

Summary
Phenotypes of haploid embryonic stem cells (haESCs) are dominant for recessive traits in mice. However, one major obstacle to their use is self-diploidization in daily culture. Although haESCs maintain haploidy well by deleting p53, whether they can sustain haploidy in differentiated status and the mechanism behind it remain unknown. To address this, we induced p53-deficient haESCs into multiple differentiated lineages maintain haploid status in vitro. Haploid cells also remained in chimeric embryos and teratomas arising from p53-null haESCs. Transcriptome analysis revealed that apoptosis genes were downregulated in p53-null haESCs compared with that in wild-type haESCs. Finally, we knocked out p73, another apoptosis-related gene, and observed stabilization of haploidy in haESCs. These results indicated that the main mechanism of diploidization was apoptosis-related gene-triggered cell death in haploid cell cultures. Thus, we can derive haploid somatic cells by manipulating the apoptosis gene, facilitating genetic screens of lineage-specific development.
Keywords: p53, haESCs, self-diploidization, apoptosis, haploidy, p73
Graphical Abstract
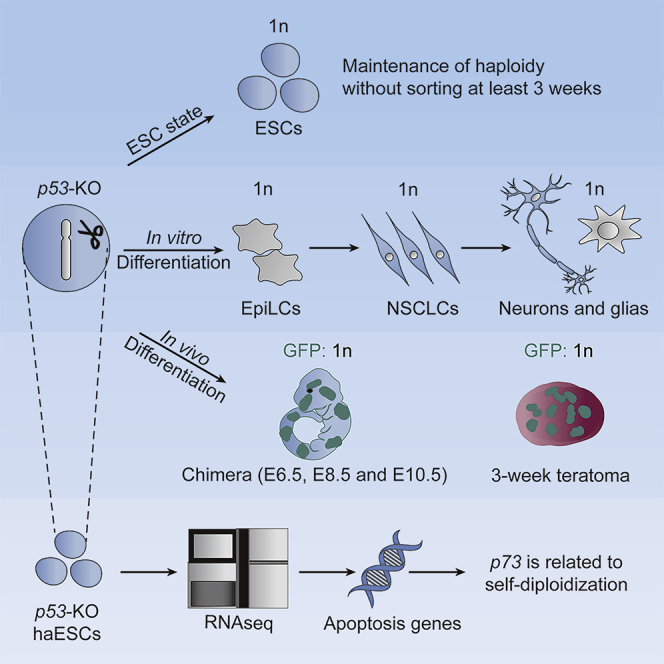
Highlights
-
•
haEpiLCs and haNSCLCs differentiated from p53-null haESCs in vitro
-
•
p53-null haESCs contributed to chimeric embryos and teratoma
-
•
Downregulation of apoptosis genes resulted in haploidy stabilization
-
•
Deletion of p73 was also of benefit for haploidy sustenance
In this article, Shuai and colleagues showed that p53-null haESCs could sustain haploidy in differentiated status both in vitro and in vivo, and the main reason for diploidization was apoptosis gene-triggered cell death in haploid cell cultures. Through modulating apoptosis genes, desired haploid somatic cells could be derived easily, which facilitated genetic screening in lineage-specific development.
Introduction
Sexually reproductive diploid animals exhibit evolutionary advantages over asexual organisms due to meiosis and the combination of two haploid gametes from different genetic backgrounds (Kondrashov and Crow, 1991, Perrot et al., 1991). Diploid genomes have allelic backups, but this hinders the elucidation of the gene function of recessive traits. Recently developed haploid cell lines, including near-haploid tumor cells (Carette et al., 2009, Gibbons et al., 1991) and haploid embryonic stem cells (haESCs) (Leeb and Wutz, 2011, Sagi et al., 2016), greatly facilitate forward and reverse genetic screening in mammalian species (Li and Shuai, 2017). It is easy to identify not only toxicant-targeting genes (Elling et al., 2011) but also crucial genes regulating critical biological processes (such as differentiation [Leeb et al., 2014] or X chromosome inactivation [Monfort et al., 2015]) through high-throughput screening in haESCs. However, daily cultured haESCs tend to revert to diploid status, and periodic sorting for haploids is necessary, which hampers the application of these cell lines in genetic screening. In addition, haESCs undergo more severe diploidization during differentiation both in vitro and in vivo, making the derivation of haploid lineage-specific somatic cells very difficult (Shuai and Zhou, 2014). Therefore, spontaneous diploidization of haESCs jeopardizes the exploration of the application of haploid technology in mammalian organ development or disease-related genetic screening despite their original single-genome status.
Many efforts have been made to clarify the mechanism underlying the diploidization of mammalian haESCs; however, no precise regulatory pathway has yet been found. A previous report showed that the reason for the diploidization of mouse haESCs was a mistake in their cell cycle rather than the fusion of two single haploid cells, as indicated by the coculturing of haESCs with different marker genes (Leeb et al., 2012). Thereafter, chemical compounds inhibiting cell-cycle-related proteins were introduced into the culture medium to reduce diploidization (He et al., 2017, Takahashi et al., 2014), which still did not solve the problem completely. Single-cell dynamic analysis of mitosis in haESCs demonstrated that the metaphase of these cells was significantly longer than that of diploid ESCs (Guo et al., 2017). Thus, delayed mitosis might be the main cause of self-diploidization in haESCs. In this case, time-consuming and complicated fluorescence-activated cell sorting (FACS) was necessary to maintain the haploidy of haESCs. Recent studies showed that knockout of p53 (Olbrich et al., 2017) or overexpression of Dnmt3b (He et al., 2018) could stabilize the haploid genome and slow down self-diploidization. In particular, p53-null haESCs could sustain haploid status well both under daily culture and in cells converting to an extraembryonic cell fate (Peng et al., 2019). p53 is an essential gene related to the cell cycle, DNA damage, and other crucial biological processes (Aladjem et al., 1998, Cross et al., 1995, Fujiwara et al., 2005). Nevertheless, whether the p53-null mutation is beneficial for the maintenance of a haploid status during differentiation is still unknown. The exact mechanism by which p53-null reduces self-diploidization is also not clear.
In this study, we deleted p53 in mouse parthenogenetic haESCs with the CRISPR/Cas9 system and performed differentiation both in vitro and in vivo to assess whether p53-null stabilized haploidy in the somatic stage. The results revealed that by deleting p53, haploid somatic cells could be easily obtained from differentiated haESCs. We further analyzed global transcriptome data and found that genes regulating apoptosis were the main reason for diploidization during the differentiation of haESCs.
Results
p53-Null HaESCs Maintain Haploidy Much More Stably Compared with Wild-Type HaESCs under Long-Term Culture
We generated two mouse parthenogenetic haESC lines from chemically activated oocytes according to a previous protocol (Shuai et al., 2014). These haESCs showed a standard mouse ESC morphology (Figure S1A) and grew well with the haploid genome assisted by FACS (Figure S1B). To introduce p53 mutations in haESCs, we designed two Cas9-mediated p53-single guide RNA (sgRNA) vectors to perform electroporation (Figure S1C). Two days after transfection, approximately 58.6% of the analyzed cells were Cas9-GFP-positive (Figure S1D), and these cells were harvested for further culturing. We randomly picked three subclones from the sorted cultures and determined whether they were gene edited. All three subclones (p53 KO-1, p53 KO-2, and p53 KO-3) carried different mutations in the p53 gene according to DNA sequencing (Figure 1A). The T7EN1 cleavage assay results also confirmed this (Figure 1B). To assess whether the P53 protein was lost, we performed western blotting in p53 KO-1, p53 KO-2, p53 KO-3, wild-type (WT) haESCs, and WT diploid ESCs (WT-diESCs). The results demonstrated that P53 was absent in all three p53-KO haESC lines (Figure 1C).
Figure 1.

p53 Deletion in haESC and DNA Analysis during Proliferation
(A). p53-deleted genotypes in subclones p53 KO-1, p53 KO-2, and p53 KO-3.
(B) T7ENI cleavage analysis of the p53-KO lines p53 KO-1, p53 KO-2, and p53 KO-3. Cleaved products (red arrowheads) indicate the presence of mutations.
(C) Western blotting to detect P53 in WT-diESCs, WT-haESCs, p53 KO-1, p53 KO-2, and p53 KO-3 cells. GAPDH was used as a loading control.
(D) Expression levels of pluripotent, p53-related, and cell-cycle-related genes (Oct4, Nanog, p53, p21, CyclinB, Cdk2, and MAPK11) in WT-haESCs, p53 KO-1, p53 KO-2, and p53 KO-3 cells determined by qPCR (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) DNA content analysis of p53-KO haESCs during culture on day 0, day 14, and day 21 without sorting, with WT-haESCs as a control. The percentages of the 1n (G0/G1) peak in WT-haESCs were 27.4%, 9.33%, and 2.4%, respectively; in p53 KO-1 cells, 23.5%, 22.8%, and 21.4%, respectively; in p53 KO-2 cells, 23.6%, 21.1%, and 20.9%, respectively; in p53 KO-3 cells, 23.7%, 21.5%, and 20.2%, respectively.
(F) Percentage of GFP+ cells in p53 KO-1-p53 OE cells after transfection. WT-haESCs without transfection were used as a negative control.
(G) Bright-field (BF) and FITC images of p53 KO-1-p53 OE cells, Scale bar, 100 μm.
(H) Expression levels of p53 in WT-haESCs, p53 KO-1-Vector, p53 KO-1, and p53 KO-1-p53 OE cells determined by qPCR (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
(I) Percentages of haploids in p53 KO-1, p53 KO-1-Vector, WT-haESCs, and p53 KO-1-p53 OE cells 10 days after sorting. Nearly 44% of sorted cells remained in haploidy in p53 KO-1 and p53 KO-1-Vector group, whereas WT-haESCs and p53 KO-1-p53 OE exhibited only 29% staying as haploid cells (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
To determine whether p53 mutation affects the pluripotency of haESCs, we examined specific markers and alkaline phosphatase activity in p53-KO haESCs. Positivity was observed not only for pluripotency markers (OCT4, NANOG, and SSEA-1) but also for alkaline phosphatase in p53-KO haESCs (Figures S1E and S1F). Karyotype analysis showed that most chromosome spreads of p53-KO haESCs contained 20 chromosomes (Figure S1G). Next, we analyzed pluripotent genes and p53-related genes in p53-KO haESCs by real-time PCR with WT-haESCs as a control. The expression levels of pluripotent markers (Oct4 and Nanog) and Cyclin B did not change significantly in p53-KO haESCs compared with WT-haESCs. However, expression of p53 and p21 decreased, meanwhile Cdk2 and MAPK11 increased significantly in all three p53-KO haESC lines (Figure 1D). This result indicated that p53 mutations did not affect pluripotency but do affect p53-related genes, including some cell-cycle genes. Since p53 deletion increased haploidy maintenance in the ESC stage, as previously described, we tested whether p53-KO haESCs exhibited advantages in haploidy maintenance in daily culture without sorting for 3 weeks. There was still a high percentage of haploid cells remaining in the 1n peak (G0/G1 phase) of p53-KO haESCs (p53 KO-1, 21.4%; p53 KO-2, 20.9%; p53 KO-3, 20.2%), whereas the percentage of the 1n peak (G0/G1 phase of haploid cells) in WT-haESCs decreased to 2.4% (Figure 1E). Here, we chose p53 KO-1 (the most stable) for subsequent experiments. We designed p53 overexpression (p53 OE) vectors (Figure S1H) and transferred them into p53 KO-1 cells having a high proportion of haploids, with an empty vector as control. After transfection, transfected cells expressing exogenous GFP (a GFP cassette was included in the p53 OE vector or empty vector) were enriched for GFP and haploidy double-positive cells by FACS (Figures 1F and 1G). Real-time PCR result revealed that p53 OE-haESCs (p53 KO-1-p53 OE) had re-expressed p53 gene compared with WT-haESCs, p53 KO-1, and p53 KO-1-vector (Figure 1H). Western blotting analysis further confirmed this result (Figure S1I). Thereafter, we checked the haploidy-maintaining ability among p53 KO-1, p53 KO-1-vector, WT-haESCs, and p53 KO-1-p53 OE cells without sorting for 10 days (all the samples started as 100% of haploid cells). DNA-content analysis demonstrated that only p53 KO-1 and p53 KO-1-vector were able to maintain haploidy stably in this period, whereas p53 KO-1-p53 OE and WT-haESCs diploidized to some degree (Figure 1I). The rescue of p53 expression in p53-KO haESCs made them unable to sustain the phenotype of stable haploidy any longer, which proved that p53 was the main trigger of the diploidization phenomenon. As Cdk2 and MAPK11 (two important cell-cycle genes) were upregulated in p53-KO haESCs according to the above qPCR analysis, we conducted overexpression (OE) of Cdk2 and MAPK11 separately in WT-haESCs to address whether either of the two genes would affect the process of diploidization. Although the OE-haESCs showed upregulation of Cdk2 and MAPK11 independently (Figures S1J and S1K), neither of them obtained an advantage in maintaining haploidy like p53-KO in long-term cell culture (Figure S1L).
p53-Null HaESCs Differentiate Reliably with a Haploid Genome In Vitro
To examine the haploid-maintaining ability of p53-KO haESCs during differentiation in vitro, we performed differentiation of p53-KO haESCs at different stages. Standard embryoid bodies (EBs) formed (Figure 2A), and 29.7% of haploid cells (at G0/G1 phase) were observed in 7-day EBs from p53-KO haESCs (Figure 2B). The expression levels of pluripotency genes and specific genes of the three germ layers showed that the haploid EBs had differentiated beyond the ESC stage (Figure 2C). Although WT parthenogenetic haESCs were not able to remain in the epiblast stem cell-like cell (EpiLC) stage with a haploid genome (Leeb et al., 2012), p53-KO haESCs differentiated into haploid EpiLCs (haEpiLCs) quite easily and stably sustained haploidy (Figures 2D and 2E). The primed state haEpiLCs expressed pluripotent markers such as OCT4 and NANOG (Figure 2F). Besides, naive-primed distinguishing marker TFE3 (Betschinger et al., 2013) was located in the cytoplasm of haEpiLCs (Figure 2G), demonstrating that p53-KO haEpiLCs could remain in the epiblast stage with a single genome in the presence of basic fibroblast growth factor (bFGF) and activin A. The expression levels of specific genes further proved that these haploid cultures expressed primed stage genes (FGF5 and Otx2), rather than naive genes (Rex1 and Klf4), with WT-haESCs and WT epiblast stem cells (WT-EpiSCs) as controls (Figures 2H and 2I). To assess differentiation potentials of haEpiLCs, we induced neural differentiation according to a previous protocol (Ying et al., 2003). Around 7–10 days post differentiation, NESTIN- and TUJ1-positive cells were observed in the cell cultures (Figure 2J), indicating that haEpiLCs possessed pluripotency to differentiate to the next stage.
Figure 2.

In Vitro Differentiation of p53-KO haESCs
(A) Morphology of 7-day haEBs derived from p53 KO-1 cells. Scale bar, 100 μm.
(B) DNA-content analysis of 7-day haEBs derived from p53 KO-1 cells. The percentage of the 1n (G0/G1) peak in haEBs was 21.7%.
(C) Expression levels of pluripotency and differentiation genes (Rex1, Hand1, Cdx2, Fgf5, Nestin, and T) in haEBs and haESCs determined by qPCR (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
(D) Morphology of haEpiLCs derived from p53 KO-1 cells. Scale bar, 100 μm.
(E) DNA-content analysis of haEpiLCs derived from p53 KO-1 cells. The percentage of the 1n (G0/G1) peak in haEpiLCs was 21.8%.
(F) Immunofluorescence of OCT4 and NANOG in haEpiLCs derived from p53 KO-1 cells. DNA was stained with DAPI. Scale bar, 50 μm.
(G) Immunofluorescence of TFE3 in haEpiLCs derived from p53 KO-1 cells and haESCs. DNA was stained with DAPI. Scale bar, 50 μm.
(H) Expression levels of pluripotency genes (Oct4, Nanog, Rex1, and Klf4) in WT-haESCs, WT-EpiSCs, and haEpiLCs determined by qPCR (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
(I) Expression levels of EpiSC-specific marker genes (Fgf5 and Otx2) in WT-haESCs, WT-EpiSCs and haEpiLCs determined by qPCR (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
(J) Immunostaining of NESTIN and TUJ1 in 10-day differentiation cell cultures of haEpiLCs. Scale bar, 50 μm.
(K) Morphology of haNSCLCs derived from p53 KO-1 cells. Scale bar, 100 μm.
(L) DNA-content analysis of haNSCLCs derived from p53 KO-1 cells. The percentage of the 1n (G0/G1) peak in haNSCLCs was 28.5%.
(M) Immunofluorescence of OCT4 and NANOG in haNSCLCs. DNA was stained with DAPI. Scale bar, 50 μm.
(N) Immunofluorescence of neural stem cell-specific markers in haNSCLCs, PAX6, SOX1, and NESTIN. DNA was stained with DAPI. Scale bar, 50 μm.
(O) Immunostaining of oligodendrocytes (O4) and astrocytes (GFAP) derived from p53 KO-1. Scale bar, 50 μm.
(P) Immunostaining of neuronal specific markers (MAP2, NEUN, and TUJ1) in neurons derived from p53 KO-1. Scale bar, 50 μm.
Although haploid neural progenitor cells (Elling et al., 2011) and haploid neurons (Xu et al., 2017) could be detected transiently, it is very difficult to obtain stable haploid neural stem cell-like cells (NSCLCs) by traditional methods. Whether p53-KO haESCs could differentiate into somatic stages and maintain haploidy required further investigation. We introduced endogenous NSCLC-specific Pax6-GFP into p53-KO haESCs according to our previous protocol (Li et al., 2018b). We performed neural differentiation of p53 KO-1 cells and found that there were 22.3% of Pax6-GFP-positive cells among the differentiated derivatives (Figure S2A). Interestingly, there were 6.2% of Pax6-GFP-positive cells in the 1n peak, whereas there were 27.8% of Pax6-GFP-negative cells in the 1n peak (Figure S2B). These findings demonstrated that diploidization occurred but was slowed down during the differentiation of p53-KO haESCs. Haploid and Pax6-GFP double-positive cells were enriched by FACS to derive haNSCLCs for further culture. The haNSCLCs showed the standard neural stem cell morphology when cultured under adherent conditions (Figure 2K), presenting the ability to aggregate into neural spheres (Figure S2C). DNA analysis revealed that the haNSCLCs could stably maintain haploidy during proliferation (Figure 2L). Next, we examined pluripotent and neural stem cell-specific markers in haNSCLCs by immunostaining. Pluripotent markers (OCT4 and NANOG) were absent in haNSCLCs (Figure 2M). Positivity for PAX6, SOX1, and NESTIN was observed in adherent haNSCLCs (Figure 2N) and their neural spheres (Figure S2D). To assess the differentiation potential of haNSCLCs, we performed neural lineage specification. The results showed that glial cells (O4 and glial fibrillary acidic protein [GFAP] positive) and multiple neurons (MAP2, NEUN, and TUJ1 positive) could be generated from haNSCLCs (Figures 2O and 2P). In another parallel experiment, we analyzed DNA contents of glial mixture and neuronal mixture independently. There was 2.0% of glial mixture and 4.5% of neuronal mixture remaining in the 1n peak, respectively (Figures S2E and S2F), indicating that haNSCLCs possessed multipotency for neural lineages with a haploid genome.
Haploid Cells Can Contribute to Chimeric Fetuses and Teratomas upon p53 Knockout
The finding that p53-KO haESCs showed excellent haploidy maintenance during differentiation in vitro led to the further investigation of whether they presented the same advantages during differentiation in vivo. We microinjected GFP-labeling p53-KO haESCs into blastocysts to construct chimeric embryos. The reconstructed embryos were transferred to the oviducts of 0.5 days post coitum (dpc) pseudopregnant mice. Chimeric embryonic day 6.5 (E6.5) embryos from GFP-labeling p53-KO haESCs and GFP-labeling WT-haESCs were obtained (Figures 3A and S3A). Around 17.2% cells of chimeric E6.5 1# embryo and 55.5% cells of chimeric E6.5 2# embryo generated from p53-KO haESCs microinjection were GFP-positive cells (Table S1). The haploid cells in the 1n peak of p53-KO haESCs-chimeric E6.5 embryos (p53 KO-1, chimeric 1#, 28.5%; p53 KO-2, chimeric 2#, 24.5%) were much higher than that in WT-haESCs-chimeric E6.5 embryo (WT-haESCs-chimeric, 4.7%) (Figures 3B and S3B; Table S1), demonstrating that p53-KO haESCs could contribute to an epiblast stage with a high proportion of haploid status. For chimeric E8.5 embryos, 12.4% cells of 1# embryo and 37.6% cells of 2# embryo from p53-KO haESCs microinjection were GFP-positive cells (Table S1). However, chimeric E8.5 embryos from p53-KO haESCs were abnormal (Figures 3C and S3C), possibly due to the high contribution of haploid cells (a haploid genome might not support full development) or deficiency of p53, with approximately 25% of these cells (p53 KO-1, chimeric 1#, 25.3%; p53 KO-2, chimeric 2#, 24.3%) remaining in the 1n peak (Figures 3D and S3D). To address the identity of these haploid cells from E8.5 embryos, we harvested them by FACS and performed qPCR analysis. Compared with WT-haESCs, the sorted haploid cells barely expressed any of the pluripotent markers (Oct4, Nanog, Stella, Rex1, and Klf4), demonstrating that they were indeed differentiated cells (Figure 3E). Thus, p53-KO haESCs could differentiate into a somite stage without diploidization. To investigate the effect of p53 deletion on development, we microinjected GFP-labeling p53-KO diploid ESCs into blastocysts as a parallel experiment. Interestingly, chimeric E8.5 embryos from p53-KO diploid ESCs were normal (Figure 3F), with GFP cells (24.9% in 1# and 40.3% in 2#) contributing to the chimeric E8.5 embryos (Table S1). Furthermore, abnormal chimeric E10.5 embryos were also able to be obtained (Figures 3G and S3E). The percentages of GFP cells in chimeric E10.5 1# and 2# embryos from p53-KO haESCs microinjection were 1.6% and 3.3%, respectively (Table S1). According to DNA analysis, there were still around 22% of haploid cells in 1n peak (p53 KO-1, chimeric 1#, 21.4%; p53 KO-2, chimeric 2#, 22.3%) from E10.5 GFP-positive cells (Figures 3H and S3F), indicating that p53-KO haESCs could further differentiate in a haploid genome with limited development potentials.
Figure 3.

In Vivo Analysis of p53-KO haESCs
(A) Morphology of chimeric embryos at E6.5 derived from GFP-labeling WT-haESCs and p53 KO-1 cells. Scale bar, 100 μm. WT-E6.5 embryo was used as a control.
(B) DNA-content analysis of GFP+ cells and GFP− cells in chimeric E6.5 embryos derived from GFP-labeling WT-haESCs and p53 KO-1 cells. The percentages of the 1n (G0/G1) peak in GFP+ cells from p53 KO-1 E6.5 and WT-haESCs E6.5 were 28.5% and 4.7%, respectively.
(C) Morphology of chimeric embryo at E8.5 derived from GFP-labeling p53 KO-1 cells. WT-E8.5 embryo was used as a control.
(D) DNA-content analysis of GFP+ cells and GFP− cells in chimeric E8.5 embryo derived from GFP-labeling p53 KO-1 cells. The percentage of the 1n (G0/G1) peak from p53 KO-1 chimeric E8.5 GFP+ cells was 25.3%.
(E) Expression levels of pluripotency genes (Oct4, Nanog, Stella, Rex1, and Klf4) in chimeric E8.5 haploid cells from GFP+ cells, with WT-haESCs as a loading control (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) Morphology of chimeric embryos at E8.5 derived from GFP-labeling p53 KO-diploid cells.
(G) Morphology of chimeric embryos at E10.5 derived from GFP-labeling p53 KO-1 cells. WT E10.5 was used as a control.
(H) DNA-content analysis of GFP+ cells and GFP− cells in chimeric E10.5 embryo derived from GFP-labeling p53 KO-1 cells. The percentage of the 1n (G0/G1) peak from p53 KO-1 chimeric E10.5 GFP+ cells was 21.4%.
(I) Teratoma formed from p53 KO haESCs was identified by H&E staining. Scale bar, 100 μm. The tissues shown were gut epithelium (endoderm), muscle (mesoderm), and neural tissue (ectoderm).
(J) DNA-content analysis of teratoma. The percentage of the 1n (G0/G1) peak from all the tissue was 3.1%.
(K) Expression levels of pluripotent genes (Oct4, Nanog, Stella, Rex1, and Klf4) in the 1n cells sorted from teratoma determined by qPCR, with WT-haESCs as a loading control (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
To check the pluripotency of p53-KO haESCs to form the three germ layers, we injected approximately 1 × 107 p53 KO-1 cells under the skin of severe combined immunodeficiency (SCID) mice. Three weeks later, a standard teratoma formed (Figures S3G and S3H) and was divided into two parts. Histological analysis was performed on one part, and DNA-content analysis was performed on the other. Gut epithelium, muscle, and neural tube tissues, representing the three germ layers, were observed by H&E staining (Figure 3I). According to the DNA-content analysis, 3.1% haploid cells (1n peak) still existed in the 3-week teratoma, demonstrating that the p53-KO haESCs show a powerful ability to maintain haploidy in vivo (Figure 3J). The haploid cells from teratoma were also sorted by FACS and checked by qPCR. The result showed that the haploid cells in teratoma did not express the pluripotent marker genes (Oct4, Nanog, Stella, Rex1, and Klf4), indicating that they had entered a differentiated state instead of a pluripotent state (Figure 3K).
Transcriptome Analysis of p53-KO HaESCs
To determine why p53-KO haESCs show great advantages in haploidy maintenance, we compared the transcriptomes between p53-KO haESCs and WT-haESCs by RNA-sequencing (RNA-seq) analysis. The results revealed a very high correlation (R2 = 0.9947) between p53-KO haESCs (p53 KO-1, p53 KO-2, and p53 KO-3) and WT-haESCs (three independent WT-haESC lines) (Figure 4A). According to comprehensive summaries with all expression data (Tables S2 and S3), there were 2,785 differentially expressed genes (p < 0.01, fold change >2) between p53-KO haESCs and WT-haESCs, which were further analyzed to elucidate the reason of haploidy stabilization. Hierarchical clustering (heatmap) showed the top 50 differentially expressed genes between p53-KO haESCs and WT-haESCs, considering a fold change of 2.0 and p < 0.05 (Figure S4A and Table S2). These differentially expressed genes were mainly related to the extracellular matrix and basic development according to gene ontology (GO) analysis (Figures S4B and S4C). GO analysis data further indicated that most of the upregulated genes were correlated with male meiotic nuclear division, Piwi-interacting RNA metabolic process, and other biological processes, whereas the downregulated genes were related to vasculature development and extracellular matrix organization (Figure 4B). Since p53 is a very important gene involved in multiple biological processes, including DNA-damage repair, cell-cycle arrest, and apoptosis (Hafner et al., 2019), we analyzed the expression of specific genes involved in these processes between p53-KO haESCs and WT-haESCs in volcano plots of p values (−log10 scale) versus fold changes (log2 scale). Interestingly, there were several differentially expressed genes related to apoptosis identified between the two groups (Figure 4C). However, we did not observe distinctly expressed genes related to the cell cycle or DNA damage in the RNA-seq data (Figure 4C). As we also found that the p53-KO haESCs showed better survival ability during differentiation compared with WT-haESCs in vitro (data not shown), we assessed carefully the differentially expressed genes correlated with apoptosis. TP73 (p73, also known as Trp73), Phlda3, Cdkn1a, and Bbc3 (downstream target genes of p53) were significantly downregulated in p53-KO haESCs (Figure 4D) and were listed as the top four downregulated genes in p53-KO haESCs related to apoptosis. We further detected the expression levels of some target genes of p53 related to apoptosis by qPCR and found that only p73 and Bbc3 were significantly downregulated (Figure 4E).
Figure 4.

Transcriptome of p53-KO haESCs
(A) Scatterplot of log2 transformed average gene expression profiles. Global gene expression of three p53-KO haESCs (p53 KO-1, p53 KO-2, and p53 KO-3) and three WT-haESC lines (WT-haESCs-1, WT-haESCs-2, and WT-haESCs-3) were analyzed by scatterplot (R2 is the Pearson correlation coefficient).
(B) Enriched gene ontologies of upregulated and downregulated genes in p53-KO haESCs.
(C) Volcano plot depicting the differential expression analysis between WT-haESCs and p53-KO haESCs. The higher location of the point of the gene, the more significant the difference. Highlighted points indicate genes correlated with apoptosis, cell cycle, and DNA damage, respectively, considering a fold change of 2.0 and p < 0.05.
(D) Top ten genes correlated with apoptosis in the differential expression analysis between WT-haESCs and p53 KO haESCs.
(E) Expression levels of apoptosis-related genes (p73, Phlda3, Bbc3, and Bax) and a proliferation-related gene (MAPK11) in WT-haESCs and p53-KO haESCs determined by qPCR (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗p < 0.05, ∗∗∗p < 0.001.
(F) Caspase-3 activity of p53 KO-1, p53 KO-2, and p53 KO-3 cells, with WT-haESCs as a control (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
To address whether apoptosis was the main reason for the diploidization of haESCs, we assessed the survival rate of p53-KO haESCs 24 h after sorting for haploids compared with WT-haESCs. Nearly 70.5% of the sorted cells survived and expanded in the p53-KO group, whereas in the control group this percentage was only 52.9% (Figure S4D). As caspase-3 is a key molecule in the process of apoptosis (Song et al., 2015), we measured the caspase-3 activity between p53-KO haESCs and WT-haESCs after sorting. The caspase-3 activities in p53 KO-1, p53 KO-2, and p53 KO-3 were significantly decreased compared with that in WT-haESCs (Figure 4F). In summary, apoptosis of p53-KO haESCs was reduced in daily culture and FACS processes.
p73 Is a Major Target of p53-Regulating Diploidization
To find out more about apoptosis-related gene-regulating diploidization, we designed p73 and Bbc3 knockout vectors (Figures S5A and S5B) and transfected them separately into WT-haESCs to introduce gene editing. Among the 60 randomly picked subclones of the p73-edited group, 25 subclones maintained haploidy (Figure S5C). We sequenced 11 subclones with high percentages of haploid cells and found that four of them were p73-KO cells (Figure 5A). To assess whether the P73 protein was lost, we performed western blotting in p73 KO-1, p73 KO-2, p73 KO-3, WT-haESCs, and diploid ESCs. The results demonstrated that P73 was absent in three p73-KO haESC lines (Figure 5B). However, in the Bbc3-edited group, 18 out of 60 randomly selected subclones exhibited haploid cells, 9 of which showed a high percentage of haploids, which were sequenced and did not contain a Bbc3 mutation (Figure S5C). We randomly checked two diploidized subclones by sequencing and found Bbc3 mutations (Figure S5D). DNA-content analysis again confirmed that the Bbc3 mutant subclones were diploid, without any haploid cells (Figure S5E). Notably, this result indicated that p73 alone was the key regulator of diploidization, rather than Bbc3. We further assessed whether p73-KO haESCs also showed advantages in haploidy maintenance similar to those of p53-KO haESCs. The results demonstrated that p73-KO haESCs exhibited better proliferation ability after sorting (Figure S5F) and a lower rate of diploidization during long-term culture (Figure 5C) compared with WT-haESCs. The caspase-3 activity in p73 KO-1, p73 KO-2, and p73 KO-3 was much lower than that in WT-haESCs, suggesting that apoptosis could be inhibited by p73-KO (Figure 5D). Next, we examined the expression of p53-targeted genes related to apoptosis in p73-KO haESCs (p73 KO-1, p73 KO-2 and p73 KO-3) and WT-haESCs and did not find any significant differences in the downstream gene (Figure 5E). This result indicated that p73 played a crucial role in affecting diploidization without interacting with its upstream genes, including p53.
Figure 5.

p73 Is a Major Target of p53-Regulating Diploidization
(A) p73-deleted genotypes in haESCs by DNA sequencing.
(B) Western blot to detect P73 in p73 KO-1 cells, p73 KO-2 cells, p73 KO-1 cells, WT-haESCs, and WT-diESCs. GAPDH was used as a loading control.
(C) Progression of the percentage of haploid cells in the p73-KO haESCs and WT-haESCs for 14 days, 28 days, and 35 days after sorting (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
(D) Caspase-3 activity of p73 KO-1, p73 KO-2, and p73 KO-3 cells, with WT-haESCs as a control (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗∗∗p < 0.001.
(E) Expression levels of apoptosis-related genes (p53, Bbc3, and Bax) and proliferation-related gene MAPK11 in p73 KO-1, p73 KO-2, and p73 KO-3 haESCs determined by qPCR, with WT-haESCs as a control (n = 3 independent experiments). Data presented as mean ± SEM. t test: ∗p < 0.05, ∗∗p < 0.01.
Discussion
Haploidy in lower organisms, including yeast, is fairly stable, which makes haploid individuals a powerful tool for genetic engineering (de Godoy et al., 2008). Medaka fish haESCs also show durable haploidy in both undifferentiated and differentiated statuses, despite being vertebrate species (Yi et al., 2009). However, mammalian haESCs tend to revert to diploidy under daily culture or differentiation, making complicated and frequent sorting of haploids necessary. Although haNSCLCs have been obtained by an optimized epiblast-based method (Shuai et al., 2015) or by introducing a ROCK inhibitor (Y27632) into the culture medium (He et al., 2017), these cells are not easy to handle, and the underlying mechanisms require further investigation. Our findings provide a simple strategy for deriving multiple haploid somatic cells by knocking out a single gene (p53-KO or p73-KO). We find that reduced apoptosis may be a major reason why p53-KO haESCs stably maintain haploidy. Additionally, knockout of p73, a target of p53, can also stabilize haploidy in haESCs.
Although p53 or p73 deficiency can promote haploidy maintenance in mouse haESCs, whether this phenomenon is applicable to other species remains unknown. Previous studies reported that primate haESCs maintain haploidy more stably than rodent haESCs (Sagi et al., 2016, Yang et al., 2013), and whether the mechanisms involved correlate with p53 or p73 warrants further investigation. Another study showed that although rhesus monkey haESCs maintained haploidy well in the ESC state, they underwent severe diploidization during differentiation for reasons related to apoptosis (Wang et al., 2018). Therefore, apoptosis may be the major cause of the diploidization of mammalian haploid cells. Generally, both haESCs and haploid somatic cells do not exhibit growth properties equivalent to those of their diploid counterparts (Olbrich et al., 2017). Our data revealed that p53-KO or p73-KO haESCs survived better after FACS (Figure S4D) and regained their proliferative ability faster (Figure S5F) than WT-haESCs. Previous studies have indicated that some haESCs experience cell-cycle errors such as mitosis escape, resulting in diploidization (Guo et al., 2017, He et al., 2017). According to our RNA-seq data, however, p53-KO haESCs did not show significant changes in the expression of cell-cycle-related genes (Figure 4C). We propose that the stabilization of haploidy in p53-KO haESCs depended on a reduction in apoptosis instead of regulation of the cell cycle. Nevertheless, both p53 and p73 are essential genes involved in many pathways, including key cell-cycle checkpoints (Melino et al., 2002, Stiewe, 2007). The exact mechanism by which p53-KO or p73-KO haESCs avoid mitosis escape has not yet been clarified.
Due to diploidization, complicated and time-consuming FACS purification is necessary for haploidy maintenance. Non-staining sorting methods have been introduced to enrich haploids (Freimann and Wutz, 2017, Qu et al., 2018), but their efficiency cannot be guaranteed. In our study, p53-KO or p73-KO haESCs could maintain haploidy without sorting for at least 3 weeks, which facilitates the expansion of large-scale haploid cell cultures rapidly for high-throughput genetic screening. However, previous reports demonstrated that p53-deficiency diploid ESCs showed limited pluripotency (Li et al., 2018a, Lin et al., 2005). In our study, p53-KO haESCs also showed nearly 2,800 differentially expressed genes from WT-haESCs at global transcriptome level. This indicated that there might be potential restriction of p53-KO haESCs in differentiation, which needs further investigation. Nevertheless, p53-KO haESCs could contribute to chimeras (up to E10.5) and teratomas in haploid status, which would mean that haploid somatic cells could be derived in a rigorous way in vivo, thus expanding the application of such a haploid system for lineage-specific genetic screening.
Experimental Procedures
Transfection
To obtain p53-KO haESC lines, we electroporated approximately 4 × 106 WT-haESCs with PX461-sgRNA1 (3 μg) and PX461-sgRNA2 (3 μg), using a Neon (Invitrogen) electroporator at 1,400 V for 10 ms with three pulses. GFP-positive cells were sorted by FACS 2 days post electroporation. For the overexpression of p53, a combination of 2 μg of the PBase plasmid (SBI, PB210PA-1) and 6 μg of the overexpressing (or empty piggyBac control) plasmid were electroporated into 4 × 106 p53-KO haESCs using the same electroporating conditions described above. After transfection for 4–5 days, GFP and haploidy double-positive cells were sorted for subsequent experiments. Overexpression of Cdk2 and MAPK11 plasmids and PBase plasmid were transfected into WT-haESCs, respectively. After transfection for 4–5 days, GFP and haploidy double-positive cells were sorted for subsequent experiments. To obtain GFP-labeling cell lines, we electroporated a combination of 2 μg of the PBase plasmid (SBI, PB210PA-1) and 6 μg of the piggyBac-GFP plasmid into 4 × 106 p53-KO haESCs, p53-KO diESCs, or WT-haESCs, independently.
Differentiation of p53-KO haESCs
To detect the DNA contents of haploid embryonic bodies (haEBs) derived from p53-KO haESCs, we cultured the cells with Ndiff medium (Takara, Y40002) in noncoated Petri dishes (Falcon) for 7 days. EB aggregates were then dissociated with 0.05% trypsin/EDTA (Gibco, 25300062), and the DNA content was analyzed by FACS. To generate haEpiLCs, we added p53 KO-1 cells to the wells of a 12-well plate coated with human fibronectin (Yeasen, 40105ES08) in EpiSC medium (Ndiff medium [Takara, Y40002] containing 20 ng/mL activin A [PeproTech, 120-14E-100] and 12 ng/mL bFGF [PeproTech, AF-100-18B-100]). The medium was changed every day, and standard EpiLC colonies emerged 2–3 days later. Fully grown haEpiLCs were passaged with 0.05% trypsin/EDTA and further cultured in EpiSC medium. After sorting for haploid cells 2–3 times (around 14 days), DNA content of haEpiSCs was analyzed. To generate haNSCLCs, we first established a Pax6-GFP p53 KO-1 cell line as described previously (Li et al., 2018b). Four-day EB aggregates derived from Pax6-GFP p53 KO-1 cells were then plated in fibronectin-precoated Petri dishes. After 9 days, Pax6-GFP-positive cells were sorted. The DNA content of the Pax6-GFP-positive/negative cells was then analyzed. Sorted Pax6-GFP-positive haploid cells were further cultured in N2B27 medium supplemented with 10 ng/mL EGF (PeproTech, AF-315-09-100) and 10 ng/mL bFGF (PeproTech, AF-100-18B-100) for proliferation.
For neuronal differentiation, haNSCLCs were plated into dishes precoated with poly-D-lysine (Sigma, P4957) and laminin (Sigma, L2020) and cultured in N2B27 medium supplemented with 20 ng/mL brain-derived neurotrophic factor (PeproTech, AF-450-02) and 20 ng/mL neurotrophin-3 (PeproTech, AF-450-03) to generate neurons. After 2 weeks, the differentiated haNSCLCs were examined for the expression of neuronal (TUJ1, MAP2, and NEUN) and oligodendroglial (O4) markers. For astrocyte differentiation, haNSCLCs were exposed to 1% fetal bovine serum and 10 ng/mL bone morphogenetic protein 4 (PeproTech, AF-120-05ET) in N2B27 medium for 5 days, followed by GFAP staining.
Chimeric Assay and Teratoma Formation
E6.5, E8.5, and E10.5 chimeric embryos were generated by injecting GFP-labeling p53-KO haESCs, p53-KO diploid ESCs, or WT-haESCs cells into CD-1 background blastocysts. Reconstructed embryos were transferred to the oviducts of pseudopregnant CD-1 mice at 0.5 dpc. At E6.5, E8.5, and E10.5, pseudopregnant mice were sacrificed to dissect chimeric fetuses. Chimeric embryos were trypsinized and GFP-positive and DNA-content analysis performed by FACS.
For teratoma analysis, approximately 1 × 107 p53 KO-1 cells were injected subcutaneously into the limbs of 8-week-old male SCID mice. Fully formed teratomas were dissected 3 weeks later, then fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with H&E for further analysis. Alternatively, fully formed teratomas were dissected to analyze the DNA contents.
Data and Code Availability
The Gene Expression Omnibus accession number for the RNA-seq data reported in this paper is GEO: GSE148118.
A complete description of the methods can be found in Supplemental Information.
Author Contributions
L.S. conceived and designed the study. W.Z., Y.T., and Q.G. performed most of the experiments. X.L., J.Z., C.Y., Y.W., H.W., Y.Z., and Q.Z. participated in some of the cell culture and molecular experiments. Y.L. analyzed the bioinformatics data. L.L., Y.Y., Y.F., and L.S. wrote the paper.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2019YFA0109901 and 2018YFC1004101 to L.S., 2019YFA0110804 to Y.F.), “the Fundamental Research Funds for the Central Universities,” Nankai University (63191731 to L.S.), the National Natural Science Foundation of China (31671538 and 31872841 to L.S., 81971381 and 81771580 to Y.Y.), and the Strategic Collaborative Research Program of the Ferring Institute of Reproductive Medicine, Ferring Pharmaceuticals, and Chinese Academy of Sciences (FIRMD181102 to L.S.).
Published: June 4, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.05.004.
Contributor Information
Yang Yu, Email: yuyang5012@hotmail.com.
Yong Fan, Email: yongfan011@gzhmu.edu.cn.
Ling Shuai, Email: lshuai@nankai.edu.cn.
Supplemental Information
References
- Aladjem M.I., Spike B.T., Rodewald L.W., Hope T.J., Klemm M., Jaenisch R., Wahl G.M. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol. 1998;8:145–155. doi: 10.1016/s0960-9822(98)70061-2. [DOI] [PubMed] [Google Scholar]
- Betschinger J., Nichols J., Dietmann S., Corrin P.D., Paddison P.J., Smith A. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell. 2013;153:335–347. doi: 10.1016/j.cell.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J.E., Guimaraes C.P., Varadarajan M., Park A.S., Wuethrich I., Godarova A., Kotecki M., Cochran B.H., Spooner E., Ploegh H.L. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- Cross S.M., Sanchez C.A., Morgan C.A., Schimke M.K., Ramel S., Idzerda R.L., Raskind W.H., Reid B.J. A p53-dependent mouse spindle checkpoint. Science. 1995;267:1353–1356. doi: 10.1126/science.7871434. [DOI] [PubMed] [Google Scholar]
- de Godoy L.M., Olsen J.V., Cox J., Nielsen M.L., Hubner N.C., Frohlich F., Walther T.C., Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- Elling U., Taubenschmid J., Wirnsberger G., O'Malley R., Demers S.P., Vanhaelen Q., Shukalyuk A.I., Schmauss G., Schramek D., Schnuetgen F. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell. 2011;9:563–574. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimann R., Wutz A. A fast and efficient size separation method for haploid embryonic stem cells. Biomicrofluidics. 2017;11:054117. doi: 10.1063/1.5006326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T., Bandi M., Nitta M., Ivanova E.V., Bronson R.T., Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Gibbons B., MacCallum P., Watts E., Rohatiner A.Z., Webb D., Katz F.E., Secker-Walker L.M., Temperley I.J., Harrison C.J., Campbell R.H. Near haploid acute lymphoblastic leukemia: seven new cases and a review of the literature. Leukemia. 1991;5:738–743. [PubMed] [Google Scholar]
- Guo A., Huang S., Yu J., Wang H., Li H., Pei G., Shen L. Single-cell dynamic analysis of mitosis in haploid embryonic stem cells shows the prolonged metaphase and its association with self-diploidization. Stem Cell Reports. 2017;8:1124–1134. doi: 10.1016/j.stemcr.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A., Bulyk M.L., Jambhekar A., Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019;20:199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- He Z.Q., Xia B.L., Wang Y.K., Li J., Feng G.H., Zhang L.L., Li Y.H., Wan H.F., Li T.D., Xu K. Generation of mouse haploid somatic cells by small molecules for genome-wide genetic screening. Cell Rep. 2017;20:2227–2237. doi: 10.1016/j.celrep.2017.07.081. [DOI] [PubMed] [Google Scholar]
- He W., Zhang X., Zhang Y., Zheng W., Xiong Z., Hu X., Wang M., Zhang L., Zhao K., Qiao Z. Reduced self-diploidization and improved survival of semi-cloned mice produced from androgenetic haploid embryonic stem cells through overexpression of Dnmt3b. Stem Cell Reports. 2018;10:477–493. doi: 10.1016/j.stemcr.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov A.S., Crow J.F. Haploidy or diploidy: which is better? Nature. 1991;351:314–315. doi: 10.1038/351314a0. [DOI] [PubMed] [Google Scholar]
- Leeb M., Wutz A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011;479:131–134. doi: 10.1038/nature10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M., Walker R., Mansfield B., Nichols J., Smith A., Wutz A. Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development. 2012;139:3301–3305. doi: 10.1242/dev.083675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M., Dietmann S., Paramor M., Niwa H., Smith A. Genetic exploration of the exit from self-renewal using haploid embryonic stem cells. Cell Stem Cell. 2014;14:385–393. doi: 10.1016/j.stem.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Shuai L. A versatile genetic tool: haploid cells. Stem Cell Res. Ther. 2017;8:197. doi: 10.1186/s13287-017-0657-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Yu J.S.L., Tilgner K., Ong S.H., Koike-Yusa H., Yusa K. Genome-wide CRISPR-KO screen uncovers mTORC1-mediated Gsk3 regulation in naive pluripotency maintenance and dissolution. Cell Rep. 2018;24:489–502. doi: 10.1016/j.celrep.2018.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Li X., Wang H., Gao Q., Zhang J., Zhang W., Zhang Z., Li L., Yu Y., Shuai L. CRISPR/Cas9-edited Pax6-GFP reporter system facilitates the generation of mouse neural progenitor cells during differentiation. J. Genet. Genomics. 2018;45:277–280. doi: 10.1016/j.jgg.2018.03.002. [DOI] [PubMed] [Google Scholar]
- Lin T., Chao C., Saito S., Mazur S.J., Murphy M.E., Appella E., Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- Melino G., De Laurenzi V., Vousden K.H. p73: friend or foe in tumorigenesis. Nat. Rev. Cancer. 2002;2:605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- Monfort A., Di Minin G., Postlmayr A., Freimann R., Arieti F., Thore S., Wutz A. Identification of spen as a crucial factor for Xist function through forward genetic screening in haploid embryonic stem cells. Cell Rep. 2015;12:554–561. doi: 10.1016/j.celrep.2015.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbrich T., Mayor-Ruiz C., Vega-Sendino M., Gomez C., Ortega S., Ruiz S., Fernandez-Capetillo O. A p53-dependent response limits the viability of mammalian haploid cells. Proc. Natl. Acad. Sci. U S A. 2017;114:9367–9372. doi: 10.1073/pnas.1705133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K., Li X., Wu C., Wang Y., Yu J., Zhang J., Gao Q., Zhang W., Zhang Q., Fan Y. Derivation of haploid trophoblast stem cells via conversion in vitro. iScience. 2019;11:508–518. doi: 10.1016/j.isci.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot V., Richerd S., Valero M. Transition from haploidy to diploidy. Nature. 1991;351:315–317. doi: 10.1038/351315a0. [DOI] [PubMed] [Google Scholar]
- Qu C., Yan M., Yang S., Wang L., Yin Q., Liu Y., Chen Y., Li J. Haploid embryonic stem cells can be enriched and maintained by simple filtration. J. Biol. Chem. 2018;293:5230–5235. doi: 10.1074/jbc.RA118.002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi I., Chia G., Golan-Lev T., Peretz M., Weissbein U., Sui L., Sauer M.V., Yanuka O., Egli D., Benvenisty N. Derivation and differentiation of haploid human embryonic stem cells. Nature. 2016;532:107–111. doi: 10.1038/nature17408. [DOI] [PubMed] [Google Scholar]
- Shuai L., Zhou Q. Haploid embryonic stem cells serve as a new tool for mammalian genetic study. Stem Cell Res. Ther. 2014;5:20. doi: 10.1186/scrt409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai L., Li W., Wan H., Zhao X.Y., Wang L., Zhou Q. Generation of Mammalian offspring by haploid embryonic stem cells microinjection. Curr. Protoc. Stem Cell Biol. 2014;31:1A 6 1–15. doi: 10.1002/9780470151808.sc01a06s31. [DOI] [PubMed] [Google Scholar]
- Shuai L., Wang Y., Dong M., Wang X., Sang L., Wang M., Wan H., Luo G., Gu T., Yuan Y. Durable pluripotency and haploidy in epiblast stem cells derived from haploid embryonic stem cells in vitro. J. Mol. Cell Biol. 2015;7:326–337. doi: 10.1093/jmcb/mjv044. [DOI] [PubMed] [Google Scholar]
- Song J., Li J., Hou F., Wang X., Liu B. Mangiferin inhibits endoplasmic reticulum stress-associated thioredoxin-interacting protein/NLRP3 inflammasome activation with regulation of AMPK in endothelial cells. Metab. Clin. Exp. 2015;64:428–437. doi: 10.1016/j.metabol.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Stiewe T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer. 2007;7:165–168. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- Takahashi S., Lee J., Kohda T., Matsuzawa A., Kawasumi M., Kanai-Azuma M., Kaneko-Ishino T., Ishino F. Induction of the G2/M transition stabilizes haploid embryonic stem cells. Development. 2014;141:3842–3847. doi: 10.1242/dev.110726. [DOI] [PubMed] [Google Scholar]
- Wang H., Zhang W., Yu J., Wu C., Gao Q., Li X., Li Y., Zhang J., Tian Y., Tan T. Genetic screening and multipotency in rhesus monkey haploid neural progenitor cells. Development. 2018;145:dev160531. doi: 10.1242/dev.160531. [DOI] [PubMed] [Google Scholar]
- Xu H., Yue C., Zhang T., Li Y., Guo A., Liao J., Pei G., Li J., Jing N. Derivation of haploid neurons from mouse androgenetic haploid embryonic stem cells. Neurosci. Bull. 2017;33:361–364. doi: 10.1007/s12264-017-0110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Liu Z., Ma Y., Zhong C., Yin Q., Zhou C., Shi L., Cai Y., Zhao H., Wang H. Generation of haploid embryonic stem cells from Macaca fascicularis monkey parthenotes. Cell Res. 2013;23:1187–1200. doi: 10.1038/cr.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M., Hong N., Hong Y. Generation of medaka fish haploid embryonic stem cells. Science. 2009;326:430–433. doi: 10.1126/science.1175151. [DOI] [PubMed] [Google Scholar]
- Ying Q.L., Stavridis M., Griffiths D., Li M., Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Gene Expression Omnibus accession number for the RNA-seq data reported in this paper is GEO: GSE148118.
A complete description of the methods can be found in Supplemental Information.