

Summary
Small cell lung cancer (SCLC) is a highly aggressive and lethal neoplasm. To identify candidate tumor suppressors we applied CRISPR-Cas9 gene inactivation screens to a cellular model of early-stage SCLC. Among the top hits was MAX, the obligate heterodimerization partner for MYC family proteins that is mutated in human SCLC. Max deletion increases growth and transformation in cells and dramatically accelerates SCLC progression in an Rb1/Trp53-deleted mouse model. In contrast, deletion of Max abrogates tumorigenesis in MYCL-overexpressing SCLC. Max deletion in SCLC resulted in de-repression of metabolic genes involved in serine and one carbon metabolism. By increasing serine biosynthesis, Max deleted cells exhibit resistance to serine depletion. Thus, Max loss results in metabolic rewiring and context-specific tumor suppression.
Introduction
Small cell lung cancer (SCLC) is a deadly cancer type with a 5-year survival rate of only 6% (Pietanza et al., 2015). Recent genomic analyses have provided invaluable insights into the etiology of SCLC (Augert et al., 2017; George et al., 2015; Peifer et al., 2012; Rudin et al., 2012). Inactivation of RB1 and TP53 tumor suppressor genes and amplification of MYC family members, including MYC, MYCL and MYCN are among the most common genetic alterations. Mouse models have validated MYC and MYCL function as SCLC oncogenes, and CREBBP and PTEN as tumor suppressors (Cui et al., 2014; Huijbers et al., 2014; Jia et al., 2018; McFadden et al., 2014; Mollaoglu et al., 2017), but the significance of most genomic alterations present in SCLC remains unclear. Genome-wide functional screens have been successfully employed to reveal genes that promote or suppress cell proliferation/viability, and many such genes act in a tissue-specific manner (Sack et al., 2018). Application of functional screens to cellular models of SCLC could help us understand how early-stage lung neuroendocrine cancer cells can become fully transformed, thus revealing genes with tissue-specific tumor suppressor activity in SCLC.
Of relevance to this report is the well-established association of the MYC gene family (MYCL1, MYC, MYCN) with SCLC (Peifer et al., 2012; Sos et al., 2012). MYC proteins heterodimerize with MAX to bind E-Box elements and activate broad programs of gene expression (Diolaiti et al., 2015). MAX can also heterodimerize with transcriptional repressors in the MXD family (MXD1-4, MNT) and MGA (Carroll et al., 2018). Paradoxically, MAX was found to undergo homozygous deletions or mutations in 6/98 SCLC cell lines and tumors (Romero et al., 2014) as well as in other neuroendocrine tumors (Burnichon et al., 2012; Comino-Mendez et al., 2011; Hopewell and Ziff, 1995; Schaefer et al., 2017). To date there is no model system to rigorously examine putative tumor suppressive function of MAX. Here we performed functional screens to reveal genes, including MAX, with tumor suppressor activity in cellular and in vivo models of SCLC.
Results
Genome-wide CRISPR inactivation screens identify tumor suppressive genes and pathways in SCLC
To identify genes with tumor suppressor activity relevant to SCLC, we employed preSC cells, which are immortal cells derived from early lesions of an Rb1/Trp53/Rbl2-deleted mouse model of SCLC (Kim et al., 2016). The preSC cellular model exhibits increased proliferation and transformation upon over-expression of oncogenes such as MYCL (Kim et al., 2016) and thus represents a sensitized model to identify genes that may functionally contribute to SCLC tumorigenesis. We generated Trp53-null mouse embryonic fibroblasts (MEFs) to use in our screens as non-neuroendocrine cell controls. Immunoblots confirmed lack of P53 in the various MEF isolates (Figure S1A). For genome-scale CRISPR-Cas9 screens, we utilized a pooled mouse sgRNA library (Mouse GeCKOv2), comprised of 130,209 sgRNAs targeting a total of 20,611 protein coding genes and 1,175 microRNA precursors (Sanjana et al., 2014). We transduced three replicates each of 250x106 cells for both preSCs (n=3) and Trp53−/− MEFs (n=3). Viral titers were optimized to result in <30% transduction rate (MOI<1) and 66x106 transduced cells were maintained throughout the screens in order to preserve a 500X library representation. A reference point (population doubling 0, PD0) for each screen was collected shortly after puromycin selection. Cells were expanded for 12 population doublings after which genomic DNA was extracted, libraries generated, and high throughput sequencing performed (Figure 1A).
Figure 1. A whole genome CRISPR screen identifies candidate SCLC tumor suppressor genes and pathways.
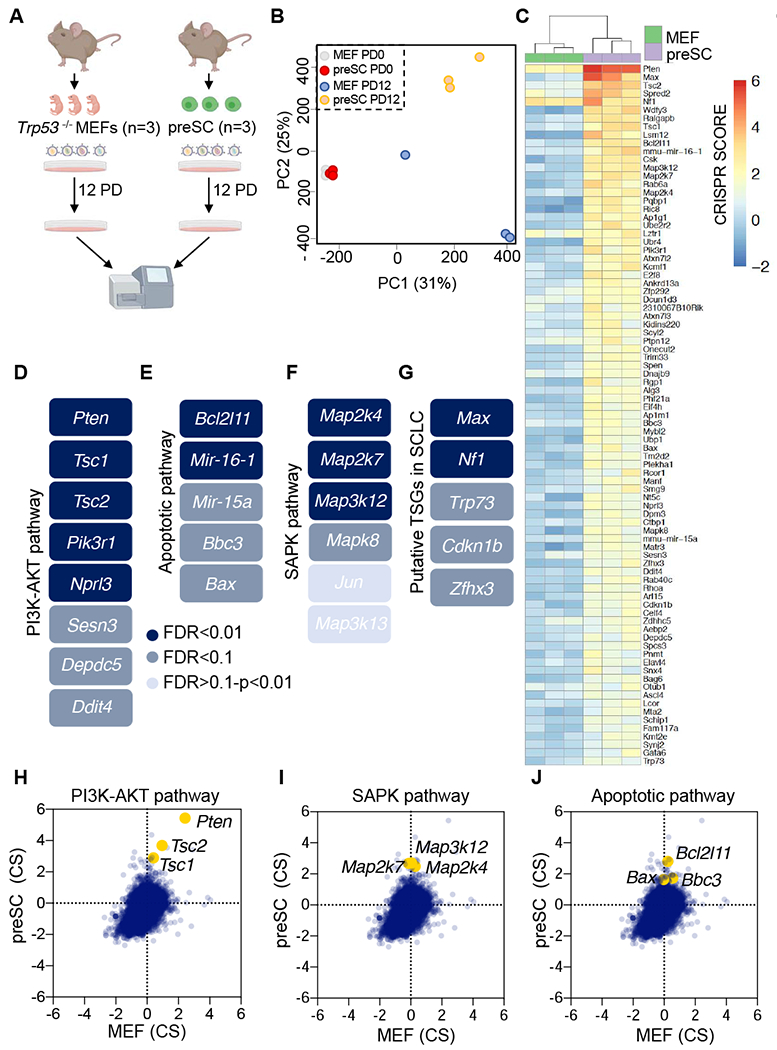
A, Schematic of CRISPR screening strategy. B, Principal component analysis (PCA) of the individual libraries generated from each replicate (PD0 and PD12) for MEFs and preSCs. C, Heat map of the top screen hits (MAGeCK FDR<0.1) enriched in preSCs as compared to MEFs. CRISPR scores are shown. D-G, Schematic of the PI3K-AKT pathway (D), the apoptotic pathway (E), the SAPK pathway (F) and putative tumor suppressor genes (G) identified in preSCs. FDR values are indicated and color coded. H-J, Scatter plot using CRISPR scores for the selected genes in the PI3K-AKT pathway (H), SAPK pathway (I) and the apoptotic pathway (J). For D-G, FDR values were calculated using MAGeCK VISPR. See also Figures S1 and S2 and Tables S1 and S2.
Principal component analysis (PCA) revealed preSCs and Trp53−/− MEFs clustered similarly at PD0 but displayed cell type specific clustering after expansion for 12 population doublings (Figure 1B). A CRISPR score, defined as the average log2 fold change in abundance of all sgRNAs targeting a given gene at final versus initial reference (Wang et al., 2015), was computed (Table S1). We also applied MAGeCK VISPR (Li et al., 2015) to identify genes with statistically significant enrichment in our screens considering the performance of all sgRNAs (Table S2). A heatmap shows CRISPR scores for genes with MAGeCK FDR<0.1 that promote preSC growth when inactivated (Figure 1C). These analyses revealed known SCLC tumor suppressor genes such as Pten (Cui et al., 2014; McFadden et al., 2014) and also uncovered regulators of early stage SCLC growth. Three major pathways were prominent among enriched “hits” as assessed by MAGeCK VISPR. First, while PTEN and PIK3CA are frequently affected in SCLC by inactivating and activating mutations, respectively, (Shibata et al., 2009), our screen suggests that other components of the PTEN-PIK3CA-AKT-mTOR pathway also exhibit tumor suppressor function. Indeed, the tumor suppressor genes Tsc1 and Tsc2, which are critical negative regulators of mTORC1, were identified (Figure 1D). Additional negative regulators of mTORC1 signaling were also uncovered including Sesn3, Ddit4 (Brugarolas et al., 2004; Parmigiani et al., 2014) and genes encoding two subunits of GATOR1: Depdc5 and Nprl3 (Bar-Peled et al., 2013; Panchaud et al., 2013). A second axis identified was the pro-apoptotic pathway including Bax, Bcl2l11 (BIM), Bbc3 (PUMA) and microRNA genes miR-15A and miR16-1 (Figure 1E). Finally, we identified many members of the stress-activated-protein-kinase pathway (SAPK), including Map3k12 (DLK), Map3k13 (MLK), Map2k4 (MKK4), Map2k7 (MKK7), Mapk8 (JNK1) and the downstream transcriptional effector Jun (Figure 1F). Genomic position of top hits is plotted in Figure S1B.
Inactivation of members of the PTEN-PIK3CA-AKT-mTOR (Figure 1H), the SAPK (Figure 1I) and the apoptotic (Figure 1J) pathways conferred relative growth advantage in preSCs compared to MEFs as judged by CRISPR scores. Volcano plots show an enrichment for individual guides targeting identified genes in these pathways in preSC cells (Figure S1C–S1E) but not in Trp53−/− MEFs, except for Pten and Tsc2 (Figure S1F–S1H). In addition, we also identified genes that are known tumor suppressors previously identified in other cancer types, such as Trp73, Nf1, Zhfx3 and Cdkn1b (Figure 1G). Among the strongest hits, based on the magnitude of CRISPR score and statistical significance assessed by MAGeCK analyses (Tables S1 and S2) was Max, with 5/6 sgRNAs enriched in preSCs after 12 population doublings (Figure S1I). No other members of the MYC network were identified in the screen (Figure S1J). Examination of published genomic data for 110 SCLC patients (predominantly primary SCLC) (George et al., 2015) and data from Project Genie, a tumor mutation database that includes targeted resequencing data for select genes from >400 SCLC patients with primary and metastatic disease (Consortium, 2017), reveals that screen hits NF1, TSC1, TSC2, PIK3R1, DEPDC5, TP73, CDKN1B, ZFHX3 and MAP2K4 undergo recurrent truncating mutations and deletions in human SCLC (Figure S2A and S2B).
Validation of candidate tumor suppressor genes in SCLC
We next validated a subset of our hits. For 9 genes of interest among the 96 with an FDR <0.1 (MAGeCK analyses) we cloned two guides per gene into the lentiCRISPRV2 vector. Following lentiviral infection and selection, gene knockouts were validated by immunoblots (Figure 2A). Expression of sgRNAs targeting Pten, Tsc1, Tsc2, Map3k12, Nf1 and Max resulted in increased preSC cell proliferation over an eight-day growth experiment, while sgRNAs targeting Bcl2l11 and Bax did not increase proliferation (Figure 2B). We next assessed gene knockouts for survival at low density and anchorage independent growth. Although Bcl2l11 and Bax KO preSCs did not grow faster than controls, they formed more colonies at low density and showed increased colony forming capability in agar (Figure 2C and 2D). Map3k12, Nf1, Max and PTEN-PIK3CA-AKT-mTOR pathway KOs (Pten, Tsc1 and Tsc2) all outgrew controls in both survival at low density and anchorage independent growth (Figure 2C and 2D). These data functionally validate major screen hits as candidate tumor suppressor genes in SCLC, supporting the utility of our experimental approach as a means to assess the importance of genes mutationally inactivated or epigenetically silenced in SCLC.
Figure 2. Validation of candidate tumor suppressor genes in SCLC.
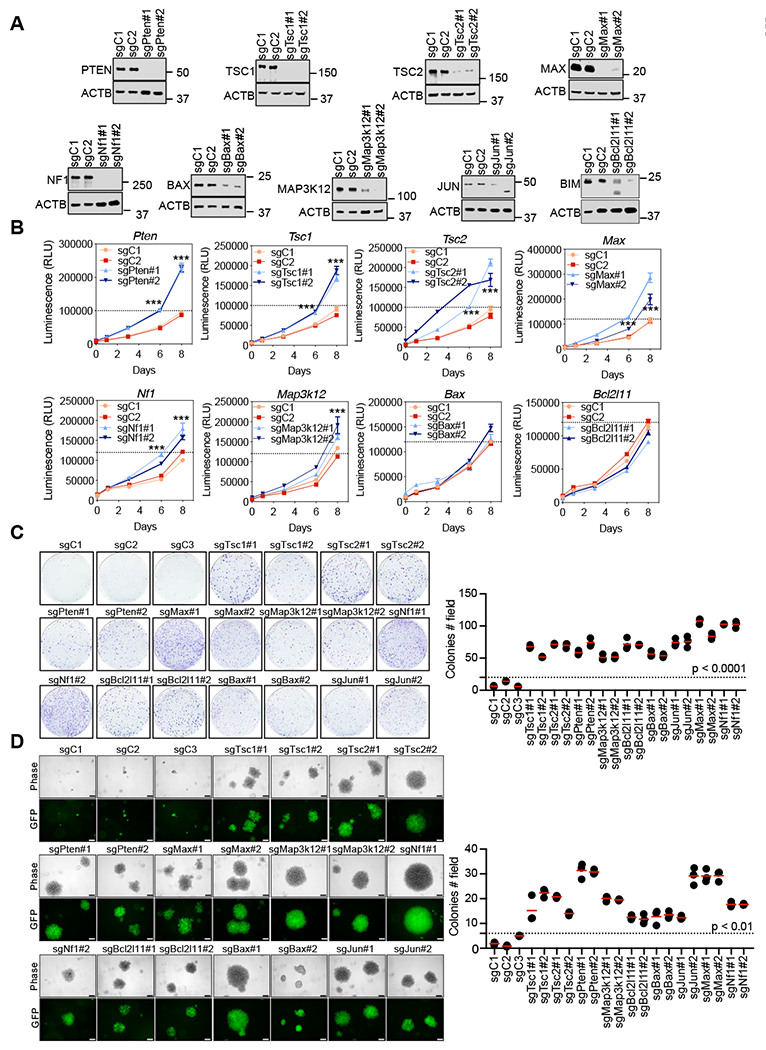
A, Immunoblotting results following lentiviral sgRNA expression for the indicated genes in preSCs. ACTB was used as a loading control. B, CellTiter-Glo viability assay for the indicated genes. Two sgRNAs were used for each gene. Representative experiments from at least 2 independent experiments are shown. Error bars represent mean ± SD (n=3) ***p<0.001, unpaired Student’s t test. RLU, relative luminescence units. C, Colony formation assays by crystal violet staining after 2 weeks expansion of cells (6 well plates) were performed for the indicated genes. The number of colonies per field were counted. Error bars represent the mean (n= 3 independent experiments), p<0.0001, unpaired Student’s t test. D, Anchorage-independent assay upon the inactivation for the genes of interest in soft agar. n = 3 independent experiments. Error bars represent mean ± SD, p<0.01, unpaired Student’s t test. Scale bar: 100 μM.
Max inactivation accelerates SCLC
Max was among the top enriched genes in the screen and we confirmed that Max loss increases transformation and produces a pro-proliferative advantage in preSC cells, comparable to deleting the potent SCLC tumor suppressor Pten (data not shown). Importantly, in contrast to what we observed in preSCs, Max loss did not result in increased growth or survival at low density in Trp53−/−, Trp53−/−;sgRb1 or Trp53−/−;sgRb1;sgRb12 MEFs (Figure S3A–S3N). This underscores the cell type-specific effect of MAX loss. Max was a particularly intriguing hit owing to a previous report of distinct, inactivating truncating MAX mutations in human SCLC (Romero et al., 2014). Thus, to determine whether Max functions as an SCLC tumor suppressor gene, we generated an autochthonous SCLC mouse model. We crossed a Max floxed allele (Mathsyaraja et al., 2019) into an Rb1/Trp53-deleted mouse model of SCLC which develops lung tumors with molecular and histopathological features that resemble human SCLC (Meuwissen et al., 2003).
Two cohorts were infected using intratracheal instillation of Ad-CMV-Cre virus:Rb1lox/lox;Trp53lox/lox(herein RP) and Rb1lox/lox ;Trp53lox/lox ;Max lox/lox (herein RPMax) cohorts. We analyzed mice at a fixed 18-week post infection time point and also performed a long-term analysis to assess the impact of Max deletion on overall survival (Figure 3A). Magnetic resonance imaging (MRI) and histological analyses revealed a striking increase in lung tumor volume and in the number of lung tumors in the RPMax animals at 18 weeks post Cre delivery (Figure 3B). Long-term analysis revealed that RPMax mice succumbed to lung tumors more rapidly, with a median lung tumor-free survival of 152 days compared to 313 days for the RP control mice (Figure 3C). Histopathology review by a lung cancer pathologist (A.F. Gazdar) confirmed SCLC histology in RP and RPMax models (Figure 3D). A subset of lung tumors that arose from RPMax mice were classified as large cell neuroendocrine (LNEC) tumors (Figure 3D). Tumors from both groups stained positive for the neuroendocrine marker CGRP (Figure 3E) and immunoblot analysis confirmed loss of MAX protein in the tumors from the RPMax cohort (Figure 3F).
Figure 3. Max inactivation accelerates SCLC.
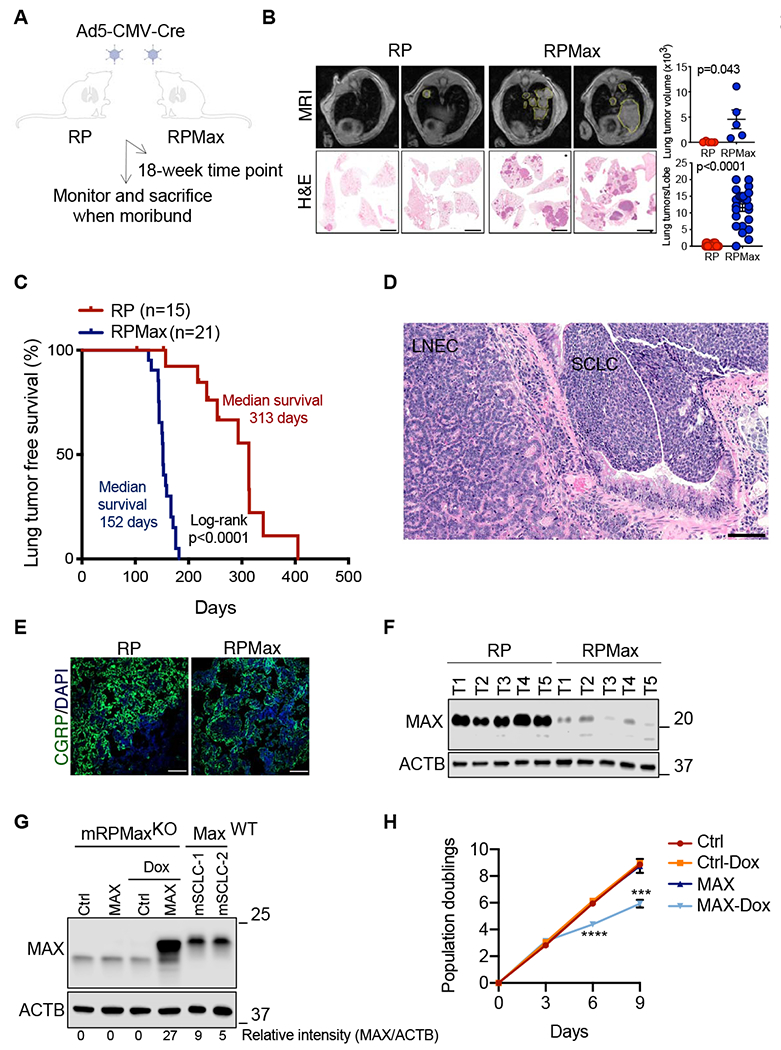
A, Schematic of the strategy to follow effects of Max deletion in a mouse model of SCLC. B, Magnetic resonance imaging (MRI) and Hematoxylin and Eosin (H&E) stains for the indicated genotypes 18 weeks post Ad5-CMV-Cre infections. Representative images of mice are shown. Error bars represent mean ± SD (n=5 mice per genotype). Unpaired Student’s t test was performed and p values are shown. Scale bar: 2 mm. C, Kaplan-Meier tumor-free survival curves of Rb1/Trp53 mutant (red, n=15) and Rb1/Trp53/Max mutant (blue, n=21) mice from autochthonous model infected with Ad5-CMV-Cre (Day 0). Statistical significance for the overall survival of the cohorts was calculated using log-rank (Mantel-Cox) test. D, Representative H&E stained section of SCLC and LNEC from the Rb1/Trp53/Max cohort. Scale bar: 100 mM. E, Representative immunofluorescence for the SCLC marker CGRP in each cohort (Rb1/Trp53 vs. Rb1/Trp53/Max). DAPI was used as a nuclear stain. Scale bar: 50 mM. F, Representative immunoblotting results of MAX protein levels in 5 lung tumor tissues from each cohort (Rb1/Trp53 vs. Rb1/Trp53/Max). ACTB was used as a loading control. G, Representative immunoblotting of MAX protein levels upon doxycycline-inducible Max restoration in a mRPMaxKO (Rb1/Trp53/Max-deleted) mSCLC cell line. ACTB was used as a loading control and two RP mSCLCs expressing endogenous MAX levels were used as internal controls. Protein levels were quantified using the LI-COR software. H, Growth curve analysis of a mRPMaxKO cells upon Max restoration at the indicated times following doxycycline addition. Error bars represent mean ± SD (n=3 independent experiments). ***, p<0.005; ****, p<0.0001, unpaired Student’s t test. See also Figure S3.
While Max inactivation promoted both SCLC and LCNEC, we focused all subsequent molecular and cellular analyses on SCLC, given MAX mutations in human SCLC. We used lentiviral transduction of a doxycycline inducible vector to restore MAX expression in a mouse SCLC cell line that we derived from a RPMax lung tumor (herein mRPMaxKO). Doxycycline treatment restored MAX expression (Figure 3G) and significantly decreased cell growth (Figure 3H). Of note, doxycycline treatment resulted in approximately 4-fold higher expression relative to MAX-intact mouse SCLCs (Figure 3G). Taken together, these in vivo and cell line data show that Max functions as a bona fide tumor suppressor gene in SCLC.
MAX is required for MYCL-driven SCLC
The potent tumor suppressor activity of MAX that we observe in vivo is intriguing given that Mycl deletion in a mouse SCLC model suppresses tumorigenesis (Kim et al., 2016). These findings raise the question of how MAX acts as a strong tumor suppressor in the same tumor type in which it is presumed to be required by MYCL. It is possible that MYCL could exhibit activities independent of MAX (Steiger et al., 2008; Wert et al., 2001). To rigorously test the impact of MAX loss on tumorigenesis driven by MYCL, we employed a Cre-activated MYCL overexpression allele, herein referred to as MyclOE (Huijbers et al., 2014). With this allele, we can overexpress MYCL in the context of Max deletion and compare the kinetics of SCLC emergence across four genotypes: RP; RPMax; Rb1/Trp53/MyclOE (RPMyc1OE) and Rb1/Trp53/Max/Myc1OE (RPMaxMyc1OE). Intratracheal instillation of Ad-CGRP-Cre virus, which expresses Cre recombinase under the control of a neuroendocrine-specific CGRP promoter (Sutherland et al., 2011) was used to infect the four cohorts. Long-term analysis revealed that RPMax mice became moribund from lung tumors much faster than RP mice, with a median lung tumor-free survival of 259 vs. 601 days (p<0.0001, log-rank test; Figure 4A). Of note, longer latency observed with Ad-CGRP-Cre (Figure 4A) relative to Ad-CMV-Cre (Figure 3C) is consistent with other recent observations (Yang et al., 2018). Max deletion or Myc1 overexpression in neuroendocrine cells using Ad-CGRP-Cre resulted in the development of SCLC tumors with “classic” histology (Figure 4B). The RPMyc1OE cohort exhibited a significant decrease in lung tumor free survival as compared to RP mice and to the RPMax cohort (p<0.0001) (Figure 4A) suggesting greater potency of activating Mycl versus inactivating Max in driving SCLC tumorigenesis. The RPMaxMyclOE cohort did not exhibit faster tumorigenesis relative to the RPMyc1 OE mice (Figure 4A). Instead, there was a 22-day delay in median survival associated with Max deletion, suggesting that MYCL oncogenic function is at least partially suppressed in the absence of MAX. When comparing the RPMaxMyc1OE to the RPMax cohorts, we found faster tumorigenesis in the RPMaxMyc1OE group. While these data raise the possibility that MYCL exhibits MAX-independent oncogenic activity, it was first necessary to determine whether the floxed Max alleles were recombined in the tumors that arose. Analyses of tumor derived RNA and protein indicated that 7 of 21 (33%) of RPMaxMyc1OE tumors analyzed escaped homozygous deletion and retained MAX expression (Figure 4C and 4D). PCR analysis confirmed that tumors with MAX protein expression evident by western blot recombined one but not both alleles of Max (Figure S4). These data suggest that tumor cells overexpressing MYCL are under selection to retain MAX. Thus, faster growth of the MYCL-overexpressing MAX positive tumors explains reduced tumor free survival of the RPMaxMyc1OE compared to the RPMax groups. Interestingly, the MAX-intact tumors exhibited high MYCL expression but the tumors with MAX loss exhibited low levels of MYCL protein and RNA (Figure 4C and 4D). Thus, lungs from RPMaxMyc1OE mice exhibit tumors that sustain Max deletion and exhibit low MYCL protein and tumors that escape Max deletion and support transformation by high MYCL. These data suggest that MYCL-driven and Max deletion-driven SCLCs are mutually exclusive transformation events.
Figure 4. MAX is required for SCLC that is driven by MYC family members.
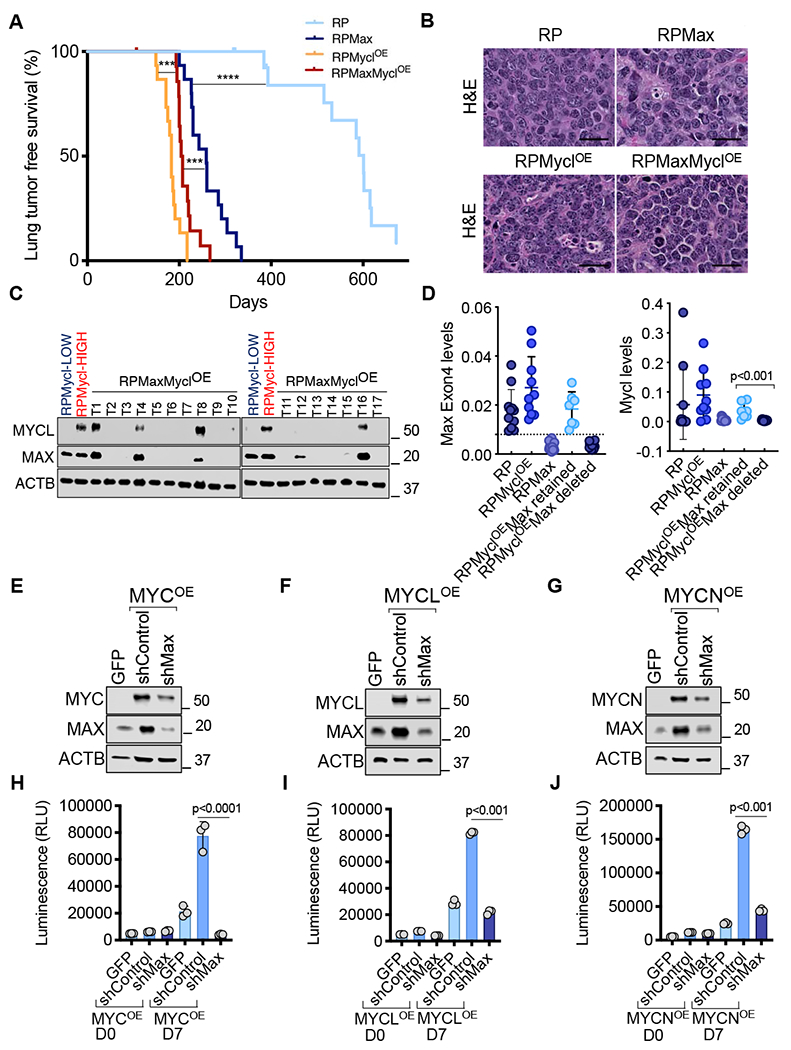
A, Kaplan-Meier tumor-free survival of RP (n=15), RPMax (n=15), RPMyc1OE (n=15) and RPMaxMyc1OE cohorts (n=15) from autochthonous model infected with Ad-CGRP-Cre (Day 0). Statistical significance for the overall survival of the different cohorts was calculated using log-rank (Mantel-Cox) test. ***, p<0.001; ****p<0.0001. B, Representative H&E stained section of SCLC for the indicated cohorts. Scale bar: 50 μM. C, Immunoblotting of MYC and MAX protein levels. ACTB was used as a loading control. D, Quantitative PCR analyses of Max (Exon 4) and Mycl levels relative to Gapdh as a loading control. Error bars represent mean ± SD (n≥7 tumors per group). Unpaired Student’s t test was used. E-G, Immunoblotting of MYC (E), MYCL (F), MYCN (G) and MAX protein levels upon Max knockdown in MYC (E), MYCL (F) and MYCN (G) over-expressing preSCs. ACTB was used as a loading control. H-J, CellTiter-Glo viability assay upon Max knockdown in MYC (H), MYCL (I) and MYCN (J) overexpressing preSCs. Error bars represent mean ± SD (n=3 independent experiments); p values are indicated, unpaired Student’s t test. RLU, relative luminescence units. See also Figure S4.
To further assess the dependency of MYC family function on MAX we next tested the effect of depleting Max in preSCs harboring lentiviral overexpression of MYC, MYCN and MYCL (Figure 4E, 4F and 4G). preSC cell lines overexpressing MYC family members grew faster than their control counterparts (Figure 4H–4J). Upon lentiviral shRNA-mediated Max depletion, MYC (Figure 4E), MYCL (Figure 4F) or MYCN (Figure 4G) levels decreased and Max depletion abrogated cell growth (Figure 4H–4J). Together, the cellular and in vivo data reveal that MAX does not act as a tumor suppressor in SCLC cells overexpressing MYC family members. We surmise that there is a window in development of SCLC during which Max loss and Rb1/Trp53 deletion cooperate to initiate SCLC tumorigenesis. However, SCLC cells overexpressing MYC paralogs are dependent on MAX and represent a distinct evolutionary path of carcinogenesis.
Transcriptional analyses of MAX altered SCLC
We next performed molecular analyses of tissues and cell lines derived from the RPMax vs RPAd-CMV-Cre mouse models as well as matched Max-WT vs KO preSC cells. MAX can enable transcriptional activation through heterodimerization with MYC members and transcriptional repression through heterodimerization with proteins of the MXD family, MNT and MGA (Diolaiti et al., 2015). To identify genes regulated by MAX in SCLC we performed RNA sequencing (RNA-seq) analyses using the different MAX-perturbed models mentioned above. First, we compared gene expression in RP versus RPMax mouse SCLC tumors. Second, we compared Max-WT versus Max-KO preSCs. Finally, we restored Max expression in the mRPMaxKO mSCLC cell line using an inducible vector (as in Figure 3G) and then examined transcriptional changes. We used EdgeR (Robinson et al., 2010) to identify genes differentially expressed upon either Max inactivation or Max restoration. An FDR<0.05 cut off revealed 2355 and 3231 differentially expressed genes in mSCLC tumors or preSCs deleted for Max respectively (Figure 5A and Figure S5A). A similar cut off applied to Max restored mRPMaxKO cells revealed 8055 differentially expressed genes (Figure 5A and Figure S5A). A comparable number of genes were found to be upregulated and downregulated upon MAX alteration in these comparisons (Figure 5A and Figure S5A). We reasoned that key functionally important MAX target genes would be consistently differentially expressed across all three comparisons. Cross model analyses identified a core set of 113 genes, upregulated upon Max loss and repressed upon Max restoration (Figure 5A). These genes were used as input for enrichment analyses using Enrichr (Kuleshov et al., 2016). ChIP Enrichment Analysis (ChEA) and Encyclopedia of DNA Elements (ENCODE) analyses for the 113 genes revealed a significant enrichment for MAX and MYC binding sites (FDR=5.53x10−16 and FDR=2.09x10−12 respectively) (Figure 5B). Also enriched were binding sites for E2F6 and SIN3A (FDR=3.95x10−5 and FDR=1.02x10−4 respectively) (Figure 5B). E2F6 is a component of an atypical polycomb complex containing MAX and MGA (Gao et al., 2012; Ogawa et al., 2002; Stielow et al., 2018) while MXD-MAX repressive complexes include SIN3A (Ayer et al., 1995; Carroll et al., 2018; Hurlin et al., 1997). In contrast to the data on genes upregulated upon Max loss, the same analyses on the core set of 56 genes found downregulated upon Max loss and upregulated upon Max restoration did not identify significant pathway enrichment (Figure S5A). Overall, our data are consistent with MAX loss primarily causing derepression of genes via MAX- repressive complexes with MXD, MNT or MGA.
Figure 5. Transcriptional analyses of MAX altered SCLCs.
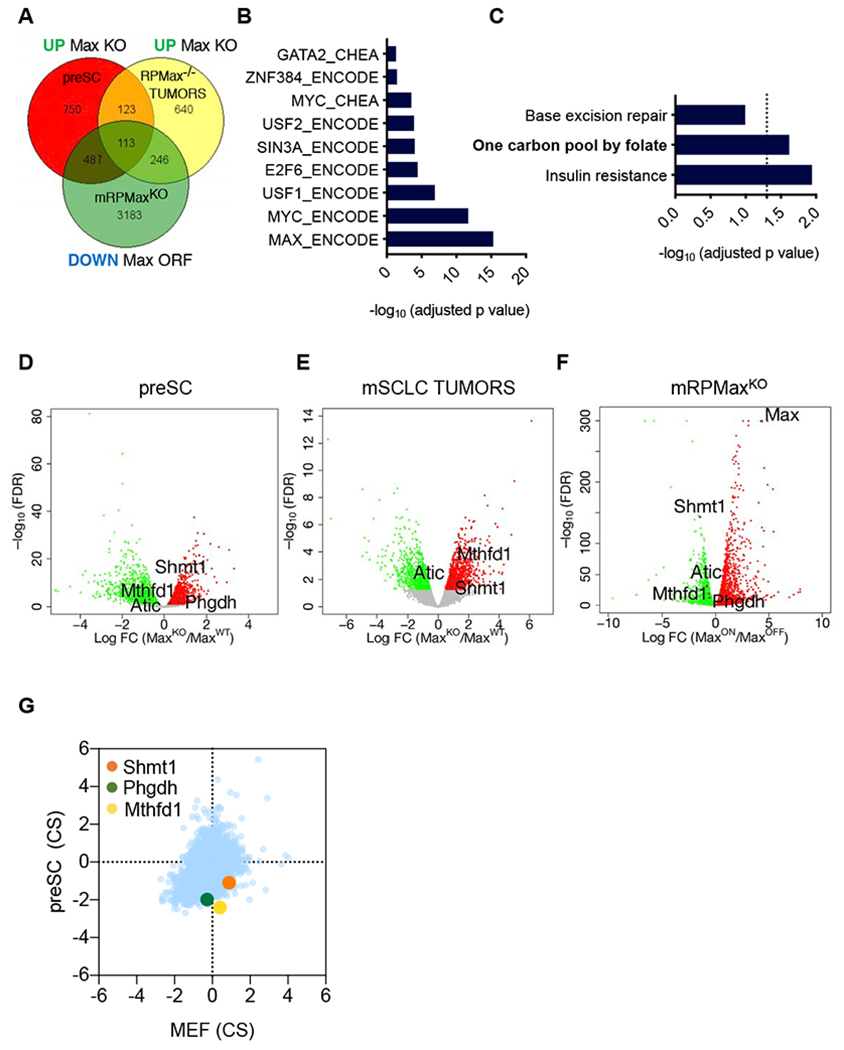
A, Venn diagram of the upregulated genes upon Max loss in preSCs and mSCLCs tumors and downregulated upon Max restoration in the mRPMaxKO Max-null mSCLC line. The list of genes used to generate the Venn diagram were selected from EdgeR analysis with a cut off of FDR<0.05. B, ENCODE or CHEA binding analysis for the 113 significant genes from (A) shared across the 3 models, upregulated upon Max loss and downregulated upon Max restoration. An adjusted p value of p<0.05 was considered significant. C, KEGG pathway analysis for the 113 genes from (A) shared across the 3 models, upregulated upon Max loss and downregulated upon Max restoration. An adjusted p value of p<0.05 was considered significant. D-F, Volcano plots from the Max-KO vs Max-WT preSC cell comparison (D), Max deleted vs Max control mSCLC tumors (E) and inducible MAX restoration in mRPMaxKO cell line (F) are shown. Significant genes upregulated upon Max perturbation are in red or downregulated in green. An FDR<0.05 was considered significant. Individual genes of interest are depicted. G, CRISPR score plot of the metabolic hits of interest showing increased depletion of guide RNAs targeting these genes in preSCs as compared to MEFs following 12 population doublings. See also Figure S5.
Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of the 113 genes indicated significant enrichment for metabolic pathways, more specifically one carbon pool by folate (FDR<0.05) (Figure 5C). The KEGG analyses highlighted the derepression of metabolic genes upon Max deletion. Considering that RP tumors were collected at much later time points than RPMax mSCLC and may have undergone selection for similar changes that Max loss induced, we next performed an analysis in which we reduced the stringency of our filters. We identified genes both upregulated upon Max deletion in preSCs and downregulated upon Max re-expression in the RPMaxKO SCLC cell line. Similar to the 3-model comparison described above, ChEA and ENCODE analyses for the shared 600 genes that changed in the opposite direction compared to MAX (Figure S5B) revealed a significant enrichment for MAX and MYC as well as E2F6 and SIN3A binding sites (FDR<0.05) (Figure S5C). KEGG analyses of the same 600 genes identified significant enrichment for the one carbon by folate pathway as well as other metabolic pathways such as alanine, aspartate and glutamate metabolism and amino sugar and nucleotide sugar metabolism (FDR<0.05) (Figure S5D). The overlap of the downregulated genes upon Max deletion in preSCs and upregulated upon Max re-expression in the RPMaxKO SCLC cell line included 770 genes (Figure S5E). ChEA and ENCODE analyses for the shared 770 genes again did not show MYC/MAX binding enrichment (Figure S5F) suggesting MAX functionally drives repression in these systems. Consistent with MAX driving transcriptional repression, meiotic genes Stag3 and Meil known to be repressed by the MAX-associated atypical polycomb complex (Endoh et al., 2017; Suzuki et al., 2016), were strongly activated upon Max deletion in the mouse SCLC model and repressed upon Max restoration (Figure S5G–I).
Among the metabolic genes up-regulated upon Max loss and repressed upon Max restoration, we noted several that encode enzymes involved in one carbon metabolism: the serine hydroxymethyltransferase 1 (Shmt1), the methylenetetrahydrofolate dehydrogenase (NADP+ dependent) (Mthfd1) and the 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase (Atic). We also identified the 3-phosphoglycerate dehydrogenase (Phgdh) (Figure 5D–5F), which is the rate limiting enzyme for conversion of glucose into serine, a major fuel for one carbon metabolism. Importantly, enzymes of the one carbon metabolism have been linked to oncogenic properties such as growth, metastasis and survival (Ducker and Rabinowitz, 2017). Upon reexamination of our CRISPR screen data, we found that sgRNAs targeting Mthfdl, Shmtl and Phgdh were preferentially negatively selected in preSC cells vs MEFs (Figure 5G). Taken together, cross model transcriptional analyses revealed a core set of MAX-regulated genes enriched for metabolic enzymes involved in serine and one carbon metabolism that are functionally important for the growth of preSC cells.
Genomic occupancy studies of MAX in SCLC
We next performed genomic occupancy analyses to identify genes that are direct targets of MAX in both preSCs and upon Max restoration in the Max-null mRPMaxKO SCLC cell line. As mentioned previously, CHEA analysis on our gene expression data indicated that promoters of genes that are subject to upregulation upon loss of MAX are significantly enriched for repressors such as SIN3A and E2F6. We reasoned that MAX directly represses these genes via heterodimerization with MXD proteins. MNT, one of the most extensively studied MXD proteins, associates with SIN3A and class 1 histone deacetylases, and co-occupies promoters with MAX in several systems, including B lymphocytes, K562 leukemia cells, HepG2 hepatocellular carcinoma cells and 3T3 fibroblasts (ENCODE data, Mathsyaraja et al, 2019, unpublished data). We performed ChIP-seq to interrogate MAX, MYC, and MNT occupancy in preSCs. In addition, we also assessed whether increased expression of MAX target genes upon MAX loss corresponds with enhanced association of RNA pol II with these genes. Heat maps showing global analyses of binding revealed a decrease in MAX, MNT and MYC occupancy at the TSS in Max-KO preSCs when compared to controls (Figure 6A, Figure S6A, B). In addition, HOMER analysis of MAX bound motifs revealed a significant enrichment for E-boxes (Figure 6C, Figure S6C). The decrease in MAX and MYC occupancy is consistent with both the loss of heterodimer formation and the strong reduction in their protein levels in Max-KO preSCs (Figure S6D). The loss of MNT binding occurs despite an increase in MNT protein levels in Max-KO preSCs (Figure S6D). Importantly, MAX and MNT binding decreased at promoters of genes involved in one carbon metabolism such as Mthfd1, Atic and Shmt1, supporting these genes as direct targets of MAX-mediated repression (Figure 6B, Figure S6E). MYC is also present at these promoters and its binding is also diminished following MAX loss. We frequently observe co-occupancy of MYC, MAX and MXD family proteins at promoters (Figure 6B, Figure S6E) (Mathsyaraja et al., 2019 and unpublished data). MAX-MXD heterodimers are generally thought to antagonize MYC function (Carroll et al., 2018; Hurlin et al., 1997; Hurlin et al., 1999), suggesting that such co-occupancy by MAX, MNT, and MYC serves to maintain a balanced transcriptional output that determines tonic expression levels. We hypothesize that MAX loss in SCLC removes this balanced regulation and fuels tumor growth via de-repression of a group of growth-related genes, perhaps mediated by binding of other transcriptional activators. Moreover, when we compared our gene expression and occupancy data, we observed that around 36% of genes upregulated in Max KO preSCs are directly bound by MAX in WT preSCs (Figure 6E). The upregulation was accompanied by a significant increase in RNA pol II association with MAX-bound genes in Max-KO preSCs when compared to Max-WT (Figure 6F). This gene activation occurs despite the general decrease in MYC binding at these promoters, further suggesting that MAX-MXD heterodimer activity predominates over or balances MYC-MAX activating heterodimers in the preSC system.
Figure 6. Genomic occupancy of MAX altered SCLC.
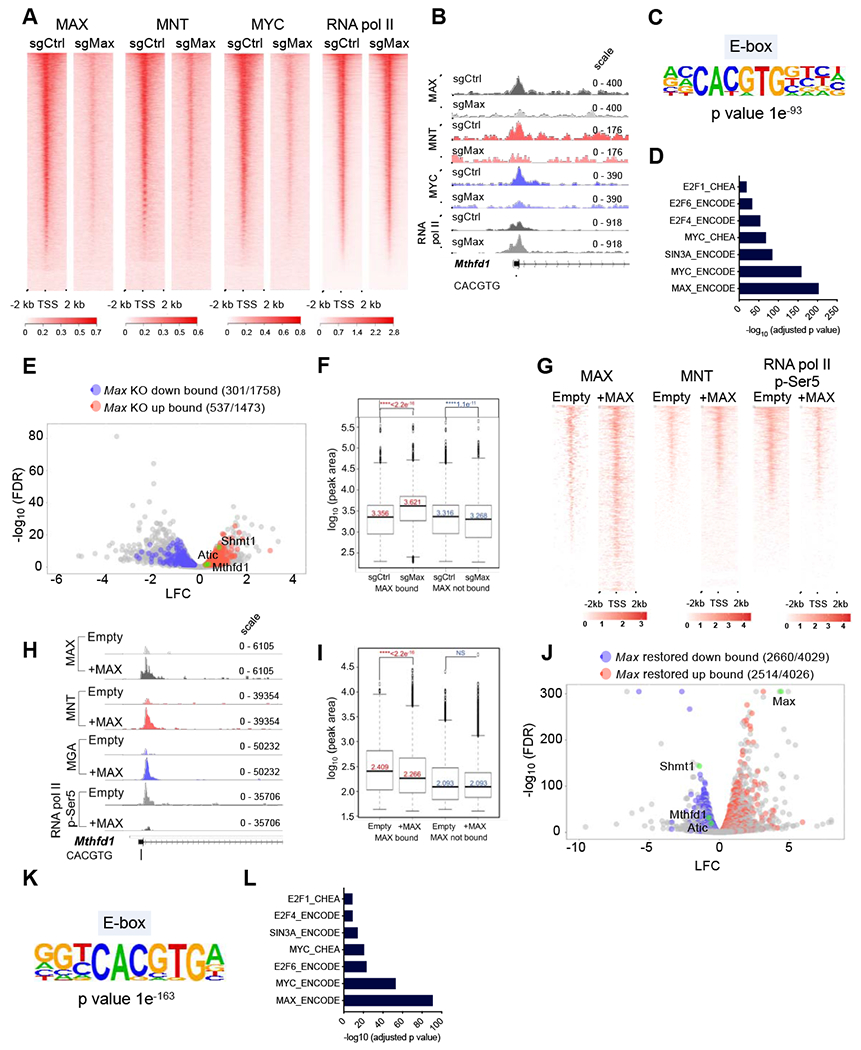
A, Heat maps depicting promoter enrichment (+/− 2 kb of TSS) of MAX, MNT, MYC and RNA pol II in control and Max-deleted preSCs. B, Representative tracks for MAX, MNT, MYC and pol II binding at the Mthfd1 promoter in preSCs. C, Enriched de novo motifs from HOMER analysis on MAX-bound sequences in preSCs. D, ENCODE and CHEA analysis on MAX-bound genes in preSCs. E, Volcano plot depicting overlap between expression and genomic occupancy analyses (one carbon genes highlighted in green). F, Box plots of RNA pol II occupancy at TSS +/− 2 kb at MAX bound vs. non-bound genes in Max-null and control preSCs. Box boundaries indicate 1st and 3rd quartiles, whiskers represent 1.5 times the interquartile range, and dots indicate datapoints that fall outside that region. Genes considered MAX-bound if peak occurred −5 kb to TSS. p values computed using Wilcox test (p < 0.01 considered significant). G, Heatmap of MAX, MNT and RNA pol II phospho-Ser5 binding in Max-null and MAX restored mRPMaxKO SCLC cells. H, Representative peaks for MAX, MNT, MGA and pol II phospho-Ser5 binding at the Mthfd1 promoter in mRPMaxKO SCLC cells. I, Box plots depicting RNA pol II phospho-Ser5 occupancy levels at MAX bound vs. unbound genes in MAX restored and Max-null mRPMaxKO SCLC cells. Box boundaries mark 1st and 3rd quartiles, whiskers indicate 1.5 times the interquartile range, and dots represent data points that fall outside that region. Genes considered MAX-bound if peak occurs at TSS to −5 kb. p values computed using Wilcox test. J, Volcano plot showing overlap between expression and CUT&RUN analyses in MAX restored mRPMaxKO cells (one carbon genes and Max highlighted in green). K, HOMER analysis showing significant de novo motifs identified from MAX bound sequences in mRPMaxKO SCLC cells. L, ENCODE and CHEA analysis on MAX bound genes in MAX restored mRPMaxKO cells. See also Figure S6.
Next, we performed genomic occupancy analyses using CUT& RUN (Janssens et al, 2018) upon restoration of Max expression in a RPMax-deleted mSCLC cell line (mRPMaxKO) using an inducible vector. We observed a strong enrichment for MAX binding in Max-expressing cells when compared to Max-null controls (Figure 6G). This was accompanied by an overall increase of MNT occupancy at the TSS and decrease of RNA pol II occupancy in MAX restored preSCs (Figure 6G–6I). To further confirm the direct binding of repressive heterodimers at key genes, we examined MAX, MNT, MGA and RNA pol II phospho-Ser5 occupancy at key one carbon genes such as Mthfd1 and Atic. We noted a significant increase in MAX, MNT and MGA occupancy in MAX restored cells, suggesting that MAX is capable of directly repressing these genes via heterodimerization with multiple MXD proteins, causing their downregulation in this system (Figure 6H, Figure S6F). Concomitantly, we observed a decrease of RNA pol II phospho-Ser5 binding at these promoters and an overall reduction of Pol II phospho-Ser5 occupancy at genes bound by MAX in MAX restored cells when compared to controls (Figure 6H, I). Overall, these data complement our findings from the preSC system suggesting that MAX predominantly acts as a repressor in the absence of elevated MYC levels. We then overlapped our occupancy data in the MAX restored system with our transcriptional analyses. About 60% of genes that are upregulated or downregulated upon MAX restoration were bound by MAX, indicating that MAX directly regulates a major part of the transcriptional re-wiring that occurs in Max-null SCLC (Figure 6J). Consistent with this idea, we observed enrichment for MAX, MYC, SIN3A and E2F6 and E-boxes when we performed CHEA and HOMER analyses on MAX-bound genes, respectively (Figure 6K, L). Overall, our data strongly suggest that MAX-MXD heterodimers functionally predominate over activating MAX-MYC heterodimers in early stage preSCs. Therefore, loss of MAX leads to the up-regulation of growth promoting genes, such as those involved in one carbon metabolism. Consistent with this, restoration of MAX to a Max-null mSCLC cell line reinstates repression of genes at several thousand loci, including key one carbon metabolic regulators such Mthfdl, Shmtl and Atic. Importantly, several of the 113 genes commonly regulated by MAX in these systems, including Mthfdl, Shmtl and Atic are directly bound by MAX in both preSCs and the MAX restored system (Figure S6G).
Max deletion results in the activation of one carbon and serine biosynthesis metabolic pathways
Our data so far suggests that MAX directly represses many metabolic genes including key regulators of one carbon (1C) metabolism, an observation that may be highly significant in light of recent studies linking upregulation of 1C metabolism to pro-growth phenotypes (Labuschagne et al., 2014; Maddocks et al., 2017). Flux through the 1C pathway can help to meet high demand for nucleic acid production to fuel cancer cell growth. We therefore sought to further elucidate the function of the 1C pathway in SCLC. Immunoblot analyses confirmed upregulation of MTHFD1, SHMT1, ATIC and PHGDH expression upon Max loss in preSCs (Figure 7A, 7B). To compare the effects of Max loss in preSC cells to that of deleting another SCLC tumor suppressor, Pten, which also regulates metabolic targets and increases growth/transformation, we performed western blot analyses for key 1C enzymes. While Pten deletion in preSC cells also led to upregulation of PHGDH, upregulation of MTHFD1, ATIC, and SHMT1 was specific to Max deletion (Figure S7A). To determine whether Max loss could further enhance the increased proliferation seen with Pten loss in the preSC system, we deleted Max in Pten-null preSC cells. Max deletion in Pten null preSC cells resulted in increased proliferation that was additive to the effects of Pten loss alone (Figure S7B–D).
Figure 7. Max deletion results in increased serine and one carbon metabolism.
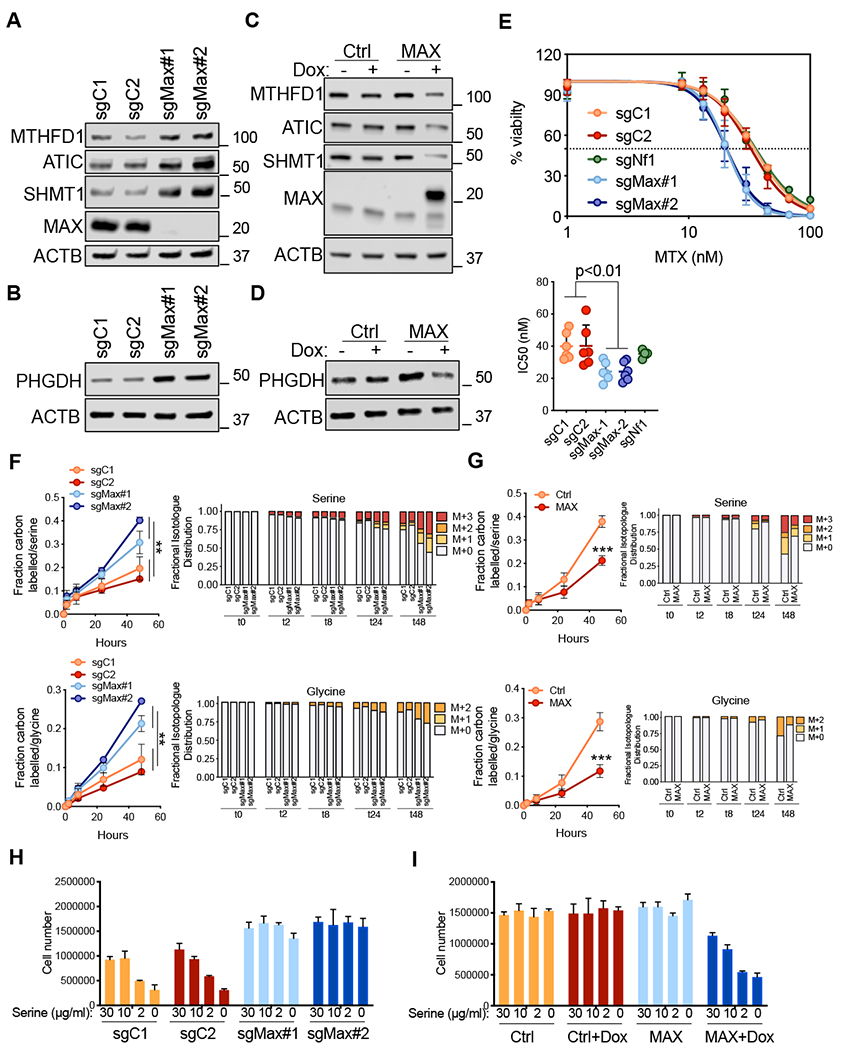
A and B, Immunoblotting for MTHFD1, SHMT1, ATIC (A) and PHGDH (B) proteins upon Max loss in preSCs. ACTB was used as a loading control. C and D, Immunoblotting for MTHFD1, SHMT1, ATIC (C) and PHGDH (D) upon MAX restoration in mRPMaxKO cells. ACTB was used as a loading control. E, Dose-response curves of preSCs treated with methotrexate for 96 h. Viability was assessed with the CellTiter-Glo assay and calculated relative to the vehicle control. Data are mean ± SEM from at least n = 3 biological replicates. IC50s are shown for each genotype and each single data point represents an independent experiment. ** p<0.01, unpaired student’s t-test. F, U-13C glucose tracing experiments in preSCs upon Max loss followed across the indicated time points. Fraction carbon labelled and isotopologues for both serine and glycine are shown. ** p<0.01, unpaired student’s t-test. G, U-13C glucose tracing experiments in mRPMaxKO cell upon MAX restoration followed across the indicated time points. Fraction carbon labelled and isotopologues for both serine and glycine are shown. *** p<0.001, unpaired student’s t-test. H-I, Growth in serine depleted media for preSCs upon Max loss (H) and for mRPMaxKO upon MAX restoration (I). Error bars represent mean ± SD (n=3). See also Figure S7.
We next restored MAX in mRPMaxKO SCLC cells and observed decreases in MTHFD1, SHMT1, ATIC and PHGDH protein levels (Figure 7C, 7D). If upregulation of 1C metabolic processes are important for Max-deleted preSC cells, we reasoned that these cells would exhibit increased sensitivity to methotrexate, a dihydrofolate reductase (DHFR) inhibitor (Farber and Diamond, 1948). Methotrexate acts in part by depleting tetrahydrofolate (THF), an essential co-factor required for folate reactions (Ducker and Rabinowitz, 2017). Indeed, Max-KO preSCs exhibited significantly increased sensitivity to methotrexate compared to preSCs expressing Max (Figure 7E). Importantly, this effect was not simply due to augmented growth of the Max-KO preSCs alone, as the increased sensitivity was not observed in Nfl-KO preSCs which grow at a similar rate to Max-KO preSC (Figure 7E and data not shown).
Our data also identified PHGDH, which was upregulated following Max loss in preSC cells and downregulated following MAX restoration, as a potential MAX target in SCLC cells (Figure 7B, 7D). PHGDH is the first, and rate limiting, enzyme in the de novo serine synthesis pathway from glucose, catalyzing 3-phosphoglycerate into 3-phosphohydroxy-pyruvate. Several cancer types show an upregulation in the expression of the serine synthesis pathway correlated with PHGDH amplification or over-expression. PHGDH upregulation leads to increased tumor growth and its depletion abrogates cancer growth (DeNicola et al., 2015; Locasale et al., 2011; Possemato et al., 2011). More recently, PHGDH has been shown to be critical for tumor growth in environments where serine is limiting and is sufficient to promote tumorigenesis in mouse models of cancer (Sullivan et al., 2019). The upregulation of PHGDH uponMax deletion led us to hypothesize that Max loss would increase the production of serine from glucose. Increased serine production from glucose would also potentially result in increased glycine synthesis through SHMT1, an enzyme that uses THF to convert serine into glycine (Ducker and Rabinowitz, 2017). To test this hypothesis, we set up a time course experiment in which we examined the fate of uniformly 13C-labelled glucose (U-13C-glucose) in Max-WT and Max-KO preSCs. As expected, the percentage of carbon labelled serine and glycine increased over time regardless of genotype (Figure 7F). Interestingly, the rate of label incorporation from U-13C-glucose into serine and glycine from U-13C-glucose increased in Max-KO preSCs compared to Max-WT preSCs, consistent with increased PHGDH expression (Figure 7F). Isotopologue distribution revealed an increase in serine (M+1-3) and glycine (M+1-2) in Max-KO preSCs (Figure 7F). Notably, partial labeling of serine (M+1 and M+2) also increased in Max-KO preSCs relative to M+3 serine. This finding is consistent with enhanced label scrambling though exchange with glycine by the action of SHMT1, which exhibits increased expression upon Max deletion in these cells (Figure 7A). In contrast to the effects of Max deletion in preSC cells, MAX restoration in mRPMaxKO SCLC cells resulted in a decrease in carbon labelling of serine and glycine from U-13C-glucose (Figure 7G). U-13C glucose derived carbon is incorporated into AMP through ribose production (M+5) and through serine contribution to the purine ring via 10-formyl-tetrahydrofolate (M+1 or M+2) and glycine (M+2). An increase in higher order labeled species of AMP (M+6-9) from U-13C glucose, which require contribution from labeled serine, was observed upon Max loss (Figure S7E). Conversely, a significant decrease of AMP labelled carbons (M+6-9) was observed upon MAX restoration (Figure S7F). Collectively, these results suggest increased capacity of Max- deficient cells for serine synthesis and one carbon pathway metabolism.
We next tested the capability of MAX altered cells to grow in environments where serine is a limiting factor. We found that Max-KO preSCs were strikingly resistant to limiting serine in the media as compared to control preSCs (Figure 7H). Conversely, MAX restoration in the mRPMaxKO SCLC line re-sensitized cells to loss of viability upon serine depletion (Figure 7I). Taken together, our data demonstrate that Max loss upregulates a panel of genes that control serine biosynthesis and one carbon metabolism, with several of these, namely Atic, Mthfdl and Shmtl direct targets of MAX-mediated repression (Figure 6 and Figure S6). Functionally, this enables Max-deficient cells to survive and proliferate in the absence of serine. Thus, Max loss can lead to growth promoting metabolic rewiring.
Discussion
Applying genome-scale CRISPR-Cas9 screens to a cellular model derived from early-stage mouse SCLC cells, revealed MAX as a candidate tumor suppressor gene in SCLC. Because MAX is an obligate dimerization partner for the entire MYC oncoprotein family, it was paradoxical that MAX loss in preSC cells so dramatically increased cell growth and transformation. Because there is a lack of in vivo evidence validating MAX as a tumor suppressor gene for any tumor type, we deleted Max in mouse models sensitized towards developing SCLC. Max inactivation cooperated with Rb1/Trp53 loss to result in dramatic increases in lung tumor size and numbers, a magnitude of effect comparable to deletion of potent tumor suppressors such as Pten. Although a previous in vitro study identified genetic MAX inactivation in 6 % of human SCLC cells (Romero et al., 2014) our work provides a rigorous in vivo model to demonstrate tumor suppression conferred by MAX.
Importantly, we observed strong selective pressure to retain Max alleles in the context of Myc1 transgenic overexpression. Our data suggest that the expression/activity of MYC members determines whether MAX loss results in pro or anti-growth phenotypes. In agreement with this hypothesis, we demonstrate that Max deletion in preSCs, where MAX-MXD function predominates, leads to increased growth. In contrast, preSCs over-expressing MYC paralogs shifts the stoichiometry towards activating MYC-MAX heterodimers. In this context, we show that the growth of MYC-family overexpressing preSCs was abrogated upon MAX loss. Of note, we performed CRISPR inactivation screens in mouse SCLC cell lines derived from late stage tumors and, unlike our results in preSC cells, have not found enrichment of MAX targeting sgRNAs (unpublished results). We speculate that inactivating mutations in MAX occur at an early stage of SCLC tumor initiation, where MYC paralog activity may be low. Our use of SCLC mouse models and the preSC cellular model allowed targeting of early stage SCLC cells and uncovered the potent context-dependent tumor suppressive activity of MAX. However, once selection for incipient SCLC cells with high expression of MYC/MYCL/MYCN occurs, the pro-proliferative effects of MAX loss are unable to overcome the acute dependence of these cells on MYC function. This notion is consistent with data showing that Max deletion in normal B cells, which express low levels of MYC, has only a modest impact on B cell development in mice (Mathsyaraja et al., 2019; Perez-Olivares et al., 2018). In contrast, in Eμ-Myc mice, where high levels of MYC drive lymphomagenesis, loss of MAX completely abrogates lymphoma incidence (Mathsyaraja et al., 2019).
Our analyses reveal repressive MAX-MNT/MXD activity predominates over MYC-MAX in both matched models studied. This is consistent with the significant enrichment we see for SIN3A and E2F6 (part of ncPRC1.6) genomic occupancy at genes upregulated upon Max deletion. Loss of MAX-associated repression can have important biological consequences. For example, loss of MAX-MGA re-activates meiotic gene expression programs in embryonic stem and germ cells via displacement of PRC1.6 complexes (Endoh et al., 2017; Suzuki et al., 2016). Indeed, we observed strong activation of meiotic genes upon Max deletion in the mouse SCLC model. Moreover, deletion of Mnt has been associated with tumorigenesis and disrupted cell cycle control (Hurlin et al., 2003). Across three different transcriptional analyses, the genes upregulated in the absence of MAX were highly enriched for MYC/MAX binding sites, but the downregulated genes exhibited no such enrichment. We hypothesize that tumor promoting activity associated with MAX loss occurs via derepression of key pro-growth genes caused by loss of MAX-MXD/MGA-family heterodimers. The shared MAX bound genes in both models included the one carbon regulators MTHFD1, ATIC and SHMT1. We provide several lines of evidence supporting the notion that regulation of serine and one carbon metabolic enzymes regulated by MAX are functionally important in Max-null SCLC: (i) our CRISPR screen data shows that SHMT1, MTHFD1 and PHGDH are required for preSC growth; (ii) Max-deleted preSC cells exhibit increased sensitivity to folate pathway inhibition using methotrexate; (iii) serine and glycine production from labeled U-13C-glucose increased upon MAX loss in preSC cells, while restoration of MAX expression in RPMaxKO SCLC cells triggered the opposite effects; (iv) we found an increased growth ability for Max-null preSCs in serine deprived media and, conversely, sensitization to serine depletion upon reintroduction of MAX in RPMaxKO SCLC cells. Our data demonstrate that genetic perturbation of MAX alters the expression of multiple metabolic genes in addition to those encoding the one carbon enzymes and consider it likely that many of these also contribute to MAX-dependent tumorigenesis.
Germline MAX mutations in humans are associated with neuroendocrine tumorigenesis beyond SCLC, including the development of pheochromocytomas, paragangliomas and pituitary tumors (Burnichon et al., 2012; Comino-Mendez et al., 2011; Daly et al., 2018). We speculate that a common circuitry among these tumors makes their growth particularly sensitive to activation or suppression of serine/one carbon pathways. In prostate neuroendocrine cancer, for example, a metabolic switch to activate serine and one carbon pathways occurs not with MAX deletion but via activation of mTORC1/ATF4 signaling (Reina-Campos et al., 2019). An in-depth analysis of whether de-repression of similar metabolic programs is commonly observed upon MAX inactivation across multiple neuroendocrine tumor types will shed light on why these tumor types undergo selection for MAX deletion. The generation of new Max-deleted mouse models for other neuroendocrine cancer types, including those tumor types that develop in patients with germline mutations in MAX will be critical.
Since the discovery of MAX nearly three decades ago as the heterodimerization partner required for MYC DNA binding (Blackwood and Eisenman, 1991), we have learned a great deal about how MYC-MAX promotes tumorigenesis across many cancer types. Recent findings that germline and somatic inactivation mutations in MAX result in neuroendocrine cancer types and our own findings that Max deletion dramatically promotes SCLC highlight a pressing need to better understand the broader functions of MAX beyond interactions with MYC/MYCN/MYCL. Our genomic occupancy and gene expression studies reveal that MAX and MNT repress multiple genes, including those involved in the one carbon pathway, that may contribute to the tumor suppressive function of MAX. We expect that MAX heterodimers with other MXD proteins and/or MGA will also mediate repression at other loci involved in growth and proliferation. Recent analyses of TCGA data across many cancer types highlight frequent mutations and deletions in genes encoding MAX dimerization partners such as MNT, MGA, MXD3 and others (Schaub et al., 2018). MGA for example is inactivated in a subset of SCLC (Romero et al., 2014). Surprisingly, MAX was the only member of the MYC transcription factor network to promote cell growth when deleted in our preSC CRISPR screen. Therefore, simultaneous genetic deletion of multiple MXD family proteins may be required to phenocopy the pro-growth effects of MAX loss. Our work underscores context dependent functions of MAX and how MAX heterodimers repress transcription of thousands of genes to mediate tumor suppressive functions in SCLC and in a broad range of malignances.
STAR*METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents including plasmids, cell lines and mouse models should be directed to and will be fulfilled by the Lead Contact, David MacPherson (dmacpher@fredhutch.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Studies
We thank Dr. Tyler Jacks for the Rb1lox/lox and Dr. Anton Berns for the Trp53lox/lox and the frt-invCAG-Mycl1-Luc strains. We employed either Ad5-CMV-Cre, which expresses Cre under a CMV promoter and cell type specific Ad5-CGRP-Cre, which uses a neuroendocrine-specific promoter to drive Cre expression. Cre expressing adenoviruses were obtained from the University of Iowa Gene Vector Core, with permission of Dr. Anton Berns. The floxed Max mice were bred to compound Rb1lox/lox ;Trp53lox/lox mice to obtain Rb1lox/lox ;Trp53lox/lox ;Max mice. Rb1lox/lox;Trp53lox/lox ;Maxlox/lox mice were bred to Rb1lox/lox ;Trp53lox/lox ;invCAG-Mycl1-Luc to obtain Rb1lox/lox;Trp53lox/lox;Maxlox/lox;invCAG-Mycl1-Luc mice. Genotypes were confirmed by PCR. Littermates controls of indicated genotypes were infected following intratracheal instillation of 3x108 plaque-forming units (pfu) of either Ad5-CMV-Cre or Ad5-CGRP-Cre viruses as described (DuPage et al., 2009). Mice were monitored every week following viral infection and euthanized when moribund with labored breathing. Tumor tissues were used to generate cell lines, frozen for molecular analyses and fixed for histologic analyses as well as immunofluorescence experiments. After excising tumors, the whole lung was inflated with neutral buffered formalin (NBF) and processed for histologic analyses. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the Fred Hutchinson Cancer Research Center.
Cell Lines
MEFs were isolated as described above. preSC cell lines were provided by Dr. Kwon Park, isolated as described (Kim et al., 2016). The mRPMaxKO cell line was isolated from a Rb1/Trp53/Max deleted mouse SCLC tumor. mRPMaxKO and preSC(s) were cultured in RPMI 1640 (11875-093, ThermoFisherScientific) supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) (15140122, ThermoFisherScientific) and 10 % fetal bovine serum (FB-01, Omega Scientific). MEFs were grown in DMEM (11965-092, ThermoFisherScientific) supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) (15140122, ThermoFisherScientific), and 10 % fetal bovine serum (FB-01, Omega Scientific). Cells were maintained at 37 °C in a humidified atmosphere containing 5 % CO2 and 95 % air.
METHOD DETAILS
Chemicals
Methotrexate was purchased from Cayman chemical (13960), resuspended according to the manufacturer’s recommendations and used at concentrations ranging from 0-100 nM as described in the figure legend. Doxycycline was purchased from Sigma (44577) and used at concentrations ranging from 500-1000 nM as described in figure legends.
Magnetic resonance imaging
Mice from each cohort infected with Ad5-CMV-Cre or Ad5-CGRP-Cre on the same date were monitored by MRI scan (ICON small animal MRI system, Bruker Biospin) to determine lung tumor burden. Mice were anesthetized with isoflurane during imaging. Respiratory gating was employed, and a total of 15 slices of 1 mm thickness was acquired. Tumor volume was quantified using 3D ImageJ Suite with manual quantification of consecutive axial image slices.
Generation of Trp53flox/flox MEFs
Trp53flox/flox MEFs were generated from embryos at dpc13.5 under a cell culture hood (sterile conditions). Briefly, each embryo was placed in a sterile 10 cm cell culture dish and covered in sterile PBS (1X). Placental and other maternal tissues were removed along with the head and all other innards. The embryo body was moved into a different 10 cm cell culture dish and minced with a sterile razor blade in presence of warm 1 ml trypsin/EDTA (0.05 %) (25300054, ThermoFisherScientific) and incubated for 1 hour at 37 °C. The trypsin was then quenched using DMEM (11965-092, ThermoFisherScientific) and the digested cells mixed and plated into a 10 cm cell culture dish. Cells were briefly cultured (3-4 days), split an additional time to allow for cell expansion and frozen down into multiple vials as early passage stocks. Each embryo was processed separately. In order to inactivate Trp53, Cre recombination was performed using Ad5-CMV-Cre viruses (MOI=100) (University of Iowa Gene Vector Core). Two rounds of infections were performed in order to obtain a high recombination rate.
CRISPR–Cas9 based screen
More than 250x106 Trp53-null MEFs (n=3) and preSCs (n=3) were transduced with the V2 GeCKO mouse library (1000000052, Addgene) at a MOI<1 (30 % transduction rate), puromycin selected for 3-4 days after which 66x106 cells, approximately 500X library coverage, were collected (reference point, PD0) and the remaining cells (at least 66x106) were seeded, expanded for a total of 12 population doublings (end point, PD12) and collected for subsequent analyses. Genomic DNA extraction, library amplification and high throughput sequencing is described below. Sequencing data was analyzed using MAGeCK-VISPR (Li et al., 2015). Alternatively, CRISPR scores were calculated as described in (Wang et al., 2015). CRISPR scores are defined as the average log2 fold-change in the final versus initial abundance of all sgRNAs targeting a given gene.
Genomic DNA Extraction
For genomic DNA extraction, a salt precipitation method previously described was used (Chen et al., 2015). In a 15 ml conical tube, 6 ml of NK Lysis Buffer (50 mM Tris, 50 mM EDTA, 1 % SDS, pH 8.0) and 30 μl of 20 μg/ml Proteinase K (19131, Qiagen) were added to the cell sample (30x106-70x106 frozen cells) and incubated at 55 °C overnight. The next day, 30 μl of 10 μg/ml RNase A (19101, Qiagen) was added to the lysed sample, which was then inverted 25 times and incubated at 37 °C for 30 min. In order to precipitate proteins, samples were cooled on ice before addition of 2 ml of pre-chilled 7.5 M ammonium acetate/dH2O (A1542, Sigma). Samples were vortexed at high speed and centrifuged at 4,000 g for 10 min at 4 °C. After the spin, a tight pellet was visible in each tube and the supernatant was carefully decanted into a new 15 ml conical tube. Isopropanol (6 ml) was used to precipitate the DNA by inverting 50 times and centrifugation at 4,000 g for 10 min. Genomic DNA pellets were washed with 70 % ethanol and centrifuged at 4,000 g for 5 min. After air-drying the pellets for at least 10 min, the DNA was resuspended in 500 μl of 1X TE buffer (T9285, Sigma) and incubated at 65 °C for 1 hour and subsequently at room temperature overnight to fully resuspend the DNA. The next day the gDNA was measured using a Nanodrop 2000 (Thermo Fisher Scientific).
V2 GeCKO mouse library propagation and PCR amplification and high throughput sequencing
The V2 GeCKO mouse library was propagated as described in (Sanjana et al., 2014). The sgRNA library readout was performed using a two steps PCR protocol as described in (Chen et al., 2015). The first PCR (12 cycles) was performed to amplify and preserve full library complexity and the second PCR (16-19 cycles) adds appropriate sequencing adapters to the products from the first PCR. To maintain a 500X library representation, 430 μg of gDNA per condition was PCR amplified. For each 50 μl reaction, 2 μg of genomic DNA was used. The second PCR products were migrated on a 2 % agarose gel, pooled and purified using PureLink quick gel extraction kit (K210012, Invitrogen). The purified libraries were quantified using Kapabiosystems Library Quantification Kit (KK4824, Roche) and sequenced on a HiSeq 2500 (Illumina) according to the manufacturer’s recommendations. All PCR were performed using Phusion Flash High Fidelity Master Mix (F548L; ThermoFisherScientific) according to the manufacturer’s recommendations. PCR primers can be found in (Table S3).
Lentivirus vector production, concentration, and generation of stable lines
For lentiviral experiments, two sgRNA sequences targeting the genes of interest were selected based on enrichment scores from the whole genome CRISPR screens and cloned into the lentiCRISPR v2 (Addgene plasmid no. 52961) according to the protocol provided by the Zhang Lab. A complete list of CRISPR sequences can be found in (Table S3). Lentiviral vectors were produced by cotransfecting 293TN producer cells (LV900A-1, System Bioscience) with the lentiviral vectors and helper plasmids psPAX2 (Addgene plasmid no. 12260) and pMD2.G (Addgene plasmid no. 12259; ratio, 1:1:0.67) using lipofectamine 2000 (11668019, ThermoFisherScientific) according to the manufacturer’s recommendations. Viral supernatants were collected at 48 hours and 72 hours (second collection) after transfection, filtered through a 0.45 pm PVDF syringe filter (F5510, Denville). Viral transductions were performed for 8 hours in the presence of polybrene (4 μg/ml) for preSCs and 16-24 hours in the presence of polybrene (8 μg/ml) for other cell lines. Equal viral titers were used. Puromycin selection (A1113803, ThermoFisherScientific) was performed for 3 to 4 days at 0.8 μg/ml (preSCs), 1 μg/ml (MEFs) and 1.5 μg/ml (mRPMaxKO). preSCs over-expressing MYC members were generated by stable transduction of a lentiviral vector, pLX304 expressing either MYCL, MYC or MYCN. The empty pLX304 vector was used as a control (Addgene plasmid no. 25890). pLX304 expressing either MYCL, MYC or MYCN were generated by Gibson assembly (E5510S, New England Biolabs). Blasticidin (ant-bl-05, Invivogen) selection was performed at 2,5 μg/ml. A MAX inducible lentiviral vector was generated by PCR amplification of the MAX ORF Isoform 2 (NM_145112.2, short isoform) using the Phusion® High-Fidelity PCR Kit (E0553, New England Biolabs) and cloned into the pCW57-RFP-P2A-MCS (Addgene plasmid no. 78933) using EcoR1 and Agel restriction sites.
RNA extraction and RNA-seq analyses
RNA was extracted using TRIZOL according to the manufacturer’s recommendations (15596018, ThermoFisherScientific). Tumor samples were homogenized in TRIZOL using the AgileGrinder™ Tissue Grinder (ACT-AG3080, Thomas Scientific-ACTGene). For RNA-seq analyses, the Ultra RNA Library Prep Kit for Illumina (E7530L, New England BioLabs) was used to generate libraries from total RNA (500 ng). All library preparation was conducted according to the manufacturer’s instructions. Single-end sequencing (50 bp) was performed using an Illumina HiSeq 2500, and reads of low quality were filtered before alignment to the mm9 genome build using TopHat v2.0.12 (Trapnell et al., 2009). Cuffdiff v2.1.1 (Trapnell et al., 2013) was used to generate FPKM expression values. Counts were generated from TopHat alignments using the Python package HTSeq v0.6.1 (Anders et al., 2015) using the “intersection-strict” overlap mode. Genes with low counts across conditions were discarded before identification of differentially expressed genes using the Bioconductor package edgeR, v3.16.5 (Robinson et al., 2010). An FDR method (Reiner et al., 2003) was used to correct for multiple testing, where differentially expressed genes were identified with the FDR set at 5 %. Reads from the mRPMaxKO cell line were aligned to the mm9 genome build using STAR v2.5.2a (Dobin et al., 2013) in 2-pass mode, followed by generation of counts for each gene using featureCounts from the Subread package v1.6.0 (Liao et al., 2014).
Genomic Occupancy Studies
For conventional crosslinked ChIP-seq experiments, sgCtrl or sgMax preSC chromatin was used for IP (anti-MYC, MAX, MNT, RNA pol II). Normalization was performed based on protein amounts and ChIP was performed as previously described (Skene and Henikoff, 2015). Libraries were generated using the NEB Ultra II kit. 50X50 paired end sequencing was performed on an Illumina HiSeq 2500 instrument. For CUT&RUN experiments, MAX expression was induced in SCLC tumor lines by doxycycline treatment for 4 days. Cells were bound to ConA beads and permeabilized using a digitonin containing buffer. 1 million cells were used per IP and incubated overnight with antibodies against MYC, MAX and MNT, MGA and RNA polII phosphor Ser-5. The CUT& RUN protocol was followed for library prep (Janssens et al, 2018). Libraries were sequenced using 25x25 paired-end sequencing was performed on an Illumina HiSeq 2500 instrument. 5-10 million reads were obtained per antibody. For both ChIP seq and CUT&RUN experiments, sequences were aligned to the mm10 reference genome assembly using Bowtie2. Spike-in normalization was performed using yeast DNA spike in for CUT&RUN studies whereas library size normalization was done for ChIP-seq experiments. In all cases, peaks were called using MACS at different thresholds. Post peak calling, processing was carried out with bedtools, custom R scripts defining genome position, and the GenomicRanges R package. For MAX, peaks were identified as being associated with a gene if they were within + or − 5 kb from the TSS. For MAX peak calling in preSCs, reads from two independent experiments were aggregated. For CUT&RUN experiments, peak calls from two independent experiments were intersected following IgG subtraction to derive a gene list. For RNA poll II and RNA pol II phosphor-Ser5 peak area, genomic regions +/− 2 kb of the TSS were considered. Genomic plots and heatmaps were made using ngs.plot (Shen et al., 2014) or the R package ggplot2. Heatmaps for genomic binding were made ranking genes according to log fold change in expression from RNA-seq datasets associated with each experiment. Volcano plots were generated using the R package ggplot2. Enrichr was utilized to overlap peak calls with existing ENCODE and ChEA data. De novo and known motif enrichment for sequence specificity was determined using HOMER (Heinz et al., 2010).
Western blot analysis
Whole-cell protein extracts were prepared in cold cell lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % NP-40, 1 % sodium deoxycholate] supplemented with protease and phosphatase inhibitors (78441, ThermoFisherScientific). Tumor samples were homogenized in cold cell lysis buffer using the AgileGrinder™ Tissue Grinder (ACT-AG3080, Thomas Scientific-ACTGene). Samples were sonicated for 5 minutes, 30 seconds ON/OFF using a Bioruptor (UCD-200, Diagenode). Proteins were quantified using Pierce™ BCA Protein Assay Kit (23227, ThermoFisherScientific), resolved on 4–20 % Mini-PROTEAN®TGX™ Precast Protein Gels (4561096, BioRad), and transferred to Amersham Protran 0.45 NC nitrocellulose membranes (10600002, GE healthcare life science). Protein samples were normalized to ACTB or HSP90. Imaging was performed using the LI-COR Odyssey Fc.
Drug treatments, cell proliferation and viability
Cells were seeded at 7,500 cells per well in 96-well plates and live cell content was estimated using a CellTiter-Glo assay (G7573, Promega) according to the manufacturer’s protocol at the times indicated in the figure legends. Alternatively, for growth in serine deprived media, cells were plated in 12 well plates at 200,000 (preSCs) and 100,000 (mRPMaxKO) and counted using an automated Z2 Series Coulter Counter (6605700, Beckman Coulter). Methotrexate studies were performed in 96-well plates (15,000 cells per well) for 96 hours. CellTiter-Glo was used to determine the relative cell content and viability.
Growth curve analyses
Cells were plated in 6-well plates (200,000 cells/well) on day 0 in the presence or absence of doxycycline (0.5 μg/ml). Doxycycline was replenished every 3 days. The number of cells was counted using an automated Z2 Series Coulter Counter (Beckman Coulter), and the same number of cells was seeded for further counts. Experiments were carried out for a total of 9 days. Population doublings were calculated using the following formula: n = 3.32 (log UCY – log l) + X, where n is the final population doubling at end of a given subculture, UCY is the cell yield at that point, l is the cell number used as inoculum to begin that subculture, and X is the doubling of the inoculum used to initiate the subculture being quantitated.
Colony formation assays
Cells were seeded at 3x103 cells/well (6 well plate), expanded for 2 weeks, fixed with 4 % PFA for 15 minutes and stained with crystal violet solution (0.05 %). The number of colonies per field were counted. Representative wells for each condition are shown.
Anchorage independent growth
Cells (1.5x105/well of a 6 well plate) were seeded in 0.4 % low-melting-point SeaPlaqueTM agarose (Lonza; Catalog no: 50101) on top of 0.8 % low-melting-point SeaPlaqueTM agarose layer. Low-melting-point agarose was premixed with DMEM 2X (Fisher Scientific; SLM202B) complemented with 20 % FBS, sodium pyruvate (2 mmol/L), 200 U/mL penicillin, and 200 μg/mL streptomycin. Cells were allowed to grow at 37 °C with 5 % CO2 for 3 weeks. For each well, colonies from at least 5 random fields were counted. Representative microscopic images are displayed.
Glucose tracing experiments
Cells were incubated in the presence of Glucose, Glycine and Serine free RPMI (R9660-02, TEKNOVA) supplemented with 10 % dialyzed serum (26400044, ThermoFisherScientific), 10 μg/L Glycine (G5417-100G, Sigma), 30 μg/L L-Serine (S4311-25G, Sigma) and 2 g/L D-Glucose (U-13C6, 99 %) (CLM-1396-PK, Cambridge Isotope Laboratories) for the indicated times. At the time of collection, cells were washed three times in cold saline and the metabolites extracted by scraping the cells in ice cold Methanol (80 %). Chloroform was added and the tubes vortexed at 4 °C for 10 min. The tubes were centrifuged at 4 °C at 16,000 g and the aqueous phase collected. Samples were dried using a speedvac and subsequently ran on the LC-MS.
LC-MS
Metabolite quantitation was performed using a QExactive HF-X Hybrid Quadrupole-Orbitrap Mass Spectrometer equipped with an Ion Max API source and H-ESI II probe, coupled to a Vanquish Flex Binary UHPLC system (Thermo Scientific). Mass calibrations were completed a minimum of once per week using LTQ Velos ESI Calibration Solution (Pierce). Samples were chromatographically separated by injecting a sample volume of 1-3 μL into a SeQuant ZIC-pHILIC Polymeric column (2.1 × 150 mm 5 μM, EMD Millipore). The flow rate was set to 150 μL/min, autosampler temperature set to 10 °C, and column temperature set to 30 °C. Mobile Phase A consisted of 20 mM ammonium carbonate and 0.1 % (v/v) ammonium hydroxide, and Mobile Phase B consisted of 100 % acetonitrile. The sample was gradient eluted (%B) from the column as follows: 0-20 min.: linear gradient from 85 % to 20 % B; 20-24 min.: hold at 20 % B; 24-24.5 min.: linear gradient from 20 % to 85 % B; 24.5 min.-end: hold at 85 % B until equilibrated with seven column volumes. Mobile Phase was directed into the ion source with the following parameters: sheath gas = 45, auxiliary gas = 15, sweep gas = 2, spray voltage = 2.9 kV, capillary temperature = 300 °C, RF level = 40 %, auxiliary gas heater temperature = 325 °C. Mass detection was conducted with a resolution of 240,000 in full scan mode or 120,000 in SIM mode, with an AGC target of 3,000,000 and maximum injection time of 100 msec for the full scan mode, or 100,000 and 150 msec for the SIM mode. Metabolites were detected over mass range of 70-1050 m/z in full scan positive mode, or SIM in positive mode using a quadrupole isolation window of 0.7 m/z. Quantitation of metabolites was performed using Tracefinder 4.1 (Thermo Scientific) referencing an in-house metabolite standards library, permitting 5 ppm mass error. Data from stable isotope labeling experiments included a natural abundance correction.
Histology and Immunofluorescence
Mouse lungs were fixed in NBF for 24 hours and then transferred to 70 % ethanol before paraffin embedding. Tissue sections (4 μM thick) were stained with Haemotoxylin and Eosin (H&E). Immunofluorescence was performed on fresh lung tumor tissues fixed in 4% paraformaldehyde (PFA) overnight at 4°C, transferred to 30 % sucrose at 4°C overnight, and then embedded in Tissue-Tek OCT solution and stored at −80 °C. Serial sections (8 μM thick) were cut using a Leica CM1950 cryostat at −20 °C. The following primary antibody was used: anti-CGRP (Rabbit polyclonal; Sigma C8198). Fluorescence images were obtained using a Zeiss LSM 700 confocal microscope. For preserving fluorescence and nuclear counter staining, ProLong Gold Antifade Mountant with DAPI (ThermoFisherScientific, P36935) was used. H&E and immunostained images were acquired using Nikon Eclipse E800 microscope.
Illustration Tool
The graphical abstract image is created with BioRender.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism 7.0 was used to perform statistical analyses. Lung tumor free survival analyses were analyzed using log-rank (Mantel-Cox) test. For the statistical analysis of lung tumor volume (MRI), number of lung tumors, proliferation assays, growth curves, colony formation assays, soft agars experiments and tracing experiments, statistical analyses were performed by Student’s unpaired t test. A p value of < 0.05 was considered statistically significant. Error bars represent mean ± SD unless otherwise indicated in the figure legends.
DATA AND CODE AVAILABILITY
The accession number for the raw and processed data of RNA sequencing, CHIP sequencing and CUT&RUN generated and reported in this paper is GEO: GSE138955, GSE146385 and GSE146388.
Supplementary Material
Table S3, related to STAR Methods: Oligonucleotides used in the study.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| JUN | Cell Signaling | 9165; RRID: AB_2130165 |
| PTEN | Cell Signaling | 9188; RRID: AB_2253290 |
| BAX | Cell Signaling | 2772; RRID: AB_10695870 |
| TSC1 | Cell Signaling | 6935; RRID: AB_10860420 |
| TSC2 | Cell Signaling | 4308; RRID: AB_10547134 |
| NF1 | Bethyl Labs | A300-140A-T; RRID: AB_2779035 |
| BIM | Cell Signaling | 2933; RRID: AB_1030947 |
| DLK/MAP3K12 | GeneTex | GTX124127; RRID: AB_11170703 |
| PHGDH | Bethyl Labs | A304-732A-T; RRID: AB_2782127 |
| ATIC | Bethyl Labs | A304-271A-T; RRID: AB_2781794 |
| SHMT1 | Cell Signaling | 80715; RRID: AB_2799957 |
| MTHFD1 | Bethyl Labs | A305-285A-M; RRID: AB_2631678 |
| MAX | Santa Cruz | sc-197; RRID: AB_2281783 |
| ACTIN | Sigma | A3854; RRID: AB_262011 |
| HSP90 | Santa Cruz | sc-13119; RRID: AB_675659 |
| CGRP | Sigma | C8198-.2ML; RRID: AB_259091 |
| MAX | Proteintech | 10426-1-AP; RRID: AB_2141660 |
| MNT | Bethyl Labs | A303-627A; RRID: AB_11205638 |
| MYC | Cell Signaling | 13987; RRID: AB_2631168 |
| MYC | Cell Signaling | 5605; RRID: AB_1903938 |
| RNApol II | Active Motif | 39097; RRID: AB_2732926 |
| RNA pol II phospho Ser5 | Cell Signaling | 13523; RRID: AB_2799721 |
| MGA | Suske Lab | Stielow et al., 2018 |
| TP53 | Cell Signaling | 2524; RRID: AB_331743 |
| RB1 | Abcam | [EPR17512]; ab181616 |
| GFP | Cell Signaling | 2956S; RRID: AB_1196615 |
| MYCL1 | R&D SYSTEMS | AF4050; RRID: AB_2282440 |
| Bacterial and Virus Strains | ||
| Ad5-CMV-Cre | University of Iowa Gene Vector Core | VVC-U of Iowa-5;RRID: SCR_015417 |
| Ad5-CGRP-Cre | University of Iowa Gene Vector Core | VVC-Berns-1160 ;RRID: SCR_015417 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NBF (10 %) | ThermoFisherScientific | 5701TS |
| Trypsin/EDTA (0.05 %) | ThermoFisherScientific | 25300054 |
| DMEM | ThermoFisherScientific | 11965-092 |
| Puromycin | ThermoFisherScientific | A1113803 |
| Proteinase K | Qiagen | 19131 |
| RNase A | Qiagen | 19101 |
| Ammonium acetate/dH2O | Sigma | A1542 |
| TE buffer | Sigma | T9285 |
| RPMI 1640 | ThermoFisherScientific | 11875-093 |
| Penicillin/ streptomycin | ThermoFisherScientific | 15140122 |
| Fetal bovine serum | Omega Scientific | FB-01 |
| Methotrexate | Cayman chemical | 13960 |
| Doxycycline | Sigma | 44577 |
| Lipofectamine 2000 | ThermoFisherScientific | 11668019 |
| Hexadimethrine bromide | Sigma | H9268 |
| Crystal violet | Sigma | 3886 |
| DAPI | ThermoFisherScientific | P36935 |
| Paraformaldehyde | Fisher | AA433689M |
| Blasticidin | Invivogen | ant-bl-05 |
| Phusion® High-Fidelity PCR Kit | New England Biolabs | E0553 |
| Trizol | ThermoFisherScientific | 15596018 |
| Protease and phosphatase inhibitors | ThermoFisherScientific | 78441 |
| 4–20 % Mini-PROTEAN®TGX™ Precast Protein Gels | BioRad | 4561096 |
| Amersham Protran 0.45 NC nitrocellulose membranes | GE healthcare life science | 10600002 |
| Glucose, Glycine and Serine free RPMI | TEKNOVA | R9660-02 |
| Dialyzed serum | ThermoFisherScientific | 26400044 |
| Glycine | Sigma | G5417-100G |
| L-Serine | Sigma | S4311-25G |
| D-Glucose (U-13C6, 99 %) | Cambridge Isotope Laboratories | CLM-1396-PK |
| D-(+)-Glucose | Sigma | G7021-100G |
| Methanol HPLC grade | Sigma | 34860 |
| RPMI 1640 2X | Wisent Bioproducts | 350-200-CL |
| SeaPlaque™ Agarose | ThermoFisherScientific | 50101 |
| Critical Commercial Assays | ||
| Nanodrop 2000 | ThermoFisherScientific | N/A |
| PureLink quick gel extraction | Invitrogen | K210012 |
| Kapabiosystems Library Quantification Kit | Roche | KK4824 |
| HiSeq 2500 | Illumina | N/A |
| Phusion Flash High Fidelity Master Mix | ThermoFisherScientific | F548L |
| AgileGrinder™ Tissue Grinder | Thomas Scientific-ACTGene | ACT-AG3080 |
| Ultra RNA Library Prep Kit for Illumina | New England BioLabs | E7530L |
| NEB Ultra II kit | New England BioLabs | E7645S |
| Pierce™ BCA Protein Assay Kit | ThermoFisherScientific | 23227 |
| CellTiter-Glo assay | Promega | G7573 |
| Z2 Series Coulter Counter | Beckman Coulter | 6605700 |
| iScript reverse transcription supermix for RT-qPCR | Bio-rad | 1708840 |
| all-in-One qPCR mix | GeneCopoeia | AOPR-4000 |
| CFX384 Touch Real-Time PCR Detection System | Bio-rad | N/A |
| Deposited Data | ||
| RNA seq | This paper | Accession Number GEO: GSE138955 |
| CHIP-seq | This paper | Accession Number GEO: GSE146385 |
| CUT&RUN | This paper | Accession Number GEO: GSE146388 |
| Experimental Models: Cell Lines | ||
| Human: 293TN | System Bioscience | LV900A-1 |
| Mouse: preSCs | Kim et al., 2016 | N/A |
| Mouse: mRPMaxKO | This paper | N/A |
| Mouse: Trp53flox/flox MEFs | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Rb1lox/lox GEMM | Sage et al., 2003 | N/A |
| Trp53lox/lox GEMM | Meuwissen et al., 2003 | N/A |
| frt-invCAGMyc1 1 -Luc GEMM | Huijbers et al., 2014 | N/A |
| Maxlox/lox GEMM | Mathsyaraja et al., 2019 | N/A |
| Oligonucleotides | ||
| See Table S3 | N/A | N/A |
| Recombinant DNA | ||
| V2 GeCKO mouse library | Sanjana et al., 2014 | Addgene; Cat# 1000000052 |
| psPAX2 | Gift from Didier Trono | Addgene; Cat# 12260 |
| pMD2.G | Gift from Didier Trono | Addgene; Cat# 12259 |
| pLX304 | Yang et al., 2011 | Addgene; Cat# 25890 |
| pLX304-MYC | This paper | N/A |
| pLX304-MYCN | This paper | N/A |
| pLX304-MYCL | This paper | N/A |
| pCW 5 7 -RFP-P2A-MC S | Barger et al., 2019 | Addgene; Cat# 78933 |
| pCW57-MAX | This paper | N/A |
| lentiCRISPR v2 | Sanjana et al., 2014 | Addgene; Cat# 52961 |
| lentiCRISPR v2-sgCtrl# 1 | This paper | N/A |
| lentiCRISPR v2-sgCtrl#2 | This paper | N/A |
| lentiCRISPR v2-sgMax# 1 | This paper | N/A |
| lentiCRISPR v2-sgMax#2 | This paper | N/A |
| lentiCRISPR v2-sgBcl2l11#1 | This paper | N/A |
| lentiCRISPR v2-sgBcl2l11#2 | This paper | N/A |
| lentiCRISPR v2-sgTsc 1 # 1 | This paper | N/A |
| lentiCRISPR v2-sgTsc1#2 | This paper | N/A |
| lentiCRISPR v2-sgPten# 1 | This paper | N/A |
| lentiCRISPR v2-sgPten#2 | This paper | N/A |
| lentiCRISPR v2-sgMap3k12#1 | This paper | N/A |
| lentiCRISPR v2-sg Map3k12#2 | This paper | N/A |
| lentiCRISPR v2-sgJun#1 | This paper | N/A |
| lentiCRISPR v2-sgJun#2 | This paper | N/A |
| lentiCRISPR v2-sgTsc2# 1 | This paper | N/A |
| lentiCRISPR v2-sgTsc2#2 | This paper | N/A |
| lentiCRISPR v2-sgNf1#1 | This paper | N/A |
| lentiCRISPR v2-sgNf1#2 | This paper | N/A |
| lentiCRISPR v2-sgBax# 1 | This paper | N/A |
| lentiCRISPR v2-sgBax#2 | This paper | N/A |
| lentiCRISPR v2-sgRb1#1 | This paper | N/A |
| lentiCRISPR v2-sgRb1#2 | This paper | N/A |
| lentiCRISPR v2-sgRb12 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software Inc. | http://www.graphpad.com |
| ImageStudio Light | ImageStudio Software | https://www.licor.com/bio/image-studio-lite/ |
| Fiji NIH | ImageJ | https://imagej.nih.gov/ij/docs/guide/146-2.html |
| FlowJo | FlowJo Software | https://www.flowjo.com/ |
| BioRender | BioRender Software | https://biorender.com/ |
| Tracefinder4.1 | Tracefinder Software | N/A |
| ClustVis | Metsalu et al.; 2016 | https://biit.cs.ut.ee/clustvis/ |
| MAGeCK-VISPR | MAGeCK-VISPR Software |
http://bitbucket.org/liulab/mageck-vispr |
| TopHat v2.0.12 | Trapnell et al., 2009 | N/A |
| Cuffdiff v2.1.1 | Trapnell et al., 2013 | N/A |
| Python package HTSeq v0.6.1 | Anders et al., 2015 | N/A |
| Bioconductor package edgeR, v3.16 | Robinson et al., 2010 | N/A |
| STAR v2.5.2a | Dobin et al., 2013 | N/A |
| Subread package v1.6.0 | Liao et al., 2014 | N/A |
Highlights.
CRISPR-Cas9 screens identify candidate tumor suppressor genes and pathways in SCLC
MAX is a context-dependent tumor suppressor in early stage SCLC
MAX is required for MYC-driven SCLC and MAX tumor suppression is independent of MYC
MAX represses metabolic genes including regulators of one carbon metabolism
Highlights.
Augert et al. identify MAX as a context-dependent tumor suppressor in small cell lung cancer (SCLC) that can regulate serine and one carbon metabolism. MAX loss accelerates SCLC progression in a Rb1/Trp53-deficient mouse model, while abrogating tumorigenesis in MYCL-overexpressing SCLC.
Significance.
MAX is an obligate heterodimerization partner for MYC oncoproteins. Genomic binding and transformation activities driven by dysregulated MYC in many cancers are known to require MAX. SCLC, a neuroendocrine tumor type, frequently exhibits amplification of MYC family genes. Paradoxically however, a subset of human SCLC exhibits deletions or inactivating mutations in MAX, and germline inactivating mutations in MAX predisposes to other neuroendocrine cancers. Here we have developed both cell-based and murine models demonstrating accelerated SCLC progression upon inactivation of Max. We show that widespread gene expression changes and metabolic alterations in cells transformed by Max loss are independent of MYC and that MYC overexpression is unable to transform Max-null cells. Therefore, MAX inactivation constitutes a MYC-independent pathway to SCLC.
Acknowledgements
We thank members of the MacPherson, Eisenman, Sullivan and Paddison labs for sharing reagents and providing advice. We would especially like to thank Andrew Snyder, Emily Eastwood, Justin Norton and Eli Grunblatt for their technical help. We thank the Fred Hutch Genomics and Bioinformatics Shared Resource for help regarding the generation and analysis of next generation sequencing data. We also acknowledge support from the Fred Hutch Histopathology and small animal imaging Shared Resources. We are grateful to David Hockenbery for critical reading of the manuscript. These studies were supported in part by NCI R00CA218679 (to LS), NIH/NCI R35CA231989 (to RNE), U01CA235625 and R01CA200547 (to DM) and by NIH/NCI Cancer Center Support Grant P30 CA015704.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
R.N.E is a member of the Scientific Advisory Boards of Kronos Bio Inc. and Shenogen Beijing.
REFERENCES
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, and Land H (1993). Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 72, 233–245. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augert A, Zhang Q, Bates B, Cui M, Wang X, Wildey G, Dowlati A, and MacPherson D (2017). Small Cell Lung Cancer Exhibits Frequent Inactivating Mutations in the Histone Methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J Thorac Oncol 12, 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer DE, Lawrence QA, and Eisenman RN (1995). Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 80, 767–776. [DOI] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, and Sabatini DM (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger CJ, Branick C, Chee L, and Karpf AR (2019). Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu ME, Jauset T, Masso-Valles D, Martinez-Martin S, Rahl P, Maltais L, Zacarias-Fluck MF, Casacuberta-Serra S, Serrano Del Pozo E, Fiore C, et al. (2019). Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, and Vogt PK (2002). Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci U S A 99, 3830–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, and Eisenman RN (1991). Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, and Kaelin WG Jr. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18, 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnichon N, Cascon A, Schiavi F, Morales NP, Comino-Mendez I, Abermil N, Inglada-Perez L, de Cubas AA, Amar L, Barontini M, et al. (2012). MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 18, 2828–2837. [DOI] [PubMed] [Google Scholar]
- Carroll PA, Freie BW, Mathsyaraja H, and Eisenman RN (2018). The MYC transcription factor network: balancing metabolism, proliferation and oncogenesis. Front Med 12, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ , Weissleder R, et al. (2015). Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, Landa I, Leandro-Garcia LJ, Leton R , Honrado E, Ramos-Medina R, Caronia D, Pita G, et al. (2011). Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43, 663–667. [DOI] [PubMed] [Google Scholar]
- Consortium APG (2017). AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Augert A, Rongione M, Conkrite K, Parazzoli S, Nikitin AY, Ingolia N, and MacPherson D (2014). PTEN is a potent suppressor of small cell lung cancer. Mol Cancer Res 12, 654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AF, Castermans E, Oudijk L, Guitelman MA, Beckers P, Potorac I, Neggers S, Sacre N, van der Lely AJ, Bours V, et al. (2018). Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr Relat Cancer 25, L37–L42. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diolaiti D, McFerrin L, Carroll PA, and Eisenman RN (2015). Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim Biophys Acta 1849, 484–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M, Endo TA, Shinga J, Hayashi K, Farcas A, Ma KW, Ito S, Sharif J, Endoh T, Onaga N, et al. (2017). PCGF6-PRC1 suppresses premature differentiation of mouse embryonic stem cells by regulating germ cell-related genes. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber S, and Diamond LK (1948). Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 238, 787–793. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, and Reinberg D (2012). PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45, 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopewell R, and Ziff EB (1995). The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol Cell Biol 15, 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon MC, Proost N, Song JY, de Vries H, Badhai J, Sutherland K, et al. (2014). Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO Mol Med 6, 212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, Queva C, and Eisenman RN (1997). Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev 11, 44–58. [DOI] [PubMed] [Google Scholar]
- Hurlin PJ, Steingrimsson E, Copeland NG, Jenkins NA, and Eisenman RN (1999). Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J 18, 7019–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, Zhou ZQ, Toyo-oka K, Ota S, Walker WL, Hirotsune S, and Wynshaw-Boris A (2003). Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J 22, 4584–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens DH, Wu SJ, Sarthy JF, Meers MP, Myers CH, Olson JM, Ahmad K, and Henikoff S (2018). Automated in situ chromatin profiling efficiently resolves cell types and gene regulatory programs. Epigenetics Chromatin 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, Kim KB, Dunn CT, Pillai SPS, Gazdar AF, et al. (2018). Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov 8, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, Dunn CT, Lee AY, Kim K, Lee CS, et al. (2016). Genetic requirement for Myc1 and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev 30, 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, and Maddocks OD (2014). Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 7, 1248–1258. [DOI] [PubMed] [Google Scholar]
- Li W, Koster J, Xu H, Chen CH, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30(7):923–30. [DOI] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- Mathsyaraja H, Freie B, Cheng PF, Babaeva E, Catchpole JT, Janssens D, Henikoff S, and Eisenman RN (2019). Max deletion destabilizes MYC protein and abrogates Emicro-Myc lymphomagenesis. Genes Dev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, et al. (2014). Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 156, 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, and Berns A (2003). Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189. [DOI] [PubMed] [Google Scholar]
- Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 43, W566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al. (2017). MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 31, 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, and Nakatani Y (2002). A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296, 1132–1136. [DOI] [PubMed] [Google Scholar]
- Panchaud N, Peli-Gulli MP, and De Virgilio C (2013). Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal 6, ra42. [DOI] [PubMed] [Google Scholar]
- Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, and Budanov AV (2014). Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep 9, 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. (2012). Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 44, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Olivares M, Trento A, Rodriguez-Acebes S, Gonzalez-Acosta D, Fernandez-Antoran D, Roman-Garcia S, Martinez D, Lopez-Briones T, Torroja C, Carrasco YR, et al. (2018). Functional interplay between c-Myc and Max in B lymphocyte differentiation. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietanza MC, Byers LA, Minna JD, and Rudin CM (2015). Small cell lung cancer: will recent progress lead to improved outcomes? Clin Cancer Res 21, 2244–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, Bhangoo MS, Thorson PK, Richards A, Rooslid T, et al. (2019). Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 35, 385–400 e389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, and Benjamini Y (2003). Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero OA, Torres-Diz M, Pros E, Savola S, Gomez A, Moran S, Saez C, Iwakawa R, Villanueva A, Montuenga LM, et al. (2014). MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov 4, 292–303. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. (2012). Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. (2018). Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 173, 499–514 e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, and Jacks T (2003). Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424, 223–228. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer IM, Wang Y, Liang CW, Bahri N, Quattrone A, Doyle L, Marino-Enriquez A, Lauria A, Zhu M, Debiec-Rychter M, et al. (2017). MAX inactivation is an early event in GIST development that regulates p16 and cell proliferation. Nat Commun 8, 14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, Zhao W, Zhang X, Ventura A, Liu Y, et al. (2018). Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst 6, 282–300 e282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Li H, O’Hagan RC, Hou H Jr., Horner JW 2nd, Lee HW, and DePinho RA (2000). Essential role for Max in early embryonic growth and development. Genes Dev 14, 17–22. [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Kokubu A, Tsuta K, and Hirohashi S (2009). Oncogenic mutation of PIK3CA in small cell lung carcinoma: a potential therapeutic target pathway for chemotherapy-resistant lung cancer. Cancer Lett 283, 203–211. [DOI] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2015). A simple method for generating high-resolution maps of genome-wide protein binding. Elife 4, e09225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Dietlein F, Peifer M, Schottle J, Balke-Want H, Muller C, Koker M, Richters A, Heynck S, Malchers F, et al. (2012). A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A 109, 17034–17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger D, Furrer M, Schwinkendorf D, and Gallant P (2008). Max-independent functions of Myc in Drosophila melanogaster. Nat Genet 40, 1084–1091. [DOI] [PubMed] [Google Scholar]
- Stielow B, Finkernagel F, Stiewe T, Nist A, and Suske G (2018). MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet 14, e1007193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, Ramirez MA, Liang T, Caballero F, Wildschut MHE, et al. (2019). Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem Biol 26, 711–723 e714. [DOI] [PubMed] [Google Scholar]
- Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, Meeth K, Muir A, Darnell AM, Bosenberg MW, et al. (2019). Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab 29, 1410–1421 e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, and Berns A (2011). Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Hirasaki M, Hishida T, Wu J, Okamura D, Ueda A, Nishimoto M, Nakachi Y, Mizuno Y, Okazaki Y, et al. (2016). Loss of MAX results in meiotic entry in mouse embryonic and germline stem cells. Nat Commun 7, 11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wert M, Kennedy S, Palfrey HC, and Hay N (2001). Myc drives apoptosis in PC12 cells in the absence of Max. Oncogene 20, 3746–3750. [DOI] [PubMed] [Google Scholar]
- Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y, Granja JM, Jahchan NS, Lim JS, Kwok S, et al. (2018). Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov 8, 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C, Knudson CM, Korsmeyer SJ, and Van Dyke T (1997). Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 385, 637–640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3, related to STAR Methods: Oligonucleotides used in the study.
Data Availability Statement
The accession number for the raw and processed data of RNA sequencing, CHIP sequencing and CUT&RUN generated and reported in this paper is GEO: GSE138955, GSE146385 and GSE146388.