

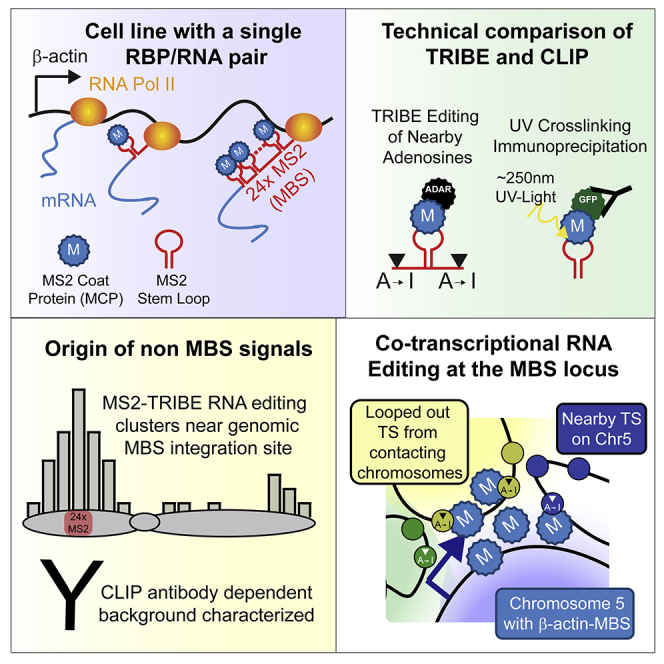
Summary
Both UV-cross-linking and immunoprecipitation (CLIP) and RNA editing (TRIBE) can identify the targets of RNA-binding proteins. To evaluate false-positives of CLIP and TRIBE, endogenous β-actin mRNA was tagged with MS2 stem loops, making it the only bona fide target mRNA for the MS2 capsid protein (MCP). CLIP and TRIBE detected β-actin, albeit with false-positives. False-positive CLIP signals were attributed to nonspecific antibody interactions. In contrast, putative false-positive TRIBE targets were genes spatially proximal to the β-actin gene. MCP-ADAR edited nearby nascent transcripts consistent with interchromosomal contacts observed in Hi-C. The identification of nascent contacts implies RNA regulatory proteins (e.g., splicing factors) associated with multiple nascent transcripts, forming domains of post-transcriptional activity. Repeating these results with an integrated inducible MS2 reporter indicated that MS2-TRIBE can be applied to a broad array of cells and transcripts to study spatial organization and nuclear RNA regulation.
Subject Areas: Molecular Biology Experimental Approach, Transcriptomics
Graphical Abstract
Highlights
-
•
Using the β-actin-MBS system, we identify the false-positives of TRIBE and CLIP
-
•
Mammalian TRIBE has fewer putative false-positives than CLIP
-
•
The non-MS2 targets for TRIBE are neighboring nascent transcripts
-
•
We identify a nuclear domain where nascent RNAs and physically interact
Molecular Biology Experimental Approach; Transcriptomics
Introduction
RNAs are regulated throughout all aspects of their lives (Vera et al., 2016). Their regulation by RNA-binding proteins (RBPs) provides spatiotemporal control over processing, export, localization, translation, and decay. RBP genes make up approximately 10% of all protein coding genes in the human genome (Gerstberger et al., 2014), and RBP families have coevolved with their RNA targets to provide control over functionally related transcripts (Hogan et al., 2015). Many RBPs contain more than one RNA-binding domain, with each domain contributing to the final specificity of the protein (Gerstberger et al., 2014). Accurately defining the targets of a given RBP has been a longstanding challenge in biology due to their complex and multivalent interactions.
In vitro approaches have determined the sequence specificity of single domains within RBPs. Approaches such as SELEX and “RNA bind-n-seq” (Lambert et al., 2014) have determined the consensus sequence preference for RBPs using short RNA fragments (<40 nucleotide [nt]). Complementary approaches have found consensus binding sites in vivo, using cross-linking and immunoprecipitation (CLIP).
Cell lysis before immunoprecipitation promotes adventitious interactions such as those between cytoplasmic RBPs and nuclear RNAs (Mili and Steitz, 2004). To overcome these limitations, CLIP sequencing and its derivatives use UV to irreversibly cross-link protein and RNA within cells (Ule et al., 2003). These interactions can be defined with single nucleotide resolution (Zhang and Darnell, 2011). However, CLIP relies on antibodies and suffers from high background due to nonspecific antibody-antigen interactions (Friedersdorf and Keene, 2014). Therefore, an orthogonal approach is important to obtain high-confidence targets for RBPs of interest. It should be highly specific, antibody independent, retain information about the total RNA present for a given species, and be limited in the amount of sample and its handling steps.
Two approaches have recently been developed, RNA editing in Drosophila (“TRIBE”; McMahon et al., 2016; Xu et al., 2018) and RNA tagging in yeast (Lapointe et al., 2015). Both approaches utilize enzymes fused to an RBP of interest to deposit marks on their RNA targets. We chose to focus on RNA editing because of its straightforward approach utilizing standard RNA sequencing (RNA-seq) library preparation.
Conspicuously missing from previous transcriptome-wide studies of RBPs is a positive control, which could resolve rigorously the extent of false-positives: the best-case scenario would be an RBP that recognizes only a single transcript within the cell. Thus, any targets identified outside of the single transcript would be defined as false-positives. The specificity of the MCP-MBS system provides such a tool: its capsid protein (MCP) should only bind the MS2 stem loops (MBS) inserted into a gene of interest. We used cells derived from a mouse that has the MBS inserted into the 3′ UTR of the endogenous β-actin gene and used the MCP as a means to evaluate various approaches to identifying targets of RBPs (Lionnet et al., 2011).
To validate this approach, both CLIP and TRIBE were performed on cells containing the MCP-MBS system. The single β-actin mRNA labeled with the MCP-MBS system allowed for unambiguous determination of background or false-positives for each technique. Nonspecific antibody interactions contributed significantly to MCP-CLIP signals, whereas there were fewer non-β-actin target mRNAs with MCP-TRIBE (Tables S1 and S2). Moreover, these reproducible, non-random targets correlated with their proximity to the β-actin locus on the chromosome rather than manifesting some association with β-actin mRNA. This result led to the unexpected conclusion that MCP-ADAR had edited nearby transcribing RNAs. The tethered editing enzyme can therefore define a nuclear transcriptional domain with implications for how the transcriptome may be organized in the nucleus.
Results
MCP-ADAR Edits Targets Consistent with Binding the β-Actin-MBS
We evaluated both CLIP and TRIBE with an RBP that should recognize a single target within the transcriptome. We used mouse fibroblasts containing 24xMBS integrated into the endogenous β-actin locus (Figure 1A, Lionnet et al., 2011). mRNA was imaged with single-molecule fluorescence in situ hybridization (smFISH) (Figure S1A), and live cells were imaged with MCP-GFP (Figure S1B). Both approaches demonstrated a sufficient signal-to-noise ratio for single molecule detection. The sub-nanomolar Kd of MCP for the β-actin-MBS in vitro (Figures S1C and S1D) was consistent with the high specificity of MCP for its stem-loop target in vivo.
Figure 1.

Transcriptome-wide Studies Show CLIP Peaks and TRIBE Editing at β-Actin-MBS
(A) Schematic of MS2 stem loops that have been integrated into the endogenous β-actin locus being actively transcribed by RNA polymerase II. (Right arrows, top) Schematic of MCP-ADAR binding to MS2 stem loops and editing adjacent nucleotides from adenosine to inosine (which is then recognized as guanosine upon cDNA conversion). After background subtraction, editing sites that are conserved across both replicates are used for further analysis. (Right arrows, bottom) Schematic of MCP-GFP binding to MS2 stem loops. UV cross-linking and immunoprecipitation (CLIP) isolates RNA fragments (red horizontal bars), which are then computationally analyzed and fit to peaks (red peaks).
(B) β-Actin gene, focusing on the MBS array showing (from top to bottom) the following. MS2 genome-wide sequence search: bioinformatic search with blue horizontal bars depicting discovered sites containing the MS2 consensus sequences. CLIP alignment: CLIP sequencing reads that remain after mapping to the genome are depicted in gray (scale bar to right). MS2 sites at genomic locus (ground truth): blue horizontal bars represent the known location of the MS2 stem loops. MCP-TRIBE alignment with multimapping: reads are depicted in gray (scale bar for number of reads on right); editing sites are depicted as red bars. Uniquely mapped read alignment: mRNA coverage without multimapping depicted in blue (scale bar for number of reads on right). MCP-TRIBE sites' both replicates uniquely mapped: editing events as indicated by dark blue bars where height corresponds to the average editing percentage across both replicates at that nucleotide (scale to right). Light blue shading indicates location of the stem-loop nucleotides.
To determine if the tight binding of MCP to the β-actin-MBS array would lead to RNA editing of the β-actin transcript, MCP fused to ADAR was introduced into cells containing the β-actin-MBS mRNA. We also performed CLIP on cells stably expressing MCP-GFP and β-actin-MBS. In addition, the MCP consensus binding sequence was utilized to perform an in silico search for MS2 binding sites across the transcriptome (Tutucci et al., 2018). All three approaches were successful in predicting MCP binding to the target sites (Figures 1B, S2A, and S2B).
Reproducible MCP-CLIP and MCP-TRIBE Define MCP Binding to β-Actin-MBS
In both approaches, the β-actin-MBS array was the top ranked site (by total peak height in CLIP or by total number of editing sites in TRIBE, [Tables S1 and S2]). For example, it had a TRIBE signal 4× greater than the next most highly edited transcript in the transcriptome (Table S1). This robust binding was expected because the β-actin-MBS array has the highest density of MCP binding motifs of any transcript in the transcriptome.
There were also 139 other transcripts identified with TRIBE and 105 with CLIP. One possibility is that they contained stem-loop structure resembling MS2 and were due to direct binding of MCP. To address this possibility, transcripts were examined for the presence of the MCP consensus sequence and the RNAs detected by MCP-TRIBE were compared with those cross-linked by MCP-CLIP.
Only two of the 139 MCP-TRIBE transcripts showed an MCP consensus sequence, and only three transcripts were shared between MCP-CLIP and MCP-TRIBE in addition to β-actin (Figure 2A, Tables S1, S2, S3). The three transcripts shared between MCP-CLIP and TRIBE showed no resemblance to either the sequence or structure of MBS (Table S3), suggesting that they were coincidental. Therefore, β-actin is the only transcript identified using all approaches (TRIBE, CLIP and consensus sequence).
Figure 2.

Comparison of MCP-CLIP and MCP-TRIBE Targets
(A) Venn diagram showing significant peaks discovered in MCP-CLIP (blue oval) and MCP-TRIBE (red oval) recognize different target pools. Four RNAs overlap between both techniques (intersection with line to names of RNAs), with only one RNA containing evidence of the MS2 consensus sequence (β-actin-MBS).
(B) Venn diagram showing intersection of RNAs found by MCP-CLIP (blue oval) and CLIP without MCP (background CLIP, gray oval). 41/106 significant CLIP peaks are reproduced in the cells that do not express MCP, representing antibody background.
(C) Venn diagram showing intersection of RNAs found with MCP-MBS-TRIBE (red circle) and MCP without MBS TRIBE (background TRIBE, blue circle).
(D) Dot plot of data from (C). Individual genes from MCP-TRIBE in cells with β-actin-MBS plotted on x axis (red circle), MCP-TRIBE in cells without β-actin-MBS plotted on y axis (blue circle). The RNAs found in both experiments are plotted on the diagonal (purple circle).
Off-Target Characterization for CLIP and TRIBE
To characterize these targets further, we performed the CLIP and TRIBE without MCP or without the stem loops. CLIP of cells without MCP-GFP generated nearly 200 significant peaks, reproducing 40% of the peaks present in cells with MCP-GFP (Figure 2B). These peaks most likely reflect nonspecific antibody interactions. In support of this interpretation, there was a 5-fold increase in transcript targets when polyclonal antibodies rather than monoclonal antibodies were used (Biswas et al., unpublished data).
In contrast, MCP-TRIBE in cells without the β-actin-MBS reproduced only 13% of the MBS peaks (Figure 2C), and there were no transcripts with editing levels comparable to the 27 β-actin-MBS sites, e.g., more than 4 editing sites (Figure 2D). Interestingly TRIBE showed far fewer off-target peaks compared with CLIP; only 28 transcripts in the entire transcriptome had at least two editing sites.
As the MCP β-actin-MBS TRIBE target transcripts were highly reproducible, did not have an MCP consensus sequence, were not found in MCP-CLIP, and were dependent on the presence of the MBS array (they did not appear in MCP-ADAR-expressing cells without MS2 stem loops), we considered explanations for these particular off-target transcripts.
Origin of MCP-TRIBE Off-Target Signals Can Be Correlated with the Genetic Locus
One possibility for these reproducible transcripts detected by TRIBE was that they could be spatially proximal to the β-actin-MBS-bound ADAR. For instance, if the mRNAs were packaged with β-actin mRNAs in a granule, the mRNAs might come close enough together to facilitate trans-editing. If this were the case, we would expect the transcripts to share a common feature, such as a consensus sequence similar to the β-actin mRNA zipcode (Patel et al., 2012). However, interrogation of the targets did not find any common consensus sequence. In addition, gene ontology analysis failed to find transcripts that code for proteins related to β-actin's role in the cytoplasm (e.g., cell motility). We then considered the possibility that the targets we identified could be close enough together to undergo trans-editing in the nucleus, perhaps in some nuclear domain. To this end, the off-target transcripts edited by MCP-TRIBE were interrogated to determine if β-actin-MBS mRNA could be within spatial proximity within the nucleus (Figure 3A).
Figure 3.

Increased Presence of Editing Sites on Chromosome Five and Increased Density of Editing around the β-Actin Locus
(A) Schematic of cell nucleus (large circle) containing individual chromosomes within their territories (smaller black circles with chromosome five highlighted in blue). Enlarged view of chromosome five territory showing a snapshot of DNA (blue line), as well as region of flexible movement (blue circle). Enlarged view of β-actin gene locus, showing transcription of β-actin mRNA and loading of MCP-ADAR near the site of transcription and chromatin contacts. Schematic showing increased loading of MCP-ADAR onto actively transcribing RNA.
(B) Bar graph showing enrichment (red) versus expected (blue) number of sites for chromosome five across experiments. Two-tailed chi-square test was used to determine significance for each group.
(C) Bar graph showing number of editing sites (y axis) along chromosome five (x axis, 10-Mb bins). Editing at the bin containing the β-actin-MBS is represented as a hashed box.
(D) Bar graph showing number of RNAs edited organized by chromosome of origin. Chromosomes are colored by Hi-C contacts with chromosome five (data from Battulin et al., 2015). Chromosomes enriched for contacts are colored in red; chromosomes depleted in contacts are colored in blue.
(E) Number of intronic editing sites organized by chromosome of origin. Chromosomes are colored by Hi-C contacts with chromosome five (data from Battulin et al., 2015). Chromosomes enriched for contacts are colored in red; chromosomes depleted in contacts are colored in blue.
(F) Data in (D) represented as individual points; mean of the data summarized as a bar graph with error bars representing standard deviation. Chromosomes were grouped by their relationship to chromosome five in MEFs, either into contact enriched (red) or contact depleted (blue). Unpaired, two-tailed t test was used to determine significance.
(G) Data in (E) represented as individual points; mean of the data summarized as a bar graph with error bars representing standard deviation. Chromosomes were grouped by their relationship to chromosome five in MEFs, either into contact enriched (red) or contact depleted (blue). Unpaired, two-tailed t test was used to determine significance.
(H) Schematic of proposed model for chromosome contacts. Transcription sites (TS, angled arrows) often loop away from chromosome of origin and intermingle with other active loci. The β-actin-MBS (blue star) recruits MCP-ADAR at the time of transcription and marks nearby active loci.
Surprisingly, many of these transcripts that were edited were on chromosome five, the same chromosome as the β-actin-MBS. Chromosome five genes were edited significantly more often than expected in MCP-TRIBE data (Figure 3B), but not in TRIBE without MBS or MCP-CLIP. This suggested that most of the RNAs edited by β-actin-MBS TRIBE were originating not due to direct MCP binding to those RNAs but rather due to indirect editing by ADAR.
Spatial interactions would be expected to affect regions proximal to the β-actin locus more than distal parts of chromosome five. Indeed, higher numbers of chromosome editing sites were nearer to the β-actin-MBS array (Figure 3C). After addressing cis-contacts on chromosome five, transcripts from other chromosomes were queried to determine if trans-interactions with chromosome five could explain some of the other identified targets. Previously published Hi-C inter-chromosomal contacts from MEFs found chromosome five to have enriched contacts with chromosomes, one, two, four, five through nine, and eleven and depleted for contacts with chromosomes three, twelve through sixteene, eighteen, nineteen, and X (Battulin et al., 2015). The most enriched chromosome contacts in this dataset were two, seven, and eleven with chromosome three being notable as an example of a large chromosome with depleted contacts. Consistent with these Hi-C data, edited RNAs originated more frequently from chromosomes predicted to have more chromosome five contacts (Figures 3D, 3F, and S3A–S3C) and less frequently from chromosome three. The latter also indicated that chromosome size was not a major confounding factor.
We postulated that if editing of transcripts was occurring on nascent pre-mRNAs, including those transcribed from other chromosomes, they should all be enriched in intronic sequences. Indeed, chromosome five transcripts proximal to the MS2 insertion had more intronic editing sites as did transcripts on contacted chromosomes (Figures 3E and 3G). To validate our observations independently, we used published data provided by the intron seq-FISH approach (Shah et al., 2018). The genes that were identified as being in proximity to the β-actin-MBS locus also had their transcription sites in spatial proximity. Of the RNAs that were edited by MCP-TRIBE, 85 of 86 transcription sites interacted with chromosome five as determined by the reported seq-FISH results (within 500 nm). Strikingly, 56 of the 100 genes found to be transcribing RNA by intron seq-FISH closest to the β-actin locus on chromosome five were MCP-TRIBE targets (Figure S3D). This substantial correlation indicated that edited RNAs in the MCP-TRIBE experiment resulted from intra- and inter-chromosomal contacts with the transcripts from the β-actin-MBS locus.
Hence, editing could be a proximity labeling approach that can define a nuclear domain wherein transcripts and their processing factors spatially interact. We analyzed Hi-C data and determined that chromosomes likely to be near the β-actin-MBS locus also correlated with the chromosomes that we found to contain edited nascent transcripts (Figures 3H and S3A). Notably, as technical advantage, in contrast to chromosome conformation capture, where high sequencing depth is required to achieve megabase resolution, RNA editing is able to detect the likelihood of inter-chromosomal interactions with single gene resolution (Figures S3A, S5A–S5C, and S6).
Extending the Utility of MS2-TRIBE Using a Randomly Integrated, Inducible MBS Cassette
To explore the generality of this approach, we asked if similar results on chromatin contacts could be obtained within other cell lines containing an integrated MBS cassette. By using human cancer cells and a randomly integrated reporter cassette rather than an endogenous targeted gene, possible biases that were present due to the cell type or integration were mitigated. We previously published the use of a human osteosarcoma cell line (U2OS-2-6-3) containing an inducible MBS array integrated into chromosome one (specifically near the1p36 locus, Figure 4A) (Janicki et al., 2004).
Figure 4.

Increased Presence of Editing Sites on Chromosome One and Increased Density of Editing around the MBS Reporter Locus
(A) Schematic of the reporter locus on chromosome 1p36. Lac operator repeats (LacO) are present upstream of 96 repeats of the tet response element (TRE); when bound by reverse tet transactivator, the cytomegalovirus (CMV) promoter is inactive. Upon doxycycline induction the RNA containing the MBS array becomes actively transcribed (right arrow) at which point, MCP-ADAR (blue circles) can bind to the RNA at the site of transcription.
(B) Reporter MBS gene, focusing on the MBS array showing (from top to bottom) the following. MS2 sites at genomic locus (ground truth): blue horizontal bars represent the known location of the MS2 stem loops. MCP-TRIBE sites: editing events as indicated by dark blue bars where height corresponds to the average editing percentage across both replicates at that nucleotide (scale to right). Light blue shading indicates location of the stem-loop nucleotides.
(C) Bar graph showing number of editing sites (y axis) along chromosome one (x axis, 10-Mb bins) at 24 h post-doxycycline induction.
(D) Bar graph showing number of editing sites (y axis) along chromosome one (x axis, 10-Mb bins) at 48 h post-doxycycline induction.
(E) Bar graph showing number of genes edited (y axis) organized by chromosome of origin (x axis). Relative number of editing sites calculated by normalizing genes edited per chromosome to the total number of genes edited in the experiment. Chromosomes are colored by experiment, and colors correspond to (B). Gray bars representing cells with no doxycycline induction (low expression), blue bars represent 24 h of doxycycline induction, and red bars represent 48 h of doxycycline induction.
When transfected with the TRIBE constructs, RNA containing the MBS was edited in MCP-ADAR samples and not in the mCherry controls (Figure 4B). We found that genes proximal to the MBS transcription site on chromosome one were more highly edited, consistent with cis-chromosomal transcripts being identified by MS2-TRIBE (Figures 4C and 4D). When plotted along the length of chromosome one, the majority of edited transcripts occurred near the known 1p36 integration locus, validating that the signal originated from transcription of the MBS. The signal appeared to broaden along the chromosome at 48 h after induction, relative to 24 h, possibly reflecting increased RNA-RNA interactions over time.
When looking at editing sites across all chromosomes, the second highest abundance of RNA editing occurred from genes on chromosome 17 (Figure 4E). Two possibilities exist for the chromosome 17 edits. There could be a translocation between chromosome 1 and 17 leading to the presence of the MS2 array physically close to the chromosome 17 transcripts. Alternatively, this could reflect transient chromosome interactions that are preferred. Further experiments with karyotyping the cell line may clarify this.
To determine if MS2-TRIBE was dependent on high expression of the stem-loop arrays we used several conditions to induce their expression. Cells transfected with reverse tetraycline transactivator (rtTA) but without doxycycline repression showed 5–6× higher levels of RNA expression than untransfected cells, similar to previously observed leaky expression of tet-inducible systems (Raj et al., 2006). Cells could then be strongly induced 10× further using 1 μM doxycycline (Figures S5A and S5B). Together, these experiments allowed us to test the dynamic range of MS2-TRIBE and determine that it provided similar results for both modestly and highly expressed transcripts.
TRIBE editing results from continuous exposure of ADAR to the target of interest. Therefore TRIBE modification of transcripts is different from methods that rely on fixation to capture a snapshot of mRNA associations. To determine whether MS2-TRIBE signal changed over time, mRNA expression was induced with doxycycline for either 24 or 48 h before RNA-seq library preparation. We found no significant difference in overall editing between 24 and 48 h of induction, suggesting that both conditions had reached steady state (Figures S5A and S5B) and that differential expression of the reporter mRNA was not be a confounding factor.
We investigated inter-chromosomal contacts over time. There was a modest 12% increase in the total number of genes edited at the 48 h time point; chromosomes one and seventeen increased the number of genes edited by 33% and 27% respectively. This suggested that integration of chromosome contacts over time provided higher editing signal of TRIBE (Figures 4E and S5C, Table S4).
Discussion
By adapting TRIBE to mammalian cells and converting it to a single plasmid system, a number of advantages were evident. Mammalian TRIBE is an antibody independent approach that can be performed on 1000× fewer cells (samples were generated from fewer than 2,000 cells) than CLIP (2–20 million cells required) or chromosome conformation capture-based approaches (requiring >10 million cells). The sequencing depth required for TRIBE is similar to either CLIP or chromatin conformation capture variants, but by relying on a minimum number of processing steps for RNA-seq library preparation, TRIBE leads to more efficient sequencing (Figures S4A and S4B). By retaining the sequence complexity of the cellular transcriptome, fewer PCR cycles are required for library amplification, thus leading to increased sequencing fidelity. Significantly more sequenced TRIBE reads are usable than the most efficient CLIP approaches (Figures S4A and S4B).
During the process of gel excision, CLIP tags are limited in size due to the additional mass of the protein-RNA complex, 100 nt of RNA add approximately or 32 kDa mass to the protein-RNA smear. Post processing, most CLIP tags are ∼50 nt; in contrast, TRIBE can utilize longer RNA-seq reads (150 nt here), which increases its ability to uniquely map reads (Figure S4A). This allows TRIBE to discover the entire repertoire of RNA targets, as evidenced by saturated target discovery and RNA editing (Figures S4B and S4C). In addition, the molecular biology required for TRIBE is radiation free and limited to cell sorting, RNA isolation, and standard RNA library preparation, processes that are routine in many laboratories and can be completed in as few as 3 days.
Definition of Background
Until recently, limited alternative approaches to CLIP existed. One prior approach was pertinent to the study of splicing factors. In this context, CLIP was assisted by correlating binding sites with sites of alternative splicing, as determined by RNA-seq. Ultimately, multiple transcriptome-wide approaches should be integrated to evaluate the targets of RBPs (Herzog et al., 2020; Lapointe et al., 2018; Rahman et al., 2018).
Prior work comparing CLIP with RNA tagging in yeast found ∼50% overlap between the two approaches (Lapointe et al., 2015, 2018). Similar percentages were found when comparing CLIP and TRIBE in Drosophila (McMahon et al., 2016). However, most comparisons have been performed on RBPs that bind to a significant percentage of cell transcripts (often >50%), thus confounding the value of percentage overlap. The large number of bound targets also makes it challenging to identify the origin of these differences. By using the MCP-MBS system we were able to perform CLIP and TRIBE with only one true target within the cell. The small number of discovered targets in each technique (<200 of 15,000 transcripts) avoided spurious overlap and readily identified overlap as well as false-positives.
Prior work showed that UV cross-linking occurred at the site of RBP binding to RNA. We investigated the origin of off-target CLIP signal using multiple methods. We were unable to find the same RNAs when assayed by TRIBE (Figure 2A), and off-target RNAs discovered by CLIP did not contain matches to the well-defined MS2 consensus sequence (Table S3). Finally, for CLIP, the use of control cells not expressing MCP reveals that nonspecific RNA binding to antibodies contributes to CLIP signal. A similar issue has been described in chromatin immunoprecipitation sequencing data from both Drosophila and yeast, leading to the presence of “phantom peaks” that are present even when the antigen is not (Jain et al., 2015; Teytelman et al., 2013).
There are a number of advantages to CLIP such as the nucleotide-level identification of RBP binding sites and its application to endogenous targets and tissues. CLIP has clear merits that support its widespread adaptation and use by the scientific community. However, it is necessary to consider antibody-dependent background issues that can dominate the CLIP data of RBPs. These findings provide a comparison of relative strengths and limitations of the two techniques using a definitive positive control experiment. Defining a single positive control, and the insights gained from this, provides a standard test for any methodology for characterizing their signal and noise components.
Considerations and Limitations when Using MS2-TRIBE
The small number of MCP-TRIBE targets and higher signal to noise allowed us to discover the cause of off-target transcripts. When multiple copies of MCP-ADAR were tethered to a highly transcribed 24xMS2 locus, editing also affected nearby transcripts, both on its own chromosome and on nearby chromosomes. This is likely assisted by frequent transcription of both the β-actin gene and our reporter gene (Kalo et al., 2015). The high levels of transcription may promote assembly of “transcription hubs” and further promote inter-chromosomal interactions. As 13 molecules of MCP are on average loaded onto each molecule of mRNA containing the MBS reporter (Wu et al., 2012) this likely enhances the amount of RNA editing of nearby transcripts.
The 24xMS2 stem loops in the MBS create high local concentration of ADAR loaded onto the nascent chains. We do not expect that this will affect the use of TRIBE to define binding sites when fused to an RBP. This is based on two observations from our MS2-TRIBE data, the first being that (as detailed in Figure 4D) the direct interactions of ADAR with the stem loop containing RNA causes far more editing sites (27 cis-editing sites for β-actin-MBS) than those in trans (the majority having one or two sites). Consistent with this finding, we also observe that when performing TRIBE with other well-known RBPs such as ZBP1, its targets have a high number of RNA editing sites, far more than could be garnered from trans interactions (Biswas et al., 2020, Biswas et al., in preparation). As many sequence-specific RBPs bind with a 1:1 stoichiometry (Wu et al., 2015) we anticipate that a single ADAR enzyme will not be able to act with high efficiency upon nearby targets when fused to RBPs that are not highly multiplexed on their targets. This is further supported by a paucity of RNA edits when the MBS scaffold is absent. When MCP-ADAR is introduced into cells but cannot multiplex given the lack of MBS, the background of TRIBE is far lower than CLIP. Future experiments may modify the number of MS2 repeats on the MBS to characterize the sensitivity of a single enzyme.
Identification of Chromatin Contacts and Transcription Domains
Editing that spans inter-chromosomal transcription sites would only occur if the transcribing loci from nearby chromosomes shared intimate contacts with the β-actin nascent chains. Inter-chromosomal contacts have been identified by Hi-C (Battulin et al., 2015; Lieberman-Aiden et al., 2009) and further supported by transcriptome-wide RNA FISH studies in MEFs that observed nascent transcripts looping away from the chromosomal DNA (on average 0.8 ± 1.1 μm away) (Shah et al., 2018). This implies a possible intermingling with other nascent transcripts even from multiple chromosomes (Shah et al., 2018).
Owing to the limited resolution of ligation-based approaches (Hi-C) and optical microscopy (intron seq-FISH), specific interactions at the nucleotide level could not be previously resolved. In addition, the requirement for edited RNA focuses on loci that are simultaneously transcribing and should reduce contributions from many other sources of DNA/DNA interactions. This functional filter may simplify the extensive interchromosomal contacts that have been observed with other approaches.
Recently, proteins that interact with the β-actin-MBS were profiled using MCP fused to the biotin ligase BirA. After controlling for background, nuclear proteins as well as chromatin components were discovered to interact with β-actin-MBS, suggesting similar co-transcriptional RNA-protein interactions when the MBS array is used (Mukherjee et al., 2019). However, TRIBE differs in the mechanism of transcriptional marking: the enzyme is affixed to the transcript rather than an interaction that occurs by chance diffusion of a reactive intermediate. Therefore, the transcripts must be in actual physical contact rather than simply nearby.
Regulatory Considerations
The fact that nascent transcripts intermingle makes it likely that nearby transcripts can share factors; for instance, the effective concentration of splicing factors would be increased by the proximity of the transcripts undergoing splicing. This should significantly increase reaction rates, an explanation for why in vivo splicing is so efficient. ADAR may possibly edit excised introns, but as these are short-lived compared with the exonic sequences, it is more likely that the editing occurs before or during splicing. Effects on splicing rates may be testable by assaying a splicing reporter placed near highly transcribed and rapidly spliced genes.
The distance from the MCP (PDB: 2BU1) bound to the stem loops to the editing pocket of the ADAR (PDB: 5ED1) is only a few nanometers, indicating that transcripts come very close to one another. However, nascent chains may explore a much larger volume so they eventually come into contact with each other; this may occur when nascent chains of different transcripts are looped into a constrained volume. Calculations using the MS2 stem loop, the MBS, and β-actin-MBS suggest that this distance be can up to 200 nm away from where the nascent chain is anchored at the transcription site (Figure S7). In either case, collisions of these RNA strands do not result in entanglements and examples of trans-splicing are rare. This could best be accomplished if the nascent chains assume a minimal spherical volume rather than a linear dimension, so editing would then occur where these surface regions of exposed nucleotides come into contact.
Because the ADAR is an enzyme, it requires some time to find and modify the appropriate adenosine. This has been estimated to be about 24 s (Kuttan and Bass, 2012). Therefore, ADAR must stay in contact with its substrate longer than would be required for CLIP, where cross-linking is instantaneous. This would provide a filter for more persistent interactions and possibly more physiologically relevant ones (McMahon et al., 2016). In addition, the integration of RNA editing signal over time is an advantage compared with techniques requiring cellular fixation. Expression of RBP fusions (such as in MS2-TRIBE and also in other recently reported methods, Medina-Muñoz et al., 2019) may allow for RBPs to bind to a larger proportion of their targets when integrated over time, leading to more efficient identification of targets.
The discovery of chromatin contacts by MCP-TRIBE suggests a number of future directions. Large nuclear foci consisting of nascent transcripts have been previously observed (Fay et al., 1997), and more recent studies have proposed a transcriptional hub model where multiple transcribing loci spatially organize (Cho et al., 2018; Hnisz et al., 2017). Several theories have been proposed for the function of transcriptional hubs; in particular these data support the role of transcription factors in hub formation (Liu and Tjian, 2018). Consistent with a transcription factor-driven process for RNA-RNA clustering, half of the MCP-TRIBE targets contain upstream binding sites for serum response factor (Roider et al., 2009; Table S5). Further work will determine if additional parameters such as processing and splicing rate are correlated across contacts and if this coordination is mediated by specific transcription factors.
Transcription hubs change during stem cell differentiation and have been shown to contain multiple copies of both mediator and RNA polymerase II (Cho et al., 2018). To date, it is still not known which genes are actively transcribing within these transcriptional hubs. The highly multivalent nature of proteins within the hub, e.g., Mediator, makes it an attractive target for future studies of chromatin contacts with TRIBE. In addition, the discovery of enhancer RNAs makes them an attractive target for MS2 tagging and subsequent TRIBE to determine a list of functional RNA targets by their possible co-editing. The development of MS2-tagged gene libraries and CRISPR-mediated integration will further allow chromatin contacts to be studied by MCP-TRIBE.
Limitations of the Study
Here we show that MS2-TRIBE provides a standard for determining false-positives across different techniques and show that it has a low false-positive rate compared with CLIP. The low levels of RNA editing in cells without the β-actin-MBS reveal that the observed editing of interacting RNAs near the MBS requires these binding sites for the MCP-ADAR. The use of the MS2 approach has both strengths and limitations. It provides a benchmark for approaches such as CLIP (Ule et al., 2003), Hi-C (Lieberman-Aiden et al., 2009), TSA sequencing (Chen et al., 2018; Zhang et al., 2020), and APEX sequencing (Fazal et al., 2019; Padrón et al., 2019), and it uniquely can interrogate RNA-RNA contacts originating from a single locus. A limitation of the approach is the requirement for MBS to be integrated into a site of interest. Thus, MS2-TRIBE cannot be applied to patient samples to ascertain important protein RNA contacts related to disease, as can be done with CLIP (Luna et al., 2017). However, the genetic engineering required to perform MS2-TRIBE on cultured cells has been made easier with the widespread adaptation of CRISPR insertions (Spille et al., 2019) and coding sequence tagging where hundreds of lines have been made to express MS2 stem loops (Sheinberger et al., 2017; Wan et al., 2019). In addition, a mouse expressing the β-actin gene homozygously tagged with MS2 loops provides a resource for these studies in vivo (Lionnet et al., 2011) and hence the transcriptional environment around the actin locus in a variety of tissues.
TRIBE when fused to an RBP of interest can complement CLIP (Ule et al., 2003) to determine RBP binding targets both cis and trans. Because TRIBE represents a proximity approach to editing, the location of the cis-editing sites correlates with the location of RBP binding, but does not provide the exact nucleotides for RBP binding as does CLIP. Additional applications of MS2-TRIBE are being developed; for instance, MS2-based RNA editing within single cells (Rodriques et al., 2020) provides a way to interrogate mRNA age by assessing the increase of edits with time and may be used to determine how cis-editing sites evolve with changes in developmental state.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Robert H. Singer (robert.singer@einsteinmed.org).
Material Availability
Plasmids encoding mCherry-ADAR (Addgene plasmid #154786) and MCP-ADAR (Addgene plasmid #154787) are available at Addgene.
Data and Code Availability
All raw sequencing data and identified RNA editing sites have been deposited in NCBI's Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152855) and are accessible through accession number GSE152855. Code used to process and analyze the data is publically available at (https://github.com/rosbashlab/HyperTRIBE/). All other relevant data are available from the authors upon request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors would also like to thank Charles Query (CCQ), members of the Singer Lab, and the Rosbash Lab for their helpful discussions and comments on the manuscript. We would also like to thank the members of the Cai and Fishman labs for access to their chromosome interaction data in MEFs. J.B. was supported with funding from an MSTP Training Grant T32GM007288 and predoctoral fellowship F30CA214009. V.G. was supported by RO1GM57829 to CCQ. M.R. was supported by the Howard Hughes Medical Institute and R01DA037721. R.H.S. was supported by National Institutes of Health grant R01NS083085 and the NIH Common Fund 4D Nucleome Program U01DA047729. FACS sorting was performed with core support from NIH P30CA013330.
Author Contributions
Conceptualization, J.B. and R.H.S.; Methodology, J.B., V.G., and R.R.; Investigation, J.B., V.G., and R.R.; Writing – Original Draft, J.B. and R.H.S.; Writing – Review & Editing, J.B., R.R., M.R., and R.H.S.; Funding Acquisition, J.B., R.H.S., and M.R.; Resources, M.R. and R.H.S.; Supervision, M.R. and R.H.S.
Declaration of Interests
The authors have no competing interests.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101318.
Supplemental Information
Target lists for MCP-TRIBE in cells with (left) or without (right) MBS stem loops. Gene symbols are listed alongside the number of editing sites per transcript.
Target lists for CLIP in cells with (left) or without (right) MCP-GFP. Gene symbols are listed alongside the peak height per transcript. Traditional peak calling (top) as well as CIMS analysis (bottom) were performed to determine the total number of targets.
Summary of all MS2 consensus sequence containing transcripts within mm10 reference. BED format on left includes Chromosome number, Start nucleotide, End nucleotide, Consensus Sequence, Strand (+or -), Gene ID (official gene symbol).
Target lists for MCP-TRIBE in cells where the MS2 stem-loop construct was induced for 24 h (left) or 48 h (right) with doxycycline. Gene symbols are listed alongside the number of editing sites per transcript.
MCP-TRIBE sites were input into PASTAA website, and the list of putative transcription factors that bound were generated; highlighted in yellow is the serum response factor. On right, MCP-TRIBE sites as official gene symbols, ENSEMBL Gene IDs, and common names. MCP-TRIBE sites that are bound by serum response factor are highlighted in yellow.
References
- Battulin N., Fishman V.S., Mazur A.M., Pomaznoy M., Khabarova A.A., Afonnikov D.A., Prokhortchouk E.B., Serov O.L. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015;16:77. doi: 10.1186/s13059-015-0642-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas J., Nunez L., Das S., Yoon Y.J., Eliscovich C., Singer R.H. Zipcode binding protein 1 (ZBP1; IGF2BP1): a model for sequence-specific RNA regulation. Cold Spring Harb. Symp. Quant. Biol. 2020:039396. doi: 10.1101/sqb.2019.84.039396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zhang Y., Wang Y., Zhang L., Brinkman E.K., Adam S.A., Goldman R., van Steensel B., Ma J., Belmont A.S. Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J. Cell Biol. 2018;217:4025–4048. doi: 10.1083/jcb.201807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W.-K., Spille J.-H., Hecht M., Lee C., Li C., Grube V., Cisse I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361:412–415. doi: 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay F.S., Taneja K.L., Shenoy S., Lifshitz L., Singer R.H. Quantitative digital analysis of diffuse and concentrated nuclear distributions of nascent transcripts, SC35 and poly(A) Exp. Cell Res. 1997;231:27–37. doi: 10.1006/excr.1996.3460. [DOI] [PubMed] [Google Scholar]
- Fazal F.M., Han S., Parker K.R., Kaewsapsak P., Xu J., Boettiger A.N., Chang H.Y., Ting A.Y. Atlas of subcellular RNA localization revealed by APEX-Seq. Cell. 2019;178:473–490.e26. doi: 10.1016/j.cell.2019.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedersdorf M.B., Keene J.D. Advancing the functional utility of PAR-CLIP by quantifying background binding to mRNAs and lncRNAs. Genome Biol. 2014;15:R2. doi: 10.1186/gb-2014-15-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog J.J., Xu W., Deshpande M., Rahman R., Suib H., Rodal A.A., Rosbash M., Paradis S. TDP-43 dysfunction restricts dendritic complexity by inhibiting CREB activation and altering gene expression. Proc. Natl. Acad. Sci. U S A. 2020;117:11760–11769. doi: 10.1073/pnas.1917038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A. A phase separation model predicts key features of transcriptional control. Cell. 2017;169:13–23. doi: 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan G.J., Brown P.O., Herschlag D. Evolutionary conservation and diversification of Puf RNA binding proteins and their mRNA targets. PLoS Biol. 2015;13:e1002307. doi: 10.1371/journal.pbio.1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D., Baldi S., Zabel A., Straub T., Becker P.B. Active promoters give rise to false positive “Phantom Peaks” in ChIP-seq experiments. Nucleic Acids Res. 2015;43:6959–6968. doi: 10.1093/nar/gkv637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki S.M., Tsukamoto T., Salghetti S.E., Tansey W.P., Sachidanandam R., Prasanth K.V., Ried T., Shav-Tal Y., Bertrand E., Singer R.H. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalo A., Kanter I., Shraga A., Sheinberger J., Tzemach H., Kinor N., Singer R.H., Lionnet T., Shav-Tal Y. Cellular levels of signaling factors are sensed by β-actin alleles to modulate transcriptional pulse intensity. Cell Rep. 2015;11:419–432. doi: 10.1016/j.celrep.2015.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttan A., Bass B.L. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U S A. 2012;109:E3295–E3304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert N., Robertson A., Jangi M., McGeary S., Sharp P.A., Burge C.B. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell. 2014;54:887–900. doi: 10.1016/j.molcel.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe C.P., Wilinski D., Saunders H.A.J., Wickens M. Protein-RNA networks revealed through covalent RNA marks. Nat. Methods. 2015;12:1163–1170. doi: 10.1038/nmeth.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe C.P., Stefely J.A., Jochem A., Hutchins P.D., Wilson G.M., Kwiecien N.W., Coon J.J., Wickens M., Pagliarini D.J. Multi-omics reveal specific targets of the RNA-binding protein Puf3p and its orchestration of mitochondrial biogenesis. Cell Syst. 2018;6:125–135.e6. doi: 10.1016/j.cels.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T., Czaplinski K., Darzacq X., Shav-Tal Y., Wells A.L., Chao J.A., Park H.Y., de Turris V., Lopez-Jones M., Singer R.H. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat. Methods. 2011;8:165–170. doi: 10.1038/nmeth.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Tjian R. Visualizing transcription factor dynamics in living cells. J. Cell Biol. 2018;217:1181–1191. doi: 10.1083/jcb.201710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna J.M., Barajas J.M., Teng K., Sun H.-L., Moore M.J., Rice C.M., Darnell R.B., Ghoshal K. Argonaute CLIP defines a deregulated miR-122 bound transcriptome that correlates with patient survival in human liver cancer. Mol. Cell. 2017;67:400–410.e7. doi: 10.1016/j.molcel.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A.C., Rahman R., Jin H., Shen J.L., Fieldsend A., Luo W., Rosbash M. TRIBE: Hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell. 2016;165:742–753. doi: 10.1016/j.cell.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Muñoz H.C., Lapointe C.P., Porter D.F., Wickens M. Records of RNA localization through covalent tagging. BioRxiv. 2019 doi: 10.1101/785816. [DOI] [Google Scholar]
- Mili S., Steitz J.A. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee J., Hermesh O., Eliscovich C., Nalpas N., Franz-Wachtel M., Maček B., Jansen R.-P. β-Actin mRNA interactome mapping by proximity biotinylation. Proc. Natl. Acad. Sci. U S A. 2019;116:12863–12872. doi: 10.1073/pnas.1820737116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrón A., Iwasaki S., Ingolia N.T. Proximity RNA labeling by APEX-seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol. Cell. 2019;75:875–887.e5. doi: 10.1016/j.molcel.2019.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V.L., Mitra S., Harris R., Buxbaum A.R., Lionnet T., Brenowitz M., Girvin M., Levy M., Almo S.C., Singer R.H. Spatial arrangement of an RNA zipcode identifies mRNAs under post-transcriptional control. Genes Dev. 2012;26:43–53. doi: 10.1101/gad.177428.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman R., Xu W., Jin H., Rosbash M. Identification of RNA-binding protein targets with HyperTRIBE. Nat. Protoc. 2018;13:1829. doi: 10.1038/s41596-018-0020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., Peskin C.S., Tranchina D., Vargas D.Y., Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriques S.G., Chen L.M., Liu S., Zhong E.D., Scherrer J.R., Boyden E.S., Chen F. Recording the age of RNA with deamination. BioRxiv. 2020 doi: 10.1101/2020.02.08.939983. [DOI] [Google Scholar]
- Roider H.G., Manke T., O’Keeffe S., Vingron M., Haas S.A. PASTAA: identifying transcription factors associated with sets of co-regulated genes. Bioinform. Oxf. Engl. 2009;25:435–442. doi: 10.1093/bioinformatics/btn627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S., Takei Y., Zhou W., Lubeck E., Yun J., Eng C.-H.L., Koulena N., Cronin C., Karp C., Liaw E.J. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell. 2018;174:363–376.e16. doi: 10.1016/j.cell.2018.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinberger J., Hochberg H., Lavi E., Kanter I., Avivi S., Reinitz G., Schwed A., Aizler Y., Varon E., Kinor N. CD-tagging-MS2: detecting allelic expression of endogenous mRNAs and their protein products in single cells. Biol. Methods Protoc. 2017;2:1–14. doi: 10.1093/biomethods/bpx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spille J.-H., Hecht M., Grube V., Cho W.-K., Lee C., Cissé I.I. A CRISPR/Cas9 platform for MS2-labelling of single mRNA in live stem cells. Methods San Diego Calif. 2019;153:35–45. doi: 10.1016/j.ymeth.2018.09.004. [DOI] [PubMed] [Google Scholar]
- Teytelman L., Thurtle D.M., Rine J., Oudenaarden A.van. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci. U S A. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutucci E., Vera M., Biswas J., Garcia J., Parker R., Singer R.H. An improved MS2 system for accurate reporting of the mRNA life cycle. Nat. Methods. 2018;15:81–89. doi: 10.1038/nmeth.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B. CLIP Identifies nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Vera M., Biswas J., Senecal A., Singer R.H., Park H.Y. Single-cell and single-molecule analysis of gene expression regulation. Annu. Rev. Genet. 2016;50:267–291. doi: 10.1146/annurev-genet-120215-034854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Anastasakis D.G., Rodriguez J., Palangat M., Gudla P., Zaki G., Tandon M., Pegoraro G., Chow C.C., Hafner M. Social Science Research Network; 2019. Dynamic Imaging of Nascent RNA Reveals General Principles of Transcription Dynamics and Stochastic Splice Site Selection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Chao J.A., Singer R.H. Fluorescence fluctuation spectroscopy enables quantitative imaging of single mRNAs in living cells. Biophys. J. 2012;102:2936–2944. doi: 10.1016/j.bpj.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Buxbaum A.R., Katz Z.B., Yoon Y.J., Singer R.H. Quantifying protein-mRNA interactions in single live cells. Cell. 2015;162:211–220. doi: 10.1016/j.cell.2015.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Rahman R., Rosbash M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA. 2018;24:173–182. doi: 10.1261/rna.064691.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Darnell R.B. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Zhang Y., Chen Y., Gholamalamdari O., Wang Y., Ma J., Belmont A.S. TSA-Seq reveals a largely “hardwired” genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. BioRxiv. 2020:607–614. doi: 10.1101/gr.266239.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Target lists for MCP-TRIBE in cells with (left) or without (right) MBS stem loops. Gene symbols are listed alongside the number of editing sites per transcript.
Target lists for CLIP in cells with (left) or without (right) MCP-GFP. Gene symbols are listed alongside the peak height per transcript. Traditional peak calling (top) as well as CIMS analysis (bottom) were performed to determine the total number of targets.
Summary of all MS2 consensus sequence containing transcripts within mm10 reference. BED format on left includes Chromosome number, Start nucleotide, End nucleotide, Consensus Sequence, Strand (+or -), Gene ID (official gene symbol).
Target lists for MCP-TRIBE in cells where the MS2 stem-loop construct was induced for 24 h (left) or 48 h (right) with doxycycline. Gene symbols are listed alongside the number of editing sites per transcript.
MCP-TRIBE sites were input into PASTAA website, and the list of putative transcription factors that bound were generated; highlighted in yellow is the serum response factor. On right, MCP-TRIBE sites as official gene symbols, ENSEMBL Gene IDs, and common names. MCP-TRIBE sites that are bound by serum response factor are highlighted in yellow.
Data Availability Statement
All raw sequencing data and identified RNA editing sites have been deposited in NCBI's Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152855) and are accessible through accession number GSE152855. Code used to process and analyze the data is publically available at (https://github.com/rosbashlab/HyperTRIBE/). All other relevant data are available from the authors upon request.