

Abstract
To further assess the scale and level of parental somatic mosaicism, we queried the CMA database at Baylor Genetics. We selected 50 unrelated families where clinically relevant apparent de novo CNV-deletions were found in the affected probands. Parental blood samples screening using deletion junction-specific PCR revealed four parents with somatic mosaicism. Droplet digital PCR (ddPCR), qPCR, and amplicon-based next-generation sequencing (NGS) were used. Using ddPCR levels of mosaicism ranged from undetectable to 18.5%. Amplicon-based NGS and qPCR for the father with undetectable mosaicism was able to detect mosaicism at 0.39%. In one mother, ddPCR analysis revealed 15.6%, 10.6%, 8.2%, and undetectable levels of mosaicism in her blood, buccal cells, saliva, and urine samples, respectively. Our data suggest that more sensitive and precise methods, e.g. CNV junction-specific LR-PCR, ddPCR, or qPCR may allow for a more refined assessment of the potential disease recurrence risk for an identified variant.
Keywords: copy-number variant (CNV), parental somatic mosaicism, clinical diagnostic testing, mosaicism carrier, recurrence risk
INTRODUCTION
Single nucleotide variants (SNVs) or copy-number variants (CNVs) that arise as post-zygotic errors during cell divisions can result in different cell populations with distinct genotypes, leading to somatic mosaicism1–3. The importance of somatic mosaicism has been demonstrated in the etiology of many human genetic disorders, including cancer and Mendelian conditions4–21. If a mosaic pathogenic variant is also present in germline cells, both affected and unaffected carriers can transmit it to their offspring22–24.
In clinical diagnostics settings, testing for CNVs is completed using chromosomal microarray analysis (CMA) with either single nucleotide polymorphism (SNP) arrays or array comparative genomic hybridization (aCGH) or fluorescence in situ hybridization (FISH). Currently, depending on its size, the minimum detectable level of a mosaic CNV using these methods ranges from 5% to 30%25. If a somatic CNV deletion is below this level in a parent, the deletion in the proband may be interpreted as likely representing a de novo mutational event.
In 2014, we reported the identification of low-level (<10%) parental somatic mosaicism for CNV deletions in four out of 100 unrelated families with children affected by various genetic conditions26. Several subsequent studies, investigating somatic mosaic SNVs in large cohorts, have corroborated our original findings27–31. Importantly, it has been shown that the level of somatic and germline mosaicism in the parents positively correlates with the overall recurrence risk26–33.
Here, we have re-evaluated the inheritance status of 50 additional families with CNV deletions previously determined by clinical CMA in the probands as apparently de novo. We propose an approach for more effective detection of low-level parental somatic mosaicism for CNV deletions.
MATERIALS AND METHODS
Subjects
By querying the CMA database at Baylor Genetics (BG) Laboratories at Baylor College of Medicine (BCM), we have randomly selected 50 unrelated family trios for which clinically relevant CNV deletions were determined by CMA in the probands and their parents to be apparent de novo events in the affected individuals. These CNV deletions were classified as pathogenic, likely pathogenic, or variants of uncertain significance with recurrent CNV deletions or terminal deletions excluded from this study. The CNV sizes ranged from 1.3 kb to 1,172,091.8 kb with a mean size 24,503.0 kb. All samples were de-identified using the Institutional Review Board (IRB) for Human Subject Research at BCM waiver protocols H-41191 and H-42680. Written informed consent under BCM IRB approved protocol H-28088 was obtained from one parent for the study of mosaicism in different somatic tissues.
DNA extraction
Peripheral blood DNA was extracted using the Gentra Puregene Blood kit (Qiagen, Germantown, MD, USA). Saliva or buccal cells were collected using the ORAgene Discover OGR-500 kit or ORAcollect OC-175 kit, respectively (DNA Genotek, Ottawa, Canada). Both saliva and buccal cell DNA were extracted using the prepIT-L2P reagent (DNA Genotek). DNA from urine was isolated using the Quick-DNA Urine Kit (Zymo Research, Irvine, CA, USA). All procedures followed the manufacturers’ instructions.
Characterization of deletion junction fragments
For each affected individual, long-range PCR (LR-PCR) primers, flanking the deletion, were designed based on the CMA data. Two-step PCR using LA Taq DNA polymerase (Takara Bio USA, Inc., Mountain View, CA, USA) was performed to amplify the deletion junction fragment in the proband’s whole blood DNA sample. When the density of array probes at the deletion breakpoint region is too low, the breakpoint location cannot be precisely determined and deletion junction-specific primers may be targeted too far away from the actual breakpoint, preventing the amplification of the junction fragment by LR-PCR. In such cases, a customized 60K array (Agilent Technologies, Santa Clara, CA, USA) targeting the deletion regions was designed for aCGH studies. Based on these results, LR-PCR with re-designed junction-specific primers was performed. When a deletion junction fragment was amplified in the affected proband, the parental samples were examined using the same primer set with 40 cycles of LR-PCR amplification. The precise deletion breakpoint coordinates were determined by Sanger sequencing of the LR-PCR products by Sanger sequencing.
Droplet digital PCR (ddPCR)
In families with parents carrying a mosaic CNV deletion identified by LR-PCR, two sets of ddPCR primers were designed: one set was either specific for the deletion junction or mapping within the deleted region, and another set amplified a diploid segment close to the deletion region. Design details are described in Liu et al34 and are shown in Supplemental Figure 1. Briefly, in each 20 μl reaction, 10 μl of DX200™ ddPCR™ EvaGreen Supermix (Bio-Rad, Hercules, CA, USA), 0.5 μM of each forward and reverse primer, 5 units of HindIII-HF (New England Biolabs, Ipswich, MA, USA), and 100 ng of genomic DNA were added. For each family, the proband’s DNA sample and an unrelated control DNA blood sample was utilized as a positive and negative control, respectively, with each reaction run in triplicate. The ddPCR reactions were carried out using the QX200 AutoDG Droplet Digital PCR System (Bio-Rad). Concentrations of the target alleles were analyzed using QuantaSoft Analysis Pro software (Bio-Rad) according to the manufacturer’s instructions. Variant allele fraction (VAF) representing the level of mosaicism for a CNV deletion was calculated as a proportion of the alleles with CNV deletion relative to the diploid alleles.
Amplicon-based next-generation sequencing (NGS)
An amplicon harboring a heterozygous SNP within a deletion region was amplified by PCR using recombinant Taq DNA Polymerase (ThermoFisher Scientific, Waltham, MA, USA) in the parental mosaic sample. The PCR product was purified by QIAquick PCR Purification Kit (Qiagen) followed by quantification using the Qubit 4 Fluorometer with dsDNA BR Assay (ThermoFisher Scientific). The amplicons of ~ 200 bp were sequenced using the HiSeq X system (Illumina, San Diego, CA, USA) with PE150 reads at CloudHealth Genomics (Shanghai, China)34. Integrative Genomics Viewer (IGV) software was used to analyze the data35,36. In a parental sample, the genotype with fewer reads represents the allele carrying the somatic deletion, while the genotype with more reads represents the allele without the deletion. The relative level of mosaicism for the CNV deletion was estimated as the difference in the number of reads between the two SNP genotypes divided by twice the value of the more frequent genotype reads.
Quantitative PCR (qPCR)
Deletion junction-specific primers were designed to amplify the junction fragment of ~ 400 bp. The GAPDH gene was used as the internal reference. Each 20 μl reaction contains 10 μl of PowerUp™ SYBR™ Green Master Mix (ThermoFisher Scientific), 0.25 μM of each forward and reverse primer, and 50 ng blood DNA with each sample run in duplicate. The qPCR reactions were carried out by CFX Connect Real-Time PCR Detection System (Bio-Rad) and the quantitation cycle (Cq) value for each reaction was read by Bio-Rad CFX Maestro software (Bio-Rad). Relative quantification of the deletion junction fragments (ΔCq) in the proband, the mosaic parent, and an unrelated wild-type control was calculated by comparing it to the internal reference gene. The default quantity of the deleted allele is 50% in the proband’s sample. By comparing the fold-change differences between the parental and the proband’s samples (ΔΔCq), the relative level of mosaicism in the parental sample was determined as 50% (2−ΔΔCq)34.
RESULTS
Characterization of the CNV deletions in 50 probands
Using LR-PCR with primers designed solely based on the results of CMA performed at BG, we were able to amplify the CNV deletion junction fragment in 34 out of 50 (68%) of the affected probands. Of note, we have observed a 90.1% (10 out of 11, Supplemental Table 1) detection rate of the junction-specific products of probands when using CMA V11.2, which had higher oligo probe resolution than the other versions. In the remaining samples (16/50), a lower resolution was insufficient to design primers allowing for amplification of deletion junction and the new custom-designed aCGH had to be used to narrow the CNV deletion breakpoint regions. All CNV deletion breakpoint coordinates determined by Sanger sequencing are listed in Supplemental Table 1. The substantial fraction, 20 of the 50 (40%) junctions studied, of the identified microhomologies and insertions at the breakpoint sites further indicates the DNA replication errors as a major mechanism of CNV formation. In the remainder, non-homologous end joining might have played an important role37,38.
Parental somatic mosaicism identified in four parental samples
Somatic mosaic CNV deletions were identified by junction-specific LR-PCR in four (8%) parental blood samples of the 50 families, including the father of proband 12, father of proband 26, father of proband 32, and mother of proband 38. As proband 38 is a male with chromosome X deletion, only the mother was examined for this deletion (Figure 1). In each proband, VAF assessed by ddPCR was close to 50%, as would be expected for a normal heterozygous variant and 0% in an unrelated control sample. As shown in Table 1, in the four available parental samples, VAFs were assessed as 18.5% (father of proband 12), undetectable (father of proband 26), 6.2% (father of proband 32), and 15.6% (mother of proband 38). Whereas SNP arrays can detect mosaicism as low as 5%7, oligonucleotide array CGH detect 20–30% mosaicism8,39,40. Thus, it is not unexpected that, in some cases, even double digit-mosaicisms were not detected in routine clinical diagnostics using oligo array CGH. Results are also shown in Supplemental Figure 1.
Figure 1.
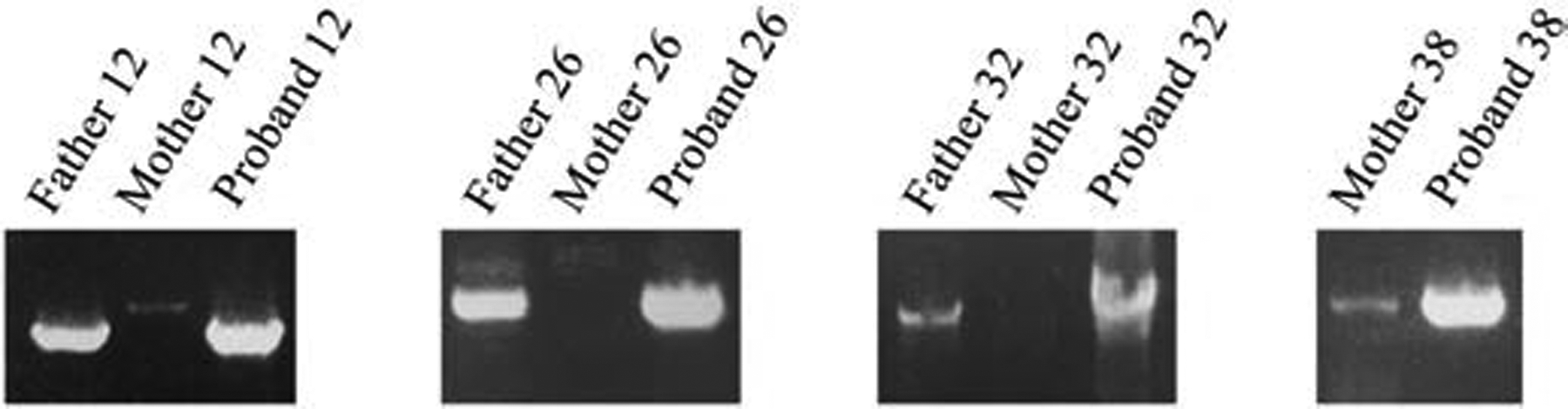
Mosaic CNV deletions detected by LR-PCR in four parental samples. The familial deletion-specific amplicon detected in the patient is clearly visible from the LR-PCR reaction performed on paternal or maternal DNA.
Table 1.
Summary of the ddPCR and amplicon-based NGS results for the detected mosaicism.
| Parent | Sample | ddPCR | Amplicon-based NGS | qPCR |
|---|---|---|---|---|
| father of proband 12 | blood | 18.5% | rs6727415: 13.9% (A 1259, G 910) | Not tested |
| rs1234405: 17.3%(A 1444, C 943) | ||||
| rs1234413: 15.9% (C 1203, T 820) | ||||
| Average VAF: 15.7% | ||||
| father of proband 26 | blood | undetectable | rs1023367: 7.3% (C 1162, T 993) | 0.39% |
| rs8132955: 2.3% (A 636, T 607) | ||||
| rs218652: 0.7% (A 1470, G 1450) | ||||
| father of proband 32 | blood | 6.2% | no informative SNPs | Not tested |
| mother of proband 38 | blood | 15.6% | no informative SNPs | Not tested |
| buccal | 10.6% | |||
| saliva | 8.2% | |||
| urine | undetectable |
The scale and levels of low-level parental somatic mosaicism are incompletely understood.
We propose an approach for more effective detection of low-level parental somatic mosaicism for CNV deletions for a more accurate assessment and estimate of disease recurrence risk.
To estimate the level of mosaicism for the CNV deletion, we calculated the proportions of two genotypes of a heterozygous SNP within a deletion region41. Using amplicon-based NGS, we analyzed three SNPs in each of the tested parental samples. The mosaicism level in father of proband 12 was determined as 15.7% (average of the three genotyped SNPs). In the father of proband 26, it varied between 0.7–7.3% likely due to sequence context, and we elected not to average its value (Table 1). Using qPCR, the mosaicism level in the father of proband 26 was determined as 0.39% (Figure 2).
Figure 2.
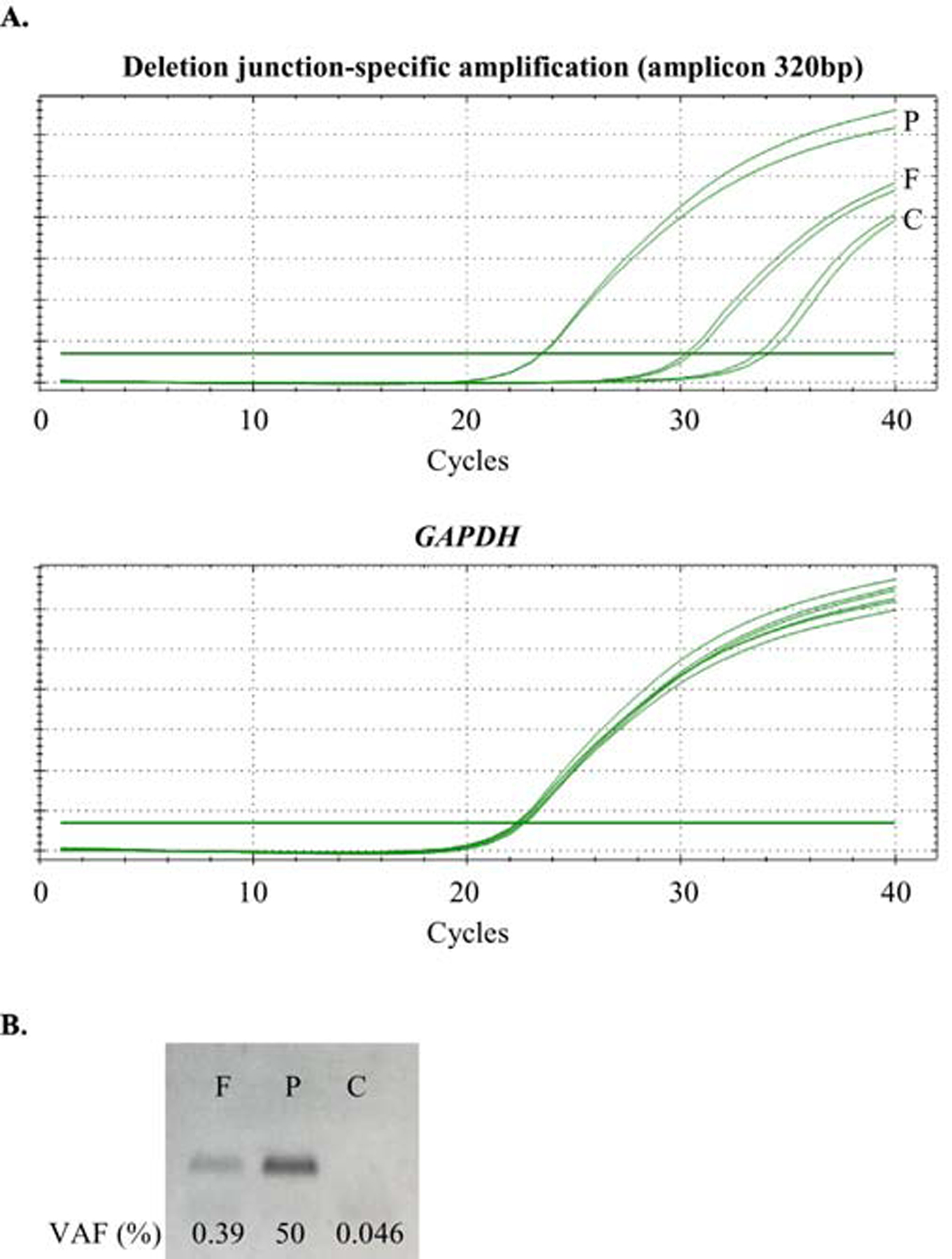
Results of qPCR study in family 26. F: father 26; P: proband 26; C: unrelated control. A) Plots of qPCR amplifications. For deletion junction-specific amplification, the average Cq in the proband is 23.48, in the father is 30.38, and in the unrelated control is 33.81. The average Cq of GAPDH amplifications are similar among the three samples. B) Agarose gel electrophoresis image of the deletion junction-specific qPCR amplification.
Distribution of VAFs among different somatic tissues
To evaluate the distribution of mosaicism in various somatic tissues, the levels of mosaicism in the mother of proband 38’s blood, buccal cells, saliva, and urine samples were assessed by ddPCR. VAFs in these tissues were estimated as 15.6%, 10.6%, 8.2%, and undetectable, respectively (Table 1). Unfortunately, different somatic samples were not available in other families.
DISCUSSION
Although somatic and germline variants in parents have significant implications for family planning, testing for parental somatic mosaicism for both SNVs and CNV are not routinely offered by the clinical diagnostic laboratories. Thus far, the importance of the role of the transmitted somatic mosaic variants has been indicated in only a few large cohort studies26–31.
The detection rate of parental mosaicism is limited by technical challenges, the cost of the analysis, and somatic tissue availability. Here, we have used three quantitative methods to determine the level of parental somatic mosaicism, including ddPCR, which is a robust method to precisely assess the level of somatic mosaicism42,43. The mosaicism level in father 12 was determined as 18.5% using ddPCR, which was comparable to 15.7% assessed by screening of heterozygous SNPs within the deleted region using amplicon-based NGS. However, in the father of proband 26, there was no significant difference in the amount levels between the amplification of regions with and without the deletion, indicating the level of mosaicism is too low to be detected by ddPCR. Using amplicon-based NGS, the VAFs of the three SNPs mapped within the mosaic deletion in father 26 varied from 0.7% to 7.3% (Table 1), indicating that even ultra-deep screening of amplicons for heterozygous SNPs located within the deleted region may not be reliable for identification and quantitation of very low-level mosaicism of CNVs. While the ratio of mosaic CNV deletion in father of proband 26 remained either undetectable by ddPCR or undetermined by amplicon-based NGS, the level of parental mosaicism for the CNV deletion was estimated at 0.39% using qPCR. This suggests that, in some cases, qPCR may be a more efficient tool for the assessment of low-level mosaicism for CNV deletions than other methods. Unfortunately, we were not able to test samples sourced at different times to reproduce the results due to sample unavailability.
Each method used for the estimation of the level of parental somatic mosaicism has its own advantages and limitations. ddPCR or qPCR experiments can be performed within a few days, whereas results using amplicon-based NGS may be obtained within a few weeks if the NGS is performed commercially. The study design can be hampered by high GC-content in ddPCR experiments, while the absence of informative heterozygous SNPs can be a limiting factor for investigation of small CNVs in amplicon-based NGS. An advantage of qPCR is its low cost compared to ddPCR. In addition, qPCR does not require the use of expensive equipment and reagents, unlike ddPCR. Due to the small sample size, we cannot conclude which method is the most precise method for the assessment of the level of mosaicism for CNV deletion.
As somatic mosaic variants may arise at different developmental stages, the distribution may vary substantially among various somatic tissues representing the three primary germ layers. In the clinical diagnostic setting, a whole blood sample is typically tested for somatic mosaicism. However, growing evidence suggests that peripheral blood cells may demonstrate skewed percentages of mosaic cell lines due to gradual clonal expansion of hematopoietic stem and progenitor cells, especially in older subjects44. Thus, mosaic variants may be under- or over-represented in DNA derived from blood. We and others have observed that non-blood tissues usually exhibited different VAFs from blood samples, suggesting that tissues other than blood may be a better source to test somatic mosaicism45,46. However, a larger sample set would be needed to confirm this conclusion.
Our findings corroborate previous results that true inheritance status can be accurately determined by examining the parental samples using LR-PCR, which is much more sensitive than CMA or FISH for mosaicism detection26. Our data implies that more sensitive and precise methods e.g. CNV junction-specific PCR, ddPCR, or qPCR, are needed in clinical diagnostic settings for detecting parental somatic mosaicism for CNV deletions. It is also important for a more accurate assessment and estimate of disease recurrence risk.
Supplementary Material
ACKNOWLEDGEMENTS
This work was financially supported by the NIH grant R01HD087292 (P.S.). We are thankful to our colleagues who provided their expertise that greatly assisted this research work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
BCM and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories (BMGL), which performs chromosomal microarray analysis and clinical exome sequencing. JAR, CAB, SRL, AP, JLS, SWC and WB are current employees of BCM and derive support through a professional service agreement with BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The remaining authors declare that they have no competing interests.
REFERENCES
- 1.Iourov IY, Vorsanova SG, and Yurov YB. Somatic genome variations in health and disease. Curr. Genomics 11, 387–396 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lupski JR. Genetics. Genome mosaicism--one human, multiple genomes. Science 341, 358–359 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Acuna-Hidalgo R, Bo T, Kwint MP, et al. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am. J. Hum. Genet 97, 67–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson RP. Somatic gene mutation and human disease other than cancer: an update. Mutat. Res 705, 96–106 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Biesecker LG and Spinner NB. A genomic view of mosaicism and human disease. Nat. Rev. Genet 14, 307–320 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Goriely A, Lord H, Lim J, et al. Germline and somatic mosaicism for FGFR2 mutation in the mother of a child with Crouzon syndrome: Implications for genetic testing in ‘paternal age-effect’ syndromes. Am. J. Med. Genet. A 152A, 2067–2073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conlin LK, Thiel BD, Bonnemann CG, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet 19, 1263–1275 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boone PM, Bacino CA, Shaw CA, et al. Detection of Clinically Relevant Exonic Copy-Number Changes by Array CGH. Hum Mutat. 31, 1326–1342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartnik M, Derwinska K, Gos M, et al. Early-onset seizures due to mosaic exonic deletions of CDKL5 in a male and two females. Genet Med. 13, 447–52 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Delhanty JD. Inherited aneuploidy: germline mosaicism. Cytogenet. Genome Res 133, 136–140 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Poduri A, Evrony GD, Cai X, et al. Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ansari M, Poke G, Ferry Q, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet 51, 659–668 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King DA, Jones WD, Crow YJ, et al. Mosaic structural variation in children with developmental disorders. Hum. Mol. Genet 24, 2733–2745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papavassiliou P, Charalsawadi C, Rafferty K, et al. Mosaicism for trisomy 21: a review. Am. J. Med. Genet. A 167A, 26–39 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Nathan N, Keppler-Noreuil KM, Biesecker LG, et al. Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol. Clin 35, 51–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xin B, Cruz Marino T, Szekely J, et al. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin. Genet 91, 623–628 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Stosser MB, Lindy AS, Butler E, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. Off. J. Am. Coll. Med. Genet 20, 403–410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King DA, Sifrim A, Fitzgerald TW, et al. Detection of structural mosaicism from targeted and whole-genome sequencing data. Genome Res. 27, 1704–1714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krupp DR, Barnard RA, Duffourd Y, et al. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet 101, 369–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim ET, Uddin M, De Rubeis S, et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci 20, 1217–1224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic mutations in early-onset genetic diseases. Genet. Med. Off. J. Am. Coll. Med. Genet 18, 746–749 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers CT, Hollingsworth G, Muir AM, et al. Parental Mosaicism in ‘De Novo’ Epileptic Encephalopathies. N. Engl. J. Med 378, 1646–1648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell IM, Shaw CA, Stankiewicz P, et al. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 31, 382–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldmann JM, Veltman JA, and Gilissen C. De Novo Mutations Reflect Development and Aging of the Human Germline. Trends Genet. 35, 828–839 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Notini AJ, Craig JM, and White SJ. Copy number variation and mosaicism. Cytogenet. Genome. Res 123, 270–277 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet 95, 173–182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahbari R, Wuster A, Lindsay SJ, et al. Timing, rates and spectra of human germline mutation. Nat. Genet 48, 126–133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krupp DR, Barnard RA, Duffourd Y, et al. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am J Hum Genet. 101, 369–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jónsson H, Sulem P, Arnadottir GA, et al. Multiple transmissions of de novo mutations in families. Nat. Genet 50, 1674–1680 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Wright CF, Prigmore E, Rajan D, et al. Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nat. Commun 10, 2985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao Y, Tokita MJ, Chen ES, et al. A clinical survey of mosaic single nucleotide variants in disease-causing genes detected by exome sequencing. Genome Med. 11, 48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell IM, Stewart JR, James RA, et al. Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am. J. Hum. Genet 95, 345–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breuss MW, Antaki D, George RD, et al. Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nat. Med 26, 143–150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Grochowski CM, Bi W, et al. Quantitative assessment of parental somatic mosaicism for CNV deletions. Curr Protoc Hum Genet. 106, e99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative Genomics Viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thorvaldsdóttir H, Robinson JT, and Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arlt MF, Mulle JG, Schaibley VM, et al. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet 84, 339–350 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carvalho CM and Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet 17, 224–238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheung SW, Shaw CA, Scott DA, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 143A, 1679–86 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Scott SA, Cohen N, Brandt T, et al. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet Med. 12, 85–92 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Summerer A, Schäfer E, Mautner VF, et al. Ultra-deep amplicon sequencing indicates absence of low-grade mosaicism with normal cells in patients with type-1 NF1 deletions. Hum. Genet 138, 73–81 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem 83, 8604–8610 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinheiro LB, Coleman VA, Hindson CM, et al. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem 84, 1003–1011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shlush LI Age-related clonal hematopoiesis. Blood. 131, 496–504 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Yang X, Liu A, Xu X, et al. Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Sci. Rep 7, 15677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karolak JA, Liu Q, Xie NG, et al. Highly sensitive blocker displacement amplification and droplet digital PCR reveal very low-level parental FOXF1 somatic mosaicism in families with ACDMPV. J Mol Diagn. 22, 447–456 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.