

A dominant D252N mutation in TMEM106B is linked to hypo-myelinating leukodystrophy. However, how TMEM106B regulates myelination is unclear. Feng et al. report that TMEM106B deficiency in mice results in myelination defects and show that TMEM106B regulates lysosome pH, positioning and exocytosis in oligodendrocytes.
Keywords: myelination, lysosome, TMEM106B, oligodendrocytes
Abstract
TMEM106B encodes a lysosomal membrane protein and was initially identified as a risk factor for frontotemporal lobar degeneration. Recently, a dominant D252N mutation in TMEM106B was shown to cause hypomyelinating leukodystrophy. However, how TMEM106B regulates myelination is still unclear. Here we show that TMEM106B is expressed and localized to the lysosome compartment in oligodendrocytes. TMEM106B deficiency in mice results in myelination defects with a significant reduction of protein levels of proteolipid protein (PLP) and myelin oligodendrocyte glycoprotein (MOG), the membrane proteins found in the myelin sheath. The levels of many lysosome proteins are significantly decreased in the TMEM106B-deficient Oli-neu oligodendroglial precursor cell line. TMEM106B physically interacts with the lysosomal protease cathepsin D and is required to maintain proper cathepsin D levels in oligodendrocytes. Furthermore, we found that TMEM106B deficiency results in lysosome clustering in the perinuclear region and a decrease in lysosome exocytosis and cell surface PLP levels. Moreover, we found that the D252N mutation abolished lysosome enlargement and lysosome acidification induced by wild-type TMEM106B overexpression. Instead, it stimulates lysosome clustering near the nucleus as seen in TMEM106B-deficient cells. Our results support that TMEM106B regulates myelination through modulation of lysosome function in oligodendrocytes.
Introduction
The myelin sheath is a multilamellar, spirally wrapping extension of the plasma membrane of oligodendrocytes that insulates axons in the CNS (Salzer and Zalc, 2016). Myelination is essential not only for action potential propagation but also axon maintenance. The formation of the myelin sheath around axons is an intricate process but the cellular mechanisms involved in myelination are still poorly understood. Understanding the proper development and maintenance of myelin sheaths in the CNS is imperative in order for us to develop strategies to treat demyelinating diseases, such as the hereditary leukodystrophies and the acquired inflammatory demyelinating disorder, multiple sclerosis (Alizadeh et al., 2015).
Recently lysosomes have been demonstrated to play a critical role in myelination. Lysosomes are highly acidic cellular organelles traditionally viewed as the degradation and recycling centre of the cell, and are now known as signalling hubs that integrate nutrient information to promote cellular and organismal homeostasis (Lie and Nixon, 2019). Lysosomal dysfunction often leads to myelination defects, as observed in the lysosomal storage diseases resulting from lysosome enzyme deficiencies or misregulation (Faust et al., 2010). Mutations in several lysosomal proteins, including cathepsin D (Guo et al., 2018), Cln5 (Schmiedt et al., 2012), sialin (Prolo et al., 2009), and LIMP-2 (Gamp et al., 2003), cause severe myelination defects. Proteolipid protein (PLP), a multi-pass transmembrane protein and the main protein found in CNS myelin, is known to be stored in the late endosome/lysosome and is delivered to the myelin sheath upon neuronal signalling. This is achieved through a process called lysosome exocytosis, which involves fusion of the lysosome with the plasma membrane and subsequent release of lysosomal contents (Trajkovic et al., 2006). Furthermore, compaction of the myelin sheath requires extensive remodelling of the plasma membrane and removal of extraneous materials from the sheath itself, which is likely to be dependent on endocytosis and subsequent lysosomal degradation. One such example is the removal of the repulsive glycocalyx between adjacent extracellular leaflets through the activities of lysosomal enzymes (Garcia-Mateo et al., 2018).
Hypomyelinating leukodystrophy (HLD) is a group of heritable neurodevelopmental disorders characterized by abnormal myelination in the CNS (Charzewska et al., 2016). Recently, a dominant mutation (D252N) in the lumenal domain of TMEM106B, was found to cause HLD (Simons et al., 2017; Yan et al., 2018). TMEM106B, originally identified as a genetic risk factor for frontotemporal lobar degeneration (FTLD) with GRN mutations (Van Deerlin et al., 2010; van Blitterswijk et al., 2014), has recently been associated with many other neurodegenerative diseases, including FTLD-C9orf72 (Deming and Cruchaga, 2014; Gallagher et al., 2014; Lattante et al., 2014; van Blitterswijk et al., 2014), amyotrophic lateral sclerosis (ALS) (Vass et al., 2011), Parkinson’s disease (Tropea et al., 2019), Alzheimer’s disease (Rutherford et al., 2012), and limbic-predominant age-related TDP-43 encephalopathy (LATE) (Nelson et al., 2019). TMEM106B is also associated with neuronal proportion (Li et al., 2020) and is a known determinant of brain ageing (Rhinn and Abeliovich, 2017). At molecular and cellular levels, TMEM106B is a type II transmembrane protein located within the late endosome/lysosome (Chen-Plotkin et al., 2012; Lang et al., 2012; Brady et al., 2013) and has been shown to affect lysosomal morphology and function (Chen-Plotkin et al., 2012; Lang et al., 2012; Brady et al., 2013; Stagi et al., 2014), lysosome pH (Chen-Plotkin et al., 2012; Klein et al., 2017; Kundu et al., 2018), lysosome exocytosis (Kundu et al., 2018) and lysosome trafficking in neuronal dendrites (Schwenk et al., 2014). Furthermore, lysosome dysfunction was observed in patient-derived fibroblasts with the TMEM106B D252N mutation (Ito et al., 2018). Thus, the association of the TMEM106B D252N mutation with HLD further reinforces the connection between lysosome and myelination.
However, currently little is known about how TMEM106B regulates myelination. Here we show that TMEM106B is expressed in oligodendrocytes and that TMEM106B-deficient mice show myelination defects. TMEM106B ablation leads to lysosome clustering near the nucleus and decreased lysosome exocytosis and cell surface PLP levels in the Oli-neu oligodendroglial precursor cell line. The D252N mutation abolishes normal lysosomal functions of TMEM106B and induces peri-nuclear clustering of lysosomes. Taken together, our results show that TMEM106B regulates myelination through modulating lysosome function in oligodendrocytes.
Material and methods
Primary antibodies and reagents
The following antibodies were used in this study: The monoclonal antibody O10, which recognizes an epitope in the extracellular domain of PLP were provided by Dr Eva-Maria Krämer-Albers [Institute of Developmental Biology and Neurobiology (IDN), Johannes Gutenberg University Mainz, Mainz, Germany], M2 mouse anti-FLAG (Sigma, F1804), mouse anti-Myc (9E10, Hybridoma bank), mouse anti-GAPDH (Proteintech Group, 60004-1-Ig), rat anti-mouse LAMP1 (BD Biosciences, 553792), goat anti-CathB (R&D Systems, AF965), goat anti-CathD (R&D Systems, AF1029), goat anti-CathL (R&D Systems, AF1515), mouse anti-PLP (Millipore, MAB388), mouse anti-MBP (Millipore, SMI-99), mouse anti-MOG (Abcam, ab203058), mouse anti-MAG (Proteintech Group, 14386-1-AP), mouse anti-CNPase (Millipore, MAB 326R), rabbit anti-UGT8 (Aviva System), Goat anti-OLIG2 (R&D Systems, AF2418), mouse anti-GFAP (GA5) (Cell Signaling, 3670S), rabbit anti IBA-1 (Wako, 01919741), and goat anti-AIF-1/Iba1 (Novus Biologicals, NB100-1028). Myc-Trap and GFP-Trap were from ChromoTek. Rabbit anti-GFP antibodies were a gift from Professor Anthony Bretscher and rabbit anti cathepsin D antibodies were a gift from Dr William Brown at Cornell University. Rabbit anti-TMEM106B antibodies were characterized previously (Brady et al., 2013). Alexa Fluor® conjugated secondary antibodies (488/594/647 nm) were from Invitrogen. IRDye® 680RD/800CW secondary antibodies were from Invitrogen and LI-COR Biosciences.
The following reagents were also used in the study: Dulbecco’s modified Eagle’s media (DMEM) (Cellgro, 10-017-CV), N1 medium supplement (100×) (Sigma, N6530), 0.25% Trypsin (Corning, 25-053-CI), Papain (Worthington, LS003120), DNase I (Sigma D5025), Neurobasal™ Media (Invitrogen, 21103-049), B27 Supplement (Invitrogen, 17504044), poly-d-lysine (Sigma, P0899), apo-transferrin (Sigma, T2252), murine PDGF-AA (Shenandoah, 200-54), murine FGF (Peprotech, 450-33), insulin (Sigma, I6634), D-biotin (Amresco, 0340), Poly-d, l-ornithine (Sigma P0421), Odyssey blocking buffer (LI-COR Biosciences, 927-40000), protease inhibitor (Roche, 05056489001), Hochest 33342 (Invitrogen), Black-Gold II Stain kit (Millipore, AG400), rhodamine phalloidin (Biotium, 00027), Pierce BCA Protein Assay Kit (Thermo scientific, 23225), O.C.T. compound (Electron Microscopy Sciences, 62550-01) and Quetol 651/NSA Embedding Kit (Electron Microscopy Sciences, 14640).
Plasmids
Human TMEM106B cDNA inserted in the pEGFP and pCMV3XFLAG7.1 vectors was obtained as previously described (Brady et al., 2013). In addition, TMEM106B was cloned in the lentiviral vector pCDH-puro (System Biosciences) using restriction enzymes XbaI and EcoRI, and into the mApple-C1 vector using HindIII and SmaI. The D252N mutation was created via site directed mutagenesis. Human PLP1 cDNA was obtained from GenScript [pcDNA3.1-hPLP-(K)DYK, OHu25924] and subcloned in the pEGFP-N3 vector through restriction sites NheI and KpnI. Human cathepsin D cDNA was inserted in pcDNA3.1-myc/hisA through restriction sites BamHI and XhoI. The pLenti-CRISPR v2 vector was obtained from Addgene.
Mouse strains
TMEM106B knockout mice were produced using CRISPR/Cas9 genome editing with one guide (g) RNA (5′-AGTGAAGTGCACAACGAAGA-3′) or two gRNAs (5′-AGTGAAGTGCACAACGAAGA-3′, 5′-ACCCTATGGGATATATTTAC-3′) targeting exon 3 of mouse Tmem106b, flanking the start codon. C57BL/6J × FVB/N mouse embryos were injected with gRNAs and Cas9 mRNA at the Cornell Transgenic Core Facility. Editing was confirmed by sequencing PCR products from genomic DNA and loss of protein products was determined by western blot of tissue lysate. Δ341bp knockout mice were backcrossed to C57BL6 background for seven generations and Δ7bp knockout mice were backcrossed to C57BL6 background for three generations at the time of the study. Δ7bp knockout mice genotyping was performed by PCR using oligos 5′‐GTGCACAACGAAGACGGAAG‐3′ and 5′-TGGCAAACCTCAGGCTCATT‐3′ to specifically detect the wild-type (WT) allele (550 bp) and oligos 5′‐TGAAGTGCACAACGAAGAAA‐3′ and 5′-TGGCAAACCTCAGGCTCATT‐3′ to specifically detect mutant allele (547 bp). Δ341bp knockout mice genotyping was performed by PCR using oligos 5′‐GTGCACAACGAAGACGGAAG‐3′ and 5′‐TGGCAAACCTCAGGCTCATT‐3′ to specifically detect the wild-type allele (550 bp), and using oligos 5′‐GTTCACCTGCAGTGCCAACT‐3′ and 5′‐TTGTTTGCTTTTGTGTTTCTGA‐3′ to detect the mutant allele (397 bp). Mixed male and female mice were used for this study.
Behavioural tests
Male and female 4.5 to 5-month-old Tmem106b−/−mice (Δ341bp) and wild-type controls in the C57/BL6 background (10–14 mice/group, males and females analysed separately) were subject to the following behavioural tests. Open field test: mice were placed in a clear plastic chamber (40 × 40 × 40 cm) for 15 min. The total distance was tracked by the Viewer III software (Biobserve, Bonn, Germany). The apparatus was thoroughly cleaned with 70% ethanol between trials. Rotarod test: the five-lane rotarod apparatus (ENV-577M, Med associates Inc.) was used to measure motor coordination and balance (Hamm et al., 1994). During the training period, each mouse was placed on the rotarod at a constant speed (16 rpm) for a maximum of 180 s. Mice received three trials per day for four consecutive days after a steady baseline was attained. Mice were then subject to three trials at 4 to 40 rpm accelerating speed levels. The latency to fall off the rotarod was recorded and analysed. Balance beam test: mice were placed on a horizontal wooden beam (55 cm in length and 12 mm in diameter) 50 cm above the ground and allowed to traverse the beam (Stanley et al., 2005). Mice received three consecutive trainings per day for 2 days. During testing, times to cross the beam were recorded by Viewer III Tracking System (Biobserve). The apparatus was cleaned with 70% ethanol after testing of each mouse. For all behavioural analyses, experimenters were blind to the genotypes of the mice.
Cell culture
HEK293T, Neuro2a, and BV2 cells were maintained in DMEM (Cellgro) supplemented with 10% foetal bovine serum (Sigma) in a humidified incubator at 37°C with 5% CO2. Oli-Neu cells were grown at 37°C in SATO medium containing 1× N1 supplement, 20 mg/ml biotin, 2.5 mg/ml insulin, 2 mM l-glutamine and 1% horse serum and maintained in a humidified incubator at 37°C with 10% CO2. Oli-Neu cells with TMEM106B deletion or controls were generated by infecting the cells with lentivirus expressing Cas9 and gRNAs (TACTTCACCTCAATAGATCG and ACTTCACCTCAATAGATCGA) targeted to mouse Tmem106b exon 3 [oligos with 5′-caccgTACTTCACCTCAATAGATCG-3′ and 5′-aaacCGATCTATTGAGGTGAAGTAc-3′ or 5′-caccgACTTCACCTCAATAGATCGA-3′ and 5′-aaacTCGATCTATTGAGGTGAAGTc-3′ were ligated to pLenti-CRISPRv2 (Addgene)] or Cas9 only. Cells were selected with puromycin (1 µg/ml) 2 days after infection and the knockout was confirmed by western blot and immunostaining. For transient overexpression, cells were transfected with polyethylenimine (PEI) as previously described (Vancha et al., 2004).
Primary cortical neurons, wild-type and Tmem106b−/− mouse fibroblasts were cultured as described (Zhou et al., 2015). Mouse primary oligodendrocytes were cultured according to published protocols (Chen et al., 2007).
Immunoprecipitation, and western blot analysis
Cells were lysed in a cold, near-neutral pH solution containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1% Triton™ X-100, 0.1% deoxycholic acid, 1× protease inhibitors (Roche). After centrifugation at 14 000g, for 15 min, at 4°C, supernatants were transferred to clean tubes on ice, to which GFP-Trap or myc-Trap beads (ChromoTek) were added, then rocked for 3–4 h at 4°C. Samples were run on 12% or 15% polyacrylamide gels, then transferred to Immobilon-FL polyvinylidene fluoride membranes (Millipore Corporation). Membranes were blocked with either 5% non-fat milk in phosphate-buffered saline (PBS) or Odyssey Blocking Buffer (LI-COR Biosciences) for 1 h at room temperature and then incubated with primary antibodies, rocking overnight at 4°C. Membranes were then washed with Tris-buffered saline with 0.1% Tween-20 (TBST) three times, 5 min each, and incubated with fluorescently tagged secondary antibodies (LI-COR Biosciences) for 1 h at room temperature, followed by three washes, 10 min each. Membranes were scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences). Densitometry was performed using Image Studio (LI-COR Biosciences) and ImageJ.
Tissue preparation for western blot analysis
Mice were perfused with PBS and tissues were dissected and snap-frozen with liquid nitrogen and kept at −80°C. On the day of the experiment, frozen tissues were thawed and homogenized on ice with bead homogenizer (Moni International) in a cold solution of RIPA buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 1% Triton™ X-100, 0.5% sodium deoxycholate, 0.1% SDS) with proteinase inhibitors. After centrifugation at 14 000g for 15 min at 4°C, supernatants were collected. Protein concentrations were determined via BCA assay, then standardized. Equal amounts of protein were analysed by western blotting using the indicated antibodies.
Immunofluorescence staining, image acquisition and analysis
Cells were fixed, permeabilized with 0.05% saponin, and visualized using immunofluorescence microscopy as previously described (Brady et al., 2013). For cell surface staining, live Oli-neu cells were incubated on ice for 45 min with antibodies recognizing extracellular domain of LAMP1, or PLP (O10) in DMEM + 10% horse serum, followed by fixation in 4% paraformaldehyde (PFA) for 15 min at 4°C, and secondary antibody incubation. The intensity of cell surface LAMP1 and the ratio between cell surface PLP to the PLP-GFP signal were quantified using ImageJ from three independent experiments (15–35 cells/experiment). For brain section staining, mice were perfused and fixed with 4% PFA. After dehydration, the brain tissues were embedded in O.C.T compound (Electron Microscopy Sciences). Ten-micrometre thick brain sections were cut with cryotome. Tissue sections were blocked and permeabilized with 0.05% saponin in Odyssey blocking buffer or 0.2% Trion™ X-100 in PBS with 10% horse serum before incubating with primary antibodies overnight at 4°C. Sections were then incubated with secondary fluorescence antibodies and Hoechst at room temperature for 1 h, washed three times with PBS, and mounted. Antigen retrieval was performed by microwaving in sodium citrate buffer (pH 6.0) for 15 min. Images were acquired on a CSU-X spinning disc confocal microscope (Intelligent Imaging Innovations) with an HQ2 CCD camera (Photometrics) using 40× and 100× objectives. The quantitative analysis of fluorescence images was performed using ImageJ. For the quantitative analysis of intracellular levels of LAMP1, the entire cell body was selected, and the fluorescence intensity was measured directly using ImageJ after a threshold application. For the quantitative analysis of lysosomal positioning within the cell, individual cells were divided into two distributions at 15% from nuclear rim (perinuclear), the rest to cell border (peripheral) by measuring the distance from the cells nuclear rim to the cell border. LAMP1 density in each rim from individual cells was measured and the density ratio (density in each rim/total density) was calculated. Forty-five or more cells were analysed per condition for quantifying lysosome distribution. For quantifying GFAP, IBA1 and PLP level in the brain sections, 5–10 different random images were captured, and the fluorescence intensity was measured directly with ImageJ after a threshold application. Data from three or more brains for each genotype were used for quantitative analysis.
Lysosomal pH measurement
HEK293 cells grown in imaging chambers coated with poly-d-lysine were transfected with mApple tagged TMEM106B (wild-type or D252N) or empty mApple vector. Next, cells were incubated with dextran, previously functionalized with ApHID, a structurally improved pH-sensitive probe related to previous ratiometric fluorescent dyes based on boron dipyrromethene (BODIPY) (Komatsu et al., 2009) and Alexa 405, at 3 mg/ml in DMEM + 10% FBS overnight. J774 macrophages were similarly incubated with dextrans for creating a pH calibration. All cells were chased for at least 3 h in fresh DMEM. Following chase, J774 macrophages were fixed in 0.5% PFA for 5 min, washed with PBS and thereafter incubated in 50 mM Tris maleate (pH 4.0 and pH 5.5) or 50 mM sodium phosphate monobasic (pH 6.5) buffers containing 2.5 µM nigericin and monensin as ionophores. Following incubation, the cells were imaged at 37°C using an LSM880 confocal microscope with a 63× objective, and an ApHID/Alexa405 versus buffer pH calibration was prepared. Finally, transfected HEK293 cells were imaged in a buffer solution adjusted to pH 7.4 containing 150 mM sodium chloride, 20 mM HEPES, 1.3 mM calcium chloride, 5 mM potassium chloride, 1 mM magnesium chloride and 0.2% glucose at 37°C. The experiment included three identical repeats, and two dishes were imaged per condition. Ratiometric pH quantification was carried out with a custom-made journal in Metamorph. The Alexa405 (pH-independent) signal for each cell was masked and transferred to the ApHID channel (pH-dependent), and the Alexa405/ApHID ratio was measured for each cell and interpolated to a pH value using the pH calibration prepared using J774 macrophages. Statistical differences between conditions were evaluated using a one-way ANOVA test with multiple comparison within groups.
Black-Gold II staining
Thirty-micrometre thick mice brain cryosections were incubated with warm (60–65°C) Black-Gold II solution (0.3%, in 0.9% NaCl solution) for 15 min, and fixed in a sodium thiosulfate solution for 3 min. After rinsing in tap water for 15 min, sections were dehydrated through gradated alcohol, cleared in xylene and mounted onto cover slips. Myelinated axons were clearly visible as brick red fibres. Images were acquired using a 20× objective on a Leica DMi8 inverted microscope. Images were captured from the cerebral cortex and corpus callosum regions of the brain sections and images from three brains per genotype were used for quantitative analysis.
Transmission electron microscopy
Isolated optic nerves were fixed in 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate/HCl buffer (pH 7.3). After washing with 0.1 M sodium cacodylate/HCl buffer (pH 7.3), tissues were post-fixed in 1% osmium tetroxide, 1.5% potassium ferricyanide, followed by dehydration in 25%, 50%, 70%, 95%, 100% and 100% alcohol series. Finally, the nerves were embedded using the Quetol 651/NSA Embedding Kit (Electron Microscopy Sciences), followed by polymerizing in a 60°C oven for 36 h. After polymerization, blocks were sectioned at 70 nm, transferred onto 3 mm copper grids and stained for 30 min in 1.5% aqueous uranyl acetate, followed by 10 min in lead citrate. Images were acquired on Morgnani 268 transmission electron microscope (FEI).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8. Statistical significance was assessed by paired or unpaired t-test (for two group comparisons). P-values ≤ 0.05 were considered statistically significant: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Ethical approval and consent to participate
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. The work under animal protocol 2017-0056 is approved by the Institutional Animal Care and Use Committee at Cornell University.
Data availability
The data supporting the findings of this study are available from the corresponding author on request.
Results
Generation of TMEM106B-deficient mice
To understand the physiological function of TMEM106B, we generated TMEM106B-deficient mice using the CRISPR/Cas9 technique (Cong et al., 2013; Mali et al., 2013). Two independent lines were characterized. In one line, a 341-bp fragment including the Tmem106b start codon (Δ341bp) was removed using two gRNAs (Fig. 1A). In another line, a 7-bp deletion (Δ7bp) after amino acid 39 resulted in a frame shift and an early termination of the TMEM106B protein (Fig. 1B). In both lines, gene editing was verified by sequencing, PCR analysis (Fig. 1C and data not shown) and western blot analysis using antibodies specific to TMEM106B (Fig. 1D and E). These mice were backcrossed to C57/BL6 mice for seven generations (Δ341bp) or three generations (Δ7bp) to rule out any off-target effects from gene editing. TMEM106B-deficient mice do not have any obvious growth phenotypes or brain pathology (data not shown). However, there is a slight increase in astrogliosis with elevated GFAP signals at 5 months of age in the Δ341bp line (Fig. 2A–D) and at 10 months of age in the Δ7bp line (Fig. 2E and F), consistent with a previous study (Nicholson et al., 2018). No obvious differences in microglia were observed (Fig. 2A–F). In addition, TMEM106B-deficient mice do not show any obvious motor defects in the open field or the rotarod behavioural test (Fig. 2G and H), but exhibit a mild deficit in the balance beam test (Fig. 2I), suggesting a defect in motor coordination.
Figure 1.

Generation of TMEM106B-deficient mice. (A and B) Illustration of mouse TMEM106B genomic locus and guide RNAs used to generate TMEM106B-deficient mice. Red box indicates deleted region. (C–E) Ablation of TMEM106B gene product was confirmed by PCR (C) and western blot analysis using antibodies specific to TMEM106B (D and E).
Figure 2.
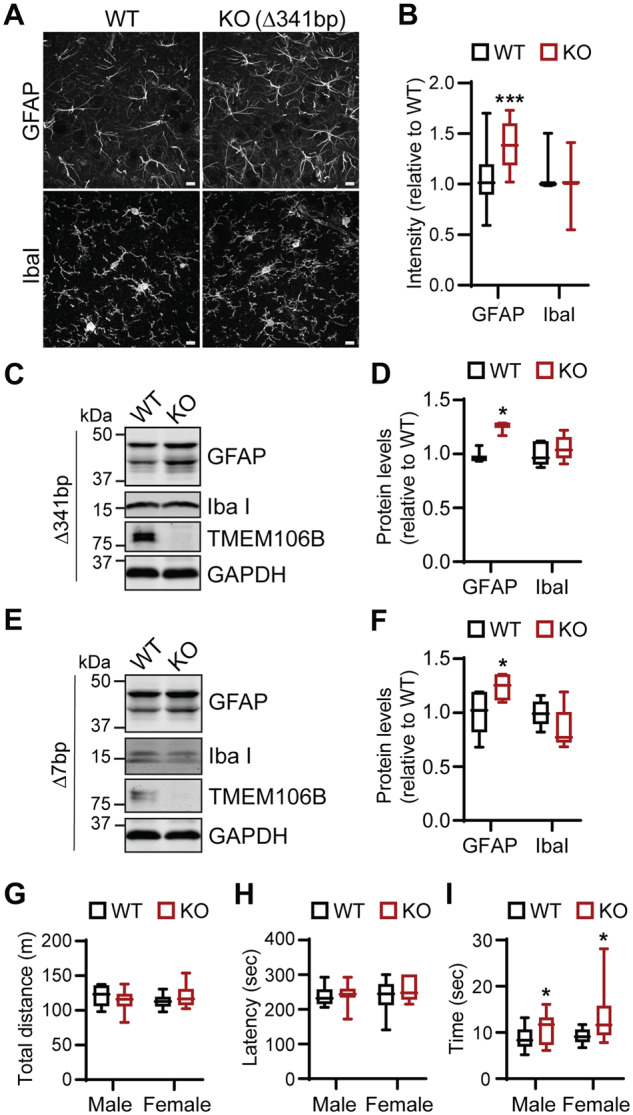
Increased gliosis and mild behavioural defects in TMEM106B-deficient mice. (A and B) Increased astrocyte activation in 5-month-old Tmem106b−/− (Δ341bp) mice. Brain sections were stained with anti-GFAP or IbaI antibodies, and images were captured in hippocampus region. Scale bar = 10 µm. n = 3 (two females, one male each genotype), ***P < 0.001, Student’s t-test. (C–F) Western blot analysis of GFAP and Iba1 protein levels in wild-type and Tmem106b−/− frontal cortex lysates (C and D: 5 months old, Δ341bp; E and F: 10 months old, Δ7bp). n = 4 (two females, two males each genotype), *P < 0.05, paired t-test with littermate wild-type controls. (G–I) Tmem106b−/− mice show normal behaviour in the open field test (G) and rotor rod test (H), but show a slight delay in passing balance beam (I). n = 12–17 (male), 10–12 (female); *P < 0.05, Student’s t-test. KO = knockout; WT = wild-type.
Myelination defects in TMEM106B-deficient mice
Because of the genetic link between TMEM106B and HLD (Simons et al., 2017; Yan et al., 2018), we investigated potential myelination defects in TMEM106B-deficient mice. In comparison to wild-type mice, Tmem106b−/− mice have reduced myelin intensity in the cerebral cortex and corpus callosum indicated by Black-Gold II staining (Fig. 3A–C). Furthermore, the levels of PLP, the main membrane protein of the myelin sheath, are significantly decreased in 5-month-old Δ341bp (Fig. 3D–G) and 10-month-old Δ7bp Tmem106b−/− mouse brains (Fig. 3Hand I). Myelin basic protein (MBP), another main protein component of the myelin sheath is also reduced in both strains, but the difference is only significant in the 10-month-old Δ7bp Tmem106b−/− mouse brains. The slight variation seen between the two mouse strains might be due to the difference in the strain background (the Δ7bp and Δ341bp strains were backcrossed to C57BL6 for three and seven generations, respectively) and the age of the mice used. MOG, another transmembrane protein found in outer loop (abaxonal) region of the myelin sheath (Winterstein et al., 2008), is also affected by TMEM106B loss (Fig. 3D–I). However, the levels of myelin-associated glycoprotein (MAG), which localizes to periaxonal loops (Winterstein et al., 2008), the levels of 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase), and 2-hydroxyacylsphingosine 1-beta-galactosyltransferase (UGT8), two early markers for oligodendrocyte differentiation, and the levels of OLIG2, a transcription factor required for oligodendrocyte differentiation, are not affected by TMEM106B ablation (Fig. 3F–I). These results indicate the specific effect of TMEM106B on PLP, MOG and MBP. Ultrastructural analysis of optic nerves by transmission electron microscopy showed separation and vacuolation of the myelin sheath in Tmem106b−/− mice at 12 months of age (Fig. 3J and K). These findings support that TMEM106B deficiency leads to myelination defects in mice.
Figure 3.

Myelination defects in TMEM106B-deficient mice. (A–C) Tmem106b−/− mice show reduced myelination in the corpus callosum and cortex as indicated by Black-Gold staining. Scale bar = 100 µm. n = 3 (two females, one male each genotype), *P < 0.05, Student’s t-test. Signals from the corpus callosum and cortex regions were quantified separately in C. (D and E) Tmem106b−/− mice show a significant reduction in PLP and MOG fluorescent intensity in frontal cortex region. Scale bar = 10 µm. n = 3 (two females, one male each genotype), *P < 0.01, Student’s t-test. (F–I) Western blot analysis of myelin proteins in wild-type (WT) and Tmem106b−/− frontal cortex lysates (F and G: 5 months old, Δ341bp; H and I: 10 months old, Δ7bp) n = 4 (two females, two males each genotype), *P < 0.05; **P < 0.01; paired t-test with wild-type littermate control. (J) Electron microscope images of myelin sheath in optic nerve isolated from wild-type and Tmem106b−/− mice (Δ7bp) at 12 months of age; scale bar = 200 nm. Magnified images show the separation and vacuolation of myelin sheath (arrowheads). Optic nerves from three male mice of each genotypes were analysed and representative images are shown. (K) Quantification of vacuoles per axon for experiment in J. Optic nerves from three mice per group (n = 3) were analysed. Vacuoles with size >13 nm were manually counted by experimenter blinded to genotypes. Total 196 axons (wild-type, WT) and 165 axons (KO) were analysed. **P < 0.01, Student’s t-test.
TMEM106B is expressed in oligodendrocytes
Proper myelination is dependent on the crosstalk between axons and oligodendrocytes (Salzer and Zalc, 2016). TMEM106B was found to be highly expressed in oligodendrocytes including precursors at mRNA levels (https://web.stanford.edu/group/barres_lab/brain_rnaseq.html) (Simons et al., 2017). Western blot analysis of TMEM106B protein levels in various cell lines, including mouse neuroblastoma cell line Neuro2a (N2a), mouse oligodendroglial precursor cell line Oli-Neu (Jung et al., 1995) and microglia cell line BV2 (Blasi et al., 1990), and mouse primary cortical neurons showed similar expression levels of TMEM106B in N2a and Oli-Neu and much lower levels in BV2 (Fig. 4A and B), consistent with RNAseq analysis data. Immunostaining with antibodies specific to TMEM106B (Brady et al., 2013) showed that TMEM106B is expressed in oligodendrocytes and co-localizes with the lysosome marker LAMP1 in adult brain sections (Fig. 4C) and in cultured immature (Fig. 4D) and mature (Fig. 4E) oligodendrocytes. In mature oligodendrocytes, co-localization between TMEM106B, LAMP1 and the myelin membrane protein PLP was observed (Fig. 4D). This is consistent with a previous report that PLP is mainly stored in lysosomes in oligodendrocytes (Trajkovic et al., 2006). Taken together, our data suggest that TMEM106B is expressed in oligodendrocytes at a similar level as in neurons and is localized in the lysosome compartment in these cells. However, TMEM106B expression does not seem to exhibit significant changes during oligodendrocyte differentiation at the mRNA level (https://web.stanford.edu/group/barres_lab/brain_rnaseq.html). While we cannot assess changes in TMEM106B protein levels during oligodendrocyte differentiation in vitro because of the impurity of oligodendrocyte culture, the protein levels of TMEM106B in the whole brain lysates does not appear to change significantly at different stages of oligodendrocyte differentiation during development (Supplementary Fig. 1).
Figure 4.

TMEM106B is expressed in oligodendrocytes and localizes in the lysosome compartment. (A and B) Western blot analysis of TMEM106B proteins levels in various cell types as indicated. TMEM106B levels relative to GAPDH were quantified from three independent samples and plotted relative to its levels in day in vitro 14 primary cortical neurons. n = 3. (C) Immunostaining of TMEM106B, LAMP1 and OLIG2 in brain sections from a 5-month-old wild-type mouse to show that TMEM106B is expressed and localized in the lysosome compartment in oligodendrocytes. Scale bar = 10 µm. (D and E) Immunostaining of TMEM106B, LAMP1 and OLIG2 in immature (C) and mature (D) oligodendrocytes cultured from postnatal Day 0–3 pups. Scale bar = 10 µm.
TMEM106B affects lysosome function and positioning, and PLP trafficking in oligodendrocytes
As TMEM106B is a lysosome protein, we examined the effect of TMEM106B loss on the levels of lysosome proteins in cortical lysates. Contrary to previous studies (Klein et al., 2017), a slight decrease in LAMP1 protein levels was detected in cortical lysates from 5- and 10-month-old Tmem106b−/− mice compared to wild-type littermate controls (Fig. 5A–D). This decrease was significant in the 5-month-old Δ341bp mice. A slight increase in levels of mature lysosomal cathepsins (D and L) was also observed in the cortical lysates of 10-month-old Δ7bp mice. The level of mature cathepsin L was also slightly increased in the cortical lysates of 5-month-old mice Δ341bp mice, but the increase did not reach statistical significance. This suggests a mild lysosomal defect in Tmem106b−/− mouse brain.
Figure 5.

Changes in the levels of lysosome proteins in TMEM106B-deficient mice. Western blot analysis of lysosomal proteins in wild-type and Tmem106b−/− frontal cortex lysates (A and B: 5-month-old, Δ341bp; C and D: 10-month-old, Δ7bp). n = 3 (two females, one male each genotype), *P < 0.05, paired t-test with wild-type littermate controls.
Next we examined lysosomal changes more specifically in the Oli-Neu oligodendrocyte cell line. TMEM106B gene products were removed from these cells using the CRISPR/Cas9 technique (Fig. 6A) and the changes in the levels of lysosome proteins were determined by western blot analysis. A significant reduction in LAMP1, cathepsin D and L protein levels was observed in TMEM106B-deficient Oli-Neu cells (Fig. 6A and B).
Figure 6.

TMEM106B deficiency results in lysosome and PLP trafficking defects in oligodendrocyte cells. (A and B) Western blot analysis of lysosome protein levels in control and Tmem106b−/− Oli-Neu cells generated using CRISPR/Cas9. n = 3–4, *P < 0.05, **P < 0.01; Student’s t-test. (C) Flag-tagged TMEM106B and control vector or myc-tagged Cath D construct were co-transfected into HEK293T cells as indicated. Cell lysates were subjected to anti-myc immunoprecipitation (IP) and the IP products were analysed using western blot with anti-myc and anti-Flag antibodies. (D–F) Tmem106b−/− Oli-Neu cells show increased lysosome clustering in the perinuclear region (D and E) and decreased LAMP1 intensity per cell (D and F). Scale bar = 10 µm. Data were analysed from four independent experiments (n = 4), ***P < 0.001, Student’s t-test. (G and H) Live control and Tmem106b−/− Oli-Neu cells were stained with antibodies against extracellular domain of LAMP1. LAMP1 intensity was quantified in H. Scale bar = 10 µm. Data were analysed from three independent experiments (n = 3), ****P < 0.0001, Student’s t-test. (I–K) Control and Tmem106b−/− Oli-Neu cells were transfected with GFP-PLP1 and live cells were subject to immunostaining using the O10 antibody, which recognizes the extracellular domain of PLP. The ratio of cell surface PLP/PLP-GFP was quantified in J. The percentage of cells with intracellular punctate GFP-PLP accumulation was quantified in K. Scale bar = 10 µm. Data were analysed from three independent experiments (253–485 cells/experiment, n = 3), ****P < 0.0001, Student’s t-test. (L) Control and Tmem106b−/− Oli-Neu cells were transfected with GFP-PLP1 and stained with anti-CathD antibody. The representative images from three independent experiments are shown. Scale bar = 10 µm.
Since cathepsin D deficiency has been shown to affect PLP trafficking and demyelination (Guo et al., 2018), we wondered whether TMEM106B could affect myelination through cathepsin D. Co-immunoprecipitation studies demonstrated a physical interaction between TMEM106B and cathepsin D in HEK293T cells with overexpressed TMEM106B and cathepsin D (Fig. 6C and Supplementary Fig. 2). No interaction between TMEM106B and cathepsin B or L was detected under similar conditions (data not shown), indicating a specific interaction between TMEM106B and cathepsin D.
To visualize any changes in lysosome morphology or positioning, we immunostained LAMP1 in control and Tmem106b−/− Oli-Neu cells. While there are no obvious changes in lysosome size upon TMEM106B loss, the positioning of lysosomes appears to be altered by TMEM106B ablation. Lysosomes are more clustered in the perinuclear region in Tmem106b−/− Oli-Neu cells (Fig. 6D and E), along with a decrease in LAMP1 signal (Fig. 6F). Similar phenotypes were observed in fibroblasts (Supplementary Fig. 3). These data suggest that TMEM106B regulates lysosome trafficking within the cells.
PLP has been shown to be transported to the myelin sheath from the lysosome compartments through lysosome exocytosis (Trajkovic et al., 2006). Since TMEM106B deletion affects lysosome positioning and PLP levels, we hypothesized that TMEM106B might regulate PLP trafficking via lysosome exocytosis. To determine lysosome exocytosis defects, the levels of cell surface LAMP1 were measured in live control and Tmem106b−/− Oli-Neu cells. A significant reduction in cell surface LMAP1 levels was observed in Tmem106b−/− Oli-Neu cells (reduced to 49.6% compared to controls) (Fig. 6G and H). Although there is a decrease of total LAMP1 signal in Tmem106b−/− Oli-Neu cells (reduced to 67.5% compared to controls), a greater decrease in the cell surface LAMP1 levels indicate a defect in lysosome exocytosis in TMEM106B-deficient cells to deposit LAMP1 onto plasma membrane. Furthermore, a drastic decrease in cell surface PLP levels was also detected in Tmem106b−/− Oli-Neu cells expressing GFP tagged PLP compared to control cells (Fig. 6I and J). A concomitant increase of intracellular punctate PLP signal in the lysosome compartment was also detected (Fig. 6I, K and L). These data suggest that TMEM106B regulates PLP trafficking through lysosome exocytosis within the oligodendrocytes.
The D252N mutation affects TMEM106B function and alters lysosome positioning
The D252N mutation in TMEM106B is directly associated with HLD (Simons et al., 2017; Yan et al., 2018). However, how this mutation affects TMEM106B function is still unclear. To determine whether this mutation alters TMEM106B protein stability, GFP-tagged wild-type and D252N mutant TMEM106B were transfected into HEK293T cells. Western blot analysis showed that the D252N mutant is expressed at similar levels as wild-type TMEM106B with no obvious difference in molecular weight (Fig. 7A and B), indicating that the D252N mutation does not affect the posttranslational modification or stability of TMEM106B. Previously, we have shown that TMEM106B is subject to cleavage first by lysosome proteases in the luminal domain to generate an N-terminal fragment and then by the SPPL2a family protease in the transmembrane region to generate a intracellular cytosolic domain (ICD) (Brady et al., 2014). The D252N mutation appears to decrease the amount of the N-terminal fragment generated compared to wild-type controls (Fig. 7A and B), suggesting that D252N mutation might affect the processing of TMEM106B by lysosomal proteases. In addition, TMEM106B tends to form SDS-resistant dimers, which is not affected by the D252N mutation (Fig. 7C and D). The D252N mutant is localized properly in the lysosome compartment when expressed in Oli-Neu cells (Fig. 7E and H) or fibroblasts (data not shown), indicating that this mutation does not affect lysosomal trafficking of TMEM106B.
Figure 7.

The D252N mutant abolishes TMEM106B induced lysosome enlargement but induces lysosome clustering in the perinuclear regions. (A and B) The D252N mutant of TMEM106B is expressed at levels comparable to wild-type TMEM106B but shows reduced cleavage. GFP tagged wild-type or D252N mutant of TMEM106B were transfected into HEK293T cells and lysates were collected 2 days later for western blot analysis. n = 4, n.s = not significant, *P < 0.05, Student’s t-test. (C and D) D252N does not affect TMEM106B dimerization. Untagged wild-type or D252N mutant of TMEM106B were transfected into HEK293T cells as indicated and lysates were collected 2 days later for western blot analysis under non-reducing conditions. n = 4, n.s = not significant, Student’s t-test. (E–G) Wild-type Oli-Neu cells were transfected with GFP vector or GFP tagged wild-type or D252N mutant TMEM106B and stained with anti-LAMP1 antibodies. Lysosome size and distribution were measured and quantified. Scale bar = 10 µm. Data were analysed from three independent experiments (n = 3), *P < 0.05, **P < 0.01; ***P < 0.001, ****P < 0.0001, n.s = not significant, Student’s t-test. (H and I) Tmem106b−/− Oli-Neu cells were transfected with GFP vector or GFP tagged wild-type or D252N mutant TMEM106B and stained with anti-LAMP1 antibodies. Lysosome distribution were quantified. Scale bar = 10 µm. Data were analysed from three independent experiments (n = 3), *P < 0.05, **P < 0.01, n.s = not significant, Student’s t-test.
TMEM106B overexpression has been shown to result in enlarged lysosomes and decreased number of lysosomes within the cell (Chen-Plotkin et al., 2012; Lang et al., 2012; Brady et al., 2013). This phenotype is also observed in the oligodendrocyte cell line, Oli-Neu cells (Fig. 7E and F). However, when the D252N mutant is overexpressed in Oli-Neu cells, it fails to induce lysosome enlargement (Fig. 7E and F). Instead, it results in clustering of lysosomes in the perinuclear region, mimicking the phenotypes of TMEM106B deficiency (Fig. 7E and G). Furthermore, the lysosome positioning defect in TMEM106B-deficient Oli-Neu cells can be rescued by expressing wild-type TMEM106B but not the D252N mutant (Fig. 7H and I). These results indicate that the D252N mutation affects the normal function of TMEM106B on lysosome membrane.
Overexpression of wild-type but not the D252N mutant of TMEM106B results in increased lysosome acidification
Besides enzyme content and lysosome positioning within the cell, another important factor that modulates lysosomal activity is intraluminal pH (Sole-Domenech et al., 2016; Lie and Nixon, 2019). A role of TMEM106B in lysosome pH regulation has been reported by several groups (Chen-Plotkin et al., 2012; Klein et al., 2017; Kundu et al., 2018). After transfecting HEK293 cells with wild-type and D252N mutant of TMEM106B, we labelled cellular lysosomes with dextran polymers coupled with a custom-made pH-sensitive dye, ApHID, and the pH-insensitive dye Alexa405. ApHID is related to previous ratiometric fluorescent dyes based on boron dipyrromethene (BODIPY) (Komatsu et al., 2009). Transfected HEK293 cells internalized the dextrans by endocytosis and incorporate them into lysosomes, where they serve as in vivo ratiometric pH sensors (Fig. 8A and B). We found that overexpression of wild-type but not the D252N mutant of TMEM106B leads to increased lysosome acidification as shown by increased ratios of ApHID/Alexa 405 signal (Fig. 8C and D). Studies have also shown an interaction between TMEM106B and the AP1 subunit of the V-ATPase (Klein et al., 2017). However, the D252N mutation does not seem to affect this interaction in the co-immunoprecipitation assay with overexpressed proteins (Supplementary Fig. 4). Nevertheless, our results indicate that the D252N mutation might affect lysosome function through regulating lysosome pH since lysosomal acidification modulates enzymatic activity, and failure to acidify the organelles leads to reduced lysosomal proteolysis (Sole-Domenech et al., 2016; Lie and Nixon, 2019).
Figure 8.

HEK293T cells overexpressing TMEM106B-wild-type, but not TMEM106B-D252N, hyper-acidify their lysosomes. (A and B) Fixed J774 macrophages, with their lysosomes labelled with dextran-Alexa405-ApHID, were incubated in various pH-adjusted buffers containing 2.5 µM nigericin and monensin (A), and ApHID/Alexa405 signal ratios were plotted against buffer pH (B). Scale bars = 10 µm. (C and D) HEK293 cells transfected with mApple tagged wild-type or D252N TMEM106B, or empty mApple vector were incubated with dextran-Alexa405-ApHID, which was delivered to lysosomes. mApple is pseudo-coloured blue in the magnified images. The three small panels next to each image correspond to greyscale images indicating Alexa405 (top), ApHID (middle), and mApple (bottom) (D). Red arrowheads indicate enlarged lysosomes with strong ApHID fluorescence. Ratiometric pH imaging was carried out for each condition, and ApHID/Alexa405 ratios were interpolated to pH values using the calibration shown in B. Lysosomal pH values for each condition are shown in C. Scale bars = 10 µm. Error bars in the plots indicate SEM.
Discussion
Based on the genetic association between TMEM106B and HLD, we examined the role of TMEM106B in myelination using mouse models. Our results from two independent lines of TMEM106B deficiency strongly support an essential role of TMEM106B in myelination: (i) TMEM106B deficiency leads to hypomyelination in the mouse brain (Fig. 3A–C); (ii) TMEM106B deficiency results in a significant reduction in the levels of myelin proteins PLP, MBP and MOG (Fig. 3D–I); and (iii) TMEM106B deficiency leads to separation and vacuolization of the myelin sheath (Fig. 3J and K).
While the exact mechanism by which TMEM106B regulates myelination remains elusive, our data support a model in which TMEM106B modulates lysosome functions in oligodendrocytes to affect myelination: (i) TMEM106B is expressed in both immature and mature oligodendrocytes (Fig. 4); and (ii) TMEM106B is critical for proper lysosome function in oligodendrocytes (Fig. 6). Deletion of TMEM106B in oligodendrocytes not only leads to a significant reduction in the levels of many lysosome proteins but also results in clustering of lysosomes in the perinuclear region (Fig. 6). As the main myelin membrane protein PLP resides in the lysosome compartment prior to delivery to the myelin sheath via lysosome exocytosis (Trajkovic et al., 2006), our results suggest that TMEM106B regulates myelination via lysosome-dependent PLP trafficking. Indeed, we found that TMEM106B deficiency reduce lysosome exocytosis and cell surface levels of PLP and results in intracellular PLP accumulation in the lysosome compartment (Fig. 6).
Among all the myelin proteins, the levels of PLP and MOG are the two most affected by TMEM106B (Fig. 3D–I). MOG, a type I transmembrane protein, was found in outer loop (abaxonal) region of myelin sheath (Winterstein et al., 2008). MOG was internalized via clathrin-dependent endocytosis, and subsequently transported to recycling endosomes (Winterstein et al., 2008). Further studies are needed to determine whether TMEM106B plays a direct role in regulating MOG trafficking.
We demonstrated a physical interaction between TMEM106B and cathepsin D (Fig. 6C and Supplementary Fig. 2) and a significant reduction in cathepsin D protein levels was detected upon TMEM106B deficiency in oligodendrocytes (Fig. 6). Previously, cathepsin D deficiency was shown to cause profound defects in PLP trafficking and myelination (Guo et al., 2018). These results suggest TMEM106B might affect myelination process by modulation of cathepsin D.
Our data also showed perinuclear accumulation of lysosomes in TMEM106B-deficient cells. This phenotype was most obvious in the Oli-Neu oligodendrocyte cell line (Fig. 6D and E), but it is also observable in fibroblasts (Supplementary Fig. 3). TMEM106B has been shown to play a role in lysosome positioning and trafficking in neurons (Schwenk et al., 2014) and Tmem106b knockdown was shown to restore endo-lysosomal trafficking and branching defects in mutant CHMP2BIntron5 neurons (Clayton et al., 2018). Microtubule associated protein 6 (MAP6) was found to interact with TMEM106B and mediate the effect of TMEM106B on lysosome trafficking along microtubules (Schwenk et al., 2014). It remains to be determined whether the same mechanism leads to the lysosome positioning defects in TMEM106B-deficient oligodendrocytes. Lysosomes are known to cluster in the perinuclear region in many lysosome storage diseases (Xu and Ren, 2015). It is possible that the clustering seen in the Tmem106b−/− cells is secondary to lysosome dysfunction. Regardless, alteration of lysosome positioning in Tmem106b−/− cells would affect many lysosome functions, including lysosome exocytosis, which involves docking and fusion of lysosomes with the plasma membrane. Clustering of lysosomes in the perinuclear region in Tmem106b−/− oligodendrocytes would decrease the pool of lysosomes in the cell periphery available for docking and fusion with plasma membrane, leading to a defect in lysosome exocytosis and PLP trafficking.
Our results showed that the D252N mutation in TMEM106B abolishes TMEM106B induced lysosome enlargement (Fig. 7E–G) and lysosome acidification (Fig. 8C and D) but induces perinuclear lysosome clustering (Fig. 7E–G). The D252N mutation does not affect TMEM106B protein stability or dimerization, but slightly affects the processing of TMEM106B by lysosomal proteases (Fig. 7A–D). This mutation is likely to alter the interaction of TMEM106B with other proteins. However, it does not impact the interaction between TMEM106B and cathepsin D or the AP1 subunit of the V-ATPase (Supplementary Figs 2 and 4). Future proteomic studies will allow us to identify protein interactions affected by the D252N mutation.
While this study is focused on the role of TMEM106B in oligodendrocytes, we cannot rule out a possible function for TMEM106B in axons, as proper myelination relies on the interaction between oligodendrocytes and axons. Previously we and others have shown that TMEM106B is highly expressed in neurons (Chen-Plotkin et al., 2012; Lang et al., 2012; Brady et al., 2013; Stagi et al., 2014), thus it is possible that the neuronal pool of TMEM106B also affects myelination process.
TMEM106B was initially identified as a risk factor for FTLD, especially in patients with GRN mutations (Van Deerlin et al., 2010; Cruchaga et al., 2011; Finch et al., 2011; van der Zee et al., 2011). During the past decade, TMEM106B polymorphisms have also been associated with FTLD caused by C9orf72 mutations (Deming and Cruchaga, 2014; Gallagher et al., 2014; Lattante et al., 2014; van Blitterswijk et al., 2014), cognitive impairment in ALS (Vass et al., 2011) and Parkinson’s disease (Tropea et al., 2019), and pathological presentation of Alzheimer’s disease (Rutherford et al., 2012). A recent study has also identified TMEM106B and GRN as two of the five risk factors for a recently recognized disease entity, limbic-predominant age-related TDP-43 encephalopathy (LATE) (Nelson et al., 2019). TMEM106B is also associated with neuronal proportion (Li et al., 2020), and TMEM106B and GRN were found to be two main determinants of differential ageing in the cerebral cortex with genome-wide significance (Rhinn and Abeliovich, 2017). Thus, TMEM106B is intimately linked to overall brain health. Our results suggest that at least one function of TMEM106B in the brain is to ensure proper myelination through regulating lysosome function in oligodendrocytes. Meanwhile, TMEM106B has been reported to regulate lysosome positioning and trafficking in neuronal dendrites (Schwenk et al., 2014). It might also play a role in maintaining proper function of astrocytes given that TMEM106B deficiency leads to increased astrogliosis (Fig. 2A–F) (Nicholson et al., 2018). Tmem106b−/− mice do not show any obvious behavioural phenotypes, but have a slight deficit in the balance beam test (Fig. 2G–I), which requires high levels of motor coordination. More sophisticated behavioural tests might reveal additional deficits in Tmem106b−/− mice. Future studies to elucidate the detailed mechanisms by which TMEM106B regulates lysosome activities will provide greater insights into how TMEM106B functions in multiple cell types to maintain brain health.
Supplementary Material
Acknowledgements
We would like to thank Dr Maria Feltri’s lab at University of Buffalo for providing Oli-Neu cells, Dr Eva-Maria Krämer-Albers’s lab for providing O10 antibody against PLP1, Dr Tony Bretscher’s lab for anti-GFP antibodies and assistance with confocal microscope, Dr Thomas Cleland’s lab and Dr Chris Schaffer and Dr Nozomi Nishimura’s lab for assistance with behavioural tests. We would like to acknowledge Cornell Center for Materials Research for EM sample preparation (supported by NSF DmR 117985) and Jeff R. Jorgensen for help with microtome and transmission electron microscopy. In addition, we also thank Cornell Transgenic Core Facility for generating the CRISPR/Cas9 knockout mice.
Funding
This work is supported by NINDS/NIA (R01NS088448 & R01NS095954) and the Bluefield project to cure frontotemporal dementia to F.H. and Leon Levy Foundation Fellowship in Neuroscience to S.S.D.
Competing interests
The authors report no competing interests.
Glossary
- FTLD =
frontotemporal lobar degeneration
References
- Alizadeh A, Dyck SM, Karimi-Abdolrezaee S.. Myelin damage and repair in pathologic CNS: challenges and prospects. Front Mol Neurosci 2015; 8: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F.. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol 1990; 27: 229–37. [DOI] [PubMed] [Google Scholar]
- Brady OA, Zheng Y, Murphy K, Huang M, Hu F.. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 2013; 22: 685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Zhou X, Hu F.. Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J Biol Chem 2014; 289: 19670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charzewska A, Wierzba J, Izycka-Swieszewska E, Bekiesinska-Figatowska M, Jurek M, Gintowt A, et al. Hypomyelinating leukodystrophies - a molecular insight into the white matter pathology. Clin Genet 2016; 90: 293–304. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci 2012; 32: 11213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, et al. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc 2007; 2: 1044–51. [DOI] [PubMed] [Google Scholar]
- Clayton EL, Milioto C, Muralidharan B, Norona FE, Edgar JR, Soriano A, et al. Frontotemporal dementia causative CHMP2B impairs neuronal endolysosomal traffic-rescue by TMEM106B knockdown. Brain 2018; 141: 3428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339: 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 2011; 68: 581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming Y, Cruchaga C.. TMEM106B: a strong FTLD disease modifier. Acta Neuropathol 2014; 127: 419–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust PL, Kaye EM, Powers JM.. Myelin lesions associated with lysosomal and peroxisomal disorders. Expert Rev Neurother 2010; 10: 1449–66. [DOI] [PubMed] [Google Scholar]
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 2011; 76: 467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 2014; 127: 407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamp AC, Tanaka Y, Lullmann-Rauch R, Wittke D, D’Hooge R, De Deyn PP, et al. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum Mol Genet 2003; 12: 631–46. [PubMed] [Google Scholar]
- Garcia-Mateo N, Pascua-Maestro R, Perez-Castellanos A, Lillo C, Sanchez D, Ganfornina MD.. Myelin extracellular leaflet compaction requires apolipoprotein D membrane management to optimize lysosomal-dependent recycling and glycocalyx removal. Glia 2018; 66: 670–87. [DOI] [PubMed] [Google Scholar]
- Guo DZ, Xiao L, Liu YJ, Shen C, Lou HF, Lv Y, et al. Cathepsin D deficiency delays central nervous system myelination by inhibiting proteolipid protein trafficking from late endosome/lysosome to plasma membrane. Exp Mol Med 2018; 50: e457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, Jenkins LW.. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma 1994; 11: 187–96. [DOI] [PubMed] [Google Scholar]
- Ito Y, Hartley T, Baird S, Venkateswaran S, Simons C, Wolf NI, et al. Lysosomal dysfunction in TMEM106B hypomyelinating leukodystrophy. Neurol Genet 2018; 4: e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Kramer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, et al. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci 1995; 7: 1245–65. [DOI] [PubMed] [Google Scholar]
- Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, et al. Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 2017; 95: 281–96 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Urano Y, Fujikawa Y, Kobayashi T, Kojima H, Terai T, et al. Development of 2,6-carboxy-substituted boron dipyrromethene (BODIPY) as a novel scaffold of ratiometric fluorescent probes for live cell imaging. Chem Commun 2009; 7015–7. [DOI] [PubMed] [Google Scholar]
- Kundu ST, Grzeskowiak CL, Fradette JJ, Gibson LA, Rodriguez LB, Creighton CJ, et al. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat Commun (Camb) 2018; 9: 2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 2012; 287: 19355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Le Ber I, Galimberti D, Serpente M, Rivaud-Pechoux S, Camuzat A, et al. Defining the association of TMEM106B variants among frontotemporal lobar degeneration patients with GRN mutations and C9orf72 repeat expansions. Neurobiol Aging 2014; 35: 2658 e1–e5. [DOI] [PubMed] [Google Scholar]
- Li Z, Farias FHG, Dube U, Del-Aguila JL, Mihindukulasuriya KA, Fernandez MV, et al. The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol 2020; 139: 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie PPY, Nixon RA.. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis 2019; 122: 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science 2013; 339: 823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019; 142: 1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AM, Zhou X, Perkerson RB, Parsons TM, Chew J, Brooks M, et al. Loss of Tmem106b is unable to ameliorate frontotemporal dementia-like phenotypes in an AAV mouse model of C9ORF72-repeat induced toxicity. Acta Neuropathol Commun 2018; 6: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo LM, Vogel H, Reimer RJ.. The lysosomal sialic acid transporter sialin is required for normal CNS myelination. J Neurosci 2009; 29: 15355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn H, Abeliovich A.. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst 2017; 4: 404–15.e5. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA, et al. TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 2012; 79: 717–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer JL, Zalc B.. Myelination. Curr Biol 2016; 26: R971–R5. [DOI] [PubMed] [Google Scholar]
- Schmiedt ML, Blom T, Blom T, Kopra O, Wong A, von Schantz-Fant C, et al. Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol Dis 2012; 46: 19–29. [DOI] [PubMed] [Google Scholar]
- Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, et al. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J 2014; 33: 450–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons C, Dyment D, Bent SJ, Crawford J, D’Hooghe M, Kohlschutter A, et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 2017; 140: 3105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole-Domenech S, Cruz DL, Capetillo-Zarate E, Maxfield FR.. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res Rev 2016; 32: 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM.. Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci 2014; 61: 226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley JL, Lincoln RJ, Brown TA, McDonald LM, Dawson GR, Reynolds DS.. The mouse beam walking assay offers improved sensitivity over the mouse rotarod in determining motor coordination deficits induced by benzodiazepines. J Psychopharmacol 2005; 19: 221–7. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Dhaunchak AS, Goncalves JT, Wenzel D, Schneider A, Bunt G, et al. Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J Cell Biol 2006; 172: 937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J, et al. TMEM106B effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol 2019; 85: 801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 2014; 127: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010; 42: 234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 2011; 134: 808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancha AR, Govindaraju S, Parsa KV, Jasti M, Gonzalez-Garcia M, Ballestero RP.. Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol 2004; 4: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 2011; 121: 373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterstein C, Trotter J, Kramer-Albers EM.. Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J Cell Sci 2008; 121: 834–42. [DOI] [PubMed] [Google Scholar]
- Xu H, Ren D.. Lysosomal physiology. Annu Rev Physiol 2015; 77: 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Kubisiak T, Ji H, Xiao J, Wang J, Burmeister M.. The recurrent mutation in TMEM106B also causes hypomyelinating leukodystrophy in China and is a CpG hotspot. Brain 2018; 141: e36. [DOI] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bastos de Oliveira F, Qi X, Brown WJ, Smolka MB, et al. Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J Cell Biol 2015; 210: 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author on request.