterson et al., 2012; Gillette and Carr, 2013). Identifying antibodies that efficiently recover a representative range of neurofilament peptides will be a prerequisite. Development of such a mass spectrometric assay would pay dividends by enabling further neurofilament peptide characterization to test for differences in the nature of the species present in CSF or peripheral blood with ageing or diseases and characterizing post-translational modifications.
Evolution of NfL immunoassays
Interest in neurofilaments as a soluble biomarker of disease and its progression have risen dramatically in recent years, driven in part by evolution of the assay and the associated improvements in analytical sensitivity (Kuhle et al., 2016a). Improved analytical methods for detecting NfL or its constituent peptides in CSF and blood have enabled strong associations to be demonstrated between elevated NfL peptides (albeit with currently unspecified structural characteristics) and nervous system injury and disease (Table 2) (Khalil et al., 2018). Concentrations of NfL peptides in the CSF can be measured reliably by enzyme-linked immunosorbent assay (ELISA) using antibodies directed against the mid-domain rod region of the protein (Khalil et al., 2018). For a long time, there was only one ELISA for NfL available on the market (Petzold et al., 2010), but now additional assays exist (Gaetani et al., 2018). However, the analytical sensitivity of the ELISA (∼25–50 ng/l) precludes its general use for measurement of NfL in peripheral blood.
Table 2.
Major disorders reported to have associations with increased NfL concentration in peripheral blood
Advances in technology have enabled major extensions of the range of applications possible. Semi-sensitive electrochemiluminescence detection was the first approach that allowed disease-related increases in peripheral blood concentration to be measured in samples from patients with ALS (Gaiottino et al., 2013) or active multiple sclerosis (Kuhle et al., 2016a). A further major advance enabling new applications came in 2015, when the first ultrasensitive assay for NfL using single molecule array (SIMOA) technology to enhance the ELISA signal was described (Gisslen et al., 2016). This assay allowed concentrations in peripheral blood to be measured reliably even in people without PNS or CNS pathology. For the first time, correlations between CSF and peripheral blood levels of NfL could be demonstrated using this assay in patients with HIV encephalopathy (Gisslen et al., 2016). Paired CSF and peripheral blood measures showed similar dynamics following acute brain injury, suggesting relatively rapid trafficking between the compartments; concentrations of NfL reach a maximum around 40–70 days post-injury in both CSF and peripheral blood and normalize at similar rates within about 6 months (Bergman et al., 2016). However, in conditions in which central and peripheral neuronal injury or degeneration could be found, specific interpretations of blood measures in terms of these distinct pathologies may be ambiguous. Given that α-internexin is CNS-specific intermediate filament, a blood-based assay could potentially help distinguish CNS-specific pathologyin cases where the disease is associated with altered levels of this specific protein (Shaw, 2015). Whether neurofilament markers associated with central and peripheral nerve injury can be differentiated biochemically is an important topic for future research, e.g. through development of assays sensitive not just to the mid-domain, but also epitopes distributed more widely in the NfL protein.
Neurofilament peptide concentrations as biomarkers of brain injury or neurodegeneration
CSF and peripheral blood NfL are increased in most acute and chronic CNS diseases characterized by neuronal damage (Table 2) and correlate with longitudinal imaging findings of neurodegeneration (Khalil et al., 2018). Here, we briefly review applications of the assay to neuroinflammatory injury and neurodegeneration with specific reference to diseases where this approach has been most extensively researched. Serum or plasma NfL concentrations (either sample matrix works well) are moderately to strongly correlated with CSF concentration measures (correlation coefficients of 0.74 to 0.97) for diseases affecting the CNS primarily (Gaiottino et al., 2013); CSF findings with a range of neurodegenerative diseases (increased NfL concentrations in Alzheimer’s disease, frontotemporal dementia, vascular dementia and atypical parkinsonian disorders) have been replicated in peripheral blood (Zetterberg, 2016).
The potential utility of NfL as a biomarker of neuroinflammatory injury is well-illustrated by applications in multiple sclerosis, in which NfL assay results are being used as evidence routinely in some centres for continuing inflammatory disease in diagnosis, treatment monitoring or estimating prognosis if treatment is not changed. These assays also are increasingly integrated as secondary or exploratory measures in clinical trials (Sormani et al., 2019). Increased levels of NfH and NfL were first described in the CSF of patients with multiple sclerosis (Malmestrom et al., 2003; Norgren et al., 2004; Teunissen et al., 2009; Gunnarsson et al., 2011; Kuhle et al., 2013) in association with clinical relapses and proposed as biomarkers of acute inflammatory activity (Lycke et al., 1998). Moreover, it was recognized that CSF neurofilament concentrations tend to be increased across all clinical stages of multiple sclerosis (even in the absence of evidence for active inflammatory activity evident as a clinical relapse or detected using MRI) relative to healthy volunteer groups (Teunissen et al., 2009; Kuhle et al., 2011). The chronically increased levels detected with multiple sclerosis are 3–5-fold greater than those reported for some primary neurodegenerative diseases (e.g. frontotemporal dementia or ALS) (Gaiottino et al., 2013). With the introduction of the more sensitive SIMOA technology, peripheral blood NfL was been explored in several studies as a marker of otherwise occult acute disease activity, drug response or future disease progression (Khalil et al., 2018). Unlike other indirect and retrospective measures of neurodegeneration in multiple sclerosis used clinically now (e.g. MRI or magnetic resonance spectroscopy; De Stefano et al., 2007; Pini et al., 2016; Kalra, 2019), NfL measurements potentially allow neurodegeneration to be assessed in near ‘real-time’. Furthermore, NfL should be sensitive to neuronal damage in the brain and spinal cord, the latter being a CNS compartment where quantitative magnetic resonance-based imaging methods for assessment of neuronal damage are technically more difficult, less standardized and not yet able to be used routinely in the clinic (Disanto et al., 2017; Barro et al., 2018). However, there also are important limitations to the use of NfL concentrations in CSF or peripheral blood for disease monitoring of individual patients with multiple sclerosis (Berger and Stuve, 2019). For example, the lower levels of NfL that are found in most patients with multiple sclerosis outside of periods of acute inflammation still generally cannot be confidently interpreted as pathological if obtained as single time-point measurements (Kuhle et al., 2016b). While the evidence is still limited, serial peripheral blood NfL concentration measurements appear to be as sensitive as MRI for the assessment of treatment effects (Gasperini et al., 2019). Increases in peripheral blood (or CSF) NfL also might provide a potential biomarker of suboptimally controlled acute inflammatory activity in individuals at high risk who are being considered for a change in treatment but are without clinical evidence of a relapse or objective inflammatory changes on MRI. This type of information could become more important as evidence for continuing inflammatory activity is needed to stratify individuals with progressive forms of multiple sclerosis for treatment with new, highly active anti-inflammatory treatments (https://www.nice.org.uk/guidance/TA585). An analogous application is emerging for the management of children with type I spinal muscular atrophy (SMA) who are being treated with nusinersin and in whom CSF NfL provides a measure of the effectiveness of treatment (Olsson et al., 2019). Other conditions characterized by neuronal injury may also result in raised NfL measurements and therefore the clinical context or presentation must be borne in mind for their interpretation. For example, following hypoxic or traumatic brain injuries, NfL concentration increases within days, reaching a maximum weeks following the injury followed by normalization of concentration within 6–12 months (Shahim et al., 2016).
Neurofilaments are also increased in neurodegenerative diseases and neuropathies and may inform the clinical picture at different stages of disease. For example, increased serum NfL concentrations distinguished both symptomatic and presymptomatic causal gene mutation carriers from healthy controls in familial Alzheimer’s disease (Weston et al., 2017, 2019; Preische et al., 2019) and Huntington’s disease (Byrne et al., 2017), but also correlate with longitudinal measures of neurodegeneration in sporadic Alzheimer’s disease (Mattsson et al., 2017), frontotemporal dementia (Meeter et al., 2016), progressive supranuclear palsy (Donker Kaat et al., 2018) and Huntington’s disease (Johnson et al., 2018). Peripheral neuropathies also are associated with increased blood NfL concentrations; NfL concentrations in peripheral blood are higher (almost doubled) in patients with CMT peripheral neuropathy compared with healthy controls and correlate with disease severity as measured using clinical rating scales (Sandelius et al., 2018). However, just as with brain injury and neurodegeneration, the increased blood NfL must be contextualized for clinical interpretation of its significance with peripheral neuropathies. NfL biomarker concentrations do not differentiate between inflammatory and genetic peripheral neuropathies, for example.
There are other limitations to the use of neurofilament as a clinical biomarker. There is a ∼2.2% per year increase of concentration between the ages of 18 to 70 years (Disanto et al., 2017; Mattsson et al., 2017; Barro et al., 2018), but standardized, age-corrected, normative distributions of NfL in CSF and peripheral blood are not available to define values from individual subjects as being pathological. Measures of NfL concentrations in CSF or peripheral blood also do not distinguish the underlying pathology and cannot differentiate between neuronal damage arising from acute or chronic inflammatory injury and other contributions to neurodegeneration (e.g. relevant comorbidities) (Marrie, 2016). Lack of knowledge of the kinetics of NfL peptide turnover in the blood also precludes confidence in the relative timing of presumed inflammatory events giving rise to NfL increases measured (Thelin et al., 2017). Finally, not only is the precise nature of the peptide (and thus whether it might change with disease stage or other factors) unknown, but the CNS pathology being assessed itself may be uncertain: is the major release of NfL peptide with CNS injury due to synaptic damage or turnover or, as with peripheral nerve injury, does it primarily reflect axonal damage?
Fundamental to addressing any of these questions will be to develop consensus amongst analytical laboratories for a harmonized assay standard to allow uniform interpretation of results between all laboratories. While the increasing diffusion of SIMOA is contributing to this now, other assay platforms also are in development by a number of diagnostic medicine companies. This continued commercial innovation may delay harmonization of assays and definition of the kinds of normative data that are needed for confident clinical use of measures from individual patients.
Conclusions and future directions
Neurofilaments play fundamental roles in the neuronal development, organization and function in the central and peripheral nervous systems. Primary roles of neurofilament in the pathogenesis of ALS (Figlewicz et al., 1994; Tomkins et al., 1998; Gros-Louis et al., 2004; Leung et al., 2004) and CMT (Mersiyanova et al., 2000; Rebelo et al., 2016) and secondary pathogenic roles in other disorders have been discovered. Most striking to date among the latter are disorders arising from impairments in normal mechanisms for neurofilament degradation that are associated with progressive and severe axonal pathology. However, fundamental questions still remain concerning basic mechanisms regulating neurofilament expression, assembly and turnover.
The identification of functional roles for synaptic neurofilament in modulation of excitatory glutamatergic activity (Huntley et al., 1994; Ratnam and Teichberg, 2005; Yuan et al., 2018a) has opened an entirely new range of investigations of neurofilament neurobiology. The contributions of the synaptic pool to neuronal dysfunction associated with schizophrenia-like behaviours in an NfL−/− mouse suggests the potential for abnormalities in synaptic neurofilament to contribute to the genesis of neuropsychiatric diseases more generally (Yuan et al., 2018a). Although the disease relevance is still speculative, the importance of the question and the novelty of this neurobiology make better understanding of the structurally unique synaptic neurofilament, its organization and its functions a priority.
However, understanding how neurofilaments are degraded and which and how degradation products are released from neurons is a particularly urgent agenda given the degree of clinical interest in using neurofilaments in CSF and peripheral blood as biomarkers. Physiological mechanisms for more dynamic control through post-translational modifications need to be better defined. For example, how is neurofilament phosphorylation regulated relative to the targeting of neurofilament for E3 ligase and protease activity? Exactly what forms of neurofilament (short intermediate filaments, unit length filaments, tetramers or dimers) are the major substrates for degradation with normal turnover? Applications of neurofilament measures for assessment of pathology demand some appreciation for whether these mechanisms of neurofilament degradation and release are altered with disease or injury. We hypothesize that they may vary. For example, oxidative stress leads to increased protein carbonylation and degradation of carbonylated cytoskeletal proteins including NfM and NfH is largely mediated by calpains with involvement of proteasomes (Smerjac et al., 2018), suggesting a particular role for this degradation mechanism in the context of oxidative pathologies.
Technological advances in measurement of the low concentrations of neurofilaments (particularly NfL) in CSF or peripheral blood have allowed exploration of how levels increase with ageing, brain injury and neurodegenerative diseases. These suggest that neurofilament concentration measures in these compartments have potential clinical utility as indices of peripheral or central nerve damage with trauma (Shahim et al., 2016) or neurodegeneration (Weston et al., 2017, 2019; Preische et al., 2019). Serial measures can be sensitive to subclinical disease activity in multiple sclerosis in ways that suggest the potential to monitor treatment responses (Lycke et al., 1998), potentially at an individual patient level with active disease. Particular clinical impact could arise with use of NfL measures as evidence to support therapeutic decisions regarding continuing disease activity and treatment response when other clinical or imaging evidence is lacking. They may play a role in better estimating prognosis for patients or patient groups, as well.
However, at this point, increased NfL measures in CSF or peripheral blood are non-specific to disease aetiology. The molecular characteristics of the precise peptide species being measured are uncertain and the mechanisms of their release and trafficking from the parenchyma to CSF or blood are speculative. Interpretation of peripheral blood measures of NfL also can be uncertain when both CNS and PNS injury is possible, such as after some forms of trauma. Can the release of NfL peptides and their levels in blood or CSF be related quantitatively to the degree of neuronal injury or does relationship vary substantially with the nature of the pathological insult? Are there disease- or stage-specific differences between the neurofilament species detected? What is the time course of injury over which they are reporting? Do the differences seen with age or disease solely reflect neurodegenerative changes, or could they also reflect differences in transport or turnover? If medical decisions are to be made based on these measures, answering such questions will become important.
We believe that basic questions like these emphasize the need for some caution in interpretation of neurofilament measures in peripheral blood or CSF from individual patients. At the same time, they also highlight why there is currently such excitement among those interested in the neurobiology of intermediate filaments. Moreover, with a growing range of new tools for characterizing major aspects of their biology (e.g. SILK), it is timely to ask these questions. The strong foundations that have been laid for discovery, the availability of new tools and approaches and practical importance of developing confidence in understanding neurofilament better, all highlight both the clinical promise for NfL as a biomarker and the great potential for future investigation of the neurobiology of NfL and intermediate filaments more generally.
Acknowledgements
This review was written following an open workshop concerning neurofilament biology and use as biomarker including all of the co-authors that was held at Imperial College London on 18 Nov 2018 (https://www.imperial.ac.uk/dementia-research-institute/seminars–events/past-events/neurofilaments-as-a-biomarker/). The workshop was initiated and planned by Prof. Paul Matthews and Prof. Henrik Zetterberg.
Funding
Funding for the workshop was provided by the UK Dementia Research Institute at Imperial College and an educational grant provided by Biogen. P.B.’s laboratory was funded for neurofilament biology by INSERM (ATIP-Avenir program) and the Association Française contre les Myopathies (AFM). R.O.C. and R.O.W.’s laboratory was funded by Alzheimer’s Research UK (ARUK) and Biogen. The R.A.N. laboratory has been supported by National Institute on Aging (R01AG0560; P01AG017617). H.Z. is a Wallenberg Academy Fellow supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Swedish Brain Foundation (FO2019-0228) and the UK Dementia Research Institute at UCL. P.M.M. has been in receipt of generous personal and research support from the Edmond J Safra Foundation and Lily Safra, the Imperial College Healthcare Trust – National Institute for Health Research (NIHR) Biomedical Research Centre, an NIHR Senior Investigator’s Award, the Medical Research Council and the UK Dementia Research Institute, which is supported by the Medical Research Council, The Alzheimer’s Society and Alzheimer’s Research UK.
Competing interests
H.Z. has served at scientific advisory boards for Roche Diagnostics, Samumed, CogRx and Wave, has given lectures in symposia sponsored by Biogen and Alzecure, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures-based platform company at the University of Gothenburg (all outside submitted work). P.M.M. acknowledges consultancy fees from Adelphi Communications, Biogen, Celgene and Roche. He has received honoraria or speakers’ honoraria from Biogen, Novartis and Roche, and has received research or educational funds from Biogen, GlaxoSmithKline, Nodthera and Novartis. He is a paid member of the Scientific Advisory Board for Ipsen Pharmaceuticals. All of these, except the educational grant from Biogen described above, are outside of the scope of this review.
Glossary
- ALS =
amyotrophic lateral sclerosis
- ARSACS =
autosomal recessive spastic ataxia of Charvleoix-Saguenay
- CMT =
Charcot-Marie-Tooth disease
- D1R =
dopamine D1 receptor
- GAN =
giant axonal neuropathy
- GluN1 =
glutamate ionotropic receptor NMDA type subunit 1
- IPAD =
intramural peri-arterial drainage pathway
- ISF =
interstitial fluid
- LTP =
long term potentiation
- NfH/L/M =
neurofilament heavy/light/medium chain
- NMDA =
N-methyl-d-aspartate
- SILK =
stable isotope labelling kinetic
References
- Abe A, Numakura C, Saito K, Koide H, Oka N, Honma A, et al. Neurofilament light chain polypeptide gene mutations in Charcot-Marie-Tooth disease: nonsense mutation probably causes a recessive phenotype. J Hum Genet 2009; 54: 94–7. [DOI] [PubMed] [Google Scholar]
- Ackerley S, Grierson AJ, Brownlees J, Thornhill P, Anderton BH, Leigh PN, et al. Glutamate slows axonal transport of neurofilaments in transfected neurons. J Cell Biol 2000; 150: 165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adebola AA, Di Castri T, He CZ, Salvatierra LA, Zhao J, Brown K, et al. Neurofilament light polypeptide gene N98S mutation in mice leads to neurofilament network abnormalities and a Charcot-Marie-Tooth Type 2E phenotype. Hum Mol Genet 2015; 24: 2163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol 2018; 136: 139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G.. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci 2019; 11: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor S, van der Star BJ, Bosca I, Raffel J, Gnanapavan S, Watchorn J, et al. Neurofilament light antibodies in serum reflect response to natalizumab treatment in multiple sclerosis. Mult Scler 2014; 20: 1355–62. [DOI] [PubMed] [Google Scholar]
- Asbury AK, Gale MK, Cox SC, Baringer JR, Berg BO.. Giant axonal neuropathy–a unique case with segmental neurofilamentous masses. Acta Neuropathol 1972; 20: 237–47. [DOI] [PubMed] [Google Scholar]
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015; 212: 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badner JA, Gershon ES.. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry 2002; 7: 405–11. [DOI] [PubMed] [Google Scholar]
- Balastik M, Ferraguti F, Pires-da Silva A, Lee TH, Alvarez-Bolado G, Lu KP, et al. Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc Natl Acad Sci U S A 2008; 105: 12016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro C, Benkert P, Disanto G, Tsagkas C, Amann M, Naegelin Y, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018; 141: 2382–239. [DOI] [PubMed] [Google Scholar]
- Basak JM, Kim J, Pyatkivskyy Y, Wildsmith KR, Jiang H, Parsadanian M, et al. Measurement of apolipoprotein E and amyloid beta clearance rates in the mouse brain using bolus stable isotope labeling. Mol Neurodegener 2012; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM.. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med 2006; 12: 856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Julien JP.. Peripherin-mediated death of motor neurons rescued by overexpression of neurofilament NF-H proteins. J Neurochem 2003; 85: 248–56. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Nguyen MD, Julien JP.. Late onset of motor neurons in mice overexpressing wild-type peripherin. J Cell Biol 1999; 147: 531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck R, Deek J, Safinya CR.. Structures and interactions in ‘bottlebrush’ neurofilaments: the role of charged disordered proteins in forming hydrogel networks. Biochem Soc Trans 2012; 40: 1027–31. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Nogradi A, Babetto E, Garcia-Alias G, Coleman MP.. Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration. J Neuropathol Exp Neurol 2010; 69: 455–72. [DOI] [PubMed] [Google Scholar]
- Berger T, Stuve O.. Neurofilament light chain: an important step toward a disease biomarker in multiple sclerosis. Neurology 2019; 92: 451–2. [DOI] [PubMed] [Google Scholar]
- Bergman J, Dring A, Zetterberg H, Blennow K, Norgren N, Gilthorpe J, et al. Neurofilament light in CSF and serum is a sensitive marker for axonal white matter injury in MS. Neurol Neuroimmunol Neuroinflamm 2016; 3: e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet A, Berges R, Frank R, Robert P, Peterson AC, Eyer J.. Neurofilaments bind tubulin and modulate its polymerization. J Neurosci 2009; 29: 11043–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomont P. Degradation of the intermediate filament family by gigaxonin. Methods Enzymol 2016; 569: 215–31. [DOI] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 2000; 26: 370–4. [DOI] [PubMed] [Google Scholar]
- Bomont P, Koenig M.. Intermediate filament aggregation in fibroblasts of giant axonal neuropathy patients is aggravated in non dividing cells and by microtubule destabilization. Hum Mol Genet 2003; 12: 813–22. [DOI] [PubMed] [Google Scholar]
- Bradshaw TY, Romano LE, Duncan EJ, Nethisinghe S, Abeti R, Michael GJ, et al. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum Mol Genet 2016; 25: 3232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragina L, Conti F.. Expression of neurofilament subunits at neocortical glutamatergic and GABAergic synapses. Front Neuroanat 2018; 12: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne LM, Rodrigues FB, Blennow K, Durr A, Leavitt BR, Roos RAC, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: a retrospective cohort analysis. Lancet Neurol 2017; 16: 601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Melo D, Droppelmann CA, He Z, Volkening K, Strong MJ.. Altered microRNA expression profile in amyotrophic lateral sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain 2013; 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol 2008; 34: 131–44. [DOI] [PubMed] [Google Scholar]
- Cavalier L, BenHamida C, Amouri R, Belal S, Bomont P, Lagarde N, et al. Giant axonal neuropathy locus refinement to a <590 kb critical interval. Eur J Hum Genet 2000; 8: 527–34. [DOI] [PubMed] [Google Scholar]
- Chen JX, Sun YJ, Wang P, Long DX, Li W, Li L, et al. Induction of autophagy by TOCP in differentiated human neuroblastoma cells lead to degradation of cytoskeletal components and inhibition of neurite outgrowth. Toxicology 2013; 310: 92–7. [DOI] [PubMed] [Google Scholar]
- Chou SM, Wang HS, Taniguchi A, Bucala R.. Advanced glycation endproducts in neurofilament conglomeration of motoneurons in familial and sporadic amyotrophic lateral sclerosis. Mol Med 1998; 4: 324–32. [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 2005; 6: 889–98. [DOI] [PubMed] [Google Scholar]
- Corbo M, Hays AP.. Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J Neuropathol Exp Neurol 1992; 51: 531–7. [DOI] [PubMed] [Google Scholar]
- Cote F, Collard JF, Julien JP.. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 1993; 73: 35–46. [DOI] [PubMed] [Google Scholar]
- Crisp MJ, Mawuenyega KG, Patterson BW, Reddy NC, Chott R, Self WK, et al. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest 2015; 125: 2772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras BT, Crawford TO, Finkel RS, Mercuri E, De Vivo DC, Oskoui M, et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann Clin Transl Neurol 2019; 6: 932–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Favila R, Stephano S, Mamo D, Ramirez BJ, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacol 2011; 36: 1781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, Li Y, Okamura N, Tsui WH, Saint-Louis LA, Glodzik L, et al. Cerebrospinal fluid clearance in Alzheimer Disease measured with dynamic PET. J Nucl Med 2017; 58: 1471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefano N, Filippi M, Miller D, Pouwels PJ, Rovira A, Gass A, et al. Guidelines for using proton MR spectroscopy in multicenter clinical MS studies. Neurology 2007; 69: 1942–52. [DOI] [PubMed] [Google Scholar]
- Dequen F, Bomont P, Gowing G, Cleveland DW, Julien JP.. Modest loss of peripheral axons, muscle atrophy and formation of brain inclusions in mice with targeted deletion of gigaxonin exon 1. J Neurochem 2008; 107: 253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequen F, Filali M, Lariviere RC, Perrot R, Hisanaga S, Julien JP.. Reversal of neuropathy phenotypes in conditional mouse model of Charcot-Marie-Tooth disease type 2E. Hum Mol Genet 2010; 19: 2616–29. [DOI] [PubMed] [Google Scholar]
- Disanto G, Barro C, Benkert P, Naegelin Y, Schadelin S, Giardiello A, et al. Serum neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol 2017; 81: 857–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donker Kaat L, Meeter LH, Chiu WZ, Melhem S, Boon AJW, Blennow K, et al. Serum neurofilament light chain in progressive supranuclear palsy. Parkinsonism Relat Disord 2018; 56: 98–101. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, Lariviere R, Bradshaw TY, Longo F, Sgarioto N, Hayes MJ, et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum Mol Genet 2017; 26: 3130–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyakin VV, Chen Y, Branch CA, Veeranna Yuan A, Rao M, et al. The contributions of myelin and axonal caliber to transverse relaxation time in shiverer and neurofilament-deficient mouse models. Neuroimage 2010; 51: 1098–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Fung ET, O’Brien RJ, Huganir RL.. Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J Neurosci 1998; 18: 720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, Vajkoczy P, Weller RO.. The movers and shapers in immune privilege of the CNS. Nat Immunol 2017; 18: 123–31. [DOI] [PubMed] [Google Scholar]
- Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 2000; 24: 120–5. [DOI] [PubMed] [Google Scholar]
- Faure J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 2006; 31: 642–8. [DOI] [PubMed] [Google Scholar]
- Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet 1994; 3: 1757–61. [DOI] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y.. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol 2003; 5: 1001–7. [DOI] [PubMed] [Google Scholar]
- Gaetani L, Hoglund K, Parnetti L, Pujol-Calderon F, Becker B, Eusebi P, et al. A new enzyme-linked immunosorbent assay for neurofilament light in cerebrospinal fluid: analytical validation and clinical evaluation. Alzheimers Res Ther 2018; 10: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafson AR, Savva C, Thorne T, David M, Gomez-Romero M, Lewis MR, et al. Breaking the cycle: reversal of flux in the tricarboxylic acid cycle by dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm 2019; 6: e562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiottino J, Norgren N, Dobson R, Topping J, Nissim A, Malaspina A, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 2013; 8: e75091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B.. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 2012; 11: 1709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganay T, Boizot A, Burrer R, Chauvin JP, Bomont P.. Sensory-motor deficits and neurofilament disorganization in gigaxonin-null mice. Mol Neurodegener 2011; 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia ML, Rao MV, Fujimoto J, Garcia VB, Shah SB, Crum J, et al. Phosphorylation of highly conserved neurofilament medium KSP repeats is not required for myelin-dependent radial axonal growth. J Neurosci 2009; 29: 1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini C, Prosperini L, Tintore M, Sormani MP, Filippi M, Rio J, et al. Unraveling treatment response in multiple sclerosis: a clinical and MRI challenge. Neurology 2019; 92: 180–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattringer T, Pinter D, Enzinger C, Seifert-Held T, Kneihsl M, Fandler S, et al. Serum neurofilament light is sensitive to active cerebral small vessel disease. Neurology 2017; 89: 2108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentil BJ, Lai GT, Menade M, Lariviere R, Minotti S, Gehring K, et al. Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. FASEB J 2019; 33: 2982–94. [DOI] [PubMed] [Google Scholar]
- Gentil BJ, Tibshirani M, Durham HD.. Neurofilament dynamics and involvement in neurological disorders. Cell Tissue Res 2015; 360: 609–20. [DOI] [PubMed] [Google Scholar]
- Georgiou DM, Zidar J, Korosec M, Middleton LT, Kyriakides T, Christodoulou K.. A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics 2002; 4: 93–6. [DOI] [PubMed] [Google Scholar]
- Gillette MA, Carr SA.. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods 2013; 10: 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard M, Lariviere R, Parfitt DA, Deane EC, Gaudet R, Nossova N, et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Natl Acad Sci U S A 2012; 109: 1661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA, Greengard P.. The neurobiology of dopamine signaling. Arch Neurol 2004; 61: 641–4. [DOI] [PubMed] [Google Scholar]
- Gisslen M, Price RW, Andreasson U, Norgren N, Nilsson S, Hagberg L, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV Infection: a cross-sectional study. EBioMedicine 2016; 3: 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros-Louis F, Lariviere R, Gowing G, Laurent S, Camu W, Bouchard JP, et al. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem 2004; 279: 45951–6. [DOI] [PubMed] [Google Scholar]
- Gunnarsson M, Malmestrom C, Axelsson M, Sundstrom P, Dahle C, Vrethem M, et al. Axonal damage in relapsing multiple sclerosis is markedly reduced by natalizumab. Ann Neurol 2011; 69: 83–9. [DOI] [PubMed] [Google Scholar]
- Hakansson I, Tisell A, Cassel P, Blennow K, Zetterberg H, Lundberg P, et al. Neurofilament levels, disease activity and brain volume during follow-up in multiple sclerosis. J Neuroinflamm 2018; 15: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Nakamura Y, Komai S, Kashiwagi Y, Tamura K, Goto T, et al. Site-specific phosphorylation of neurofilament-L is mediated by calcium/calmodulin-dependent protein kinase II in the apical dendrites during long-term potentiation. J Neurochem 2000; 75: 373–82. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 2011; 121: 431–43. [DOI] [PubMed] [Google Scholar]
- Heo S, Diering GH, Na CH, Nirujogi RS, Bachman JL, Pandey A, et al. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc Natl Acad Sci U S A 2018; 115: E3827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PN, Lasek RJ.. The slow component of axonal transport. Identification of major structural polypeptides of the axon and their generality among mammalian neurons. J Cell Biol 1975; 66: 351–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B.. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 2007; 27: 11496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horga A, Laura M, Jaunmuktane Z, Jerath NU, Gonzalez MA, Polke JM, et al. Genetic and clinical characteristics of NEFL-related Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry 2017; 88: 575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh ST, Kidd GJ, Crawford TO, Xu Z, Lin WM, Trapp BD, et al. Regional modulation of neurofilament organization by myelination in normal axons. J Neurosci 1994; 14: 6392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley GW, Vickers JC, Janssen W, Brose N, Heinemann SF, Morrison JH.. Distribution and synaptic localization of immunocytochemically identified NMDA receptor subunit proteins in sensory-motor and visual cortices of monkey and human. J Neurosci 1994; 14: 3603–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 2012; 4: 147ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israeli E, Dryanovski DI, Schumacker PT, Chandel NS, Singer JD, Julien JP, et al. Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum Mol Genet 2016; 25: 2143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomy H, Zhu Q, Couillard-Despres S, Beaulieu JM, Julien JP.. Disruption of type IV intermediate filament network in mice lacking the neurofilament medium and heavy subunits. J Neurochem 1999; 73: 972–84. [DOI] [PubMed] [Google Scholar]
- Jacquier A, Delorme C, Belotti E, Juntas-Morales R, Sole G, Dubourg O, et al. Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta Neuropathol Commun 2017; 5: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson J, Bjerke M, Ekman CJ, Sellgren C, Johansson AG, Zetterberg H, et al. Elevated concentrations of neurofilament light chain in the cerebrospinal fluid of bipolar disorder patients. Neuropsychopharmacology 2014; 39: 2349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EB, Byrne LM, Gregory S, Rodrigues FB, Blennow K, Durr A, et al. Neurofilament light protein in blood predicts regional atrophy in Huntington disease. Neurology 2018; 90: e717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Ahmad FS, Diaz AG, Greene JP, Gray SJ, Samulski RJ, et al. Intermediate filament protein accumulation in motor neurons derived from giant axonal neuropathy iPSCs rescued by restoration of gigaxonin. Hum Mol Genet 2015. a; 24: 1420–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Garcia Diaz A, Ekins S, Wichterle H.. Kelch domain of gigaxonin interacts with intermediate filament proteins affected in giant axonal neuropathy. PloS One 2015. b; 10: e0140157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Roth L, Greene JP, Wichterle H, Sproule DM.. Giant axonal neuropathy: an updated perspective on its pathology and pathogenesis. Muscle Nerve 2014; 50: 467–76. [DOI] [PubMed] [Google Scholar]
- Jordanova A, De JP, Boerkoel CF, Takashima H, De VE, Ceuterick C, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 2003; 126: 590–7. [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE.. Multiple phosphorylation sites in mammalian neurofilament polypeptides. J Biol Chem 1982; 257: 10467–70. [PubMed] [Google Scholar]
- Julien JP, Mushynski WE.. The distribution of phosphorylation sites among identified proteolytic fragments of mammalian neurofilaments. J Biol Chem 1983; 258: 4019–25. [PubMed] [Google Scholar]
- Julien JP, Tretjakoff I, Beaudet L, Peterson A.. Expression and assembly of a human neurofilament protein in transgenic mice provide a novel neuronal marking system. Genes Dev 1987; 1: 1085–95. [DOI] [PubMed] [Google Scholar]
- Kalra S. Magnetic resonance spectroscopy in ALS. Front Neurol 2019; 10: 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Rouach N, Nicoll RA, Bredt DS.. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc Natl Acad Sci U S A 2005; 102: 5600–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 2016; 1862: 1037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavapany S, Patel V, Zheng YL, Pareek TK, Bjelogrlic M, Albers W, et al. Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. MBoC 2007; 18: 3645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018; 14: 577–89. [DOI] [PubMed] [Google Scholar]
- Kida S, Pantazis A, Weller RO.. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol 1993; 19: 480–8. [DOI] [PubMed] [Google Scholar]
- Kioumourtzoglou MA, Rotem RS, Seals RM, Gredal O, Hansen J, Weisskopf MG.. Diabetes mellitus, obesity, and diagnosis of amyotrophic lateral sclerosis: a population-based study. JAMA Neurol 2015; 72: 905–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijmeer M, Ramm G, Schuurhuis D, Griffith J, Rescigno M, Ricciardi-Castagnoli P, et al. Reorganization of multivesicular bodies regulates MHC class II antigen presentation by dendritic cells. J Cell Biol 2001; 155: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Tung VW, Aghajanian J, Xu Z.. Antagonistic roles of neurofilament subunits NF-H and NF-M against NF-L in shaping dendritic arborization in spinal motor neurons. J Cell Biol 1998; 140: 1167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH.. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry 2006; 11: 737–47. [DOI] [PubMed] [Google Scholar]
- Kriz J, Zhu Q, Julien JP, Padjen AL.. Electrophysiological properties of axons in mice lacking neurofilament subunit genes: disparity between conduction velocity and axon diameter in absence of NF-H. Brain Res 2000; 885: 32–44. [DOI] [PubMed] [Google Scholar]
- Kuhle J, Barro C, Andreasson U, Derfuss T, Lindberg R, Sandelius A, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med 2016. a; 54: 1655–61. [DOI] [PubMed] [Google Scholar]
- Kuhle J, Barro C, Disanto G, Mathias A, Soneson C, Bonnier G, et al. Serum neurofilament light chain in early relapsing remitting MS is increased and correlates with CSF levels and with MRI measures of disease severity. Mult Scler 2016. b; 22: 1550–9. [DOI] [PubMed] [Google Scholar]
- Kuhle J, Kropshofer H, Haering DA, Kundu U, Meinert R, Barro C, et al. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology 2019; 92: e1007–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhle J, Leppert D, Petzold A, Regeniter A, Schindler C, Mehling M, et al. Neurofilament heavy chain in CSF correlates with relapses and disability in multiple sclerosis. Neurology 2011; 76: 1206–13. [DOI] [PubMed] [Google Scholar]
- Kuhle J, Malmestrom C, Axelsson M, Plattner K, Yaldizli O, Derfuss T, et al. Neurofilament light and heavy subunits compared as therapeutic biomarkers in multiple sclerosis. Acta Neurol Scand 2013; 128: e33–6. [DOI] [PubMed] [Google Scholar]
- Kuhle J, Nourbakhsh B, Grant D, Morant S, Barro C, Yaldizli O, et al. Serum neurofilament is associated with progression of brain atrophy and disability in early MS. Neurology 2017; 88: 826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Timmerman V, Bomont P.. Giant axonal neuropathy In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. Seattle, WA: GeneReviews(R); 1993. [Google Scholar]
- Lachenal G, Pernet-Gallay K, Chivet M, Hemming FJ, Belly A, Bodon G, et al. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci 2011; 46: 409–18. [DOI] [PubMed] [Google Scholar]
- Lariviere R, Gaudet R, Gentil BJ, Girard M, Conte TC, Minotti S, et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum Mol Genet 2015; 24: 727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Marszalek JR, Cleveland DW.. A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 1994; 13: 975–88. [DOI] [PubMed] [Google Scholar]
- Lee S, Pant HC, Shea TB.. Divergent and convergent roles for kinases and phosphatases in neurofilament dynamics. J Cell Sci 2014; 127: 4064–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CL, He CZ, Kaufmann P, Chin SS, Naini A, Liem RK, et al. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol 2004; 14: 290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet 2003; 73: 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jung P, Brown A.. Axonal transport of neurofilaments: a single population of intermittently moving polymers. J Neurosci 2012; 32: 746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Tsien JZ.. Memory and the NMDA receptors. N Engl J Med 2009; 361: 302–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Huang YJ, Lin CJ, Lane HY, Tsai GE.. NMDA neurotransmission dysfunction in mild cognitive impairment and Alzheimer’s disease. Curr Pharm Des 2014; 20: 5169–79. [DOI] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523: 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CH, Macdonald-Wallis C, Gray E, Pearce N, Petzold A, Norgren N, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015; 84: 2247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycke JN, Karlsson JE, Andersen O, Rosengren LE.. Neurofilament protein in cerebrospinal fluid: a potential marker of activity in multiple sclerosis. J Neurol Neurosurg Psychiatry 1998; 64: 402–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, et al. Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest 2013; 123: 1964–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmestrom C, Haghighi S, Rosengren L, Andersen O, Lycke J.. Neurofilament light protein and glial fibrillary acidic protein as biological markers in MS. Neurology 2003; 61: 1720–5. [DOI] [PubMed] [Google Scholar]
- Manek R, Moghieb A, Yang Z, Kumar D, Kobeissy F, Sarkis GA, et al. Correction to: protein biomarkers and neuroproteomics characterization of microvesicles/exosomes from human cerebrospinal fluid following traumatic brain injury. Mol Neurobiol 2018; 55: 6129. [DOI] [PubMed] [Google Scholar]
- Manser C, Stevenson A, Banner S, Davies J, Tudor EL, Ono Y, et al. Deregulation of PKN1 activity disrupts neurofilament organisation and axonal transport. FEBS Lett 2008; 582: 2303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariosa D, Kamel F, Bellocco R, Ye W, Fang F.. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur J Neurol 2015; 22: 1436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrie RA. Comorbidity in multiple sclerosis: some answers, more questions. Int J MS Care 2016; 18: 271–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews PM. Chronic inflammation in multiple sclerosis - seeing what was always there. Nat Rev Neurol 2019; 15: 582–93. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Andreasson U, Zetterberg H, Blennow K.. Alzheimer’s disease neuroimaging I. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol 2017; 74: 557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Insel PS, Palmqvist S, Portelius E, Zetterberg H, Weiner M, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol Med 2016; 8: 1184–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science (New York, NY) 2010; 330: 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntee FL, Giannoni P, Blais S, Sommer G, Neubert TA, Rostagno A, et al. In vivo differential brain clearance and catabolism of monomeric and oligomeric Alzheimer’s Abeta protein. Front Aging Neurosci 2016; 8: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016; 3: 623–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK.. Nature of glutamate alterations in schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry 2016; 73: 665–74. [DOI] [PubMed] [Google Scholar]
- Mersiyanova IV, Perepelov AV, Polyakov AV, Sitnikov VF, Dadali EL, Oparin RB, et al. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet 2000; 67: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Gowing G, Corti O, Mallet J, Julien JP.. Conditional NF-L transgene expression in mice for in vivo analysis of turnover and transport rate of neurofilaments. J Neurosci 2007; 27: 4947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa I, Nakashima I, Petzold A, Fujihara K, Sato S, Itoyama Y.. High CSF neurofilament heavy chain levels in neuromyelitis optica. Neurology 2007; 68: 865–7. [DOI] [PubMed] [Google Scholar]
- Murk JL, Stoorvogel W, Kleijmeer MJ, Geuze HJ.. The plasticity of multivesicular bodies and the regulation of antigen presentation. Semin Cell Dev Biol 2002; 13: 303–11. [DOI] [PubMed] [Google Scholar]
- Nakadate K, Noda T, Sakakibara S, Kumamoto K, Matsuura T, Joyce JN, et al. Progressive dopaminergic neurodegeneration of substantia nigra in the zitter mutant rat. Acta Neuropathol 2006; 112: 64–73. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Dynamic behavior and organization of cytoskeletal proteins in neurons: reconciling old and new findings. Bioessays 1998; 20: 798–807. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci 2006; 29: 528–35. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Logvinenko KB.. Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cell neurons. J Cell Biol 1986; 102: 647–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Marotta CA.. Degradation of neurofilament proteins by purified human brain cathepsin D. J Neurochem 1984; 43: 507–16. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Paskevich PA, Sihag RK, Thayer CY.. Phosphorylation on carboxyl terminus domains of neurofilament proteins in retinal ganglion cell neurons in vivo: influences on regional neurofilament accumulation, interneurofilament spacing, and axon caliber. J Cell Biol 1994; 126: 1031–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Sihag RK.. Neurofilament phosphorylation: a new look at regulation and function. Trends Neurosci 1991; 14: 501–6. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005; 64: 113–22. [DOI] [PubMed] [Google Scholar]
- Norgren N, Karlsson JE, Rosengren L, Stigbrand T.. Monoclonal antibodies selective for low molecular weight neurofilaments. Hybrid Hybridomics 2002; 21: 53–9. [DOI] [PubMed] [Google Scholar]
- Norgren N, Sundstrom P, Svenningsson A, Rosengren L, Stigbrand T, Gunnarsson M.. Neurofilament and glial fibrillary acidic protein in multiple sclerosis. Neurology 2004; 63: 1586–90. [DOI] [PubMed] [Google Scholar]
- Novakova L, Zetterberg H, Sundstrom P, Axelsson M, Khademi M, Gunnarsson M, et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017; 89: 2230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylen K, Csajbok LZ, Ost M, Rashid A, Karlsson JE, Blennow K, et al. CSF-neurofilament correlates with outcome after aneurysmal subarachnoid hemorrhage. Neurosci Lett 2006; 404: 132–6. [DOI] [PubMed] [Google Scholar]
- Olsson B, Alberg L, Cullen NC, Michael E, Wahlgren L, Kroksmark AK, et al. NFL is a marker of treatment response in children with SMA treated with nusinersen. J Neurol 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachter JS, Liem RK.. The differential appearance of neurofilament triplet polypeptides in the developing rat optic nerve. Dev Biol 1984; 103: 200–10. [DOI] [PubMed] [Google Scholar]
- Parfitt DA, Michael GJ, Vermeulen EG, Prodromou NV, Webb TR, Gallo JM, et al. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet 2009; 18: 1556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson RW, Gabelle A, Lucey BP, Barthelemy NR, Leckey CA, Hirtz C, et al. SILK studies-capturing the turnover of proteins linked to neurodegenerative diseases. Nat Rev Neurol 2019; 15: 419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, et al. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry 2008; 13: 1102–17. [DOI] [PubMed] [Google Scholar]
- Perez-Olle R, Jones ST, Liem RK.. Phenotypic analysis of neurofilament light gene mutations linked to Charcot-Marie-Tooth disease in cell culture models. Hum Mol Genet 2004; 13: 2207–20. [DOI] [PubMed] [Google Scholar]
- Perez-Olle R, Leung CL, Liem RK.. Effects of Charcot-Marie-Tooth-linked mutations of the neurofilament light subunit on intermediate filament formation. J Cell Sci 2002; 115: 4937–46. [DOI] [PubMed] [Google Scholar]
- Perrot R, Berges R, Bocquet A, Eyer J.. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol 2008; 38: 27–65. [DOI] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ.. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 2012; 11: 1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold A, Altintas A, Andreoni L, Bartos A, Berthele A, Blankenstein MA, et al. Neurofilament ELISA validation. J Immunol Methods 2010; 352: 23–31. [DOI] [PubMed] [Google Scholar]
- Petzold A, Hinds N, Murray NM, Hirsch NP, Grant D, Keir G, et al. CSF neurofilament levels: a potential prognostic marker in Guillain-Barre syndrome. Neurology 2006; 67: 1071–3. [DOI] [PubMed] [Google Scholar]
- Piehl F, Kockum I, Khademi M, Blennow K, Lycke J, Zetterberg H, et al. Plasma neurofilament light chain levels in patients with MS switching from injectable therapies to fingolimod. Mult Scler 2018; 24: 1046–54. [DOI] [PubMed] [Google Scholar]
- Pini L, Pievani M, Bocchetta M, Altomare D, Bosco P, Cavedo E, et al. Brain atrophy in Alzheimer’s Disease and aging. Ageing Res Rev 2016; 30: 25–48. [DOI] [PubMed] [Google Scholar]
- Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med 2019; 25: 277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prineas JW, Ouvrier RA, Wright RG, Walsh JC, McLeod JG.. Gian axonal neuropathy–a generalized disorder of cytoplasmic microfilament formation. J Neuropathol Exp Neurol 1976; 35: 458–70. [DOI] [PubMed] [Google Scholar]
- Rao MV, Campbell J, Yuan A, Kumar A, Gotow T, Uchiyama Y, et al. The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J Cell Biol 2003; 163: 1021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Houseweart MK, Williamson TL, Crawford TO, Folmer J, Cleveland DW.. Neurofilament-dependent radial growth of motor axons and axonal organization of neurofilaments does not require the neurofilament heavy subunit (NF-H) or its phosphorylation. J Cell Biol 1998; 143: 171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Yuan A, Campbell J, Kumar A, Nixon RA.. The C-terminal domains of NF-H and NF-M subunits maintain axonal neurofilament content by blocking turnover of the stationary neurofilament network. PLoS One 2012; 7: e44320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnam J, Teichberg VI.. Neurofilament-light increases the cell surface expression of the N-methyl-D-aspartate receptor and prevents its ubiquitination. J Neurochem 2005; 92: 878–85. [DOI] [PubMed] [Google Scholar]
- Rebelo AP, Abrams AJ, Cottenie E, Horga A, Gonzalez M, Bis DM, et al. Cryptic amyloidogenic elements in the 3’ UTRs of neurofilament genes trigger axonal neuropathy. Am J Hum Genet 2016; 98: 597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Karydas A, Bang J, Tsai RM, Blennow K, Liman V, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol 2016; 3: 216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooke K, Figlewicz DA, Han FY, Rouleau GA.. Analysis of the KSP repeat of the neurofilament heavy subunit in familiar amyotrophic lateral sclerosis. Neurology 1996; 46: 789–90. [DOI] [PubMed] [Google Scholar]
- Roots BI. Neurofilament accumulation induced in synapses by leupeptin. Science (New York, NY) 1983; 221: 971–2. [DOI] [PubMed] [Google Scholar]
- Rosengren LE, Karlsson JE, Karlsson JO, Persson LI, Wikkelso C.. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem 1996; 67: 2013–8. [DOI] [PubMed] [Google Scholar]
- Roy S, Coffee P, Smith G, Liem RK, Brady ST, Black MM.. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci 2000; 20: 6849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainio MT, Ylikallio E, Maenpaa L, Lahtela J, Mattila P, Auranen M, et al. Absence of NEFL in patient-specific neurons in early-onset Charcot-Marie-Tooth neuropathy. Neurol Genet 2018; 4: e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelius A, Zetterberg H, Blennow K, Adiutori R, Malaspina A, Laura M, et al. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology 2018; 90: e518. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanelli T, Ge W, Leystra-Lantz C, Strong MJ.. Calcium mediated excitotoxicity in neurofilament aggregate-bearing neurons in vitro is NMDA receptor dependant. J Neurol Sci 2007; 256: 39–51. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Gotow T, Shiozaki M, Sakaue F, Saito T, Julien JP, et al. Aggregate formation and phosphorylation of neurofilament-L Pro22 Charcot-Marie-Tooth disease mutants. Hum Mol Genet 2006; 15: 943–52. [DOI] [PubMed] [Google Scholar]
- Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, et al. Tau kinetics in neurons and the human central nervous system. Neuron 2018; 97: 1284–98 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz ML, Bruce J, Shneidman PS, Schlaepfer WW.. Deletion of 3’-untranslated region alters the level of mRNA expression of a neurofilament light subunit transgene. J Biol Chem 1995; 270: 26364–9. [DOI] [PubMed] [Google Scholar]
- Schwartz ML, Shneidman PS, Bruce J, Schlaepfer WW.. Stabilization of neurofilament transcripts during postnatal development. Mol Brain Res 1994; 27: 215–20. [DOI] [PubMed] [Google Scholar]
- Scrivo A, Codogno P, Bomont P.. Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat Commun 2019; 10: 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabanzadeh AP, D’Onofrio PM, Monnier PP, Koeberle PD.. Targeting caspase-6 and caspase-8 to promote neuronal survival following ischemic stroke. Cell Death Dis 2015; 6: e1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahim P, Gren M, Liman V, Andreasson U, Norgren N, Tegner Y, et al. Serum neurofilament light protein predicts clinical outcome in traumatic brain injury. Sci Rep 2016; 6: 36791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahim P, Zetterberg H, Tegner Y, Blennow K.. Serum neurofilament light as a biomarker for mild traumatic brain injury in contact sports. Neurology 2017; 88: 1788–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G. The use and potential of pNF-H as a general blood biomarker of axonal loss: an immediate application for CNS injury. In: Kobeissy FH, editor. Brain neurotrauma: molecular, neuropsychological, and rehabilitation aspects, Chap. 21. Boca Raton, FL: CRC Press/Taylor & Francis; 2015. [PubMed] [Google Scholar]
- Shea TB, Flanagan LA.. Kinesin, dynein and neurofilament transport. Trends Neurosci 2001; 24: 644–8. [DOI] [PubMed] [Google Scholar]
- Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC.. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res 2007; 313: 2098–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siller N, Kuhle J, Muthuraman M, Barro C, Uphaus T, Groppa S, et al. Serum neurofilament light chain is a biomarker of acute and chronic neuronal damage in early multiple sclerosis. Mult Scler 2018; 25: 678–86. [DOI] [PubMed] [Google Scholar]
- Smerjac SM, Zheng J, Hu CL, Bizzozero OA.. The role of calpain and proteasomes in the degradation of carbonylated neuronal cytoskeletal proteins in acute experimental autoimmune encephalomyelitis. Neurochem Res 2018; 43: 2277–87. [DOI] [PubMed] [Google Scholar]
- Sormani MP, Haering DA, Kropshofer H, Leppert D, Kundu U, Barro C, et al. Blood neurofilament light as a potential endpoint in Phase 2 studies in MS. Ann Clin Transl Neurol 2019; 6: 1081–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproviero D, La Salvia S, Giannini M, Crippa V, Gagliardi S, Bernuzzi S, et al. Pathological proteins are transported by extracellular vesicles of sporadic amyotrophic lateral sclerosis patients. Front Neurosci 2018; 12: 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinacker P, Feneberg E, Weishaupt J, Brettschneider J, Tumani H, Andersen PM, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 2016; 87: 12–20. [DOI] [PubMed] [Google Scholar]
- Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, et al. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci 2007; 35: 320–7. [DOI] [PubMed] [Google Scholar]
- Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF.. Drainage of interstitial fluid from different regions of rat brain. Am J Physiol 1984; 246: F835–44. [DOI] [PubMed] [Google Scholar]
- Terry-Lorenzo RT, Inoue M, Connor JH, Haystead TA, Armbruster BN, Gupta RP, et al. Neurofilament-L is a protein phosphatase-1-binding protein associated with neuronal plasma membrane and post-synaptic density. J Biol Chem 2000; 275: 2439–46. [DOI] [PubMed] [Google Scholar]
- Teunissen CE, Iacobaeus E, Khademi M, Brundin L, Norgren N, Koel-Simmelink MJ, et al. Combination of CSF N-acetylaspartate and neurofilaments in multiple sclerosis. Neurology 2009; 72: 1322–9. [DOI] [PubMed] [Google Scholar]
- Thelin EP, Zeiler FA, Ercole A, Mondello S, Buki A, Bellander BM, et al. Serial sampling of serum protein biomarkers for monitoring human traumatic brain injury dynamics: a systematic review. Front Neurol 2017; 8: 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins J, Usher P, Slade JY, Ince PG, Curtis A, Bushby K, et al. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS). Neuroreport 1998; 9: 3967–70. [DOI] [PubMed] [Google Scholar]
- Tradewell ML, Durham HD, Mushynski WE, Gentil BJ.. Mitochondrial and axonal abnormalities precede disruption of the neurofilament network in a model of charcot-marie-tooth disease type 2E and are prevented by heat shock proteins in a mutant-specific fashion. J Neuropathol Exp Neurol 2009; 68: 642–52. [DOI] [PubMed] [Google Scholar]
- Trivedi N, Jung P, Brown A.. Neurofilaments switch between distinct mobile and stationary states during their transport along axons. J Neurosci 2007; 27: 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truant R, Atwal RS, Desmond C, Munsie L, Tran T.. Huntington’s disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. FEBS J 2008; 275: 4252–62. [DOI] [PubMed] [Google Scholar]
- van Elst LT, Valerius G, Buchert M, Thiel T, Rusch N, Bubl E, et al. Increased prefrontal and hippocampal glutamate concentration in schizophrenia: evidence from a magnetic resonance spectroscopy study. Biol Psychiatry 2005; 58: 724–30. [DOI] [PubMed] [Google Scholar]
- van Lieverloo GGA, Wieske L, Verhamme C, Vrancken AFJ, van Doorn PA, Michalak Z, et al. Serum neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst 2019; 24: 187–94. [DOI] [PubMed] [Google Scholar]
- Vechio JD, Bruijn LI, Xu Z, Brown RH Jr, Cleveland DW.. Sequence variants in human neurofilament proteins: absence of linkage to familial amyotrophic lateral sclerosis. Ann Neurol 1996; 40: 603–10. [DOI] [PubMed] [Google Scholar]
- Veyrat-Durebex C, Corcia P, Dangoumau A, Laumonnier F, Piver E, Gordon PH, et al. Advances in cellular models to explore the pathophysiology of amyotrophic lateral sclerosis. Mol Neurobiol 2014; 49: 966–83. [DOI] [PubMed] [Google Scholar]
- Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ.. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res 2009; 1305: 168–82. [DOI] [PubMed] [Google Scholar]
- Von Bartheld CS, Altick AL.. Multivesicular bodies in neurons: distribution, protein content, and trafficking functions. Prog Neurobiol 2011; 93: 313–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CL, Uchida A, Li Y, Trivedi N, Fenn JD, Monsma PC, et al. Local acceleration of neurofilament transport at nodes of Ranvier. J Neurosci 2019; 39: 663–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 2006; 26: 8057–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RK, Brown A.. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol 2000; 2: 137–41. [DOI] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA.. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 2012; 196: 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Hawkes CA, Carare RO, Hardy J.. Does the difference between PART and Alzheimer’s disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Abeta and propagation of tau? Acta Neuropathol 2015; 129: 763–6. [DOI] [PubMed] [Google Scholar]
- Weston PSJ, Poole T, O’Connor A, Heslegrave A, Ryan NS, Liang Y, et al. Longitudinal measurement of serum neurofilament light in presymptomatic familial Alzheimer’s disease. Alzheimers Res Ther 2019; 11: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston PSJ, Poole T, Ryan NS, Nair A, Liang Y, Macpherson K, et al. Serum neurofilament light in familial Alzheimer disease: a marker of early neurodegeneration. Neurology 2017; 89: 2167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe JT, Jung C, Chan WK, Shea TB.. Phospho-dependent association of neurofilament proteins with kinesin in situ. Cell Motil Cytoskeleton 2000; 45: 249–62. [DOI] [PubMed] [Google Scholar]
- Yaworsky PJ, Gardner DP, Kappen C.. Transgenic analyses reveal developmentally regulated neuron- and muscle-specific elements in the murine neurofilament light chain gene promoter. J Biol Chem 1997; 272: 25112–20. [DOI] [PubMed] [Google Scholar]
- Ylikallio E, Poyhonen R, Zimon M, De Vriendt E, Hilander T, Paetau A, et al. Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum Mol Genet 2013; 22: 2975–83. [DOI] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Kumar A, Julien JP, Nixon RA.. Neurofilament transport in vivo minimally requires hetero-oligomer formation. J Neurosci 2003; 23: 9452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Sasaki T, Chen Y, Kumar A, Veeranna, et al. Alpha-internexin is structurally and functionally associated with the neurofilament triplet proteins in the mature CNS. J Neurosci 2006; 26: 10006–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Veeranna, Nixon RA.. Neurofilaments at a glance. J Cell Sci 2012. a; 125: 3257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Veeranna, Nixon RA.. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb Perspect Biol 2017; 9: a018309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sasaki T, Kumar A, Peterhoff CM, Rao MV, Liem RK, et al. Peripherin is a subunit of peripheral nerve neurofilaments: implications for differential vulnerability of CNS and peripheral nervous system axons. J Neurosci 2012. b; 32: 8501–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sasaki T, Rao MV, Kumar A, Kanumuri V, Dunlop DS, et al. Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J Neurosci 2009; 29: 11316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sershen H, Veeranna Basavarajappa BS, Kumar A, Hashim A, et al. Functions of neurofilaments in synapses. Mol Psychiatry 2015. a; 20: 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sershen H, Basavarajappa BS, Kumar A, Hashim A, et al. Neurofilament subunits are integral components of synapses and modulate neurotransmission and behavior in vivo. Mol Psychiatry 2015. b; 20: 986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sershen H, Basavarajappa BS, Smiley JF, Hashim A, et al. Neurofilament light interaction with GluN1 modulates neurotransmission and schizophrenia-associated behaviors. Transl Psychiatry 2018. a; 8: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sershen H, Basavarajappa BS, Smiley J, Hashim A, et al. Neurofilament light interaction with GluN1 modulates neurotransmission and schizophrenia-associated behaviors. Transl Psychiatry 2018. b; 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum SW, Zhang J, Mo K, Li J, Scherer SS.. A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy. Ann Neurol 2009; 66: 759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanier ER, Refai D, Zipfel GJ, Zoerle T, Longhi L, Esparza TJ, et al. Neurofilament light chain levels in ventricular cerebrospinal fluid after acute aneurysmal subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry 2011; 82: 157–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H. Neurofilament light: a dynamic cross-disease fluid biomarker for neurodegeneration. Neuron 2016; 91: 1–3. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Skillback T, Mattsson N, Trojanowski JQ, Portelius E, Shaw LM, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer Disease progression. JAMA Neurol 2016; 73: 60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J, Lin H, Julien JP, Schlaepfer WW.. Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1. Hum Mol Genet 2007; 16: 3103–16. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Couillard-Despres S, Julien JP.. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol 1997; 148: 299–316. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Lindenbaum M, Levavasseur F, Jacomy H, Julien JP.. Disruption of the NF-H gene increases axonal microtubule content and velocity of neurofilament transport: relief of axonopathy resulting from the toxin beta, beta’-iminodipropionitrile. J Cell Biol 1998; 143: 183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Vorgerd M, Sindern E, Schroder JM.. The novel neurofilament light (NEFL) mutation Glu397Lys is associated with a clinically and morphologically heterogeneous type of Charcot-Marie-Tooth neuropathy. Neuromuscul Disord 2004; 14: 147–57. [DOI] [PubMed] [Google Scholar]