

Abstract
Nonalcoholic fatty liver disease (NAFLD) represents a burgeoning worldwide epidemic whose etiology reflects multiple interactions between environmental and genetic factors. Here we review the major pathways and dominant genetic modifiers known to be relevant players in human NAFLD and which may determine key components of the heritability of distinctive disease traits including steatosis and fibrosis. In addition, we have employed general assumptions which are based on known genetic factors in NAFLD to build a systems biology prediction model that includes functional enrichment. This new prediction model highlights additional complementary pathways that represent plausible intersecting signaling networks that we define here as a NAFLD-Reactome. We review the evidence connecting variants in each of the major known genetic modifiers (variants in PNPLA3, TM6SF2, MBOAT7, GCKR and HSD17B13) to NAFLD and expand the associated underlying mechanisms using functional enrichment predictions, based on both preclinical and cell based experimental findings. These major candidate gene variants function in distinct pathways, including substrate delivery for de-novo lipogenesis; mitochondrial energy utilization; lipid droplet assembly, lipolytic catabolism and fatty acid compartmentalization; and VLDL assembly and secretion. The NAFLD-Reactome model expands these pathways and allows for hypothesis testing as well as serving as a discovery platform for druggable targets across multiple pathways that promote NAFLD development and which influence several progressive outcomes. In conclusion, we summarize the strengths and weaknesses of studies implicating selected variants in the pathophysiology of NAFLD and highlight opportunities for future clinical research and pharmacologic intervention, as well as the implications for clinical practice.
Keywords: NASH, genetics, PNPLA3, HSD17B13, TM6SF2, HSD17B13
Importance of Genetic pathways in NAFLD:
Nonalcoholic fatty liver disease (NAFLD) represents the most prevalent liver disease in the world, whose health and economic impact is now widely recognized as a global burden (1). The relentlessly increasing incidence of NAFLD is associated with the burgeoning worldwide epidemic of obesity (2), but in addition, the past two decades have witnessed increasing awareness of the role of genetic factors in both the etiology and progression of this multifactorial disease. Significant advances in understanding the heritable contribution to NAFLD have been achieved through combinations of stringent patient selection (refining phenotypic variation from families and from twin studies) and with the applications of genome-wide association studies (GWAS) and exome sequencing within large populations. However, because NAFLD is common and highly prevalent among obese subjects, and because obesity and NAFLD are both independently associated with type 2 diabetes, assigning genetic linkage to distinctive traits among NAFLD subjects is influenced by modifiers associated with each of these diseases.
Earlier retrospective studies from different US centers reported familial clustering of phenotypes among kindreds in which affected members manifested advanced features of NAFLD, including nonalcoholic steatohepatitis (NASH) and cirrhosis (3, 4). Among the shared features was that greater than 80% of first-degree relatives of probands with NASH exhibited glucose intolerance and insulin resistance, with or without overt type 2 diabetes (3, 4), findings replicated in studies showing increased prevalence of insulin resistance in family members with NAFLD (5). Studies in 313 Finnish twins selected from a population cohort showed that ~60% of the variation in serum alanine aminotransferase (ALT) levels was heritable and revealed higher ALT levels in monozygotic versus dizygotic twins (6), suggesting distinctive genetic contributions to specific traits among NAFLD patients. Those findings were amplified through studies from US populations showing that both hepatic steatosis, assessed by magnetic resonance imaging proton-density fat fraction (MRI-PDFF) and also hepatic fibrosis, assessed by magnetic resonance elastography (MRE), were each significantly correlated among monozygotic twins (7). Those findings are in line with work showing that diabetes is strongly associated with the risk of NASH and advanced fibrosis in NAFLD patients (8). Cross-sectional analyses revealed that probands of patients with NAFLD-associated cirrhosis exhibited advanced fibrosis at a rate 12 times higher (17.9%) than in first degree relatives of age-matched controls without features of NAFLD (1.4%)(9). Even first-degree relatives of NAFLD probands without cirrhosis exhibited higher rates of fibrosis (12%), suggesting that heritable factors play an important role in both the etiology and progression of NAFLD. Those conclusions are substantiated by findings in pediatric populations, where steatosis was found in 17% of siblings and 37% of parents of overweight children and in 59% of siblings and 78% of parents of children with NAFLD (10). Considered together, heritability estimates for various traits associated with NAFLD (obesity, metabolic syndrome, insulin resistance, hepatic steatosis, fibrosis) range from 30–75% (11–13) although the variants identified for certain traits, for example steatosis, are not uniformly associated with other features such as fibrosis or insulin resistance, suggesting genetic heterogeneity within pathways that influence selected traits (13).
In addition and as alluded to above, because complex traits reflect the interplay between host genetic and environmental factors, a complete understanding of the genetic pathways involved in NAFLD requires that networks linking various traits to environmental modifiers (for example smoking (14)) be considered, as well as other networks that may synergize with NAFLD traits, such as obesity, cardiometabolic disorders (15, 16) and insulin resistance (17). Accordingly, the genetic contribution to NAFLD phenotypes reflects a spectrum of Mendelian monogenic and polygenic variants as well as mitochondrial variants. Among polygenic traits (such as obesity, Figure 1), variants in many genes each contribute variable but small effects and it is the combination of these effects that determines phenotypic manifestations. Conversely, monogenic disorders reflect mutations and/or variants in single genes, typically with Mendelian recessive or dominant inheritance. Pedigree analyses in monogenic disorders typically reveals multiple affected family members and the effects of these isolated mutations/variants are usually large. There are many hereditary diseases, either monogenic or syndromic, that include hepatic steatosis as a clinical manifestation. Examples include inborn errors of metabolism, familial lipodystrophies, mitochondrial diseases, glycosylation, lysosomal and oxidative phosphorylation deficiencies. Hereditary disorders associated with hepatic steatosis includes 132 genes, among which are 32 loci associated with the pathogenesis of NAFLD in different human studies (Supplementary Table 1). For the purposes of this review, we will focus on pathways and variants regulating lipid droplet (LD) biology, de-novo lipogenesis and fatty acid compartmentalization and the assembly and secretion of very low-density lipoproteins (VLDL) (Figure 1). The causal role of these variants in the pathogenesis NAFLD has been validated by functional studies and wide replication in clinical cohorts (18).
Figure 1. Overview of genetic pathways in nonalcoholic fatty liver disease (NAFLD).
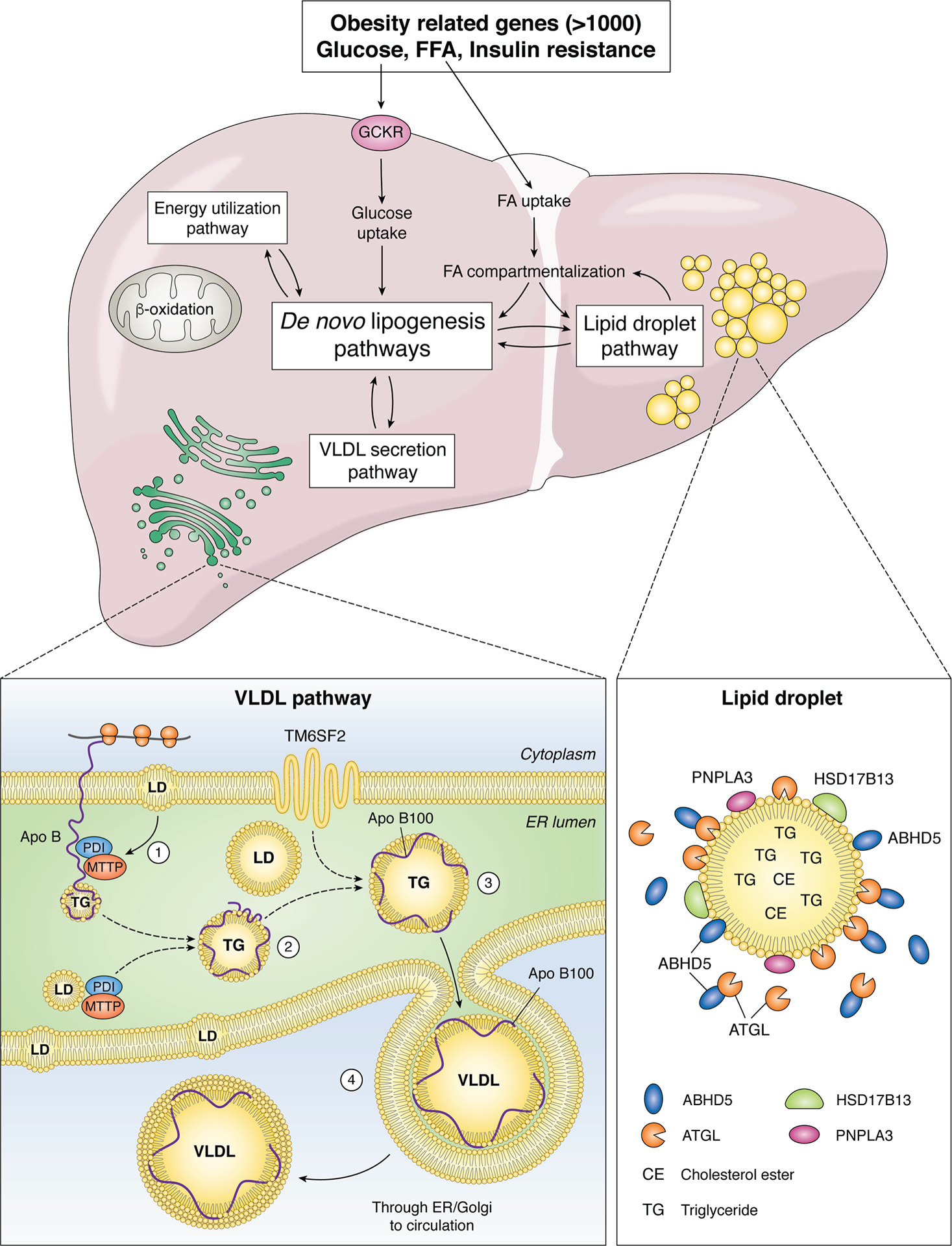
The major pathways involved in lipid trafficking, compartmentalization and utilization are represented within boxes, each of which undergo regulation through a combination of host genetic factors and environmental interactions (14). Obesity and insulin resistance are each complex genetic traits, with variants in more than 1000 genes linked to altered susceptibility (106, 107). Variations in energy metabolism are linked to variants in mitochondrial genes including uncoupling protein 1, 2 (UCP1, UCP2) as well as superoxide dismutase2 (SOD2), whose candidate genes are expressed in extrahepatic tissues (adipose, muscle) as well as the liver. Fatty acid uptake and metabolic channeling results in partitioning between de novo lipogenesis/energy utilization/lipid droplet pathways. Hepatic glucose uptake and utilization is also regulated by variants in glucokinase regulator (GCKR) (61), which in turn regulate substrate flow for de novo lipogenesis. In addition, de novo lipogenesis pathways also interact metabolically with the very low density lipoprotein (VLDL) secretion pathway. The lipid droplet (LD) pathway includes many of the candidate genes implicated in NAFLD development and progression, including patatin-like phospholipase domain-containing protein 3 (PNPLA3), abhydrolase containing domain 5 (ABDH5), adipose triglyceride lipase (ATGL), hydroxysteroid 17-β-dehydrogenase B13 (HSD17B13). Those proteins are associated with LDs which contain a core of neutral lipids (triglyceride, TG and cholesterol ester, CE). The VLDL pathway includes gatekeeper genes (microsomal triglyceride transfer protein, MTTP and apolipoprotein B, APOB, variants of which impair VLDL assembly within the endoplasmic reticulum (ER). MTTP is an endoluminal ER protein that functions as an obligate heteromeric complex with protein disulfide isomerase (PDI) and together promote lipidation and correct folding of the APOB protein around a core of neutral lipid transferred from membrane associated and intraluminal LD. In addition, variations in another transmembrane ER associated protein, transmembrane 6 superfamily 2 (TM6SF2) are associated with defective VLDL assembly and secretion (74, 75).
Role of genetic variants in LD metabolic pathways (Figure 1)
PNPLA3
The rs738409 C>G single nucleotide polymorphism (SNP), encoding for the I148M protein variant of Patatin-like phospholipase domain-containing 3 (PNPLA3), accounts for the largest fraction of genetic predisposition to NAFLD in all populations, and also accounts for the inter-ethnic variability in disease susceptibility (19). The I148M variant is specifically associated with increased hepatic fat, but without a major direct impact on adiposity and insulin resistance (20), and while mouse models show no impaired secretion of VLDL (21), there is evidence to support defective VLDL secretion in humans (19, 22). The PNPLA3 I148M variant increases susceptibility to the whole spectrum of progressive liver damage related to NAFLD (23–26) and functions as a common modifier of liver disease risk and progression (27–31). Pathophysiological studies established that during obesity and insulin resistance PNPLA3 protein expression is induced in hepatocytes, hepatic stellate cells, and adipocytes, and is localized on LDs, where it represents the dominant surface protein (32, 33). PNPLA3 has intrinsic lipase activity on triglycerides, phospholipids, and retinyl esters, mediating the hydrolytic production of oleate and other unsaturated fatty acids, including arachidonic acid (21, 33). While the wild-type protein is rapidly degraded, the mutant protein cannot be ubiquitylated and consequently accumulates on the LD surface, simultaneously altering core lipid remodeling and turnover (21, 34–36). The mutant I148M protein functions as a co-dominant negative rather than a loss-of-function allele, impairing LD hydrolysis (36, 37). The mechanisms for impaired LD lipolysis with the mutant PNPLA3 I148M include sequestration of the required cofactor, 1 abhydrolase domain containing 5 (ABHD5/CGI58) (Figure 1), that normally promotes lipolysis by adipose triglyceride lipase, ATGL/PNPLA2 (38, 39). In addition, hepatocytes expressing mutant PNPLA3 exhibit defective autophagic flux and reduced lipophagy (40). In combination, these defects promote steatosis, LD accumulation and NAFLD.
The codominant negative function model for PNPLA3 I148M is supported by human genetics data, where carriage of another PNPLA3 variant (E434K), associated with reduced PNPLA3 expression, mitigated LD accumulation produced by the 148M variant (21, 34, 35), whereas loss-of-function PNPLA3 variants were not associated with severe liver disease (35). Although the mechanism underlying steatosis development is dependent on accumulation of the PNPLA3 protein on lipid droplets with trans-repression of ATGL (21, 34, 37, 41), carriage of the I148M variant is also associated with an alteration of lipid remodeling with accumulation of poly-unsaturated fatty acids in DAG/TAGs and depletion in phospholipids, which is accounted for by reduced enzymatic activity (34, 42). Furthermore, the I148M variant impairs retinol release from hepatic stellate cells while also reducing LD abundance, a combination that promotes stellate cell activation, leading to inflammation, fibrogenesis, and carcinogenesis (33, 43, 44). Finally, the I148M variant may also modify adipocyte lipid metabolism and secretion of adipokines, which in turn may impact the liver phenotype (39, 45). Other key players in LD lipolysis and turnover degradation have also been implicated directly in genetic forms of NAFLD, including mutant forms of ABHD5 which are associated with severe NAFLD in heterozygous adult subjects. The underlying mechanisms likely reflect haploinsufficiency and progressive impairment in ABHD5 protein activity (46), a prediction based on preclinical findings that liver-specific deletion of Abhd5/CGI-58 in mouse liver is associated with progressive NAFLD, NASH and fibrosis (47).
LAL
Isoforms of lysosomal acid lipase (LAL) arising from mutations in the LIPA gene are implicated in defective lysosomal LD turnover and early onset neutral lipid storage defects, promoting accumulation of hepatic cholesteryl esters and triglycerides due to defective lysosomal hydrolysis and impaired LD autophagy, causing a severe genetic form of NAFLD (Supplemental Table 1) (48). Homozygous LAL-D in its most severe form (<1% residual activity) causes neonatal hepatosplenomegaly and early onset liver failure (Wolman disease). However, it remains unclear if heterozygous LIPA carriers are at risk for adult NAFLD and the carrier state may be much less common than previously thought. Earlier estimates indicated a carrier prevalence of 1:40,000 while a recent survey of 120 known variants found the incidence to be <1:175,000, suggesting LAL-D is an extremely rare, albeit treatable condition, that should be considered as a secondary cause of NALFD (49).
HSD17B13
Recent advances further illustrate the important role of LD remodeling in modifying the NAFLD phenotype. Specifically, variation in 17-beta hydroxysteroid dehydrogenase 13 (HSD17B13), which encodes an enzyme that localizes on LDs in hepatocytes, has been linked to protection against the development of hepatic inflammation and fibrosis in the setting of either metabolic dysfunction or alcohol abuse disorder (50–54). HSD17B13 variants which protect against NAFLD progression are associated with reduced activity of the enzyme, with the original reports linking the association to reduced NASH but not to decreased hepatic steatosis (54), suggesting a loss-of-function HSD17B13 allele impairs NAFLD progression rather than initiation (50, 51). Those conclusions are consistent with other observations suggesting that the impact of the mutant HSD17B13 on reducing ALT levels was greater in subjects carrying mutant alleles (PNPLA3, TM6SF2) predisposing to hepatic steatosis (52). The underling mechanisms linking mutant HSD17B13 with reduced progressive liver damage is unrelated to altering hepatic steatosis per se, but rather may reflect altered retinol metabolism and activation of fibrogenic and inflammatory pathways (53), although the cell-specific pathways (macrophage, stellate cell, hepatocyte) are yet to be elucidated. Nevertheless, the available information points to therapeutic targeting of HSD17B13 as a strategy to mitigate progression of NAFLD.
Role of genetic variants in de novo lipogenesis, FA compartmentalization pathways
FA compartmentalization is used loosely to reflect substrate (FA) flow across multiple pathways and includes FA uptake, FA flux in the setting of de novo lipogenesis as well as FA reutilization in the setting of LD hydrolysis and remodeling. The interconnected nature of these various settings is illustrated in Figure 1. Genetic evidence has implicated lipid synthesis pathways in the pathogenesis of NAFLD, including variants in Glucokinase regulator (GCKR) expression (55–57). GCKR regulates de novo lipogenesis by modulating the influx of glucose into hepatocytes and consequently the flow of substrate for lipogenesis. Most data point to a common SNP (rs1260326), encoding for the P446L variant, as the causal variant underlying the association with fatty liver (58–60). This amino acid substitution impairs the ability of GCKR to inhibit glucokinase in response to fructose-6-phosphate, a response which represents a negative feedback loop controlling hepatic glucose uptake (58, 60). The functional consequences for subjects harboring the P446L GCKR variant include decreased circulating glucose and heightened insulin sensitivity, which might portend net benefit, but at the same time the P446L variant increases hepatic production of malonyl-CoA, which promotes hepatic triglyceride accumulation by serving as a substrate for de novo lipogenesis and by blocking fatty acid oxidation, thus promoting hepatic steatosis (59). A rare nonsense mutation (rs149847328, p.Arg227Ter) in the glucokinase regulator (GCKR) gene has been reported in an adult patient with progressive NAFLD, morbid obesity, and type 2 diabetes (60). Hepatic GCKR protein expression was markedly decreased compared with NAFLD subjects who carry the wild-type allele. The presence of this mutation should be suspected as potentially associated with NAFLD, particularly in young adults with extreme histological phenotypes.
Gene variation at the Protein phosphatase 1 regulatory subunit 3B (PPP1R3B) locus has also been suggested to protect against hepatic fat accumulation (13, 61, 62). The mechanisms likely include increased PPP1R3B expression, which may cause metabolic redistribution of excess energy substrate towards glycogen instead of lipid synthesis (61). In particular, the rs4841132 variant also resulted in decreased risk of progressive liver disease in individuals at high risk of NASH (61). However, the overall impact of this variant on the risk of NAFLD and liver-related events remains controversial (62).
Insights into the importance of lipid remodeling in the pathogenesis of liver disease came from genome wide association studies (GWAS) evaluating susceptibility to alcohol related cirrhosis in European cohorts, highlighting the association of a variant close to (but not within) the Membrane bound O-acyltransferase domain-containing 7 (MBOAT7) locus (27). The rs641738 C>T variant was also associated with predisposition to hepatic steatosis and to develop progressive NAFLD with inflammation, fibrosis, and hepatocellular carcinoma (63, 64), although other studies concluded this variant does not confer increased NAFLD risk (65, 66). MBOAT7 is expressed in hepatocytes in the endoplasmic reticulum, and possibly lipid droplets during steatosis, and is involved in the remodeling of phosphatidylinositol and other phospholipids by incorporating arachidonic acid and other unsaturated fatty acids. The common rs641738 C>T variant linked to the 3’ untranslated region of the MBOAT7 has been linked with MBOAT7 downregulation (63). In keeping with this prediction, impaired MBOAT7 expression results in reduced levels of phophatidylinositol containing arachidonic acid, both in hepatocytes and in the circulation (63, 67). Accumulating evidence show that downregulation of MBOAT7 expression is implicated in NAFLD development during obesity and insulin resistance and reflects altered phospholipid remodeling in hepatocellular lipid droplets, specifically lysophosphatidylinositol lipids, whose administration in mice directly promotes fibrogenic and inflammatory changes in an MBOAT7-dependent manner (68, 69). Other variants have been implicated as possible modifiers of liver injury, including rs28929474, a low frequency variant in SERPINA1 (encoding alpha-1-antitrypsin Pi*Z variant) as a putative risk factor for cirrhosis in patients with NAFLD who are of European descent (70). However, earlier work showed that rs28929474 was not associated with severe liver damage in non-cirrhotic patients with NAFLD (71).
Role of genetic variants in VLDL assembly and secretion pathways (Figure 1)
Hepatic lipid export depends largely on the assembly and secretion of lipoproteins in a multistep process beginning in the rough and smooth endoplasmic reticulum (ER) with mobilization of LDs from within the bilayer into the ER lumen (Figure 1, lower inset). This initial step requires an endoluminal transfer protein, microsomal triglyceride transfer protein (MTTP) and an acceptor protein, apolipoproteinB (APOB), mutations or deletions of which eliminate VLDL assembly and result in hepatic steatosis accompanied by extremely low levels of cholesterol and triglyceride in plasma (72). MTTP is a resident endoluminal ER protein that functions as a heteromeric complex with a chaperone, protein disulfide isomerase, and transfers both neutral lipids (cholesterol and triglyceride) as well as phospholipid to the nascent APOB protein, thereby promoting assembly of a lipoprotein particle within the ER. These nascent particles become progressively enriched with neutral lipid and form a globular structure that transits through the ER and Golgi through tubulovesicular networks that eventually result in VLDL secretion into the Space of Disse. Genetic variants in APOB and MTTP cause autosomal codominant familial hypobetalipoproteinemia and autosomal recessive abetalipoproteinemia, respectively and represent rare genetic causes of NAFLD, cirrhosis and hepatocellular cancer (HCC) (73). In 2014, three groups simultaneously identified another genetic cause of hepatic steatosis and NAFLD, associated with defective VLDL assembly and secretion, namely a variant in TM6SF2 (74–76). Subjects with the rs58542926 variant TM6SF2 (E167K) exhibit a loss-of-function allele with defective VLDL secretion, reduced LDL levels (74–76), hepatic steatosis and fibrosis (77–80). These studies are consistent in suggesting a model in which defective VLDL secretion results in hepatic steatosis, NAFLD and progressive liver disease, but reduced serum lipids and an improved risk profile for cardiovascular disease (76, 81). However, basic mechanistic studies of VLDL secretion in global Tm6sf2 knockout mouse models have been inconsistent, with hepatic steatosis and decreased VLDL secretion found in some (76, 82), while others showed no change in hepatic lipid and an unexplained increase in serum triglyceride levels (83). Paradoxically, adeno-associated or transgenic Tm6sf2 overexpression increased hepatic lipid content and decreased VLDL secretion in mice (83, 84). But even those findings are inconsistent since other data show increased VLDL secretion with Ad-Tm6sf2 administration (75). The divergence in findings from these studies reflect an urgent need to understand the tissue-specific role of Tm6sf2 in hepatic VLDL secretion and fibrogenic injury. Also, because data suggest that subjects with the rs58542926 variant are at increased risk for HCC, both in alcoholic cirrhosis (85, 86) and with NAFLD (63, 78), studies to examine pathways associated with HCC in association with TM6SF2 variants are also timely and relevant.
Systems biology approaches to elucidating networks leading to NAFLD (Figure 2)
Figure 2: Mapping NAFLD onto Reactome pathways elucidates disease mechanisms.
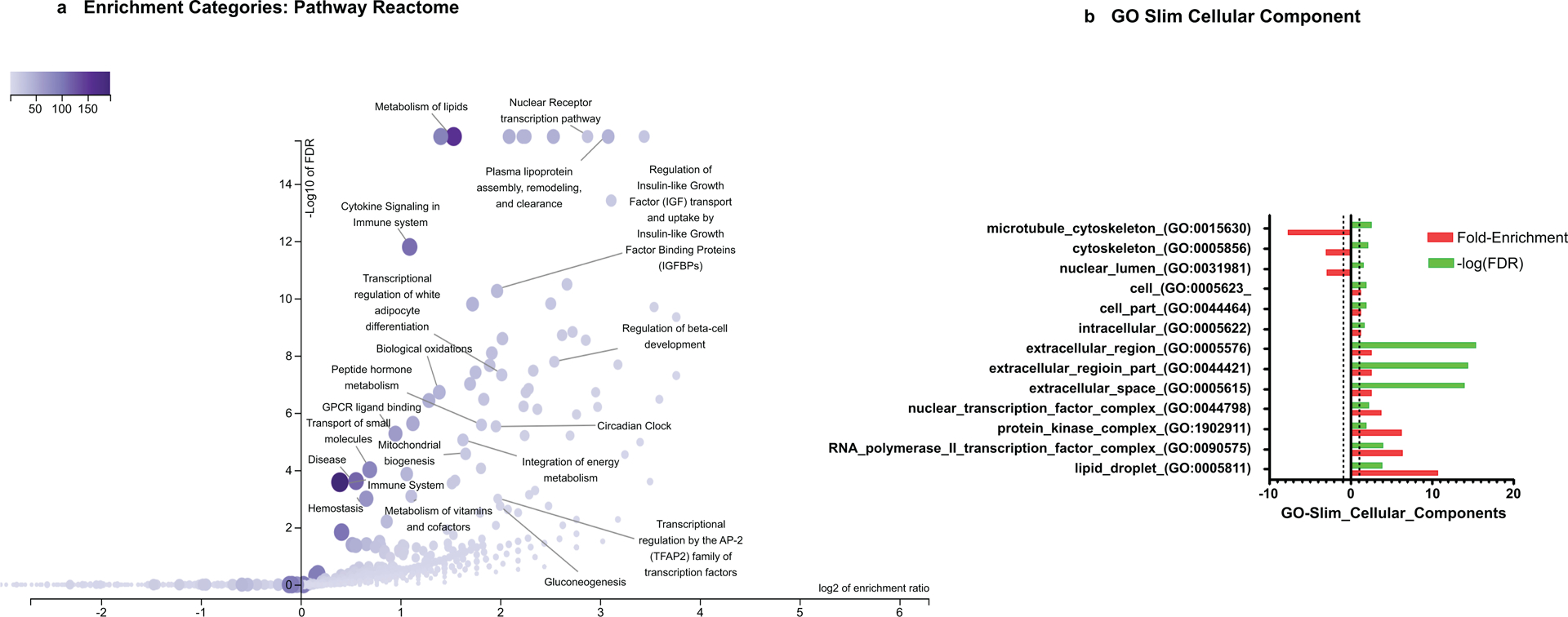
a) Volcano plot on NAFLD-predicted reactome pathways
Enrichment method: ORA (over representation analysis). Enrichment Categories: pathway_Reactome (data source: https://www.reactome.org/ and http://www.geneontology.org. Reference list: all mapped entrez gene IDs from the selected platform genome (61506entrezgene IDs and 10554 IDs are annotated to the selected functional categories that are used as the reference for the enrichment analysis.Organism: Homo sapiens. Parameters for the enrichment analysis: minimum number of IDs in the category: 5; maximum number of IDs in the category: 2000. FDR Method: Benjamini-Hochberg. Significance Level: Top 200. The size and color of the dot is proportional to the number of overlapping (for ORA) or leading edge genes (for GSEA) of the category. ORA was performed by the WebGestalt (WEB-based Gene SeTAnaLysis Toolkit) functional enrichment analysis web tool available at http://www.webgestalt.org/. Figure illustrates results of redundancy reduction (RR) of enriched gene sets; nevertheless, important biological pathways were all covered by the analysis.
To identify the most representative and statistically significant gene sets for visualization, we used redundancy reduction and weighted set cover (99)
b) Figure ORA GO Slim Cellular component
Cellular component was analyzed by PANTHER resource based on Gene ontology (GO) database. PANTHER GO-slim annotations represent only the subset of GO annotations that have been selected by curation (from available experimental annotations), and judged to be evolutionarily conserved(108)Fold-enrichment is calculated for each testing list as: (# genes for the category - # genes expected)/# genes expected; broken lines are shown at 1 and −1 (no enrichments). The negative inverse of the fold-enrichment is used to show values below 1. The -log transformation of false discovery rate (-log(FDR) is also shown.
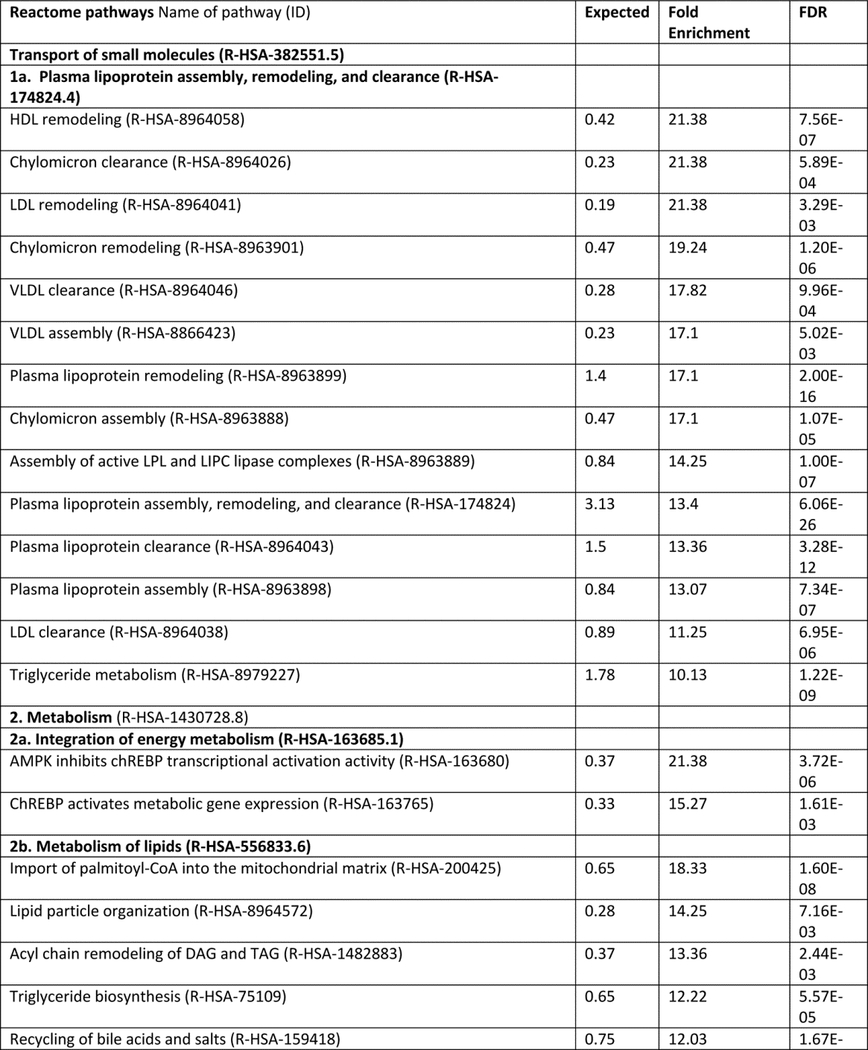
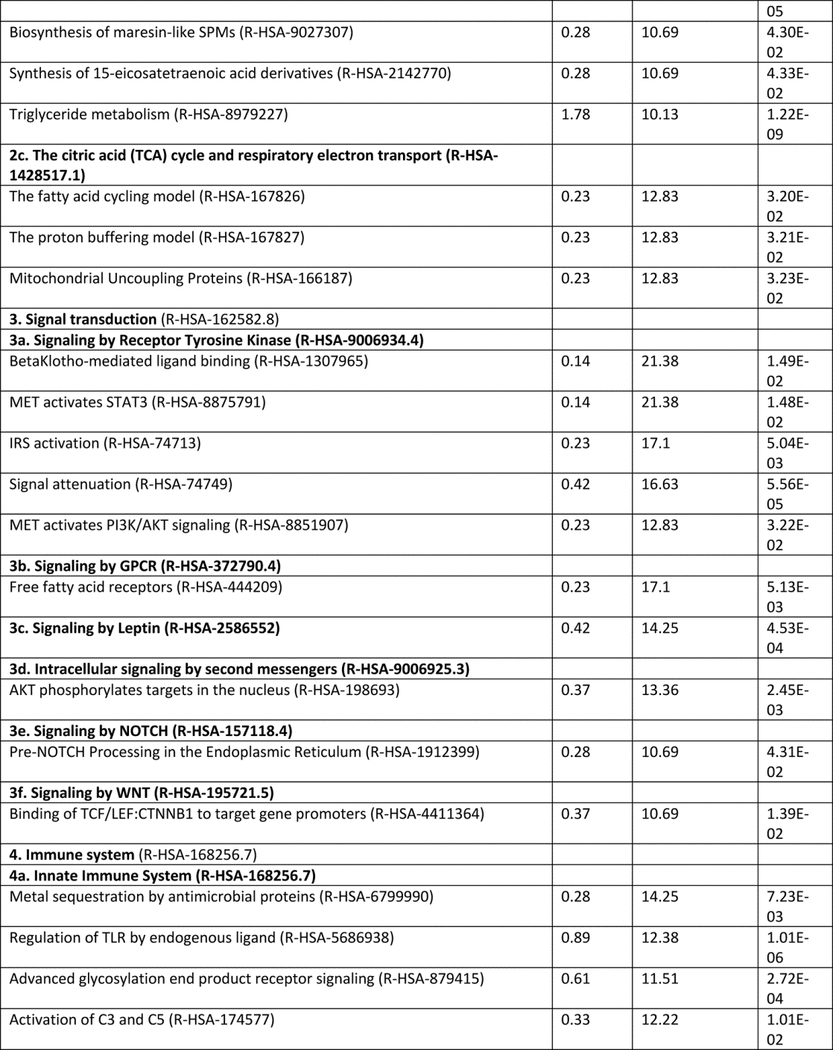
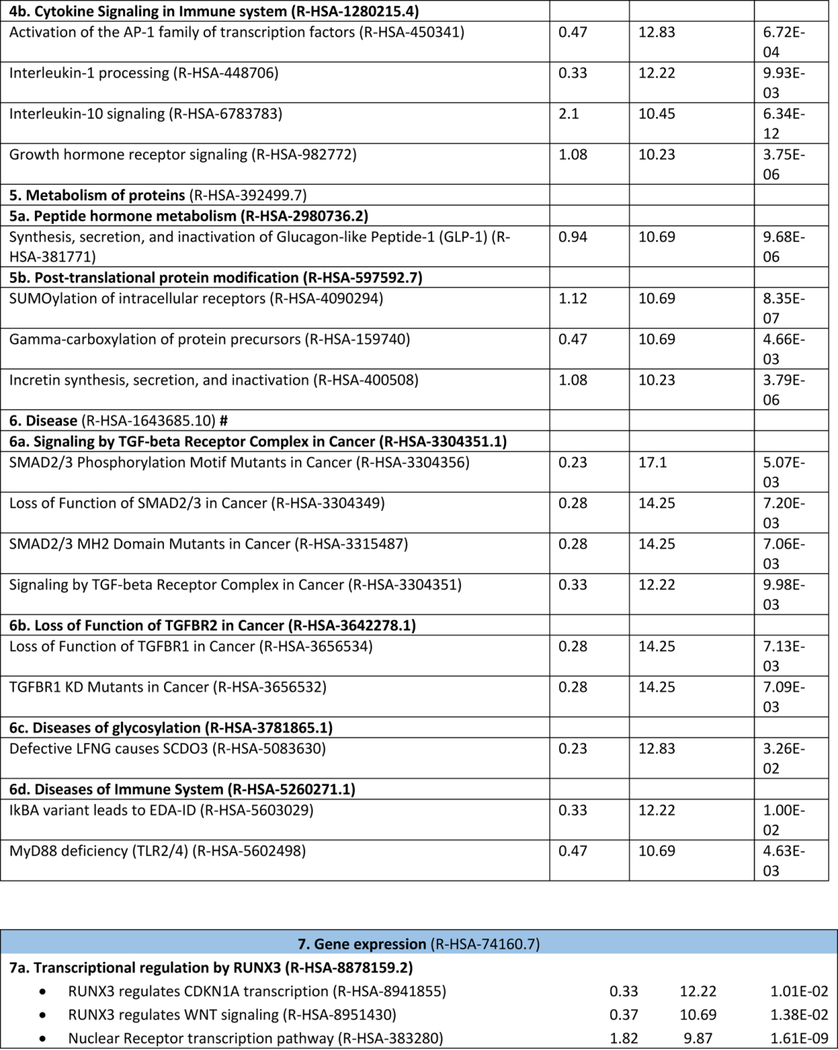
Research on NAFLD and NASH has grown exponentially in the past ten years, with ~ 20238 (updated October 2019) biomedical articles available in the Medline and PubMed query interface associated with the terms “nonalcoholic fatty liver disease”, with an increase from 656 (2010) to 2330 (2016) articles more recently (http://cbdm-01.zdv.uni-mainz.de/~jfontain/cms). Consequently, the concept of designating single mechanisms and/or single genes/proteins in NAFLD (as discussed above) has now evolved into exploration of multiple causal interactions using systems biology approaches. NAFLD represents a spectrum of histologic phenotypes with marked interindividual variability, as well as overlap with metabolic syndrome, and pleiotropy with other diseases (obesity, type 2 diabetes) as well as with several types of cancer (50, 87). These features suggest that mechanisms of pathogenesis might be elucidated by analyzing and modeling multiple NAFLD-genes and proteins into multiple, potentially overlapping networks. This notion of reclassifying disease using systems analysis or network medicine has significantly changed both diagnostic and therapeutic strategies directed towards complex traits (3, 4, 13, 88). We have taken an unbiased approach to interrogate multiple pathways and homeostatic interactions across thousands of proteins as a first step in identifying a potential role in NAFLD, a process that we refer to as the NAFLD-Reactome. The Reactome knowledgebase (https://reactome.org) provides a platform to investigate biological reactions in reactome functions (89) and systematically links human proteins to their molecular functions providing a resource for discovering unexpected functional relationships in OMICs data. Our approach was to extract information on genes and/or proteins through data mining and literature enrichment analysis. We sorted the pathways by fold enrichment values and selected for further analysis those with fold changes (FC) higher than 9.5. This yielded 11 NAFLD-Reactome pathways that were further enriched (Table 1), including transport of small molecules –particularly, “plasma lipoprotein assembly, remodeling, and clearance”, metabolism, with enrichment of the sub-pathway “integration of energy metabolism”, “metabolism of lipids”, and “the citric acid (TCA) cycle and respiratory electron transport”, signal transduction with enrichment of the sub-pathway “signaling by receptor tyrosine kinases”, immune system with enrichment of the sub-pathway “innate immune system” and “cytokine signaling in immune system”, metabolism of proteins enriched of post-translational modifications and “regulation of beta-cell development”. 8 out of the 11 NAFLD-Reactome pathways confirmed >100 terms from the original seed list, including metabolism of lipids, cytokine signaling in immune system, signal transduction, and metabolism of proteins. These findings complement and extend the genetic evidence summarized above and suggest other wide-ranging effects of altered lipid metabolism in NAFLD, including macrophage, stellate cell signaling (90).
Table 1:
NAFLD-Reactome pathway analysis. We used the following analysis types: PANTHER Overrepresentation Test (Released 20190711); annotation version and release date: Reactome version 65 released 2019–03-12; reference list: Homo sapiens (all genes in database) REFLIST (20996). Test: Fisher's exact adjusted by multiple testing (FDR: False Discovery Rate). Seed list: data mining NAFLD (1005 genes/proteins from data mining). Results are restricted to fold enrichment >9.5. #From the viewpoint of molecular pathogenesis, human disease pathways in the Reactome database have three mechanistic causes: (1) the inclusion of microbially-expressed proteins, (2) altered function of human proteins, or (3) changed expression levels of otherwise functionally normal human proteins. In The Reactome Knowledgebase (https://reactome.org), pathways are organized hierarchically, which include pathways events or steps, known as reactions. Table 1 shows the uppermost hierarchical level as pathways nodes, which are numbered consecutively from 1 to 11. Sub-pathways that rank into major nodes are shown under the corresponding node and sub-sub pathways that rank into the sub-pathways are shown under the corresponding sub-pathway. Data mining and literature enrichment analysis used the following sources: Genie web server (http://cbdm.uni-mainz.de/genie), a tool that computes associations of genes with keywords using biomedical literature annotations. The training set included 919 abstracts from PubMed in a background set that involved abstracts from the all PubMed database. Key words: NAFLD and NASH and Mesh terms. Cutoffs were set as 0.01 for abstracts, and false discovery rate<0.01 for genes. A list of 1005 terms (genes/proteins) was constructed, which was used as training list for further systems biology assessment. Reactome pathways used the following sources: PANTHER (protein annotation through evolutionary relationship http://pantherdb.org). Briefly, the statistical test for over- and underrepresentation determines whether a given (functional) class is found statistically more (or less) often in the input list, than expected by chance taking the representation of the pathways genes in the entire human genome. We used those set of terms to assess the Reactome pathways in which the NAFLD-genes/proteins are significantly enriched. The input values were compared to the distribution of all human proteins available in the dataset (~15 000 families of proteins) using Fisher's exact test. Both the overrepresentation test and the enrichment test used the Benjamini-Hochberg's False Discovery Rate (FDR) correction.
|
|
|

|
As expected, the metabolism node (R-HSA-1430728.8) yielded the highest number of terms (n =280, adjusted p value 3.08E-54), followed by signal transduction (249, adjusted p value 5.79E-23) and immune system (196, adjusted p value 1.09E-18). The large node represented by metabolism was also enriched in sub-pathways including “the citric acid/tricarboxylic acid (TCA) cycle and respiratory electron transport”, which included members of the mitochondrial transport carrier family such as uncoupling proteins, implicated in thermogenesis, fatty acid metabolism, and NAFLD-related comorbidities including obesity, diabetes and degenerative diseases. Deregulation of the chemiosmotic gradient created by the TCA cycle and electron transport chain to drive ATP synthesis, which involves the export of protons across the inner mitochondrial membrane, may be crucial to NAFLD development and severity as suggested by preclinical (91) and human studies (11, 92, 93).
Network analysis predicted a node of immune system with sub-pathways including regulation of Toll-like receptors and advanced glycosylation end-product receptor signaling, the accumulation of which has been associated with diabetes, atherosclerosis, renal failure and ageing. Similarly, scavenging by Class B receptors sub-pathway as part of the major vesicle-mediated transport node was overrepresented reinforcing the concept that host-pathogen interactions and pathogen-associated molecular patterns are important players in NAFLD pathophysiology (94) as well as in the modulation of metabolic functioning and energy balance (95). In addition, analysis of NAFLD-Reactome pathways suggests that epigenetic mechanisms (96–98), including posttranslational protein modifications, are important in disease development. More importantly, these pathways are targets of intervention because they exhibit modifications with physical activity (96). Among the sub-pathways that were overrepresented under the major node “Disease” we found overrepresentation of tumor suppressors (SMAD2/3 and TGFBR1), and Runt associated Transcription factor 3 (RUNX3) which is of interest because of findings showing hepatic RUNX3 is downregulated in NASH patients but not in alcoholic steatohepatitis (ASH) (14), a potential discriminator in differentiating of NAFLD and alcohol related fatty liver disease (99).
Pathways related to the regulation of beta-cell development were highly associated with NAFLD, particularly regulation of gene expression in beta cells and AKT-mediated inactivation of FOXO1 (also known as FOXO1A). Previous studies showed that FoxO1 (the mouse ortholog of FOXO1) regulates multiple metabolic functions in the liver, including gluconeogenic, glycolytic, and lipogenic gene expression pathways (100, 101). Finally, the node “organelle biogenesis and maintenance” was 17.1 fold-overrepresented, specifically, the sub-pathway “activation of PPARGC1A (PGC-1α) by phosphorylation”. A previous human study showed decreased PPARGC1A liver mRNA in patients with NAFLD as the consequence of gene promoter methylation (98). Tissue-specific epigenetic modifications in the PPARGC1A promoter also modulate mitochondrial mass and biogenesis, and directly impact glucose metabolism, in line with findings that patients with higher levels of promoter methylation exhibit greater insulin resistance (98).
To gain insights into the biological impact of all 344 Reactome pathways we plotted the weighted set of minimum subset NAFLD-genes/proteins, including all genes from the enriched sets with their P-value (Figure 2a). The volcano plot shows the negative log of the FDR versus the enrichment ratio for all the functional categories in the database, highlighting the degree to which the significant categories stand out from the background (Figure 2a). The NAFLD-Reactome confirmed a role for genes associated with hereditary diseases-related hepatic steatosis, including a highly significant association with mitochondrial fatty acid beta-oxidation of saturated fatty acids with a 55-fold enrichment (p value 1.5e-8), (Supplementary Figure 1). These findings underscore the utility of pathway analyses of NAFLD and provides important new insights into the role of hereditary disorders in the both the identification of causal variants and also potential pharmacological targets. In addition, we used GO Cellular Components to search for the cell/tissue where gene/proteins in the NAFLD gene list are predicted to be preferentially localized (Figure 2b and Table 2). Down-represented cell components (Table 2) reflect a paucity of hits in our data mining search which suggests these pathways are incompletely explored. Nevertheless, understanding of those downregulated pathways may offer an opportunity for future research because loci that are significantly associated with NAFLD may yet contain causative or modifier genes.
Table 2:
GO Cellular Components Associated with NAFLD Genes
| PANTHER GO-Slim Cellular Component | P value |
|---|---|
| GO Lipid droplet | |
| CIDEC (cell death activator CIDE-3) | 1.55 E-06 |
| ABHD5 (abhydrolase domain containing 5) | |
| PNPLA2 | |
| HILPDA (hypoxia-inducible lipid droplet-associated protein) | |
| PLIN4 (perilipin-4) | |
| CIDEB (cell death activator CIDE-B) | |
| CIDEA (cell death activator CIDE-A) | |
| PNPLA3 | |
| HSD17B13 (hydroxy steroid 17-beta dehydrogenase 13) | |
| GO Protein kinase complex | |
| CHUK (inhibitor of nuclear factor kappa-B kinase subunit alpha) | 3.57 E-04 |
| IGF1R (insulin-like growth factor 1 receptor) | |
| PRKAB1 (5’-AMP-activated protein kinase subunit beta-1) | |
| TGFBR1 (TGF-beta receptor type-1) | |
| PRKAB2 (5’-AMP-activated protein kinase subunit beta-2) | |
| INSR (insulin receptor) | |
| IKBKB (inhibitor of nuclear factor kappa-B kinase subunit beta) | |
| RNA polymerase II transcription II factor complex | |
| NR5A2 (nuclear receptor subfamily 5 group A member 2) | 1.03 E-06 |
| PPARG (peroxisome proliferator-activated receptor gamma) | |
| VDR (vitamin D3 receptor) | |
| NR1I2 (nuclear receptor subfamily 1 group I member 2) | |
| RARA (retinoic acid receptor alpha) | |
| PPARD (peroxisome proliferator-activated receptor delta) | |
| NR1H2 (oxysterols receptor LXR-beta) | |
| NR1H3 (oxysterols receptor LXR-alpha) | |
| PPARA (peroxisome proliferator-activated receptor alpha) | |
| NR1H4 (FXR, bile acid receptor) | |
| E2F1 (transcription factor E2F1) | |
| NR1I3 (nuclear receptor subfamily 1 group I member 3) | |
| NR1D2 (nuclear receptor subfamily 1 group D member 2) | |
| Nuclear lumen | |
| OAS1 (2’−5’-oligoadenylate synthase 1 | 8.02 E-04 |
| TP53 (cellular tumor antigen p53) | |
| NFKB1 (nuclear factor NF-kappa-B p105 subunit) | |
| MSL2 (E3 ubiquitin-protein ligase MSL2) | |
| ADARB2 (double-stranded RNA-specific editase B2) | |
| JUN (Transcription factor AP-1) | |
| SPTY2D1 (protein SPT2 homolog) | |
| ZPR1 (zinc finger protein ZPR1) | |
|
Cytoskeleton | |
| PARVB (beta-parvin) | 1.64 E-04 |
| DLG4 (lethal(2) giant larvae protein homolog 1) | |
| WDR1 (WD repeat-containing protein 1) | |
| ALMS1 (Alstrom syndrome protein 1) | |
| ADD1 (alpha-adducin) | |
| DNAH11 (dynein heavy chain 11, axonemal) | |
| MYO1B (unconventional myosin-Ib) | |
| SLC27A4 (long-chain fatty acid transport protein 4) | |
| ADD3 (gamma-adducin) | |
| Nuclear transcription factor complex (GO:0044798) | 1.24E-04 |
| Extracellular space (GO:0005615) | 8.39E-17 |
| Extracellular region part (GO:0044421) | 2.03E-17 |
| Extracellular region (GO:0005576) | 1.01E-18 |
| Intracellular (GO:0005622) | 6.08E-04 |
| Cell part (GO:0044464) | 2.72E-04 |
| Cell (GO:0005623) | 3.69E-04 |
| Microtubule cytoskeleton (GO:0015630) | 4.82E-05 |
We used PANTHER GO-slim updated with 512 cellular component terms
SUMMARY AND CONCLUSIONS
We recognize some limitations in our approach in using molecular networks to infer pathways in NAFLD pathogenesis, principally because the predicted pathways were constructed using existing data in Pubmed plus functional enrichment, both of which need to be experimentally validated. The hierarchy of the predicted pathways also requires formal experimental validation, although the available data from multiomics techniques support our major conclusions (102). Nevertheless, despite the potential constraints of data mining, the predicted NAFLD-Reactome pathways provides important new clues and insights into the genetic pathways involved in NAFLD development and its progression into advanced disease stages with fibrosis and HCC.
Supplementary Material
Perspectives and key messages.
The analysis of pathways suggests that NAFLD results from the disruption to physiological mechanisms that regulate molecular homeostasis of lipoprotein complexes (LD formation/turnover/VLDL secretion). Critical pathway nodes include: lipid particle organization, specifically the partitioning of neutral lipids from their site of synthesis at the endoplasmic reticulum to the cytosol.
The consequences of metabolic disturbances provoke adaptive changes in cellular energy levels, including TCA electron flux imbalance and impairment in proton transport capabilities.
To accommodate hepatocyte perturbations in metabolism and energy balance, complex cell signaling processes that involve phosphorylation of tyrosine residues, STAT3 dimerization and nuclear translocation, insulin and G-protein coupled receptors–signaling as well as perturbations in endoplasmic reticulum (NOTCH), may contribute to disease-specific molecular modules.
NAFLD and cancer progression. Nodes associated with cellular proliferation, differentiation, and perturbation of tissue homeostasis, including transcription factors that modulate cell cycle, as well as growth factors and inflammatory cytokines.
Nodes associated with tumor suppressors are critically involved in NAFLD pathogenesis, as well as modules that highlight the importance of the immune system.
Gaps between biology and translation.
Knowledge on genes/proteins involved in NAFLD/NASH pathogenesis as well as locus involved in the genetic disease susceptibility is not yet fully understood.
Only a few – about 20%- of the reported genetic associations explain a substantial portion of trait heritability (85). Several explanations for this missing heritability have been proposed (85), including environmental factors and host-microbe interactions. Results from Reactome analysis suggested involvement of immune system, including innate immunity and cytokine signaling.
Factors affecting epigenetic programs, including nutritional status and exposure to environmental toxins may contribute to heritability.
NAFLD-Reactome mapping may be exploited for future drug development. Enriched pathways contain gene/protein druggable targets. Also, integration of gene/protein into pathways may help to explore potential off-target effects.
Grant support:
This study was partially supported by grants PID-C2012–0061, PICT 2014–0432, PICT 2014–1816, PICT 2015–0551 and PICT 2016–0135 (Agencia Nacional de Promoción Científica y Tecnológica, FONCyT), CONICET Proyectos Unidades Ejecutoras 2017, PUE 0055 (to SS and CP); Grants from the National Institutes of Health, DK-112378, DK-119437 and DDRCC DK-52574 (to NOD); Grants AIRC n.16888, Ricerca Finalizzata Ministero della Salute RF-2016–02364358, Ricerca corrente Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, the European Union (EU) Programme Horizon 2020 (under grant agreement No. 777377) for the project LITMUS (LV).
ABBREVIATIONS:
- NAFLD
Nonalcoholic fatty liver disease
- (GWAS)
Genome-wide association studies
- NASH
Nonalcoholic steatohepatitis
- ALT
Alanine aminotransferase
- MRI-PDFF
Magnetic resonance imaging proton-density fat fraction
- MRE
Magnetic resonance elastography
- LD
Lipid Droplet
- VLDL
Very Low Density Lipoprotein
- PNPLA3
Patatin Like Phospholipase Domain Containing 3
- ABHD5
Abhydrolase domain containing 5
- ATGL
Adipose Triglyceride Lipase
- LAL
Lysosomal Acid Lipase.
- HSD17B13
Hydroxysteroid 17-Beta Dehydrogenase 13
- GCKR
Glucokinase Regulator
- PPP1R3B
Protein Phosphatase 1 Regulatory Subunit 3B
- MBOAT7
Membrane Bound O-Acyltransferase Domain Containing 7
- ER
Endoplasmic Reticulum
- MTTP
Microsomal Triglyceride Transfer Protein
- APOB
ApolipoproteinB
- HCC
Hepatocellular carcinoma
- TM6SF2
Transmembrane 6 Superfamily Member 2
- LDL
Low Density Lipoprotein
- TCA
Tricarboxylic Acid Cycle
- TLR
Toll-Like Receptor
- DAMPs
Damage Associated Molecular Patterns
- SMAD
Small worm phenotype, Drosophila Mothers Against Decapentaplegic
- TGFbeta
Transforming Growth Factor beta
- RUNX3
Runt-related transcription factor 3
- ASH
Alcoholic Steatohepatitis
- FOXO1
Forkhead box Protein O1
- PPARgamma
Peroxisome Proliferator Activated Receptor gamma
- IGF
Insulin Like Growth Factor
- GO
Gene ontology
REFERENCES
- 1.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 2.Collaborators GBDO. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med 2017;377:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willner IR, Waters B, Patil SR, Reuben A, Morelli J, Riely CA. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol 2001;96:2957–2961. [DOI] [PubMed] [Google Scholar]
- 4.Struben VM, Hespenheide EE, Caldwell SH. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am J Med 2000;108:9–13. [DOI] [PubMed] [Google Scholar]
- 5.Abdelmalek MF, Liu C, Shuster J, Nelson DR, Asal NR. Familial aggregation of insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2006;4:1162–1169. [DOI] [PubMed] [Google Scholar]
- 6.Makkonen J, Pietilainen KH, Rissanen A, Kaprio J, Yki-Jarvinen H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: a study in monozygotic and dizygotic twins. J Hepatol 2009;50:1035–1042. [DOI] [PubMed] [Google Scholar]
- 7.Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, Nguyen P, et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015;149:1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loomba R, Abraham M, Unalp A, Wilson L, Lavine J, Doo E, Bass NM, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012;56:943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caussy C, Soni M, Cui J, Bettencourt R, Schork N, Chen CH, Ikhwan MA, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest 2017;127:2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, Shiehmorteza M, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009;136:1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sookoian S, Pirola CJ. Nonalcoholic fatty liver disease and metabolic syndrome: Shared genetic basis of pathogenesis. Hepatology 2016;64:1417–1420. [DOI] [PubMed] [Google Scholar]
- 12.Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, Kahali B, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 2013;58:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentley AR, Sung YJ, Brown MR, Winkler TW, Kraja AT, Ntalla I, Schwander K, et al. Multi-ancestry genome-wide gene-smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat Genet 2019;51:636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raulerson CK, Ko A, Kidd JC, Currin KW, Brotman SM, Cannon ME, Wu Y, et al. Adipose Tissue Gene Expression Associations Reveal Hundreds of Candidate Genes for Cardiometabolic Traits. Am J Hum Genet 2019;105:773–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barata L, Feitosa MF, Bielak LF, Halligan B, Baldridge AS, Guo X, Yerges-Armstrong LM, et al. Insulin Resistance Exacerbates Genetic Predisposition to Nonalcoholic Fatty Liver Disease in Individuals Without Diabetes. Hepatol Commun 2019;3:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valenti LVC, Baselli GA. Genetics of Nonalcoholic Fatty Liver Disease: A 2018 Update. Curr Pharm Des 2018;24:4566–4573. [DOI] [PubMed] [Google Scholar]
- 19.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017;66:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Stahlman M, Taskinen MR, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276–1282. [DOI] [PubMed] [Google Scholar]
- 23.Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, Valenti L. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol 2013;19:6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011;53:1883–1894. [DOI] [PubMed] [Google Scholar]
- 25.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1209–1217. [DOI] [PubMed] [Google Scholar]
- 26.Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buch S, Stickel F, Trepo E, Way M, Herrmann A, Nischalke HD, Brosch M, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015;47:1443–1448. [DOI] [PubMed] [Google Scholar]
- 28.Valenti L, Rumi M, Galmozzi E, Aghemo A, Del Menico B, De Nicola S, Dongiovanni P, et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2011;53:791–799. [DOI] [PubMed] [Google Scholar]
- 29.Vigano M, Valenti L, Lampertico P, Facchetti F, Motta BM, D’Ambrosio R, Romagnoli S, et al. Patatin-like phospholipase domain-containing 3 I148M affects liver steatosis in patients with chronic hepatitis B. Hepatology 2013;58:1245–1252. [DOI] [PubMed] [Google Scholar]
- 30.Valenti L, Maggioni P, Piperno A, Rametta R, Pelucchi S, Mariani R, Dongiovanni P, et al. Patatin-like phospholipase domain containing-3 gene I148M polymorphism, steatosis, and liver damage in hereditary hemochromatosis. World J Gastroenterol 2012;18:2813–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stattermayer AF, Traussnigg S, Dienes HP, Aigner E, Stauber R, Lackner K, Hofer H, et al. Hepatic steatosis in Wilson disease--Role of copper and PNPLA3 mutations. J Hepatol 2015;63:156–163. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, Hobbs HH. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A 2010;107:7892–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, Burza MA, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet 2014;23:4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitsche MA, Hobbs HH, Cohen JC. Patatin-like phospholipase domain-containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem 2018;293:6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donati B, Motta BM, Pingitore P, Meroni M, Pietrelli A, Alisi A, Petta S, et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016;63:787–798. [DOI] [PubMed] [Google Scholar]
- 36.Luukkonen PK, Nick A, Holtta-Vuori M, Thiele C, Isokuortti E, Lallukka-Bruck S, Zhou Y, et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019;4:e127902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A 2019;116:9521–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019;69:2427–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang A, Mottillo EP, Mladenovic-Lucas L, Zhou L, Granneman JG. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat Metab 2019;1:560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negoita F, Blomdahl J, Wasserstrom S, Winberg ME, Osmark P, Larsson S, Stenkula KG, et al. PNPLA3 variant M148 causes resistance to starvation-mediated lipid droplet autophagy in human hepatocytes. J Cell Biochem 2019;120:343–356. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Kory N, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luukkonen PK, Nick A, Holtta-Vuori M, Thiele C, Isokuortti E, Lallukka-Bruck S, Zhou Y, et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mondul A, Mancina RM, Merlo A, Dongiovanni P, Rametta R, Montalcini T, Valenti L, et al. PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J Nutr 2015;145:1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pingitore P, Dongiovanni P, Motta BM, Meroni M, Lepore SM, Mancina RM, Pelusi S, et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum Mol Genet 2016;25:5212–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valenti L, Rametta R, Ruscica M, Dongiovanni P, Steffani L, Motta BM, Canavesi E, et al. The I148M PNPLA3 polymorphism influences serum adiponectin in patients with fatty liver and healthy controls. BMC Gastroenterol 2012;12:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youssefian L, Vahidnezhad H, Saeidian AH, Pajouhanfar S, Sotoudeh S, Mansouri P, Amirkashani D, et al. Inherited non-alcoholic fatty liver disease and dyslipidemia due to monoallelic ABHD5 mutations. J Hepatol 2019;71:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo F, Ma Y, Kadegowda AK, Betters JL, Xie P, Liu G, Liu X, et al. Deficiency of liver Comparative Gene Identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res 2013;54:2109–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pericleous M, Kelly C, Wang T, Livingstone C, Ala A. Wolman’s disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol 2017;2:670–679. [DOI] [PubMed] [Google Scholar]
- 49.Carter A, Brackley SM, Gao J, Mann JP. The global prevalence and genetic spectrum of lysosomal acid lipase deficiency: A rare condition that mimics NAFLD. J Hepatol 2019;70:142–150. [DOI] [PubMed] [Google Scholar]
- 50.Pirola CJ, Garaycoechea M, Flichman D, Arrese M, San Martino J, Gazzi C, Castano GO, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res 2019;60:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Trepo E, Nahon P, Cao Q, Moreno C, Letouze E, Imbeaud S, et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019;70:231–240. [DOI] [PubMed] [Google Scholar]
- 52.Gellert-Kristensen H, Nordestgaard BG, Tybjaerg-Hansen A, Stender S. High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase-Lowering Effect of a HSD17B13 Variant. Hepatology 2019. [DOI] [PubMed] [Google Scholar]
- 53.Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, de Boer YS, et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019;69:1504–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med 2018;378:1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol 2018;68:268–279. [DOI] [PubMed] [Google Scholar]
- 56.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, Pelusi S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283:356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez Rodriguez M, Fernandes Silva L, Vangipurapu J, Modi S, Kuusisto J, Kaikkonen MU, Laakso M. Functional Variant in the GCKR Gene Affects Lactate Levels Differentially in the Fasting State and During Hyperglycemia. Sci Rep 2018;8:15989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandes Silva L, Vangipurapu J, Kuulasmaa T, Laakso M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci Rep 2019;9:10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pirola CJ, Flichman D, Dopazo H, Fernandez Gianotti T, San Martino J, Rohr C, Garaycoechea M, et al. A Rare Nonsense Mutation in the Glucokinase Regulator Gene Is Associated With a Rapidly Progressive Clinical Form of Nonalcoholic Steatohepatitis. Hepatol Commun 2018;2:1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dongiovanni P, Meroni M, Mancina RM, Baselli G, Rametta R, Pelusi S, Mannisto V, et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol Commun 2018;2:666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stender S, Smagris E, Lauridsen BK, Kofoed KF, Nordestgaard BG, Tybjaerg-Hansen A, Pennacchio LA, et al. Relationship between genetic variation at PPP1R3B and levels of liver glycogen and triglyceride. Hepatology 2018;67:2182–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, Petta S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep 2017;7:4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, Boren J, et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016;150:1219–1230 e1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xia Y, Huang CX, Li GY, Chen KH, Han L, Tang L, Luo HQ, et al. Meta-analysis of the association between MBOAT7 rs641738, TM6SF2 rs58542926 and nonalcoholic fatty liver disease susceptibility. Clin Res Hepatol Gastroenterol 2019;43:533–541. [DOI] [PubMed] [Google Scholar]
- 66.Sookoian S, Flichman D, Garaycoechea ME, Gazzi C, Martino JS, Castano GO, Pirola CJ. Lack of evidence supporting a role of TMC4-rs641738 missense variant-MBOAT7- intergenic downstream variant-in the Susceptibility to Nonalcoholic Fatty Liver Disease. Sci Rep 2018;8:5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luukkonen PK, Zhou Y, Nidhina Haridas PA, Dwivedi OP, Hyotylainen T, Ali A, Juuti A, et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J Hepatol 2017;67:128–136. [DOI] [PubMed] [Google Scholar]
- 68.Helsley RN, Varadharajan V, Brown AL, Gromovsky AD, Schugar RC, Ramachandiran I, Fung K, et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meroni M, Dongiovanni P, Longo M, Rametta R, Badiali S, Fargion S, Romeo S, et al. Down-regulation of hepatic MBOAT7 by hyperinsulinemia favors steatosis development. J Hepatol 2018;68:S31. [Google Scholar]
- 70.Strnad P, Buch S, Hamesch K, Fischer J, Rosendahl J, Schmelz R, Brueckner S, et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019;68:1099–1107. [DOI] [PubMed] [Google Scholar]
- 71.Valenti L, Dongiovanni P, Piperno A, Fracanzani AL, Maggioni M, Rametta R, Loria P, et al. Alpha 1-antitrypsin mutations in NAFLD: high prevalence and association with altered iron metabolism but not with liver damage. Hepatology 2006;44:857–864. [DOI] [PubMed] [Google Scholar]
- 72.Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014;25:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cefalu AB, Pirruccello JP, Noto D, Gabriel S, Valenti V, Gupta N, Spina R, et al. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler Thromb Vasc Biol 2013;33:2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, Eriksson P, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 2014;111:8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W, Guo Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, Motta BM, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506–514. [DOI] [PubMed] [Google Scholar]
- 78.Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, Allison ME, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology 2015;62:1742–1756. [DOI] [PubMed] [Google Scholar]
- 80.Sookoian S, Castano GO, Scian R, Mallardi P, Fernandez Gianotti T, Burgueno AL, San Martino J, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015;61:515–525. [DOI] [PubMed] [Google Scholar]
- 81.Kahali B, Liu YL, Daly AK, Day CP, Anstee QM, Speliotes EK. TM6SF2: catch-22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology 2015;148:679–684. [DOI] [PubMed] [Google Scholar]
- 82.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem 2016;291:10659–10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fan Y, Lu H, Guo Y, Zhu T, Garcia-Barrio MT, Jiang Z, Willer CJ, et al. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 2016;150:1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, Guindi M, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet 2017;26:2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Falleti E, Cussigh A, Cmet S, Fabris C, Toniutto P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig Liver Dis 2016;48:69–75. [DOI] [PubMed] [Google Scholar]
- 86.Stickel F, Buch S, Nischalke HD, Weiss KH, Gotthardt D, Fischer J, Rosendahl J, et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am J Gastroenterol 2018;113:1475–1483. [DOI] [PubMed] [Google Scholar]
- 87.Sookoian S, Pirola CJ. Genetics of Nonalcoholic Fatty Liver Disease: From Pathogenesis to Therapeutics. Semin Liver Dis 2019;39:124–140. [DOI] [PubMed] [Google Scholar]
- 88.Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol 2007;3:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res 2018;46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petta S, Valenti L, Marra F, Grimaudo S, Tripodo C, Bugianesi E, Camma C, et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. Journal of Hepatology 2016;64:682–690. [DOI] [PubMed] [Google Scholar]
- 91.Sunny NE, Bril F, Cusi K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol Metab 2017;28:250–260. [DOI] [PubMed] [Google Scholar]
- 92.Sookoian S, Castano GO, Scian R, Fernandez Gianotti T, Dopazo H, Rohr C, Gaj G, et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am J Clin Nutr 2016;103:422–434. [DOI] [PubMed] [Google Scholar]
- 93.Sookoian S, Flichman D, Scian R, Rohr C, Dopazo H, Gianotti TF, Martino JS, et al. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J Pathol 2016;240:437–449. [DOI] [PubMed] [Google Scholar]
- 94.Jayakumar S, Loomba R. Review article: emerging role of the gut microbiome in the progression of nonalcoholic fatty liver disease and potential therapeutic implications. Aliment Pharmacol Ther 2019;50:144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Delzenne NM, Knudsen C, Beaumont M, Rodriguez J, Neyrinck AM, Bindels LB. Contribution of the gut microbiota to the regulation of host metabolism and energy balance: a focus on the gut-liver axis. Proc Nutr Soc 2019;78:319–328. [DOI] [PubMed] [Google Scholar]
- 96.Pirola CJ, Gianotti TF, Burgueno AL, Rey-Funes M, Loidl CF, Mallardi P, Martino JS, et al. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013;62:1356–1363. [DOI] [PubMed] [Google Scholar]
- 97.Pirola CJ, Scian R, Gianotti TF, Dopazo H, Rohr C, Martino JS, Castano GO, et al. Epigenetic Modifications in the Biology of Nonalcoholic Fatty Liver Disease: The Role of DNA Hydroxymethylation and TET Proteins. Medicine (Baltimore) 2015;94:e1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sookoian S, Rosselli MS, Gemma C, Burgueno AL, Fernandez Gianotti T, Castano GO, Pirola CJ. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010;52:1992–2000. [DOI] [PubMed] [Google Scholar]
- 99.Sookoian S, Pirola CJ. Systems biology elucidates common pathogenic mechanisms between nonalcoholic and alcoholic-fatty liver disease. PLoS One 2013;8:e58895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 2006;281:10105–10117. [DOI] [PubMed] [Google Scholar]
- 101.Valenti L, Rametta R, Dongiovanni P, Maggioni M, Fracanzani AL, Zappa M, Lattuada E, et al. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes 2008;57:1355–1362. [DOI] [PubMed] [Google Scholar]
- 102.Pirola CJ, Sookoian S. Multiomics biomarkers for the prediction of nonalcoholic fatty liver disease severity. World J Gastroenterol 2018;24:1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.