

Abstract
Kti12 (Kluyveromyces lactis toxin insensitive 12) is an evolutionary highly conserved ATPase, crucial for the tRNA-modification activity of the eukaryotic Elongator complex. The protein consists of an N-terminal ATPase and a C-terminal tRNA-binding domain, which are connected by a flexible linker. The precise role of the linker region and its involvement in the communication between the two domains and their activities remain elusive. Here, we analyzed all available Kti12 protein sequences and report the discovery of a subset of Kti12 proteins with abnormally long linker regions. These Kti12 proteins are characterized by a co-occurring lysine to leucine substitution in their Walker A motif, previously thought to be invariable. We show that the K14L substitution lowers the affinity to ATP, but does not affect the catalytic activity of Kti12 at high ATP concentrations. We compare the activity of mutated variants of Kti12 in vitro with complementation assays in vivo in yeast. Ultimately, we compared Kti12 to other known p-loop ATPase family members known to carry a similar deviant Walker A motif. Our data establish Kti12 of Eurotiomycetes as an example of eukaryotic ATPase harboring a significantly deviating but still functional Walker A motif.
Electronic supplementary material
The online version of this article (10.1007/s00294-020-01070-2) contains supplementary material, which is available to authorized users.
Keywords: Kti12, PSTK, Flexible linker, Active site, P-loop, ATPase
Introduction
It was noticed several years ago that yeast cells become resistant to Kluyveromyces lactis toxin, zymocin, upon removal of any of the so-called KTI genes (Butler et al. 1991b; 1994; Kishida et al. 1996). The family of Kti protein harbors various types of proteins, including Elongator core subunits (Elp1/Kti7, Elp2/Kti3, Elp3/Kti8, Elp4/Kti9, Elp6/Kti4), methyltransferases (Trm9/Kti1), zinc-binding proteins (Kti11), WD40s (Kti13), and kinases (Hrr25/Kti14) (Frohloff et al. 2001; Huang et al. 2005a; Karlsborn et al. 2014; Krutyhołowa et al. 2019b). Zymocin itself has three individual components, namely alpha-, beta- and gamma subunits (Stark and Boyd 1986). Alpha- and beta-subunits serve as auxiliary factors allowing the delivery of the gamma subunit into fungal cells (Butler et al. 1991a). The gamma subunit cleaves cellular tRNAs that specifically carry a double 5-methoxy-carbonyl-methyl-2-thio (mcm5s2U34) modification at uridines in their wobble position (Lu et al. 2005; Jablonowski et al. 2006).
Those chemically rather complicated tRNA modifications are introduced by a cascade of enzymes, including the Elongator complex (Huang et al. 2005b; Dauden et al. 2018), methyltransferases Trm9-Trm112/ALKBH8 (Studte et al. 2008; Songe-Møller et al. 2010; van Tran et al. 2018), and the Uba4/Urm1 thiolation pathway (Leidel et al. 2009; Noma et al. 2009). Despite the creation of a toxin-target site, these tRNA modifications are genuinely required for maintaining proper translation speed and preventing from co-translational protein misfolding (Nedialkova and Leidel 2015; Ranjan and Rodnina 2016; Goffena et al. 2018). The lack of these anticodon modifications in humans causes various pathophysiologies, including severe neurodegenerative diseases (Close et al. 2006; Chaverra et al. 2017; Dauden et al. 2018; Kojic et al. 2018; Hawer et al. 2018; Shaheen et al. 2019).
Recent structural analyses of the enzymatic Elp3 subunit and the Elp123 subcomplex of Elongator show that the subcomplex specifically recognizes tRNAs (Glatt et al. 2016; Dauden et al. 2019; Lin et al. 2019). The overall complex consists of six pairs of subunits and is built up by a symmetrical dimer of two Elp123 subcomplexes, decorated with a hexameric Elp456 ring (Glatt et al. 2012) on one of the Elp123 lobes (Dauden et al. 2017; Setiaputra et al. 2017). Kti12 was demonstrated to bind to the Elongator complex in vivo and in vitro (Fichtner et al. 2002; Petrakis et al. 2005), and we recently fine-mapped its binding site to the first WD40 domain of the Elp1 subunit in vitro (Krutyhołowa et al. 2019a). Kti12 harbors two domains, namely an N-terminal ATPase domain and a C-terminal tRNA-binding domain, both connected by a flexible linker region (Fig. 1a) (Schaffrath et al. 2017; Krutyhołowa et al. 2019a). Kti12 shows a very high overall sequence and structural similarity to O-phosphoseryl tRNASec kinase (PSTK), a kinase responsible for phosphorylation of Ser-tRNASec (Sherrer et al. 2008). Like PSTK (Chiba et al. 2010), Kti12 binds tRNA with its C-terminus and hydrolyzes ATP, if presented with tRNASec in vitro (Krutyhołowa et al. 2019a). Other atypical kinases, like the p-loop-containing Rio1 ATPases, are known to play key roles in ribosome biogenesis, translational control, and other cellular functions (Berto et al. 2019). However, the genuine function of Kti12 and the mechanistic consequences of its direct association with the Elongator complex remain elusive (Krutyhołowa et al. 2019a).
Fig. 1.

Identification of Kti12 proteins with elongated linker and their characterization. a Architecture of Kti12 protein. ATPase domain (grey) and tRNA-tethering domain (yellow) are connected by a flexible linker. Structural overview of Chaetomium thermophilum Kti12 ATPase domain (PDB ID: 6QP0) with a particular emphasis on nucleotide binding pocket and K14 located in a p-loop. b Distribution of linker length across fungal Kti12 proteins. Numbers on Ox indicate middle values for particular baskets, for instance, 18 ± 4. Black triangles indicate groups to which ScKti12 and CtKti12 belong to. c Scheme of supervised a machine-learning experiment that pinpointed coincidence of L14 with elongated linkers. Sequences with linkers of a regular length (grey) always carry lysine at position 14 (K14), whereas Kti12 proteins with elongated linkers (pink) can be distinguished by the presence of leucine in a p-loop (L14). d Violin plots represents distribution of Kti12 protein sequences in a two-dimensional feature space. Linker length is on Ox whereas presence of leucine or lysine qualifies sample to one of the groups on Oy axis. Number of sequences within each group is indicated next the Oy axis
Here, we systematically analyzed all annotated Kti12 and PSTK sequences and identified a subpopulation of Kti12 proteins in Eurotiomycetes with extremely elongated linker regions. Furthermore, we used a machine-learning approach to unbiasedly identify a co-occurring feature specific to those Kti12 proteins with elongated linkers. In detail, those aberrant Kti12 proteins share a substitution of a highly conserved p-loop lysine, which was previously described as an invariable element of the phosphate-binding motif in ATPases. We investigated this evolutionary coupling and dissected its potential implications for the active site of Kti12. Our study presents complementary approaches in vitro and in vivo that deepen our understanding of Kti12 and provide important insights into the variations of p-loop-based active sites.
Materials and methods
Sequence analyses and machine learning
Annotated and validated sequences of Kti12 proteins from different domains of life were obtained from the Uniprot database and aligned using Muscle web server (Edgar 2004) and Jalview software (Waterhouse et al. 2009). Alignments were manually curated, and all proteins without both ATPase domain and CTD were removed. We utilized Python, Biopython library (Cock et al. 2009), and elements of scikit-learn platform (Pedregosa et al. 2011) to conduct a machine-learning experiment. The length of the linker between ATPase domain (NTD) and tRNA-binding domain (CTD) was used for supervised learning. After randomization of multiple sequence alignment, single amino acid positions were considered as a basis for supporting vector machine classification. The significance of a linker-length difference between K14- and L14-containing groups was additionally tested using unpaired two-tailed t test and resulted in a p value of < 0.0001.
Modeling and structure visualization
The Kti12 protein structure was visualized using PyMol software (Schrödinger 2015). We used the YRB script (Hagemans et al. 2015) to analyze hydrophobicity of the nucleotide-binding pocket. Homology-based models of mu gp28 were obtained using Phyre 2.0 and AnKti12 was modelled using SWISS-MODEL web server (Kelley and Sternberg 2009; Waterhouse et al. 2018). We employed the I-TASSER server (Roy et al. 2010; Yang et al. 2015) to predict the existence of any stable secondary structure elements within the linker of AnKti12.
Strains and genetic manipulation of Saccharomyces cerevisiae
The influence of mutations on Kti12 activity was analyzed by gene replacement technology, which allows to modify specific yeast genomic loci (Rothstein 1991). The S. cerevisiae strains used and generated are listed (see Table S1). First, the genomic KTI12 gene of strain UMY2893 (Huang et al. 2005a) was disrupted by transformation with a DNA fragment containing the URA3 gene from K. lactis flanked by KTI12 homologous regions. Selection for Ura + colonies and screening for zymocin resistance yielded kti12::KlURA3 transformants, in which the WT gene was replaced with the disrupted allele by a double crossover event in vivo. The resulting strain CKY40 was used in the second step to introduce each of the mutations by generating the corresponding DNA fragments with the desired mutation restoring the KTI12 locus except for the desired changes. The corresponding DNA fragments were generated by PCR and transformed directly. Replacement of the KlURA3 cassette by the incoming DNA yielded ura3-auxotrophs selected by plating on 5-fluoroorotic acid-containing media. The proper recombination event was verified by screening colonies using PCR. The presence of the respective mutation was confirmed by sequencing of genomic DNA.
In vivo assay for Kti12 functionality by yeast phenotyping
To analyze the influence of the different kti12-alleles on tRNA modification by the Elongator, growth inhibition by exogenous zymocin toxin was assayed as previously described (Frohloff et al. 2001). Zymocin was obtained from the culture medium of Kluyveromyces lactis killer strain AWJ137 (Frohloff et al. 2001) by filtration (Klassen et al. 2011). Tenfold serial dilutions of the different strains were spotted on rich media plates (YPD, 0.5% yeast extract, 2% peptone, 2% glucose and 2% agar) containing 50% (v/v) zymocin or without the toxin and incubated for 2–3 days at 30°C. Functionally intact Kti12 confers growth inhibition due to the cleavage of three Elongator-modified tRNAs by zymocin. Functionally impaired Kti12 causes toxin resistance (Frohloff et al. 2001). In the second assay, the Kti12 function was tested via the nonsense suppression activity of the SUP4 tRNA, which requires the U34 modification in the anticodon. The kti12 mutations were transferred to the SUP4 background and the resulting strains, which contain the ade2-1 or the can1-100 allele containing ochre stop codons in the coding regions, were spotted on SD plates containing adenine (YNB), lacking adenine (Ade-), or medium without arginine but containing the toxic arginine analog canavanine. Plates were incubated for 3 days at room temperature and 3 days at 30°C. In cells lacking Kti12 activity, nonsense suppression of the ade2-1 mutation and, therefore, growth without adenine supplementation is impaired. No growth on adenine plates correlates with the growth on canavanine plates since the lack of nonsense suppression of the can1-100 allele means that the CAN1-encoded arginine permease, responsible for the uptake of canavanine, is not expressed. Hence a functional KTI12 gene product is indicated by growth on Ade− and no growth on canavanine plates. (Huang et al. 2005b).
Protein purification
The codon optimized open reading frames (ORF), encoding Chaetomium thermophilum Kti12 (CtKti12) or Aspergillus niger (AnKti12), were individually cloned into the pET24d vector using NcoI and XhoI restriction sites and purified as previously described (Krutyhołowa et al. 2019a). AnKti12 protein was additionally purified in a HEPES pH 7.2 buffer using a cation-exchange Hi-Trap SP column (GE Healthcare), followed by size-exclusion chromatography on HiLoad 16/600 Superdex 75 pg column (GE Healthcare). Proteins were stored at – 80 °C in 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM DTT buffer.
Thermal shift assay
Thermostability of CtKti12 WT protein and CtKti12 mutants was assessed using thermal shift assays. In short, storage buffer with 2 mM MgCl2 without nucleotides or with addition of 1 mM ADP or ATP was used to dissect the influence of nucleotide binding on protein stability. 10 µg of protein was heated up from 4 to 98°C at a speed of 0.6°C/min in the presence of SYPRO Orange (Sigma Aldrich) hydrophobic fluorescent dye. Data represent three independent experiments conducted in at least 2 technical replicas.
Determination of nucleotide-binding affinity by fluorescence polarization
Dissociation constant of MANT-labeled ATP was determined using fluorescence polarization. Experiments were performed in a storage buffer after addition of 2 mM MgCl2. Serial twofold dilutions of CtKti12 ranging from 100 μM (highest concentration) were prepared and mixed with 1 μM MANT-ATP (final concentration). Fluorescence was elicited using 340-nm excitation light and measured at 448 nm. Fluorescence polarization was measured using FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices) with a gain factor of 1000 and at a system temperature of 37°C for the whole duration of the experiment. Data originate from three independent experiments. Curve fitting and Kd calculation were conducted using GraphPad Prism software (La Jolla, California, USA; www.graphpad.com) assuming a single/one-site binding mode.
Production of tRNA
Human tRNASec was obtained by run-off in vitro transcription from a linearized pUC 19 vector carrying tRNASec gene under the T7 promoter. After overnight transcription reaction, RNA species were separated on a HiTrap DEAE FF column (GE Healthcare). RNA was precipitated with 1:1 v/v isopropanol overnight and subsequently denatured and refolded by heating to 80 °C and cooling to 40 °C in annealing buffer, which contained 200 mM HEPES pH 7.5, 500 mM KCl, and 500 mM NaCl. Subsequent size-exclusion chromatography on Superdex 75 10/300 GL column yielded a homogeneous population of folded tRNASec molecules that were used to elicit ATPase activity of Kti12.
ATPase activity measurements
In vitro transcribed tRNA was used to trigger the ATPase activity of different CtKti12 protein variants. Reactions were performed in the protein storage buffer after the addition of 2 mM MgCl2. In detail, 1 μM CtKti12 was mixed with 1 μM human tRNASec and either 1 mM ATP or 20 μM ATP. Reactions were incubated for 15 h or 1 h, respectively. In the case of AnKti12, the reaction was conducted in the presence of 5 μM tRNASec for 2 h at 37 °C. Reactions were monitored using malachite green ATPase assay kit (Sigma Aldrich) as per the manufacturer’s instructions. Data derived from at least three independent experiments were performed in technical duplicates.
Results
Active site mutation evolutionarily coincides with elongated linker
We harvested and aligned all annotated Kti12 protein sequences from animals, plants, and fungi to identify conserved and variable features in its domains and motifs. Protein sequences of O-phosphoseryl tRNASec kinase (PSTK) from animals, which show almost identical domain organization and structural similarity to Kti12, served as references in all analyses (Fig. S1A). First, we systematically compared the length of the flexible linker region that connects the ATPase domains with the C-terminal tRNA-binding domains of Kti12 and PSTK. In animals, Kti12 and PSTK both exhibit relatively short linker regions in the range 18–22 amino acids (Fig. S1B), whereas Kti12 proteins in plants possess longer linkers of approximately 30 amino acids (Fig. S1B). Fungal Kti12 proteins have predominantly shorter linkers of an “animal type”, but some fungi, including Saccharomyces cerevisiae, harbor long plant-like linkers. Unexpectedly, some fungal species evolved a third, clearly separated, population of Kti12 proteins with extremely elongated linkers of approximately 55–60 amino acids (Fig. 1b).
We utilized a machine-learning approach to dissect potential commonalities in the case of fungal Kti12 proteins with an elongated linker and understand what distinguishes this population from fungal Kti12 proteins with linkers of typical lengths. First, we manually curated alignments and separated sequences with elongated linkers from other fungal Kti12 sequences. This dataset was used to run a supervised machine-learning approach based on Support Vector Machine (SVM) (Fig. 1c), where we considered single amino acid positions in a multiple sequence alignment and tested if they led to a similar classification pattern. Surprisingly, SVM could achieve a completely equal classification pattern (r=1, accuracy=100%) based on the identity of a single amino acid, namely a central active site lysine residue (K14, Fig. 1d). Other identified single amino acid hallmarks of long linker Kti12 proteins neither separate the classes fully nor are located within the Kti12 active site (Fig. S1C). The identified lysine residue was previously demonstrated to be of major importance for the ATPase activity of Kti12, as a K14A mutant showed almost completely abolished ATPase activity in vitro and phenocopied Kti12- and Elongator-deficient S. cerevisiae strains (Krutyhołowa et al. 2019a). Strikingly, all sequences with an elongated linker carry leucine or isoleucine residues at the respective position. An additional analysis of the amino acid content of the linker regions revealed that long linkers, in general, harbor more threonine residues, while having noticeably decreased relative number of lysine residues (Fig. S1D). It remains to be shown, if specific residues in the linker could influence the active site of Kti12 proteins carrying the K14L substitution. Species carrying Kti12L/I14 belong to the class of Eurotiomycetes in Ascomycota (Fig. 2a). Inactivating Kti12 mutations would most likely also compromise Elongator’s tRNA modification activity (Schaffrath et al. 2017) and reduce the selective pressure to preserve a functional Elongator complex. Nonetheless, no reports indicate malfunctioning of Elongator or other tRNA modification pathways in Eurotiomycetes. Furthermore, we found that all six Elongator genes (Elp1–6) are present in several analyzed Eurotiomycetes species and do not contain any genomic alterations (e.g. internal stop codons, fused gene products or frameshift mutations) that would indicate a loss-of-function. The question immediately arises whether Eurotiomycetes that carry both Kti12L/I14 and an elongated linker have lost the regulatory link between Elongator and the ATPase activity of Kti12 or if the naturally occurring Kti12 variants remain active.
Fig. 2.

Phenotypic characterization of Kti12 K14L mutation and elongated linker in yeast. a Phylogenetic tree based on Kti12 sequence alignment displays distribution of different residues at position 14 among species of different divisions [Ascomycota (orange)/Basidiomycota (green)]. Kti12 proteins carrying L14 and long linker (purple squares) belong to Eurothiomycetes. Multiple sequence alignment of Kti12 proteins from 25 model fungi species was performed using JalView software (Waterhouse et al. 2009) and Muscle server (Edgar 2004). The phylogenetic tree was built using the neighbor-Joining method. Individual branch lengths are shown above the branches and indicate the number of amino acid substitutions per site. The fraction of replicate trees in which the associated taxa clustered together in the bootstrap test [1000 replicates (Felsenstein 1985)] are shown beneath the branching points. The evolutionary distances used to calculate the tree were calculated using the Poisson correction method with the pairwise deletion of ambiguous positions. Final dataset contained 610 positions. All evolutionary analyses were conducted in MEGA X (Kumar et al. 2018). The tree was visualized using iTOL (Letunic and Bork 2007). b Zymocin assay assessing performance of the Elongator complex and presence of U34 modifications. WT yeast strain (ScKTI12) served as a growth control, Scelp3Δ and Sckti12Δ pose negative controls. Tenfold dilutions were spotted on YEPD media with the addition of 50% Zymocin or YNB media replacing arginine by 40 µg/ml canavanine or lacking adenine (-adenine). In the different strains KTI12 was mutated (K14A/L), parts or the whole gene were exchanged with the corresponding sequences from Aspergillus niger (An knot + linker, AnKTI12) or parts of it were deleted (marked with ∆ and the deleted residues in brackets). Sckti12-NTD codes for the first 184 amino acids
Both K14L and long linker ScKti12 rescue the kti12Δ phenotype
As described previously, K14 of ScKti12 is of profound importance to proper functioning of Kti12 protein (Krutyhołowa et al. 2019a). Thus, we decided to experimentally verify if these variations in KTI12 alleles would have any functional relevance and if the linker length is indeed functionally coupled to alterations in the ATP-binding pocket. Therefore, Kti12 function was tested in S. cerevisiae by established assays for functional Elongator-dependent tRNA modifications. The degree of U34 modifications in vivo can roughly be estimated by zymocin sensitivity and suppression of ochre stop codons by the SUP4 suppressor tRNA (Frohloff et al. 2001; Huang et al. 2005a). The phenotypes of the generated Sckti12–K14L mutant resemble the WT (ScKTI12) contrasting with the Sckti12–K14A mutants (Fig. 2b), which phenocopy the null mutant lacking Kti12 protein (Sckti12Δ) or lacking the Elongator subunit Scelp3Δ (Krutyhołowa et al. 2019a). Sensitivity to zymocin and canavanine are semi-quantitative indicators of Kti12 function. Growth on media lacking adenine is complementary but sometimes less reliable due to the appearance of revertants. The phenotypes indicate that not only the K14L mutation but also shorter deletions of the N- or C-terminal part of the linker (Δ184-196 and Δ197-213) did not substantially affect Kti12 function negatively. Replacing the short linker of ScKti12 with an elongated linker from Aspergillus niger Kti12 (Sckti12–AnKnoLi) resulted in a variable phenotype on media deprived of adenine suggesting weaker Kti12 activity. Complete deletion of the linker (Δ184-213) inactivates ScKti12. The entire AnKTI12 gene from Aspergillus niger was unable to replace the function of the ScKt12, which might occur due to specific-interaction interfaces with the Elongator complex or other binding partners. Our data illustrate that the linker region in Kti12 is as important for the tRNA-modification activity of Elongator as the N-terminal ATPase domain or the C-terminal tRNA-binding domain (Schaffrath et al. 2017).
Mutation of K14 alters nucleotide binding by Kti12
Due to the poor solubility of active site mutants from S. cerevisiae Kti12 (ScKti12), we expressed and purified full-length Chaetomium thermophilum CtKti12 and the variants CtK12K14A and CtKti12K14L in bacteria (Fig. S2A, B). In addition, we generated another variant, namely CtKti12K14V, which contains a side chain with an intermediate length, allowing us to define the minimal spatial requirements for protein activity. Next, we studied thermostability, nucleotide binding, and tRNA-induced enzymatic ATP hydrolysis activity of CtKti12 and its variants at saturating and semi-saturating substrate concentrations. CtKti12 exhibits a melting point of ~ 46.0 °C (Fig. 3a), which increases to 50.0 °C upon addition of ATP. As previously shown (Krutyhołowa et al. 2019a), the substitution K14A does not affect the thermostability of CtKti12 by itself, but diminishes any nucleotide-dependent stabilization effects. The Kti12K14V mutated variant displays a thermostability profile similar to the nucleotide-bound CtKti12, even in the absence of a nucleotide, suggesting a dynamic rearrangement of the K14 carbon side chain during nucleotide binding. In contrast, the Kti12K14L variant is strongly destabilized, resulting in a decreased melting temperature of 37.5 °C, 8.5 °C less than the WT. Interestingly, neither K14V nor K14L exhibited ATP-dependent increase of the melting temperature, indicating strongly decreased nucleotide-binding properties of these p-loop variants (Fig. S2C), similar to K14A.
Fig. 3.
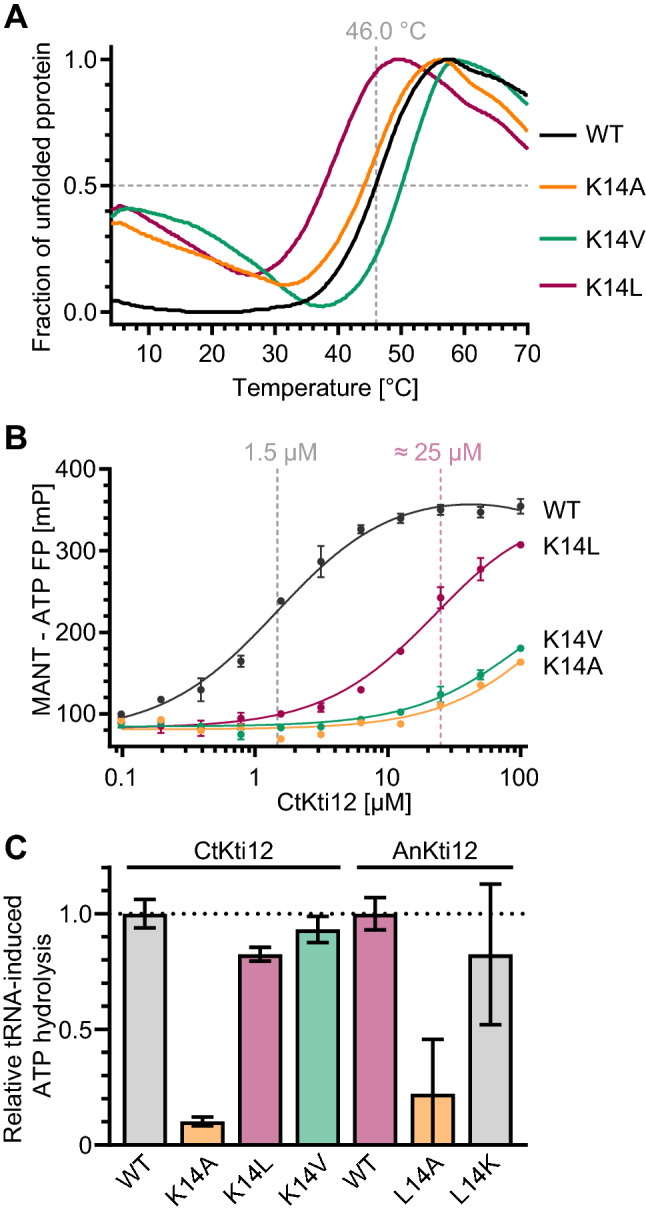
Biochemical properties of Kti12 mutants. a Thermostability profiles of CtKti12 K14 point mutants. Thermal shift assay, curves represent an average of three independent experiments. b Fluorescent polarization study of MANT-ATP bound by Kti12 mutants. CtKti12 WT Kd = 1.5 μM ± 0.2 μM, K14L Kd = 25.0 μM ± 2.4 μM. In case of CtKti12 K14A and K14V we were unable to determine Kd in a measured concentration range. c Relative tRNASec-induced ATPase activity of CtKti12 and AnKti12 mutants, compared to the respective WT versions. Free phosphate concentration was measured using malachite green assay. Bars represent an average of three independent experiments ± standard deviation. Proteins carrying different amino acids at position 14 are color coded (K14 grey, A14 orange, L14 purple and V14 green)
Although the thermal shift assay provided some initial indications regarding nucleotide binding, we decided to quantify nucleotide-binding affinity using fluorescence polarization. Using this method, we were able to confirm the previously measured Kd for CtKti12 WT and MANT-ATP (1.5 ± 0.2 μM). Apparently a very high evolutionary constraint conserved the lysine residue in the p-loop motif of yeast Kti12, PSTK, and other family members. Both K14A and K14V proteins have very low affinity towards MANT-ATP and the Kd can only be estimated to values higher than 100 μM. Interestingly, the naturally occurring K14L variant can still bind MANT-ATP (Kd 25.0 ± 2.4 μM). In summary, our results demonstrate that all tested substitutions of K14 decrease the affinity towards ATP (Fig. 3b).
Next, we examined the activity of KTI12 variants using the previously established ATPase activity assay (Krutyhołowa et al. 2019a). We observed comparable ATP hydrolysis rates in the case of CtKti12 WT, K14V, and K14L presented with physiological concentrations of ATP (1 mM; Fig. 3c). This finding supports our in vivo data showing that the K14L variant is able to rescue the S. cerevisiae Δkti12 strain. In addition, we decided to perform the same assay at more limiting ATP concentration close to the Kd of the CtKti12K14L-MANT-ATP interaction. Upon incubation with 20 μM ATP, differences in ATP hydrolysis rates between the naturally occurring mutants become unambiguous (Fig. S2D). To further validate our findings from CtKti12, we expressed and purified WT Aspergillus niger Kti12 protein and its L14K and L14A mutants (Fig. S2E). The tRNA-induced ATPase activity of WT AnKti12 naturally carrying leucine at position 14 is similar to the ATPase activity of AnKti12L14K, the common variant occurring in all other Kti12 proteins. In addition, AnKti12L14A shows strongly reduced ATPase activity which further corroborates the observed decrease in ATPase activity of CtKti12K14A and in vivo phenotyping results of the Sckti12–K14A strain. Our findings indicate that residues in the long threonine-rich linker of AnKti12 cannot contribute to the active site, since the activity of AnKti12L14A remains strongly reduced in vitro.
Discussion
Three different types of linkers are present in Kti12 proteins from fungi and the reason for this divergence currently remains unclear. In the case of animal Kti12 and PSTK, a relatively short linker can be explained by the presence of tRNASec which stimulates ATP hydrolysis by Kti12. However, apart from tRNASec, which does not exist in S. cerevisiae, ScKti12 is important for processing of 11 different tRNAs by the Elongator complex, so the relatively short 20 amino acid linkers may be preferred for tRNA-related functions. Concomitant with this, plant phenotypes caused by mutations in the Arabidopsis thaliana deformed root and leaves (DRL1) gene encoding the Kti12 homologue are similar to the phenotypes of elongata (ELO) mutants (Chen et al. 2006; Mehlgarten et al. 2010). Most of the plant genes encoding Elongator core subunits were also identified as elo mutants (Nelissen et al. 2005). All plants and some fungi, including S. cerevisiae, harbor an approximately 30-amino acid long linker composed of a putative topological knot and a disordered linker. We tried to dissect the role of these regions in S. cerevisiae by deleting individual structural elements and respective combinations in the ScKTI12 gene, but we failed to find any phenotypical difference in the wild type strains lacking the knot or the disordered linker (Fig. 2b). Complete removal of both elements, i.e. the entire linker, inactivated Kti12, whereas replacement of the ScKti12 linker by the long one of AnKti12 did not. Hence, we conclude that cooperation of the ATPase and tRNA-binding domains as described previously (Krutyhołowa et al. 2019a) is supported by a linker, which has properties found in the long AnKti12 as well as in the shorter ScKti12 linker. Attempts to model the structure of the AnKti12 linker by I-TASSER server failed, as it was not possible to find any stable secondary or tertiary structures. Taken together, these findings suggest that the linker is indeed rather flexible, most likely unstructured and not very sensitive to sequence variations. It remains to be shown experimentally, if it additionally participates in any protein–protein interactions that could be related to potential Elongator-independent “moonlighting” functions of Kti12.
Our data show that the non-canonical K14L substitution within the Walker A motif decreases nucleotide-binding affinity of Kti12, but does not result in catalytically inactive protein. Kti12 remains active under physiological conditions, as the cytoplasmatic ATP concentration in yeast is close to 2 mM (Koç et al. 2004). We have shown that the ScKTI12–K14L allele can rescue the tRNA modification phenotype of a Kti12 deficient strain in vivo (Fig. 2b) and that the CtKti12 and AnKti12 proteins have similar in vitro activities as their K14L or L14K variants, respectively (Fig. 3c). Since cytoplasmatic ATP concentration can decrease during starvation, cellular stress or other conditions the low affinity for ATP of Kti12K14Lwould allow to effectively shut down tRNA modification by Elongator via Kti12. Perhaps at a low-energy state of the cell Kti12 deactivation might be required to activate specialized translational programs and the production of Elongator-independent proteomes. If this potential regulatory circuits existed, the question arises whether it represents a unique feature of Eurotiomycetes or similar regulatory “bypasses” are present in case of other organisms and other regulatory ATPases.
We analyzed the most crucial charged and hydrophobic residues within the CtKti12 nucleotide-binding pocket and simulated the structural consequences upon mutation of K14. As the side chain of lysine is relatively long, K14A creates a significant breach which may allow water to reach the hydrophobic core of the protein. K14V and K14L substitutions would gradually revert this breach (Fig. 4a).
Fig. 4.

Influence of K14L on the nucleotide binding pocket of Kti12 protein. a Hydrophobicity of nucleotide binding pocket of Kti12 mutants. PDB ID: 6QP0. Nucleotide binding pocket consists of positively charged (blue), negatively charged (red) and hydrophobic (yellow) residues. Only surface of the residues within 5 Å from the phosphor atoms of ADP is shown. Kti12 is shown bound to ADP (standard coloring), AlF3 (cyan and pink) and magnesium (green). K14A mutation creates a relatively big gap, leaving the hydrophobic core unprotected. Subsequent K14V and K14L substitutions gradually repair the breach. b A homology-based model of gp28 terminase (based on PDB ID 3EZK) from mu phage with previously described deviant Walker-A motif shown in ball and stick representation. c Multiple sequence alignment and structural comparison of Walker A motifs in CtKti12, AnKti12 (based on 6QP0) and mu phage gp28. Shades of violet indicate conservation score. Please note that AnKti12 Walker A motif does not have additional lysine which is present in case of mu gp28, but has L14 residue (pink) which corresponds to L70 residue of mu gp28. d An overview of lysines (dark blue) present in the ATPase domain of CtKti12 (PDB ID 6QP0, top) and AnKti12 (based on PDB ID 6QP0; bottom). Walker A motif is highlighted in pink
In addition, we compared available structural information from members of the ‘DxTN’ kinase family, named after its Walker B motif (Leipe et al. 2003). Superposition of ATPase domains of eukaryotic Kti12 (PDB ID: 6QP0), archaeal PSTK from Methanocaldococcus janashii (PDB ID: 3ADB), and T4 bacteriophage PNK (PDB ID: 1LY1) revealed highly similar ATPase folds with a particularly strong conservation of the Walker A motif. While the relative position of K14 and the hydrophobic core is almost identical in the analyzed structures, we failed to identify any archaeal or viral proteins carrying L14. Furthermore, we searched the literature for described deviations from Walker A motifs that would include substitution of signature lysine residues by leucine. Gp28, a terminase from phage mu that allows the phage to pack its genome into the capsid, was described to carry a similar substitution (Mitchell and Rao 2004). The authors mentioned that the function of L70 may be successfully complemented by K64 located in close vicinity. We performed homology-based modeling to obtain a model of the gp28 structure (Fig. 4b). We compared Walker A motif of gp28 with those of CtKti12 and AnKti12 in terms of sequence and structure. Kti12 harbors no lysine in a position that would correspond to a previously described K64 residue in gp28 (Fig. 4c). Furthermore, no other lysine from the ATPase domain can perform instead of K14. According to our homology-based model, all lysine residues are located far away from the Walker A motif (Fig. 4d). We would like to highlight that a critical signature substitution within the Walker A motif of Eurotiomycetes Kti12 protein may have regulatory importance without diminishing its basal ATPase function. To systematically defy the conservation of the Walker A motif, further bioinformatical research is needed to pinpoint similar substitutions occuring in other kinases and proteomes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the MCB structural biology core facility (supported by the TEAM TECH CORE FACILITY/2017-4/6 grant from Foundation for Polish Science) for providing computational resources. This work was supported by the OPUS16 grant UMO-2018/31/B/NZ1/03559 (R.K. and S.G.) from the Polish National Science Centre, First Team Grant (FirstTEAM/2016-1/2; A.C-G, S.G.) from the Foundation for Polish Science and by the grant BR921/9 from the Deutsche Forschungsgemeinschaft (DFG) to K.D.B. Additionally, we would like to thank Katja Dieckow and Carolin Kleindienst for providing strains KDY2 and CKY40.
Author contributions
RK designed and conducted machine-learning approaches, performed all protein purifications and biochemical analyses. ACH produced and purified in vitro transcribed tRNA. ART constructed the yeast strains, performed the in vivo functional assays and did sequence alignments; RK prepared figures with input from SG. RK, KDB and SG wrote the final text of the manuscript with the input from all authors.
Compliance with ethical standards
Conflict of interest
Authors declare no conflict of interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rościsław Krutyhołowa and Annekathrin Reinhardt-Tews contributed equally.
Contributor Information
Karin D. Breunig, Email: karin.breunig@serymun.com
Sebastian Glatt, Email: sebastian.glatt@uj.edu.pl.
References
- Berto G, Feirreira-Cerca S, De Wulf P. The Rio1 protein kinases/ATPases: conserved regulators of growth, devison and cellular stability. Curr Genet. 2019;65:457–466. doi: 10.1007/s00294-018-0912-y. [DOI] [PubMed] [Google Scholar]
- Butler AR, O’Donnell RW, Martin VJ, et al. Kluyveromyces lactis toxin has an essential chitinase activity. Eur J Biochem. 1991;199:483–488. doi: 10.1111/j.1432-1033.1991.tb16147.x. [DOI] [PubMed] [Google Scholar]
- Butler AR, White JH, Stark MJ. Analysis of the response of Saccharomycescerevisiae cells to Kluyveromyces lactis toxin. J Gen Microbiol. 1991;137:1749–1757. doi: 10.1099/00221287-137-7-1749. [DOI] [PubMed] [Google Scholar]
- Butlera R, White JH, Folawiyo Y, et al. Two Saccharomycescerevisiae genes which control sensitivity to G1 arrest induced by Kluyveromyces lactis toxin. Mol Cell Biol. 1994;14:6306–6316. doi: 10.1128/MCB.14.9.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaverra M, George L, Mergy M, et al. The familial dysautonomia disease gene, Ikbkap/Elp1, is required in the developing and adult central nervous system. Dis Model Mech. 2017 doi: 10.1242/dmm.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang H, Jablonowski D, et al. Mutations in ABO1/ELO2, a subunit of Holo-Elongator, increase abscisic acid sensitivity and drought tolerance in Arabidopsis thaliana. Mol Cell Biol. 2006;26:6902–6912. doi: 10.1128/MCB.00433-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Itoh Y, Sekine S-I, Yokoyama S. Structural basis for the major role of O-Phosphoseryl-tRNA kinase in the UGA-specific encoding of selenocysteine. Mol Cell. 2010;39:410–420. doi: 10.1016/j.molcel.2010.07.018. [DOI] [PubMed] [Google Scholar]
- Close P, Hawkes N, Cornez I, et al. Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell. 2006;22:521–531. doi: 10.1016/j.molcel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauden MI, Kosinski J, Kolaj-Robin O, et al. Architecture of the yeast Elongator complex. EMBO Rep. 2017;18:264–279. doi: 10.15252/embr.201643353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauden MI, Jaciuk M, Muller CW, Glatt S. Structural asymmetry in the eukaryotic Elongator complex. FEBS Lett. 2018;592:502–515. doi: 10.1002/1873-3468.12865. [DOI] [PubMed] [Google Scholar]
- Dauden MI, Jaciuk M, Weis F, et al. Molecular basis of tRNA recognition by the Elongator complex. Sci Adv. 2019;5:1–14. doi: 10.1126/sciadv.aaw2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution (N Y) 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Fichtner L, Frohloff F, Bürkner K, et al. Molecular analysis of KTI12/TOT4, a Saccharomycescerevisiae gene required for Kluyveromyces lactis zymocin action. Mol Microbiol. 2002;43:783–791. doi: 10.1046/j.1365-2958.2002.02794.x. [DOI] [PubMed] [Google Scholar]
- Frohloff F, Fichtner L, Jablonowski D, et al. Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 2001;20:1993–2003. doi: 10.1093/emboj/20.8.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt S, Letoquart J, Faux C, et al. The Elongator subcomplex Elp456 is a hexameric RecA-like ATPase. Nat Struct Mol Biol. 2012;19:314–320. doi: 10.1038/nsmb.2234. [DOI] [PubMed] [Google Scholar]
- Glatt S, Zabel R, Kolaj-Robin O, et al. Structural basis for tRNA modification by Elp3 from Dehalococcoides mccartyi. Nat Struct Mol Biol. 2016;23:794–802. doi: 10.1038/nsmb.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffena J, Lefcort F, Zhang Y, et al. Elongator and codon bias regulate protein levels in mammalian peripheral neurons. Nat Commun. 2018;9:889. doi: 10.1038/s41467-018-03221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemans D, van Belzen IAEM, Morán Luengo T, Rüdiger SGD. A script to highlight hydrophobicity and charge on protein surfaces. Front Mol Biosci. 2015;2:56. doi: 10.3389/fmolb.2015.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawer H, Hammermeister A, Ravichandran K, et al. Roles of Elongator dependent tRNA modification pathways in neurodegeneration and cancer. Genes (Basel) 2018;10:19. doi: 10.3390/genes10010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Johansson MJO, Bystrom AS, et al. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Johansson MJO, Byström AS. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonowski D, Zink S, Mehlgarten C, et al. tRNAGlu wobble uridine methylation by Trm9 identifies Elongator’s key role for zymocin-induced cell death in yeast. Mol Microbiol. 2006;59:677–688. doi: 10.1111/j.1365-2958.2005.04972.x. [DOI] [PubMed] [Google Scholar]
- Karlsborn T, Tükenmez H, Mahmud AKMF, et al. Elongator, a conserved complex required for wobble uridine modifications in eukaryotes. RNA Biol. 2014 doi: 10.4161/15476286.2014.992276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kishida M, Tokunaga M, Katayose Y, et al. Isolation and genetic characterization of pGKL killer-insensitive mutants (iki) from Saccharomyces cerevisiae. Biosci Biotechnol Biochem. 1996;60:798–801. doi: 10.1271/bbb.60.798. [DOI] [PubMed] [Google Scholar]
- Klassen R, Wemhoff S, Krause J, Meinhardt F. DNA repair defects sensitize cells to anticodon nuclease yeast killer toxins. Mol Genet Genomics. 2011;285:185–195. doi: 10.1007/s00438-010-0597-5. [DOI] [PubMed] [Google Scholar]
- Koç A, Wheeler LJ, Mathews CK, Merrill GF. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem. 2004;279:223–230. doi: 10.1074/jbc.M303952200. [DOI] [PubMed] [Google Scholar]
- Kojic M, Gaik M, Kiska B, et al. Elongator mutation in mice induces neurodegeneration and ataxia-like behavior. Nat Commun. 2018;9:3195. doi: 10.1038/s41467-018-05765-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutyhołowa R, Hammermeister A, Zabel R, et al. Kti12, a PSTK-like tRNA dependent ATPase essential for tRNA modification by Elongator. Nucleic Acids Res. 2019 doi: 10.1093/nar/gkz190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutyhołowa R, Zakrzewski K, Glatt S. Charging the code—tRNA modification complexes. Curr Opin Struct Biol. 2019;55:138–146. doi: 10.1016/j.sbi.2019.03.014. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S, Pedrioli PG, Bucher T, et al. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature. 2009;458:228–232. doi: 10.1038/nature07643. [DOI] [PubMed] [Google Scholar]
- Leipe DD, Koonin EV, Aravind L. Evolution and classification of P-loop kinases and related proteins. J Mol Biol. 2003;333:781–815. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Lin T-Y, Abbassi NEH, Zakrzewski K, et al. The Elongator subunit Elp3 is a non-canonical tRNA acetyltransferase. Nat Commun. 2019;10:625. doi: 10.1038/s41467-019-08579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Huang BO, Esberg A, et al. The Kluyveromyces lactis γ-toxin targets tRNA anticodons. RNA. 2005;11:1648–1654. doi: 10.1261/rna.2172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlgarten C, Jablonowski D, Wrackmeyer U, et al. Elongator function in tRNA wobble uridine modification is conserved between yeast and plants. Mol Microbiol. 2010;77:1082–1094. doi: 10.1111/j.1365-2958.2010.07163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell MS, Rao VB. Novel and deviant Walker A ATP-binding motifs in bacteriophage large terminase-DNA packaging proteins. Virology. 2004;321:217–221. doi: 10.1016/j.virol.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell. 2015;161:1606–1618. doi: 10.1016/j.cell.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen H, Fleury D, Bruno L, et al. The elongata mutants identify a functional Elongator complex in plants with a role in cell proliferation during organ growth. Proc Natl Acad Sci. 2005;102:7754–7759. doi: 10.1073/pnas.0502600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Sakaguchi Y, Suzuki T. Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res. 2009;37:1335–1352. doi: 10.1093/nar/gkn1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Michel V, Grisel O, et al. Scikit-learn: Machine Learning in Python. J Mach Res. 2011;12:2825–2830. [Google Scholar]
- Petrakis TG, Søgaard TMM, Erdjument-Bromage H, et al. Physical and functional interaction between elongator and the chromatin-associated Kti12 protein. J Biol Chem. 2005;280:19454–19460. doi: 10.1074/jbc.M413373200. [DOI] [PubMed] [Google Scholar]
- Ranjan N, Rodnina MV. tRNA wobble modifications and protein homeostasis. Transl. 2016;4:e1143076. doi: 10.1080/21690731.2016.1143076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffrath R, Mehlgarten C, Prochaska H, et al. Use of a yeast tRNase killer toxin to diagnose Kti12 motifs required for tRNA modification by Elongator. Toxins (Basel) 2017;9:1–13. doi: 10.3390/toxins9090272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L. The PyMOL molecular graphics system. Version. 2015;1:8. [Google Scholar]
- Setiaputra DT, Cheng DT, Lu S, et al. Molecular architecture of the yeast Elongator complex reveals an unexpected asymmetric subunit arrangement. EMBO Rep. 2017;18:280–291. doi: 10.15252/embr.201642548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Mark P, Prevost CT, et al. Biallelic variants in CTU2 cause DREAM-PL syndrome and impair thiolation of tRNA wobble U34. Hum Mutat. 2019;40:2108–2120. doi: 10.1002/humu.23870. [DOI] [PubMed] [Google Scholar]
- Sherrer RL, O’Donoghue P, Söll D. Characterization and evolutionary history of an archaeal kinase involved in selenocysteinyl-tRNA formation. Nucleic Acids Res. 2008;36:1247–1259. doi: 10.1093/nar/gkm1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songe-Møller L, Van Den Born E, Leihne V, et al. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol. 2010;30:1814–1827. doi: 10.1128/MCB.01602-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark MJ, Boyd A. The killer toxin of Kluyveromyces lactis: characterization of the toxin subunits and identification of the genes which encode them. EMBO J. 1986;5:1995–2002. doi: 10.1002/j.1460-2075.1986.tb04455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studte P, Zink S, Jablonowski D, et al. tNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol Microbiol. 2008;69:1266–1277. doi: 10.1111/j.1365-2958.2008.06358.x. [DOI] [PubMed] [Google Scholar]
- van Tran N, Muller L, Ross RL, et al. Evolutionary insights into Trm112-methyltransferase holoenzymes involved in translation between archaea and eukaryotes. Nucleic Acids Res. 2018;46:8483–8499. doi: 10.1093/nar/gky638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, et al. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modeling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, et al. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.