

Abstract
Multiple myeloma (MM) is a hematological malignancy of monoclonal plasma cells that remains incurable. Standard treatments for MM include myeloablative regimens and autologous cell transplantation for eligible patients. A major challenge of these treatments is the relapse of the disease due to residual MM in niches that become refractory to treatments. Therefore, novel therapies are needed in order to eliminate minimal residual disease (MRD). Recently, our laboratory reported that virotherapy with oncolytic myxoma virus (MYXV) improved MM-free survival in an allogeneic transplant mouse model. In this study, we demonstrate the capacity of donor autologous murine leukocytes, pre-armed with MYXV, to eliminate MRD in a BALB/c MM model. We report that MYXV-armed bone marrow (BM) carrier leukocytes are therapeutically superior to MYXV-armed peripheral blood mononuclear cells (PBMCs) or free virus. Importantly, when cured survivor mice were re-challenged with fresh myeloma cells, they developed immunity to the same MM that had comprised MRD. In vivo imaging demonstrated that autologous carrier cells armed with MYXV were very efficient at delivery of MYXV into the recipient tumor microenvironment. Finally, we demonstrate that treatment with MYXV activates the secretion of pro-immune molecules from the tumor bed. These results highlight the utility of exploiting autologous leukocytes to enhance tumor delivery of MYXV to treat MRD in vivo.
Keywords: myxoma virus (MYXV), multiple myeloma (MM), minimal residual disease (MRD), autologous stem cells transplantation (ASCT), tumor micreoenvironment (TME), carrier cells
Graphical Abstract
McFadden and colleagues found that free MYXV, or in combination with murine autologous BM or PBMCs, has therapeutic effects against murine myeloma residual disease. Moreover, MYXV stimulates an immune response resulting in the activation of IFN-γ and TNF. Importantly, re-challenged survivor mice with fresh MM developed resistance to the disease.
Introduction
Multiple myeloma (MM) is characterized by the uncontrollable proliferation and accumulation of monoclonal plasma B cells in the bone marrow (BM), which result in the overproduction of monoclonal immunoglobulin, osteolysis, and end-organ damage.1, 2, 3, 4 In the United States, MM is the second most common hematological malignancy and is newly diagnosed in >30,000 patients each year, resulting in 12,000 deaths.5 MM currently accounts for 2.1% of all cancer-related deaths.5,6 High-dose chemotherapy, along with autologous stem cell transplantation (ASCT) for eligible patients,7,8 has been used to manage and control the progression of MM.6,9 Despite the advances in the treatment of MM, the disease is still considered incurable, and eventually patients relapse or become refractory to all available standard therapies.10 One of the major failures of the available therapies is to control or eliminate MM minimal residual disease (MRD),11, 12, 13 which occurs when myeloma cells that hide in niches within the BM become resistant to standard therapies and/or contamination of the autologous stem cell transplant graft with patient myeloma cells.14 Therefore, more effective treatments are required in order to obtain sustained cancer-free outcomes, in particular by developing strategies to eliminate the MRD remaining after standard therapies have achieved their maximal disease burden reduction.
Oncolytic viruses (OVs) have emerged as promising oncolytic agents with the potential to selectively target and eliminate different types of cancers.6,15, 16, 17 Our laboratory has developed myxoma virus (MYXV) to treat a wide variety of human and murine solid tumors and hematological malignancies.15,16,18, 19, 20, 21, 22 MYXV is a double-strand DNA virus and a member of the Poxviridae family that exhibits a very restricted host range in nature and is only pathogenic to European rabbits.16 Importantly, MYXV is not pathogenic to mice, humans, or any other vertebrate outside of leporids. In the past, our laboratory has demonstrated that ex vivo treatment of primary human MM patient samples or myeloma cell lines with MYXV eliminates these malignant cells by inducing cellular apoptosis while sparing normal CD34+ hematopoietic stem and progenitor cells.23 For hematological malignancies such as MM, one major challenge of using oncolytic virotherapy is to overcome the barriers that prevent the delivery of therapeutic virus to reach the tumor microenvironment (TME), for example, within BM niches. Some of these barriers include the neutralization of the free virus by complement-mediated pathways or anti-viral antibodies, failure of the virus to extravasate from tumor blood vessels, or the clearance of the virus by the liver.24 In order to address the limitations of this issue with virus systemic delivery, our laboratory is exploring ex vivo-loaded carrier cells to selectively transport the oncolytic MYXV into the myeloma TME, particularly within the BM. In this regard, published in vitro studies with MYXV have revealed that activated allogeneic human T cells can transport the virus and infect MM cancer cells via cell-cell contact, resulting in myeloma cell infection and killing.25 Furthermore, in vivo and in vitro studies performed with MYXV demonstrated the capacity of C57BL/6 allogeneic donor carrier leukocytes, particularly neutrophils and T cells, to bind, transport, and deliver MYXV to BALB/c-derived MOPC315.BM myeloma cells, resulting in long-term survival and the debulking of the tumor.26 In this study, it was not possible to distinguish the MM reductions reported in the recipient mice as being due to viral oncolysis, as opposed to virus-enhanced cellular cytotoxic consequences of the allo-transplant.
In this study, in order to mimic a more clinically relevant scenario, we describe the therapeutic effects of using autologous BM or peripheral blood mononuclear carrier cells pre-armed ex vivo with an oncolytic MYXV versus intravenous (i.v.) infusion of free virus to target and eliminate pre-seeded MRD of MOPC315.BM MM cells using a syngeneic murine donor BALB/c into recipient BALB/c ASCT model. The results of this study highlight the potential of exploiting ex vivo-loaded autologous carrier leukocytes to eliminate MRD by selectively delivering MYXV to myeloma tumor sites within the BM and elsewhere.
Results
Autologous Transplantation Using BM Leukocytes or Peripheral Blood Mononuclear Cells (PBMCs) Armed Ex Vivo with MYXV Improves Survival Rates and Decreases MM Disease Burden
As shown previously, the murine MOPC315.BM.DsRed MM cells are resistant to both free MYXV virion binding and infection in vitro.26 However, our published in vitro studies also demonstrated the capacity of C57BL/6 BM leukocytes ex vivo-preloaded with MYXV to bypass the need for any specific virus receptors and thereby utilize cell-cell contact to infect and eliminate “virus-resistant” MOPC315.BM.DsRed MM following co-culture.26 In these previous allo-transplant studies, we did not investigate whether autologous BALB/c BM cells or PBMCs ex vivo loaded with MYXV could target and eliminate BALB/c-derived MOPC315.BM MM cells following co-culture. Therefore, we first performed in vitro experiments in which BALB/c BM or PBMCs were isolated and either mock-treated or pre-incubated ex vivo with vMyx-M135KO-GFP at a multiplicity of infection (MOI) of 10 for 1 h at 37°C to allow virus adsorption. These MYXV-loaded leukocytes were then incubated in complete and fresh RPMI 1640 culture media at 37°C for 24 h to assess for any virus infection of the donor cells. After 24 h, donor BM cells or PBMCs with or without MYXV were co-cultured with target MOPC315.BM.DsRed MM cells at a donor cell-to-target cell ratio of 10:1. These co-cultures were then incubated at 37°C for 24 or 48 h. At the respective time points, cells were collected and stained for near-infrared (Near-IR) Live/Dead dye and fluorescently labeled anti-CD138 monoclonal antibody (mAb) to identify the MM cells (CD138+). Levels of MYXV infection of the virus-resistant myeloma cells (i.e., CD138+GFP+) and the levels of MM cell killing (CD138+Near-IR Live/Dead+) were quantified using flow cytometry. The percentages of infection and killing of MM after co-culture with BM leukocytes ex vivo preloaded with MYXV are shown in Figure 1A. For example, Figure 1A (left top panel) shows some infection (~5%) of MOPC315.BM cells (i.e., CD138+GFP+) at the indicated time points. Figure 1A (left bottom panel) indicated that BM + MYXV induces higher levels (>20%) of MM killing at 24 and 48 h post-admixture. Alternatively, Figure 1B shows the percentages of virus infection and killing of MOPC315.BM.DsRed myeloma cells after co-culture with PBMCs ex vivo-preloaded with MYXV. Similar to the co-cultures with BM, low percentages of infection of myeloma cells in co-culture with PBMCs were observed after 24 h (less than 1%) and after 48 h (~1.5%) (right top panel). In terms of myeloma killing there was a slight increase of cell killing of myeloma cells in co-culture with virus-infected PBMCs; however, it was not statistically significant as compared to mock-treated co-cultures (right bottom panel). We next investigated whether MYXV alone or in combination with autologous BM cells or PBMCs can reduce or eliminate tumor burden and increase survival rates of mice with pre-seeded MOPC315.BM.DsRed MM cells in a syngeneic BALB/c mouse model.
Figure 1.

Autologous Stem Cell Transplantation in Conjunction with Ex Vivo MYXV Virotherapy Increases Survival and Decreases Myeloma Tumor Burden
(A and B) In vitro infection and killing of target MM cells. BALB/c whole BM leukocytes were isolated from tibias and femurs. Peripheral blood was isolated via terminal cardiac puncture and subjected to Ficoll-Paque purification in order to isolate mononuclear cells (PBMCs). Both BM cells (A) and PBMCs (B) were either mock-treated (i.e., no virus) or treated ex vivo with vMyx-M135KO-GFP (MOI of 10) for 1 h at 37°C to allow virus adsorption. For in vitro studies (A and B), mock- and MYXV-treated cells were incubated overnight in complete RPMI 1640 media at 37°C to assess for virus infection. After 18 h, the MYXV-loaded donor leukocytes were mixed with target MOPC315.BM.DsRed MM cells to a donor cell-to-target cell ratio of 10:1. Levels of infection and killing of MM (CD138+) cells were accessed using flow cytometry after 24 or 48 h post-admixture. Error bars represent the mean ± standard deviation (SD). At least three independent experiments were performed in order to generate these in vitro data. (C–E) In vivo treatment of MM MRD. (C) Schematic overview of the experimental workflow including treatment timeline of the experimental cohorts. (D) Kaplan-Meier survival curves in days from the initiation of the experiment. (E) Flow cytometry plots showing the myeloma tumor burden from target organs such as BM, spleen, and CD138+/DsRed solid tumors, shown as the mean ± SD. At least five independent cohort sets were performed in order to generate the in vivo data as shown. p values are reported as non significant (ns) if p > 0.05, and significant if ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p ≤ 0.0001.
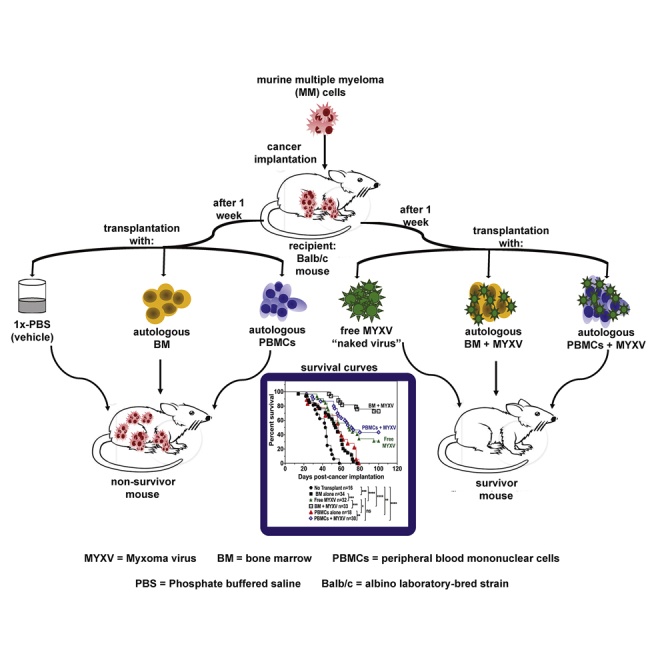
In vivo studies previously performed in our laboratory have demonstrated that allogeneic hematopoietic stem cell transplantation (allo-HSCT) using C57BL/6 leukocyte donors in conjunction with ex vivo virotherapy with MYXV improves cancer-free outcomes in BALB/c mice bearing “MYXV-resistant” MOPC315.BM.DsRed MM MRD.26 Although allo-transplantation was a successful approach to target and eliminate the MRD of MM, it lacks direct translatability to the clinic since most standard therapies used to treat MM include ablative chemotherapy/radiotherapy regimens followed by reconstitution of the immune system with ASCT for eligible patients. Based on the in vitro data shown in Figure 1, we hypothesized that ASCT in combination with virotherapy with MYXV had the potential to promote therapeutic effects in an in vivo auto-transplant setting. Therefore, we sought to investigate the therapeutic effects of ASCT in conjunction with ex vivo MYXV virotherapy in mice pre-seeded with MM MRD. Thus, we tested both BM and PBMC autografts ex vivo loaded with MYXV to assess whether these leukocyte carriers could not only transport and deliver our OV efficiently into the myeloma tumor niches in the BM and spleen (the major sites of MRD in this model), but also when compared to the use of single systemic i.v. virotherapy with free (“naked”) MYXV. The scheme shown in Figure 1C describes the experimental design, including the treatment timeline after myeloma implantation. Briefly, on day 1, 8-week-old BALB/c recipient female or male mice were sublethally irradiated with 175 cGy using an X-ray source. Irradiated mice received formulated water for 2 weeks to avoid opportunistic infections. After 24 h, mice received 1 × 105 BALB/c-derived MOPC315.BM.DsRed MM cells i.v. via tail vein. One week after cancer implantation, myeloma-bearing mice were treated as follows: vehicle control with no BM or PBMC transplant (i.e., 1× PBS) (n = 16, cohort I); BM alone (n = 34, cohort II); free MYXV (n = 32, cohort III); BM ex vivo treated with MYXV (n = 33, cohort IV); PBMCs (n = 18, cohort V); and PBMCs ex vivo pre-treated with MYXV (n = 30, cohort VI). All i.v. treatments were administrated via the retro-orbital (RO) route. Figure 1D shows the survival curves of these cohorts. Strikingly, the treatment with BM + MYXV had the highest increase in survival (72.7%) as compared to the treatment with PBMCs + MYXV (43.3%) or the free MYXV treatment (31.1%). Statistical significance of the survival curves was observed when we compared cohort I (no transplant vehicle control, 1× PBS) to cohorts II–VI, as well as when we compared cohort II to cohort III (BM alone versus free MYXV), cohort III to cohort IV (free MYXV versus BM + MYXV), cohort III to cohort V (free MYXV versus PBMCs alone), and cohort V to cohort VI (PBMCs alone versus PBMCs + MYXV). MM tumor burden (i.e., %CD138+) was assessed from myeloma-carrying organs such as BM and spleen (i.e., spleen with visible splenomegaly) and from any detected extramedullary tumors (referred to as solid tumors), using flow cytometry. Data shown in Figure 1E demonstrate a decrease in tumor burden from the BM for the cohorts exposed to MYXV (i.e., cohorts III, IV, and VI). Also, the highest decrease in tumor burden from spleen and solid tumors was observed in mice from cohort IV (BM + MYXV). Taken together, these in vivo data demonstrate the potential of ASCT in conjunction with MYXV to target and eliminate MM MRD from multiple sites and correlate with the in vitro data.
MYXV Binds to Diverse Immune Cell Populations from BM and PBMCs
Data shown in Figure 1 suggest that immune cells derived from BM or PBMCs could bind and transport MYXV to the myeloma TME compartments such as BM, spleen, and extramedullary solid tumors. Previous in vitro data indicated that, in the context of allo-HSCT, both T cells and neutrophils ex vivo pre-treated with MYXV can target and kill MOPC315.BM.DsRed myeloma cells.26 In this study, in the context of autologous leukocytes from BM or PBMCs, we looked to identify immune cell populations that can bind to MYXV and potentially traffic the virus into the myeloma microenvironment. To do this, 1 × 107 BALB/c BM cells or PBMCs were first mock-treated or incubated with fluorescently tagged vMyx-M093L-Venus at 4°C for 1 h to allow virus binding. After this, cells were washed twice with 1× PBS + 5% FBS to eliminate unbound virus. Then, mock- or MYXV-treated cells were stained with mAbs against different immune populations, including T cells (i.e., CD3+, CD4+, CD8+), neutrophils (i.e., CD11b+Gr-1+), and natural killer (NK) cells (i.e., CD3−CD49b+). Although there are more immune populations that can carry MYXV such as dendritic cells (DCs), monocytes, macrophages, and others, for these studies we have focused on T cells, neutrophils, and NK cells.
Data shown in Figures 2A and 2B and Table 1 indicate that MYXV efficiently binds to T cells (i.e., BM-derived CD3+, 3.5% ± 0.5%; CD4+, 1.0% ± 0.0%; CD8+, 1.0% ± 0.0% versus PBMC-derived CD3+, 31.6% ± 2.9%; CD4+, 69.5% ± 4.5%; CD8+, 11.5% ± 0.9%), neutrophils (i.e., BM-derived CD11b+Gr1+, 36.9% ± 3.6% versus PBMC-derived CD11b+Gr1+, 4.2% ± 0.4%), and NK cells (i.e., BM-derived CD3−CD49b+, 1.04% ± 0.04% versus PBMC-derived CD3−CD49b+, 27.2% ± 9.4%) in both BM and PBMC samples, albeit with different efficiencies. For example, neutrophils derived from BM showed a higher level of virus binding than did their counterparts from the PBMC source. Alternatively, T cells and NK cells from PBMCs are also susceptible to MYXV binding as compared to their counterparts from BM. Nevertheless, these immune cell populations all remain as potential candidate carriers to bind and transport the virus into the tumor bed in vivo. However, as mentioned before, at this point we cannot rule out the possibility that other immune cell populations from the BM or PBMC compartment can also bind and carry this OV into the myeloma tumor bed.
Figure 2.

Binding Levels of MYXV Virions to Murine BM and PBMC Populations
(A) vMyx-M093L-Venus binding to BM immune populations. (B) vMyx-M093L-Venus binding to PBMC immune populations. Fluorescently tagged MYXV particle binding levels to subcellular immune populations were quantified using flow cytometry. The mean ± SD of at least three independent experiments is shown. Briefly, 1 × 107 BALB/c BM cells or PBMCs were either mock-treated or incubated with MYXV at 4°C for 1 h to allow virus binding. Unbound virus was washed twice with cold 1× PBS + 5% FBS. Mock- or MYXV-treated cells were then stained with monoclonal antibodies against the indicated immune populations. Error bars correspond to the mean ± SD of at least three independent experiments.
Table 1.
Percentages of Different Immune Cell Populations from BALB/c BM or PBMCs
| Immune Populations | Percentage from BM (Mean ± SE) | Percentage from PBMCs (Mean ± SE) |
|---|---|---|
| CD3+ | 3.5 ± 0.7 | 31.6 ± 2.9 |
| CD4+ | 1.0 ± 0.0 | 69.5 ± 4.5 |
| CD8+ | 1.0 ± 0.0 | 11.5 ± 0.9 |
| Neutrophils | 36.9 ± 3.6 | 4.2 ± 0.4 |
| Natural killer (NK) cells | 1.04 ± 0.04 | 27.2 ± 9.4 |
SE, standard error.
Murine Autologous T Cells and Neutrophils Can Each Effectively Act as Carrier Cells to Transport MYXV into the Myeloma Tumor Bed
Our previously published in vitro data indicated that, in the context of allogeneic stem cell transplantation (allo-SCT), either BM-derived and activated T cells or neutrophils pre-armed ex vivo with MYXV can effectively target and kill MOPC315.BM.DsRed myeloma cells in co-culture.26 In this study, we investigated whether autologous neutrophils or T cells that have been ex vivo preloaded with MYXV have therapeutic effects in vivo against MOPC315.BM myeloma cells pre-seeded into BALB/c recipient mice. Figure 3A summarizes the treatment timeline after cancer implantation. Briefly, 8-week-old BALB/c mice were seeded with 1 × 105 MOPC315.BM.DsRed MM cells in identical fashion as described previously. As transplant leukocyte donors for this experiment, we used 8-week-old immunocompromised BALB/c-derived non-obese diabetic (NOD)/severe combined immunodeficiency (SCID)-γ mice that lack T cells, B cells, and have defects in NK functions. BM or PBMCs were isolated as described before. Since BM is a good reservoir of mouse neutrophils, total BM was then used to deplete or enrich neutrophils via negative selection. For the neutrophil-depleted BM sample, about 98% depletion of neutrophils was achieved, as assessed by flow cytometry. Enriched neutrophils (called “+neutrophils”) and neutrophil-depleted BM (called “BM−neutrophils”) were ex vivo adsorbed with vMyx-M135KO-GFP at an MOI of 10 for 1 h at 37°C. Recipient (wild-type [WT]) BALB/c mice bearing pre-seeded MOPC315.BM.DsRed MM cells were treated as follows: no transplant but vehicle control (i.e., 1× PBS) (n = 18, cohort I); NOD/SCID-γ BM + MYXV (n = 23, cohort II); NOD/SCID-γ PBMCs + MYXV (n = 20, cohort III); NOD/SCID-γ +neurophils + MYXV (n = 22, cohort IV); BM−neutrophils + MYXV (n = 27, cohort V) (Figure 4A). Kaplan-Meier plots in days post-cancer implantation are shown in Figure 3B. Interestingly, the most dramatic decrease in survival was observed in mice treated with BALB/c-derived NOD-SCID-γ PBMCs + MYXV (15%), suggesting the importance of lymphocytes from PBMCs to carry MYXV and mediate the therapeutic effects observed in Figure 1. Survival of mice treated with NOD/SCID-γ BM + MYXV was 52.2%, while this increased to 63.6% for mice treated with enriched neutrophils (+neutrophils) + MYXV. Finally, 40.7% of mice treated with NOD/SCID-γ BM-neutrophils + MYXV survived. These latter results with +neutrophils and −neutrophils suggest that neutrophils from BM may contribute to transport MYXV, but they are not the unique immune population that can have a role as cell carrier of the virus. Also, our results suggest that the absence of immune cell populations, such B cells or NKs, from both BM and PBMC autografts did not impair the observed therapeutic effects shown in Figure 3. Alternatively, our results also open the possibility to explore other immune cells such as DCs, macrophages, monocytes, and others from both BM and PBMC compartments as potential carrier cells of oncolytic MYXV. In terms of tumor burden, although some CD138+ cells were observed in BM, spleen, and solid tumors, comparable reductions were observed among all of the cohorts that included MYXV. Alternatively, solid tumors were mainly observed in non-treated mice (cohort I) or mice treated with NOD/SCID-γ BM depleted of neutrophils + MYXV (cohort V) (Figure 3C). Collectively, these data suggest the relevance of T lymphocytes from PBMCs to have the capacity to function as carrier cells that can transport and deliver MYXV into the MM milieu. However, in addition to BM-derived T cells, neutrophils, and NK cells, other immune cell populations, including DCs, monocytes, and macrophages, that become armed with MYXV might also exert MYXV-enhancing therapeutic effects against MM MRD in a preclinical murine autologous model.
Figure 3.

Identification of Carrier Cells from Autologous Donor BM and PBMC Compartments to Transfer MYXV into Myeloma Tumor Sites
(A) Schematic overview of the experimental workflow including the treatment timeline of the experimental cohorts using NOD/SCID-γ donors of BM or PBMCs. All samples except for the non transplant controls (cohort I) were incubated with MYXV for 1 h at 37°C prior to transplantation to allow virus adsorption. “+neutrophils” refers to enriched neutrophil samples isolated from BM, whereas “BM−neutrophils” refers to BM samples depleted of neutrophils. (B) Kaplan-Meier survival curves in days after cancer implantation. (C) Flow cytometry plots showing the myeloma tumor burden from target organs such as BM, spleen, and solid tumors. Error bars correspond to the Mean ± SD of tumor burden of representative mice. p values are reported as non significant (ns) if p > 0.05, and significant if ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p ≤ 0.0001.
Figure 4.

BM or PBMC Carrier Cells Ex Vivo Armed with Oncolytic MYXV Induce Acquired Anti-myeloma Immunity in Survivor Mice Re-challenged with Fresh Myeloma Tumor Cells
(A) Kaplan-Meier survival curves of survivor mice that had been treated with either BM or PBMC armed with MYXV (as described in Figure 1C) and after 100 days of survival each mouse was re-challenged with fresh 1 × 105 MOPC315.BM.DsRed MM cells. (B and C) Flow cytometry data showing the levels of expression of central memory T (TCM) cells or effector memory/effector T (TEM/EFF) cells. (B) Flow cytometry data of representative mouse expressions of TCM or TEM/EFF cells are shown. (C) Quantification of central memory T (TCM) versus TEM/EFF cells using flow cytometry is shown. Error bars correspond to the mean ± SD of at least three independent experiments. Significant and non-significant (ns) p values are reported.
Autologous Transplant Using BM or PBMCs Ex Vivo Pre-armed with MYXV Induces Acquired Immunity against Myeloma Tumor Cells in Re-challenged Survivor Mice
In order to determine whether ex vivo MYXV virotherapy induces long-term immunity against MM, 37 BALB/c mice from various BM or PBMC armed with MYXV cohorts that survived to 100 days following the first challenge with 1 × 105 MOPC315.BM.DsRed MM cells were then re-challenged i.v. with 1 × 105 fresh MPOC315.BM.DsRed MM cells. One-hundred days after re-challenging with fresh 1 × 105 MOPC315.BM.DsRed MM cells, 77.8% of these re-challenged mice survived. Figure 4A shows the survival curve of these re-challenged mice and compared to the survival curve of naive BALB/c recipient mice that had received only vehicle control (i.e., 1× PBS). The results strongly support the notion that ex vivo MYXV virotherapy can both eliminate MRD and induce acquired immunity against MM.
Next, we investigated the status of memory T cells from BM and spleen of survivor mice. Briefly, BM and spleen from naive and healthy mice (i.e., mice that had never been challenged with MM cells) and from mice that survived the second re-challenge with MM cells were stained with fluorescently labeled mAbs against central memory T (TCM) cells (CD62L+CD44+) or effector memory T (TEM) cells (CD62L−CD44+), both gating on live CD4+ or CD8+ T cells. Figure 4B shows the results of a representative re-challenged mouse and compared to a healthy naive mouse. Figure 4C summarizes the levels of these memory T cells (mean ± SD) from three independent experiments. By gating on live CD4+ T cells derived from BM (left top panel), we found that there was a significant increase of TCM cells in re-challenged mice as compared to naive mice. In addition, by gating on live CD4+ T cells derived from spleen, we observed a significant increase of effector memory/effector T (TEM/EFF) cells from re-challenged mice compared to naive mice (Figure 4C, left bottom panel). Alternatively, by gating on live CD8+ T cells derived from BM we did not find any significant difference for both TCM and TEM/EFF cells when compared to naive versus re-challenged mice (Figure 4C, right top panel). However, by gating on live CD8+ T cells derived from spleen we observed a significant increase in both TCM and TEM/EFF cells of re-challenged mice as compared to naive mice (Figure 4C, right bottom panel).
MYXV Therapy Regulates the Expression of Multiple Pro-immune Cytokines and Chemokines from the Myeloma TME
MOPC315.BM.DsRed MM cells are not permissive to direct MYXV binding or infection in vitro, but these MM cells can be infected and killed when the virus is delivered via cell-cell contact on carrier leukocytes of various classes. However, unexpectedly we found significant therapeutic benefits not only after MYXV was delivered after preloading of BM cells or PBMCs, but also after systemic infusion of free (“naked”) virus. This was very intriguing for several reasons and suggests that even free virus following systemic infusion likely encountered and bound circulating leukocytes, and this “cell-bound” virus was likely the active agent of the anti-myeloma effects in the BM and spleen. We also speculate that MYXV carried by mixed leukocytes can uniquely activate an effective anti-myeloma immune response that mediates the observed anti-tumor effects against MRD. In fact, it is known that different OVs can mediate the expression of multiple cytokines and immune-stimulatory molecules to enhance anti-tumor immune responses.27,28 In order to address the possibility that MYXV induces an immune response from the TME, we investigated the virotherapy-mediated immune effects in the myeloma TME, particularly within the BM. To do this, mice were implanted with 1 × 105 MPC315.DsRed MM cells and 1 week later treated as follows: no transplant vehicle control (i.e., 1× PBS) (n = 3, cohort I); BM alone (n = 3, cohort II); free MYXV (n = 3, cohort III); BM ex vivo pre-treated with MYXV (n = 3, cohort IV); PBMCs (n = 3, cohort V); PBMCs ex vivo pre-treated with MYXV (n = 3, cohort VI). After 3 or 24 h of treatment, mice were sacrificed, and BM and splenocytes were isolated to determine the levels of several soluble pro-immune molecules using mouse custom ProcartaPlex 11-plex for multiplex platform (Thermo Fisher Scientific) (Figure 5). Tables 2 and 3 summarize the mean ± standard deviation (SD) of those secreted cytokines and chemokines secreted from BM and spleen at the indicated time points, respectively, and whose expression was highly altered by the oncolytic MYXV virus. Figure 5A illustrates the secretion of T helper (Th)1 cytokines, including interferon (IFN)-γ, interleukin (IL)-12p70, and IFN-γ inducible protein 10 (IP-10) from BM or spleen after 3 or 24 h of treatment. For example, recipient mice treated with MYXV alone or in combination with autologous BM or PBMCs resulted in enhanced secretion of cytokines involved in the Th1 response such as IFN-γ and IP-10 as compared to mice treated with vehicle control (i.e., 1× PBS) or transplanted with either autologous BM cells or PBMCs alone. Because similar secretion patterns were observed among the controls (i.e., untreated mice, mice treated with BM or PBMCs alone), in the rest of this section we performed comparisons between the untreated mice and the mice treated with MYXV alone, PBMCs + MYXV, or BM + MYXV. For example, as compared to the non-treated (1× PBS) cohort, the levels of splenocyte-derived IFN-γ increased 3 and 24 h after treatment with MYXV alone (fold increase of 25.0 at 3 h versus fold increase of 20.0 at 24 h) or in combination with either PBMCs + MYXV (fold increase of 17.9 at 3 h versus fold increase of 12.0 at 24 h) or BM + MYXV (fold increase of 42.0 at 3 h versus fold increase of 14.5 at 24 h). However, enhanced secretion of BM-derived IFN-γ was observed predominantly after 24 h of treatment with naked MYXV (fold increase of 28.7), PBMC + MYXV (fold increase of 8.5), and BM + MYXV (fold increase of 6.7 at 3 h versus fold increase of 9.8 at 24 h).
Figure 5.

Virotherapy with Free MYXV, or in Combination with Autologous BM or PBMCs, Alters the Expression of Selective Cytokines and Chemokines from the Myeloma Tumor Microenvironment
(A–C) Levels of secretion of different cytokines and chemokines from BM or spleen of BALB/c mice bearing MOPC315.BM.DsRed MM after 3 or 24 h of treatment with vehicle control (1× PBS), free MYXV (“naked”) virus, or autologous BM cells or PBMCs ex vivo armed with MYXV. Briefly, 1 week after cancer implantation, vehicle control, autologous donor BM cells or PBMCs, or autologous BM cells or PBMCs ex vivo armed with MYXV at an MOI of 10 for 1 h at 37°C were delivered into recipient mice via the retro-orbital (RO) route. After 3 or 24 h, mice (n = 3) were euthanized, and myeloma target organs such as BM and spleen were collected. Supernatants derived from BM cells and splenocytes were isolated as described in Materials and Methods and used to determine the levels of different secreted cytokines and chemokines using a multiplex assay. The error bars represent the mean ± SD. Significant p values are reported.
Table 2.
Levels of Pro-immune Molecules Secreted from the Bone Marrow Myeloma Tumor Microenvironment after 3 or 24 h of Treatment
| Vehicle (1× PBS) | BM | PBMCs | MYXV | BM + MYXV | PBMCs + MYXV | |
|---|---|---|---|---|---|---|
| After 3 h | ||||||
| IFN-γ (Th1) | 15.5 ± 4.5 | 13.0 ± 0.0 | 20.0 ± 5.0 | 21.5 ± 7.3 | 103.7 ± 6.3 | 17.7 ± 1.3 |
| IP-10 (Th1) | 735.0 ± 222.0 | 1,082.5 ± 112.5 | 895.5 ± 106.0 | 981.0 ± 125.4 | 4,034.8 ± 78.3 | 1,676.3 ± 223.7 |
| IL-12p70 (Th1) | 3.5 ± 0.0 | 3.7 ± 0.2 | 4.0 ± 0.5 | 3.8 ± 1.4 | 6.1 ± 0.3 | 5.0 ± 0.3 |
| IL-4 (Th2) | 11.3 ± 1.5 | 12.8 ± 4.5 | 15.5 ± 0.7 | 10.5 ± 2.5 | 15.6 ± 3.8 | 10.5 ± 2.2 |
| IL-5 (Th2) | 7.0 ± 4.0 | 7.2 ± 0.2 | 10.7 ± 3.2 | 7.0 ± 3.1 | 11.7 ± 4.2 | 10.7 ± 0.9 |
| TNF-α | 53.5 ± 14.0 | 60.7 ± 1.2 | 53.3 ± 1.7 | 47.5 ± 5.5 | 83.5 ± 8.7 | 65.2 ± 2.3 |
| IL-1α | 38.7 ± 6.2 | 35.5 ± 8.0 | 30.2 ± 3.7 | 34.8 ± 6.0 | 66.2 ± 12.2 | 34.8 ± 3.4 |
| IL-1β | 112.0 ± 43.0 | 52.7 ± 8.7 | 83.0 ± 12.0 | 73.3 ± 5.2 | 167.2 ± 32.2 | 81.0 ± 7.0 |
| GM-CSF | 3.0 ± 0.0 | 2.5 ± 0.5 | 35.5 ± 0.5 | 3.3 ± 0.7 | 4.7 ± 0.3 | 2.7 ± 0.3 |
| MIP-1α | 341.0 ± 179.0 | 371.2 ± 2.2 | 301.5 ± 69.5 | 449.8 ± 43.0 | 1,515.8 ± 238.2 | 380.7 ± 51.8 |
| IL-6 | 17.5 ± 2.5 | 14.7 ± 1.2 | 12.0 ± 2.0 | 14.3 ± 1.9 | 20.2 ± 0.4 | 13.0 ± 1.7 |
| After 24 h | ||||||
| IFN-γ (Th1) | 20.0 ± 2.1 | 10.0 ± 1.5 | 9.0 ± 1.5 | 573.3 ± 225.2 | 196.2 ± 28.9 | 170.0 ± 5.6 |
| IP-10 (Th1) | 1,110.0 ± 71.0 | 741.3 ± 64.6 | 714.3 ± 51.6 | 2,080.0 ± 106.0 | 1,376.0 ± 187.3 | 1,670.3 ± 60.4 |
| IL-12p70 (Th1) | 2.5 ± 0.0 | 2.8 ± 0.9 | 2.2 ± 0.3 | 7.4 ± 1.1 | 5.1 ± 0.4 | 4.1 ± 0.2 |
| IL-4 (Th2) | 6.8 ± 1.5 | 5.3 ± 1.0 | 7.3 ± 1.2 | 21.0 ± 4.2 | 9.7 ± 0.7 | 14.2 ± 1.7 |
| IL-5 (Th2) | 4.5 ± 0.5 | 4.0 ± 0.3 | 5.0 ± 0.6 | 10.7 ± 1.6 | 7.4 ± 0.5 | 9.6 ± 0.4 |
| TNF-α | 32.0 ± 2.5 | 258 ± 3.8 | 22.2 ± 4.5 | 44.2 ± 6.1 | 812.6 ± 124.1 | 24.5 ± 2.1 |
| IL-1α | 41.0 ± 10.5 | 19.5 ± 2.3 | 14.5 ± 3.0 | 69.4 ± 20.0 | 35.8 ± 4.9 | 14.1 ± 1.1 |
| IL-1β | 44.0 ± 18.0 | 19.5 ± 2.3 | 127.3 ± 24.4 | 124.0 ± 80.6 | 35.8 ± 4.9 | 138.9 ± 5.7 |
| GM-CSF | 1.0 ± 0.0 | 1.3 ± 0.3 | 1.3 ± 0.3 | 7.2 ± 0.9 | 2.5 ± 0.5 | 4.3 ± 1.2 |
| MIP-1α | 422.0 ± 111.0 | 128.5 ± 16.9 | 121.7 ± 14.5 | 2,966.0 ± 853.3 | 812.6 ± 124.1 | 651.0 ± 71.5 |
| IL-6 | 4.5 ± 0.5 | 8.7 ± 5.2 | 6.2 ± 2.4 | 9.3 ± 1.3 | 9.3 ± 1.3 | 9.5 ± 1.5 |
Means ± standard deviation (SD) of levels of the secreted molecules are shown.
Table 3.
Levels of Pro-immune Molecules Secreted from Spleen Myeloma Tumor Microenvironment after 3 or 24 h of Treatment
| Vehicle (1× PBS) | BM | PBMCs | MYXV | BM + MYXV | PBMCs + MYXV | |
|---|---|---|---|---|---|---|
| After 3 h | ||||||
| IFN-γ (Th1) | 122.5 ± 54.5 | 98.5 ± 41.5 | 90.5 ± 11.5 | 3,002.0 ± 278.7 | 5,138.5 ± 810.1 | 2,191.7 ± 287.9 |
| IP-10 (Th1) | 1,046.2 ± 110.7 | 1,282.0 ± 276.0 | 1,145.7 ± 44.7 | 7,718.7 ± 324.9 | 5,791.5 ± 553.5 | 3,813.5 ± 208.6 |
| IL-12p70 (Th1) | 15.0 ± 5.0 | 8.0 ± 3.5 | 15.0 ± 1.5 | 17.5 ± 0.6 | 25.7 ± 3.2 | 12.8 ± 0.7 |
| IL-4 (Th2) | 24.5 ± 11.7 | 26.3 ± 13.0 | 19.3 ± 1.0 | 66.0 ± 10.4 | 43.8 ± 6.4 | 76.3 ± 14.4 |
| IL-5 (Th2) | 21.0 ± 4.0 | 7.0 ± 03.0 | 8.0 ± 1.0 | 17.2 ± 3.2 | 16.1 ± 1.1 | 21.7 ± 1.7 |
| TNF-α | 44.5 ± 13.0 | 32.2 ± 12.2 | 27.0 ± 3.5 | 116.5 ± 7.0 | 250.7 ± 34.0 | 71.5 ± 2.5 |
| IL-1α | 129.0 ± 7.5 | 172.5 ± 44.2 | 90.5 ± 4.0 | 510.2 ± 140.9 | 2,024.4 ± 198.8 | 325.8 ± 11.1 |
| IL-1β | 188.5 ± 4.0 | 89.0 ± 34.0 | 132.5 ± 12.5 | 437.0 ± 136.9 | 2,574.7 ± 622.7 | 204.8 ± 30.6 |
| GM-CSF | 6.7 ± 1.7 | 4.0 ± 2.0 | 5.0 ± 1.0 | 70.0 ± 8.9 | 160.9 ± 43.0 | 33.5 ± 4.8 |
| MIP-1α | 2,112.5 ± 75.5 | 2,700.5 ± 1,700.5 | 1,746.7 ± 348.7 | 10,917.7 ± 819.2 | 11,141.7 ± 1,255.4 | 5,933.8 ± 578.4 |
| IL-6 | 16.0 ± 5.5 | 18.0 ± 4.0 | 10.5 ± 1.5 | 270.8 ± 124.1 | 1,477.6 ± 438.2 | 62.0 ± 2.9 |
| After 24 h | ||||||
| IFN-γ (Th1) | 186.2 ± 4.2 | 54.5 ± 2.9 | 122.5 ± 41.5 | 3724.0 ± 9.1 | 2,703.1 ± 331.2 | 2,223.8 ± 309.1 |
| IP-10 (Th1) | 1,395.0 ± 294.0 | 867.0 ± 24.9 | 912.0 ± 98.1 | 3,788.0 ± 683.3 | 2,105.1 ± 98.2 | 2,127.8 ± 60.9 |
| IL-12p70 (Th1) | 6.5 ± 1.0 | 4.5 ± 1.2 | 3.8 ± 0.9 | 16.8 ± 3.2 | 15.2 ± 0.8 | 13.9 ± 1.7 |
| IL-4 (Th2) | 20.0 ± 7.7 | 11.3 ± 1.2 | 12.0 ± 0.3 | 21.4 ± 4.3 | 24.7 ± 2.6 | 18.5 ± 1.0 |
| IL-5 (Th2) | 5.5 ± 1.5 | 3.3 ± 0.3 | 5.7 ± 0.3 | 9.8 ± 1.0 | 12.0 ± 0.0 | 11.4 ± 1.2 |
| TNF-α | 28.0 ± 1.5 | 18.5 ± 1.5 | 17.8 ± 2.0 | 80.2 ± 9.2 | 4,692.7 ± 189.9 | 50.5 ± 7.0 |
| IL-1α | 107.6 ± 8.2 | 47.2 ± 7.8 | 42.6 ± 5.0 | 627.0 ± 206.0 | 539.5 ± 32.9 | 318.3 ± 56.8 |
| IL-1β | 95.0 ± 4.0 | 47.2 ± 7.8 | 105.2 ± 7.0 | 233.4 ± 17.7 | 538.0 ± 39.0 | 274.3 ± 40.0 |
| GM-CSF | 3.5 ± 0.5 | 4.3 ± 0.3 | 4.0 ± 0.6 | 11.7 ± 2.9 | 9.5 ± 1.2 | 7.6 ± 0.2 |
| MIP-1α | 2,514.5 ± 340.5 | 2,013.2 ± 56.5 | 790.8 ± 136.6 | 11,667.6 ± 2,233.8 | 4,692.7 ± 189.9 | 2,667.3 ± 373.6 |
| IL-6 | 14.5 ± 2.5 | 7.7 ± 3.3 | 6.3 ± 0.9 | 24.0 ± 7.5 | 20.0 ± 1.6 | 16.3 ± 1.6 |
The means ± standard deviation (SD) of levels of the secreted molecules are shown.
Secretion of BM-derived IP-10 was induced upon treatment with PBMCs + MYXV (fold increase of 2.3 at 3 h) or BM + MYXV (fold increase of 5.5 at 3 h). Spleen-derived IP-10 secretion was enhanced upon treatment with free MYXV (fold increase of 7.4 at 3 h versus fold increase of 3.0 at 24 h) versus PBMCs + MYXV (fold increase of 4.0 at 3 h) versus BM + MYXV (fold increase of 5.5 at 3 h). Regardless of the treatment and the time point, low and similar levels of secreted IL-12p70 from BM and spleen were observed. Interestingly, not much increase in the induction of Th2-associated cytokines such as IL-4 and IL-5 was observed (Figure 5B). Similar levels of secretion of IL-4 and IL-5 from either BM or spleen of untreated mice or mice treated with PBMCs or BM alone were found after 3 or 24 h. Treatment with MYXV alone enhanced induction of BM-derived IL-4 (fold increase of 3.1 at 24 h) versus spleen-derived IL-4 (fold increase 2.7 at 3 h). Treatment of PBMC + MYXV only enhanced the spleen-derived IL-4 secretion after 3 h of treatment (fold increase of 3.1). Treatment with BM + MYXV did not enhance the levels of this cytokine compared to the rest of cohorts. Regardless of the treatment, we did not find significant induction of BM- or spleen-derived IL-5. Similar cytokine secretion patterns have been shown by others. For example, oncolytic vaccinia virus bearing a thymidine kinase deletion (TK−) and expressing the mouse TRIF protein was used to treat Renca tumors.29 Overexpression of soluble murine IFN-γ and tumor necrosis factor (TNF) has also been reported upon co-cultures of allogeneic human PBMCs with mesenchymal stem cells (MSCs) infected with oncolytic adenovirus (OAdv).30 Oncolytic vesicular stomatitis virus pseudotyped with the lymphocytic choriomeningitis virus glycoprotein (VSV-GP) alone or in combination with peptide-loaded DC vaccine (DCVacc) induces a pro-inflammatory response from mouse BL6-ovalbumin (OVA) melanoma tumors characterized by the release of IFN-γ, TNF, IL-6, IL-1α, and IL-β, among others.31
In the present study, we observed that secretion of immune effector molecules such as TNF was highly induced after 24 h from BM in mice treated with autologous BM + MYXV (fold increase of 25.4). Also, enhanced levels of secreted TNF from spleen were observed upon treatment with free MYXV (fold increase of 2.6 at 3 h versus fold increase of 2.9 at 24 h) or BM + MYXV (fold increase of 5.6 at 3 h versus fold-increase of 167.6 at 24 h) (Figure 5C). Secretion of spleen-derived IL-1α was observed upon treatment with free MYXV (fold increase of 4.0 at 3 h versus fold increase of 5.8 at 24 h) versus BM + MYXV (fold increase of 15.7 at 3 h versus fold increase of 5.0 at 24 h). IL-1β secreted from BM was induced upon treatment with free MYXV (fold increase of 2.8 at 24 h) versus PBMCs + MYXV (fold increase of 3.1 at 24 h). Alternatively, IL-1β secreted from splenocytes was induced upon treatment with free MYXV (fold increase of 2.3 at 3 h versus 2.5 at 24 h), BM + MYXV (fold increase of 13.7 at 3 h versus fold increase of 5.7 at 24 h) and PBMCs + MYXV (fold increase of 2.9 at 24 h). Granulocyte macrophage colony-stimulating factor (GM-CSF) is an immunostimulatory molecule.32 We found that secreted GM-CSF from BM was primarily upregulated after treatment with free MYXV (fold increase of 7.2 at 24 h), PBMCs + MYXV (fold increase of 4.3 at 24 h), and BM + MYXV (fold increase of 2.5 at 24 h). Induction of GM-CSF from spleen was observed after treatment with MYXV (fold increase of 10.4 at 3 h versus fold increase of 3.3 at 24 h), PBMCs + MYXV (fold increase of 5.0 at 3 h versus fold increase of 2.2 at 24 h), and BM + MYXV (fold increase of 24.0 at 3 h versus fold increase of 2.7 at 24 h).
It is known that MIP-1α stimulates myeloma-mediated osteoclastogenesis.33,34 Therefore, we investigated the effects of MYXV on the levels of soluble MIP-1α. We found that MIP-1α from BM was induced upon treatment with free MYXV (fold increase of 7.0 at 24 h) and BM + MYXV (fold increase of 4.4 at 3 h versus fold increase of 2.0 at 24 h). Soluble MIP-1α from spleen was induced upon treatment with free MYXV (fold increase of 5.2 at 3 h versus fold increase of 4.6 at 24 h), PBMCs + MYXV (fold increase of 2.8 at 3 h), and BM + MYXV (fold increase of 5.3 at 3 h).
Alterations in the levels of expression of some of these immune effector molecules from established Renca tumors have been observed 4 days following single treatment with the vaccinia construct TK-TRIF.29
Soluble immunomodulatory IL-6 mediates tumor immune evasion.35 In the context of MM, IL-6 promotes myeloma growth in the TME.33,36 Interestingly, we found that soluble IL-6 from BM was induced upon treatment with free MYXV (fold increase of 2.1 at 24 h), PBMCs + MYXV (fold increase of 2.1 at 24 h), and BM + MYXV (fold increase of 2.1 at 24 h). Furthermore, secreted IL-6 from spleen was transiently upregulated 3 h after treatment of mice with free MYXV (fold increase of 16.9 at 3 h), PBMCs + MYXV (fold increase of 3.9 at 3 h), and BM + MYXV (fold increase of 92.3 at 3 h).
Taken together, the data illustrated in Figure 5 and summarized in Tables 2 and 3 demonstrate the capacity of ex vivo MYXV virotherapy to activate multiple aspects of the immune system in the BM and spleen of mice bearing MM MRD in vivo. In addition to this, these results demonstrate that treatment with free MYXV alone, or in combination with autologous BM cells or PBMCs, alters the levels of a variety of pro-inflammatory cytokines and cytokines early after treatment (i.e., 3 and 24 h). Nevertheless, it will be necessary to explore in more detail how some of these MYXV-mediated upregulated immune effector molecules and cytokines can impact the observed therapeutic effects in the long-term post-treatment. Therefore, our future efforts will focus on understanding the mechanisms by which MYXV alone or in combination with ASCT improves survival rates and decreases tumor burden in mice bearing myeloma MRD.
BM and PBMC Carrier Cells Can Transport MYXV into Sites of Myeloma MRD within the BM and Spleen In Vivo
In order to track the fate of BM or PBMC carrier cells that had been ex vivo pre-armed with MYXV, we performed in vivo experiments with leukocytes tagged with the near-infrared cell tracker reagent VivoTrack 680. This reagent has been used by others for in vivo imaging quantification of leukocyte immune responses.37 Some advantages of using this dye includes its fast diffusion into carrier cells, its long half-life after internalization in vivo, and the fact that this dye is very specific to the stained cells without transferring fluorescence to adjacent cells. In addition, this reagent is very biocompatible, keeping the fluorescently stained cells fully functional, and it fluoresces at high intensities.37 Briefly, sublethally irradiated BALB/c mice were seeded with 1 × 105 MOPC315.BM.FLuc MM cells to track the progress of the tumor in real time using luciferase signals monitored by the Xenogen in vivo imaging system (IVIS) 11 days after cancer implantation. As expected, the implanted myeloma tumor cells were localized in the long bones of the arms, legs, and along the spinal cord (Figures 6A and 6E). Next, at day 11 mice were transplanted with 2 × 106 BALB/c BM leukocytes or PBMCs fluorescently labeled with VivoTrack 680 dye and then ex vivo infected with vMyx-M135KO. Unlabeled BM leukocytes or PBMCs were used as controls. At 6, 24, and 48 h after cell infusion, mice were imaged in order to localize fluorescently labeled carrier cells loaded with MYXV. As shown in Figures 6B, 6C, 6F, and 6G, ex vivo VivoTrack 680-tagged BM or PBMCs that had been treated ex vivo with MYXV were localized in the spinal cord, spleen, and BM as well as in the liver. Figures 6D and 6H illustrate the total radiance efficiency expressed in photons per transplanted mouse. For example, Figure 6D shows the total radiance from fluorescently labeled donor BM samples mock-treated or MYXV infected, and compared to unlabeled BM mock (control) at different time points (i.e., 6 h post-transplant: control −3.4 × 106% ± 9.4 × 105% versus BM mock 1.4 × 107% ± 1.6 × 106% versus BM + MYXV 6.0 × 108% ± 1.0 × 108% photons/s; 24 h post-transplant: control −1.4 × 107% ± 1.6 × 106% versus BM mock 4.0 × 109% ± 1.0 × 109% versus BM + MYXV 5.3 × 109% ± 2.0 × 109% photons/s; 48 h post-transplant: control −2.2 × 107% ± 7.3 × 106% versus BM mock 9.0 × 109% ± 2.5 × 109% versus BM + MYXV 7.4 x 109% ± 1.8 × 109% photons/s). Alternatively, Figure 6H shows the total radiance from fluorescently labeled donor PBMCs mock-treated or MYXV infected, and compared to unlabeled PBMCs mock (control) at different time points (i.e., 6 h post-transplant: control −3.0 × 106% ± 1.7 × 106% versus PBMCs mock 1.1 × 109% ± 3.6 × 108% versus PBMCs + MYXV 7.0 × 108% ± 4.0 × 108% photons/s; 24 h post-transplant: control −5.4 × 107% ± 4.0 × 107% versus PBMCs mock 7.4 × 109% ± 2.0 × 109% versus PBMCs + MYXV 6.3 × 109% ± 1.8 × 109% photons/s; 48 h post-transplant: control −1.3 × 107% ± 6.7 × 106% versus PBMCs mock 6.2 × 109% ± 2.0 × 109% versus PBMCs + MYXV 6.0 × 109% ± 1.6 × 109% photons/s). However, using this in vivo cell tracking strategy, we could not establish major differences in the distribution of BM leukocytes versus PBMCs ex vivo preloaded with MYXV, although we were able to identify carrier cells residing in myeloma sites such as the interior of long bones and spleen. In addition to this, some carrier cells were also found in the liver. For comparison purposes, we used sublethally irradiated healthy mice without myeloma as controls. In this case autologous BM or PBMCs were first ex vivo labeled with VivoTrack 680 and then mock- or MYXV-treated in identical fashion as described before. 2 × 106 fluorescently labeled BM cells or PBMCs with and without MYXV were systemically delivered into control mice, and then we monitored the distribution of these carrier cells in vivo after 6, 24, and 48 h of delivery in identical fashion as described before. Figures 6I/6J and 6K/6L show VivoTrack 680-tagged BM with/without MYXV and PBMCs with/without MYXV, respectively. Compared to those mice bearing MM, the distribution of the carrier cells in healthy mice started to be detected in long bones after 6 h of treatment, but by 48 h most of these cells were confined into the abdominal cavity, suggesting that the presence of tumor cells in the BM regulates the final destination and retention of carrier cells with and without MYXV. When these non-myeloma control mice were examined at necropsy, we found most fluorescently labeled carrier cells exclusively in the liver. Therefore, we conclude that the use of autologous carrier leukocytes is an effective strategy to transport MYXV into sites of myeloma MRD within BM and spleen in vivo.
Figure 6.

Tracking the Fate of Fluorescently Tagged Autologous BM Cells and PBMCs with and without MYXV within Recipient Mice Pre-seeded with Myeloma MRD
(A and E) Levels of luciferase expressed by MOPC315.BM.FLuc MM cells 11 days after cancer implantation. Based on the bioluminescence signal, myeloma tumor cells were localized in spinal cord, long bones, and spleen. (B and C) Localization of VivoTrack 680 fluorescently labeled BM cells ex vivo pre-infected with MYXV or mock-treated. (F and G) Localization of VivoTrack 680 fluorescently labeled PBMCs ex vivo infected with MYXV or mock-treated. Control corresponds to unlabeled BM mock or PBMC mock. (D and H) Correspond to the quantification of the levels of VivoTrack 680 labeled BM or PBMCs with and without MYXV. (I and J) Migration of BM with and without MYXV within control healthy mice (naive mice). (K and L) Migration of PBMCs with and without MYXV within control healthy mice (naive mice). Error bars represent the mean ± standard error (SE) of at least four independent experiments.
Discussion
Despite the enormous progress reached in the treatment of MM, this disease essentially always relapses and become resistant to conventional and standard treatments, even after myeloablative chemotherapy coupled with immune rescue ASCT. A major challenge with MM is the prevalence of therapy-refractory myeloma within tissue niches that constitute MRD.11,12,38 Therefore, alternative therapeutic strategies that assure the complete elimination of MM MRD are required. The use of OVs alone or in combination with other standard therapies is becoming an attractive strategy to target and eliminate relapsed/refractory or residual solid tumors and hematological malignancies. However, a major challenge and matter of active investigation is to determine the most effective way to more efficiently deliver OVs against cancer after systemic infusion.
Our data demonstrate that ex vivo virotherapy with oncolytic MYXV, either following systemic infusion of free virus or in conjunction with ASCT using BM or PBMCs, dramatically increases survival rates and can eliminate tumor burden in a preclinical murine MM model (Figure 1). Single systemic administration of free MYXV resulted in some long-term therapeutic effect against MM MRD, but these benefits were potentiated further with the use of virus preloaded ex vivo on carrier cells from autologous BM or PBMCs. These in vivo results were somewhat unexpected and intriguing considering that MOPC315.BM.DsRed myeloma cells are fully non-permissive to MYXV binding or infection in vitro.26,39 Despite the lack of any MYXV binding receptors on these MM cells, they could nevertheless be slightly infected and killed in vitro if the virus was first preloaded onto mixed leukocytes, which can then transmit the virus via direct cell-cell contact. So, although it was not unexpected that leukocyte-loaded MYXV could be therapeutic against this myeloma in vivo, it was a relative surprise that some therapeutic effect, albeit less, was observed after systemic infusion of naked virus. Our working hypothesis is that following infusion of free MYXV, a fraction of the input virus adsorbed to circulating leukocytes and this endogenous cell-bound virus was the active therapeutic agent that migrated into sites of myeloma in the BM and spleen and cleared the MRD. Furthermore, our in vivo data demonstrate the advantage of using carrier cells preloaded with an OV like MYXV to more efficiently reach and eliminate cancer cells. We previously published that in the context of allo-HSCT, MYXV can be transported by allogeneic T cells or neutrophils as carrier cells to infect and kill MOPC315.BM.DsRed myeloma cells in vitro.26 In this study, we provide experimental evidence that, now in the context of ASCT, autologous-derived T cells or neutrophils from BM or PBMCs can also be effective carrier cells that can transport and deliver MYXV into sites of MM MRD in the BM and spleen in vivo (Figure 3). Nevertheless, the in vivo data also suggest a possible role of other immune cell populations as potential carriers of the virus, since when we used donor PBMCs derived from immune-compromised NOD/SCID-γ BALB/c mice, the therapeutic effects of these leukocytes ex vivo-treated with MYXV were compromised, but not eliminated, even when subsequently depleted of neutrophils.
The use of many different OVs in combination with carrier cells has been previously reported. For example, anti-tumor efficacy of autologous BM-derived MSCs infected with OAdv ICOVIR-5 for treatment of patients with neuroblastoma has been observed.40,41 In a different report, combination of allogeneic PBMCs and OAdv-infected MSCs enhances anti-tumor efficacy in vitro against cancer cell lines such as A431-GL and FaDu-GL and in vivo using recipient NSG mice bearing either subcutaneous A549 or A431 tumors.30 Interestingly, the A431 cell line is known to be partially resistant to adenovirus infection.30 MSCs infected with OAdv has also shown enhancing anti-tumor effects against hepatocellular carcinomas.42 Allogeneic NK cells in combination with OVs are also potential weapons for tumor eradication. For example, in a preclinical study with the B16 melanoma model, the Newcastle disease virus (NDV) activated T cells and NK cells, resulting in the induction of inflammatory responses, which led to lymphocyte infiltrates and an antitumor effect.43, 44, 45
Importantly, MYXV not only mediated long-term survival from MRD, but also survivor mice (about 80%) were now resistant to re-challenge with fresh myeloma cells, which ultimately suggests the development of acquired immune protection in these re-challenged mice. In fact, we can report that MYXV induced the development of memory anti-MM T cells in these mice (Figure 4).
OVs can mediate their anti-tumor effects through a dual mechanism of action, including direct oncolysis of tumor cells and the induction of an adaptive anti-tumor immune response.46,47 Although in this study we did not address the mechanisms by which MYXV mediated the elimination of MM MRD, we did investigate the effect of MYXV alone or in combination with autologous BM or PBMC transplantation on the early stage initiation of an immune response. This is important because the induction of systemic innate and tumor-specific adaptive immune responses plays a critical role in the eradication of tumor cells by OVs.48
We presume that the observed long-term benefits mediated by MYXV against MM MRD were likely driven by virus-mediated anti-tumor immune responses resulting in the secretion of pro-inflammatory cytokines such as INF-γ, TNF, and other early response immune molecules such as IP-10, IL-1α, IL-1β and GM-CSF (Figure 5; Tables 2 and 3). Because MM cells exquisitely depend on the BM microenvironment for growth and survival, the observation that a limited amount of IL-6 is secreted in the BM of MYXV-treated mice is important considering that this cytokine is one of the feeders of MM cells.49 Also, the fact that the secretion levels of the pro-osteoclastogenesis MIP-1α chemokine33,34 were transient is possibly relevant. In summary, it is clear that MYXV virotherapy has the capacity to activate a BM-focalized immune response in vivo that ultimately can result in the control of MM MRD progression and acquisition of anti-MM acquired immunity. At this point, we postulate that the murine MOPC315.BM myeloma cells within the TME are susceptible to the treatment with carrier cells ex vivo armed with MYXV virus but require cell-to-cell contact to initiate myeloma cell infection and killing in situ. Others have published that these MM BM cells after 20 days of implantation remain non-permissive to direct MYXV binding.39 Therefore, further studies are necessary in order to understand the mechanisms used by MYXV in vivo to control the progression of myeloma MRD.
We also demonstrated the capacity of mixed carrier leukocytes from either BM or PBMCs to selectively transport oncolytic MYXV to sites of myeloma MRD in vivo (Figure 6). In fact, we were able to show that fluorescently tagged autologous BM and PBMCs armed with MYXV become located into the myeloma TME, including the BM and spleen, at different time points post-infusion. The localization of these tagged carrier cells was rapid and revealed key differences in recipients that were MM-bearing versus MM-free. However, we could not establish obvious differences in the localization of BM leukocytes versus PBMCs, despite the former being more effective at eliminating MRD. In fact, tagged BM and PBMCs apparently migrate very similarly post-infusion and target the same organs. Taken together, these data demonstrate the capacity of mixed carrier leukocytes from either BM or PBMCs to selectively transport oncolytic MYXV to sites of myeloma MRD.
To our knowledge, this is the first study using autologous BM or PBMC carrier cells armed ex vivo with an OV to target and eliminate MM MRD in a preclinical murine model. This work suggests that standard immune rescue ASCT, commonly used for MM patients after myeloablative treatments but which is ineffective against residual myeloma in the patient, can be supplemented ex vivo with oncolytic MYXV to functionally eliminate MRD.
However, in order to make these findings translatable to clinical trials, there is the need for investigation in order to get a better understanding of the mechanisms of how OVs by themselves or in combination with other immunotherapies can eliminate myeloma cells and how carrier cells ex vivo armed with MYXV can improve delivery of virus to cancer niches, decrease tumor burden, and increase survival rates.
Materials and Methods
Cell Lines, Primary Leukocytes, and Viruses
The murine MM cell lines BALB/c-derived MOPC315.BM.DsRed and MOPC315.BM.FLuc, which were a kind gift of Dr. Bjarne Bogen (Centre for Immune Regulation, Institute of Immunology, University of Oslo and Oslo University Hospital, Oslo, Norway; and K.G. Jebsen Centre for Influenza Vaccine Research, Oslo, Norway), were used for in vivo or in vitro assays. The MOPC315.BM.DsRed and MOPC315.BM.FLuc myeloma cell lines were maintained as described previously.26 vMyx-M093L-Venus (i.e., fluorescently tagged virions) or vMyx-M135KO-GFP (i.e., unarmed but attenuated recombinant MYXV virus, in which the M135 gene has been deleted and GFP has been inserted under a poxvirus synthetic early/late promoter) virus constructs were used to perform infections for in vitro or in vivo experiments. Murine primary BM cells were isolated from femurs and tibias of donor healthy BALB/c mice, or NOD/SCID-γ immunodeficient mice, using centrifugation at 12,000 rpm for 3 min. Suspended cells were then filtered using a 40-μm nylon cell strainer (Thermo Fisher Scientific). For PBMCs, murine peripheral blood was obtained by terminal cardiac puncture and collected in 6.4% of anti-coagulant sodium citrate. Mononuclear cells were isolated using Ficoll-Plaque Plus density gradient purification (GE Healthcare) via centrifugation at 1,200 × g for 10 min at 25°C, with breaks on. All virus infections were performed in complete RPMI 1640 culture media. For virus infections, BM leukocytes or PBMCs were ex vivo incubated with the test MYXV at an MOI of 10 at 37°C for 1 h to allow virus adsorption. For in vivo studies, leukocytes were suspended in 1× PBS and systemically infused (100 μL) into the animals by the RO route as described below.
In Vivo Mouse Studies
All animal experiments were performed under the Institutional Animal Care and Use Committee (IACUC) approval (no. 171543R) of Arizona State University and conformed to all regulatory standards. For in vivo autologous leukocyte transplantations, we used 8-week-old BALB/c female or male mice (Charles River Laboratories, Wilmington, MA, USA) as donors or recipients, which were matched according to sex. For myeloma cell implantations, at 1 day prior to MM cell infusion, recipient mice were sublethally irradiated with 175 cGy total body irradiation (TBI) using an X-ray source. Irradiated mice received antibiotic-medicated water for 2 weeks in order to avoid opportunistic infections. Twenty-four hours after irradiation mice were implanted i.v. via tail vein with 1 × 105 BALB/c-derived MOPC315.BM.DsRed or 1 × 105 BALB/c-derived MOPC315.BM.FLuc myeloma cells. One week after myeloma implantation, mice were randomized to six groups (i.e., a minimum of 10 mice per group) and treated as follows: no transplant but 1× PBS vehicle control (cohort I); 1 × 107 MYXV focus-forming (i.e., infectious) units (ffu) (free virus, cohort II); 2 × 106 BM cells alone (cohort III); 2 × 106 BM cells ex vivo preloaded with MYXV at an MOI of 10 (cohort IV); PBMCs alone (cohort V); and PBMCs ex vivo preloaded with MYXV (cohort VI). Each leukocyte/virus treatment was delivered via RO injections.
For some experiments, immunocompromised NOD/SCID-γ mice (from BALB/c background), which lack T and B cell lymphocytes as well as possess deficient NK cell functions, were used as donors to investigate the role of donor T lymphocytes to carry and deliver MYXV to the recipient’s TME. The immunocompromised BALB/c-derived NOD/SCID-γ mice were obtained from the animal facility at the Biodesign Institute (Arizona State University [ASU]). Briefly, 8-week-old NOD/SCID-γ female or male mice were used to isolate BM from femurs and tibias. For PBMCs, peripheral blood was isolated via cardiac puncture and mononuclear cells isolated using Ficoll-Plaque Plus density gradient purification (GE Healthcare) as described previously in this section. Isolated NOD/SCID-γ-derived BM leukocytes or PBMCs were used for virus infection as described previously, followed by implantation of 2 × 106 cells into each recipient mouse bearing pre-seeded MM. When indicated, NOD/SCID-γ BM was used to enrich or deplete neutrophils, using the anti-Ly6G microbead kit and a magnetic cell separator (Miltenyi Biotec), according to the manufacturer’s recommendations. Briefly, enriched neutrophils from BM (i.e., +neutrophils) or BM depleted of neutrophils (i.e., BM–neutrophils) was incubated with MYXV (i.e., using the vMyx-M135KO-GFP virus construct) at an MOI of 10 for 1 h at 37°C to allow virus adsorption as described before. After this, 2 × 106 +neutrophils + MYXV or BM–neutrophils + MYXV were systemically delivered via RO infusion into wild-type BALB/c recipient mice that had been pre-seeded with MM 1 week before. At the onset of morbidity (i.e., hindlimb paralysis, hunched position, weight loss, labor breathing, solid tumors 2 cm in diameter) or at the end of the study, mice were euthanized via asphyxiation with CO2, as per institutional guidelines. Immediately after euthanasia, BM, spleen, or extramedullary solid DsRed-positive myeloma tumors, if any, were isolated and used for immunophenotyping. Survival was assessed until days 100 or 115 following cancer implantation. For re-challenge experiments, MYXV-treated mice from transplant cohorts that survived 100 days post-cancer implantation were re-infused with fresh 1 × 105 MOPC315.BM.DsRed MM cells i.v. via tail vein. Mice that survived for an additional 100 days after cancer re-challenge were considered cured and had acquired immunity to the myeloma.
In Vitro Cell Co-culture Assays
For co-culturing experiments, BM cells or PBMCs derived from BALB/c healthy mice were isolated as described elsewhere in this section. About 1 × 106 BM cells or PBMCs were either mock-treated (i.e., without adding virus) or infected with vMyx-M135KO-GFP at an MOI of 10 for 1 h at 37°C to allow virus adsorption. After this, samples were incubated in complete RPMI 1640 media (plus 20% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 μg/mL streptomycin, and 100 U/mL penicillin) at 37°C for 24 h. After 24 h, BALB/c-derived BM or PBMCs with and without MYXV were mixed with target MOPC315.BM.DsRed MM cells at an effector-to-target cell ratio of 10:1. The admixtures were incubated for 24 or 48 h at 37°C and then analyzed using flow cytometry in order to assess virus infection of target myeloma cells (i.e., CD138+GFP+). The myeloma cell killing was assessed using the fixable Near-IR Live/Dead cell stain dye (Thermo Fisher Scientific) (i.e., CD138+Near-IR Live/Dead+).
Assessment of Myeloma Burden
Primary murine BM, spleen, and solid tumors were isolated from mice at the endpoint of the study and processed. Approximately 2 × 106 cells were stained with fluorescently conjugated mAbs against murine CD138, a marker for MM. Samples were analyzed using flow cytometry (LSRFortessa, BD Biosciences) and FlowJo software.
Cytokine and Immune Effector Detection
Femurs from each mouse were collected 3 or 24 h after cohort treatments and placed in a 0.5-mL Eppendorf tube with a hole at the bottom. The tube was placed inside a 1.5-mL Eppendorf tube and centrifuged at 12,000 rpm for 3 min in order to obtain a BM cell pellet. This pellet was resuspended in 100 μL of 1× PBS and centrifuged again at 2,000 rpm for 3 min. BM supernatants were stored at −80°C until ready for analysis in duplicate with a cytokine and immune molecule mouse custom ProcartaPlex 11-plex for a multiplex bead-based platform (PPX-11-MX322uV, Thermo Fisher Scientific), according to the manufacturer’s instructions. Data were acquired on a Luminex 200 system equipped with xPONENT 3.1 software (Thermo Fisher Scientific). Lower levels of detection were in the range of 3.5–78 pg/mL. Similarly, spleens from each mouse were collected and splenocytes were isolated through a 40-μm mesh size cell strainer. Then, the obtained splenocyte suspensions were spun down at 2,000 rpm for 3 min. Splenocyte supernatants were stored at −80°C for further analysis of secreted cytokine and immune molecules as described above for BM.
In Vivo Tracking of Fluorescently Labeled Carrier BM Leukocytes or PBMCs
For tracking ex vivo MYXV-loaded carrier leukocytes after systemic infusion into MM-bearing or control mice, first recipient mice were irradiated as described before. One day later, each mouse was implanted with 1 × 105 MOPC315.BM.FLuc MM cells i.v. via tail vein. In order to image the tumor cells, mice were injected intraperitoneally (i.p.) at 1 week after cancer implantation with d-luciferin (150 mg/kg body weight) and imaged using the Xenogen IVIS. Images were acquired 10 min post-injection of d-luciferin via the i.p. route. Next, after verifying detectable myeloma burden in the BM and spleen, animals were transplanted systemically via RO injection with either BM leukocytes or PBMCs ex vivo pre-labeled with VivoTrack 680 fluorescent dye (PerkinElmer) and either mock-treated or infected ex vivo with vMyx-M135KO-GFP virus. As negative controls, mice implanted with myeloma cells received 1× PBS vehicle. Briefly, murine primary BM leukocytes or PBMCs from healthy BALB/c mice were isolated as described previously. Next, 2 × 107 cells were fluorescently labeled using the VivoTrack 680 dye for 15 min at room temperature, while protecting from light and according to the manufacturer’s recommendations. Cells were then washed three times with 1× PBS supplemented with 1× fetal bovine serum (FBS) to remove the excess of cell labeling agent. Cells were then counted and mock-treated or incubated with vMyx-M135KO-GFP virus at an MOI of 10 for 1 h at 37°C to allow virus adsorption. Two million (2 × 106) cells were infused to recipient mice using the RO route. Fluorescently labeled cells with and without MYXV were tracked at 6, 24, and 48 h, post-treatment using the fluorescent option of an Xenogen IVIS Lumina III in vivo imaging system (PerkinElmer).
Statistical Analysis
Values are represented as means ± SD or means ± SE for at least three independent experiments. Kaplan-Meier analysis of mouse survival was performed with GraphPad Prism 8 software (La Jolla, CA, USA), and a log-rank test (Mantel-Cox) was performed to compare survival curves and to perform statistical analysis. Statistical comparison between two groups was conducted using the two-tailed Student’s t test. For comparison of multiple groups, two-way ANOVA with Tukey’s multiple comparison tests were performed to compare differences within a group and between groups. Animals were assigned to treatments groups (cohorts) randomly, and the number of animals in each treatment group is reported in the figures. p values are reported as follows: not significant (ns) p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p ≤ 0.0001.
Study Approval
All animal work was conducted under the approval of the Arizona State University Institutional Animal Care and Use Committee (IACUC) (no. 171543R) in accordance with federal, state, and local guidelines.
Author Contributions
N.Y.V. designed and performed experiments, analyzed data and wrote the manuscript. M.M.R. performed experiments and analyzed data. J.M., J.D’I., E.G., J.K., K.L., J.D-V., L.T., J.C., N.A., L.H., and K.J. performed experiments. G.M. provided overall project leadership and supervised the data analysis. All authors reviewed the manuscript.
Conflicts of Interest
G.M. is co-founder of OncoMyx Therapeutics Inc., which is developing myxoma virus as an oncolytic therapeutic. The remaining authors declare no competing interests.
Acknowledgments
We thank Dr. Cameron Lilly for preliminary data that led to this study. We also thank Drs. Marta Chesi, Leif Bergsagel, and Rafael Fonseca for many helpful discussions and suggestions. This work was supported by an ASU, USA start-up grant (PG046-498) to G.M. and by a Mayo Clinic Developmental Research Award from the Myeloma SPORE grant (GR35414), USA awarded to G.M. and Leif Bergsagel.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.011.
Supplemental Information
References
- 1.Kyle R.A., Rajkumar S.V. Multiple myeloma. N. Engl. J. Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 2.Brigle K., Rogers B. Pathobiology and diagnosis of multiple myeloma. Semin. Oncol. Nurs. 2017;33:225–236. doi: 10.1016/j.soncn.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Palumbo A., Anderson K. Multiple myeloma. N. Engl. J. Med. 2011;364:1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 4.Korde N., Kristinsson S.Y., Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood. 2011;117:5573–5581. doi: 10.1182/blood-2011-01-270140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Institutes of Health. SEER cancer statistics review (CSR) 1975–2014. https://seer.cancer.gov/archive/csr/1975_2014/.
- 6.Calton C.M., Kelly K.R., Anwer F., Carew J.S., Nawrocki S.T. Oncolytic viruses for multiple myeloma therapy. Cancers (Basel) 2018;10:198. doi: 10.3390/cancers10060198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patriarca F. Frontline therapy in multiple myeloma: fast start for a long game. Lancet Haematol. 2019;6:e600–e601. doi: 10.1016/S2352-3026(19)30199-1. [DOI] [PubMed] [Google Scholar]
- 8.Merz A.M.A., Merz M., Hillengass J., Holstein S.A., McCarthy P. The evolving role of maintenance therapy following autologous stem cell transplantation in multiple myeloma. Expert Rev. Anticancer Ther. 2019;19:889–898. doi: 10.1080/14737140.2019.1674142. [DOI] [PubMed] [Google Scholar]
- 9.Soekojo C.Y., Kumar S.K. Stem-cell transplantation in multiple myeloma: how far have we come? Ther. Adv. Hematol. 2019;10 doi: 10.1177/2040620719888111. 2040620719888111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mikhael J. Treatment options for triple-class refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2020;20:1–7. doi: 10.1016/j.clml.2019.09.621. [DOI] [PubMed] [Google Scholar]
- 11.Avet-Loiseau H., Ludwig H., Landgren O., Paiva B., Morris C., Yang H., Zhou K., Ro S., Mateos M.V. Minimal residual disease status as a surrogate endpoint for progression-free survival in newly diagnosed multiple myeloma studies: a meta-analysis. Clin. Lymphoma Myeloma Leuk. 2020;20:e30–e37. doi: 10.1016/j.clml.2019.09.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romano A., Palumbo G.A., Parrinello N.L., Conticello C., Martello M., Terragna C. Minimal residual disease assessment within the bone marrow of multiple myeloma: a review of caveats, clinical significance and future perspectives. Front. Oncol. 2019;9:699. doi: 10.3389/fonc.2019.00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bal S., Weaver A., Cornell R.F., Costa L.J. Challenges and opportunities in the assessment of measurable residual disease in multiple myeloma. Br. J. Haematol. 2019;186:807–819. doi: 10.1111/bjh.16130. [DOI] [PubMed] [Google Scholar]
- 14.Kumar S., Paiva B., Anderson K.C., Durie B., Landgren O., Moreau P., Munshi N., Lonial S., Bladé J., Mateos M.V. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e346. doi: 10.1016/S1470-2045(16)30206-6. [DOI] [PubMed] [Google Scholar]
- 15.Stanford M.M., McFadden G. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert Opin. Biol. Ther. 2007;7:1415–1425. doi: 10.1517/14712598.7.9.1415. [DOI] [PubMed] [Google Scholar]
- 16.Chan W.M., Rahman M.M., McFadden G. Oncolytic myxoma virus: the path to clinic. Vaccine. 2013;31:4252–4258. doi: 10.1016/j.vaccine.2013.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson D.B., Puzanov I., Kelley M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7:611–619. doi: 10.2217/imt.15.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahman M.M., Madlambayan G.J., Cogle C.R., McFadden G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with myxoma virus. Cytokine Growth Factor Rev. 2010;21:169–175. doi: 10.1016/j.cytogfr.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pisklakova A., McKenzie B., Zemp F., Lun X., Kenchappa R.S., Etame A.B., Rahman M.M., Reilly K., Pilon-Thomas S., McFadden G. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro-oncol. 2016;18:1088–1098. doi: 10.1093/neuonc/now006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartee E. Potential of oncolytic viruses in the treatment of multiple myeloma. Oncolytic Virother. 2018;7:1–12. doi: 10.2147/OV.S136644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bais S., Bartee E., Rahman M.M., McFadden G., Cogle C.R. Oncolytic virotherapy for hematological malignancies. Adv. Virol. 2012;2012:186512. doi: 10.1155/2012/186512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahman M.M., McFadden G. Oncolytic virotherapy with myxoma virus. J. Clin. Med. 2020;9:171. doi: 10.3390/jcm9010171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartee E., Chan W.M., Moreb J.S., Cogle C.R., McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol. Blood Marrow Transplant. 2012;18:1540–1551. doi: 10.1016/j.bbmt.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munguia A., Ota T., Miest T., Russell S.J. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene Ther. 2008;15:797–806. doi: 10.1038/gt.2008.45. [DOI] [PubMed] [Google Scholar]
- 25.Villa N.Y., Wasserfall C.H., Meacham A.M., Wise E., Chan W., Wingard J.R., McFadden G., Cogle C.R. Myxoma virus suppresses proliferation of activated T lymphocytes yet permits oncolytic virus transfer to cancer cells. Blood. 2015;125:3778–3788. doi: 10.1182/blood-2014-07-587329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lilly C.L., Villa N.Y., Lemos de Matos A., Ali H.M., Dhillon J.S., Hofland T., Rahman M.M., Chan W., Bogen B., Cogle C., McFadden G. Ex vivo oncolytic virotherapy with myxoma virus arms multiple allogeneic bone marrow transplant leukocytes to enhance graft versus tumor. Mol. Ther. Oncolytics. 2016;4:31–40. doi: 10.1016/j.omto.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melzer M.K., Lopez-Martinez A., Altomonte J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: defeating cancer with a “hammer” and “anvil”. Biomedicines. 2017;5:8. doi: 10.3390/biomedicines5010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuster P., Lindner G., Thomann S., Haferkamp S., Schmidt B. Prospect of plasmacytoid dendritic cells in enhancing anti-tumor immunity of oncolytic herpes viruses. Cancers (Basel) 2019;11:651. doi: 10.3390/cancers11050651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rojas J.J., Sampath P., Bonilla B., Ashley A., Hou W., Byrd D., Thorne S.H. Manipulating TLR signaling increases the anti-tumor T cell response induced by viral cancer therapies. Cell Rep. 2016;15:264–273. doi: 10.1016/j.celrep.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreno R., Fajardo C.A., Farrera-Sal M., Perisé-Barrios A.J., Morales-Molina A., Al-Zaher A.A., García-Castro J., Alemany R. Enhanced antitumor efficacy of oncolytic adenovirus-loaded menstrual blood-derived mesenchymal stem cells in combination with peripheral blood mononuclear cells. Mol. Cancer Ther. 2019;18:127–138. doi: 10.1158/1535-7163.MCT-18-0431. [DOI] [PubMed] [Google Scholar]
- 31.Koske I., Rössler A., Pipperger L., Petersson M., Barnstorf I., Kimpel J., Tripp C.H., Stoitzner P., Bánki Z., von Laer D. Oncolytic virotherapy enhances the efficacy of a cancer vaccine by modulating the tumor microenvironment. Int. J. Cancer. 2019;145:1958–1969. doi: 10.1002/ijc.32325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fajardo C.A., Guedan S., Rojas L.A., Moreno R., Arias-Badia M., de Sostoa J., June C.H., Alemany R. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017;77:2052–2063. doi: 10.1158/0008-5472.CAN-16-1708. [DOI] [PubMed] [Google Scholar]
- 33.Hideshima T., Mitsiades C., Tonon G., Richardson P.G., Anderson K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer. 2007;7:585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 34.Roodman G.D. Pathogenesis of myeloma bone disease. Leukemia. 2009;23:435–441. doi: 10.1038/leu.2008.336. [DOI] [PubMed] [Google Scholar]
- 35.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 36.Chesi M., Mirza N.N., Garbitt V.M., Sharik M.E., Dueck A.C., Asmann Y.W., Akhmetzyanova I., Kosiorek H.E., Calcinotto A., Riggs D.L. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 2016;22:1411–1420. doi: 10.1038/nm.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swirski F.K., Berger C.R., Figueiredo J.L., Mempel T.R., von Andrian U.H., Pittet M.J., Weissleder R. A near-infrared cell tracker reagent for multiscopic in vivo imaging and quantification of leukocyte immune responses. PLoS ONE. 2007;2:e1075. doi: 10.1371/journal.pone.0001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Agostino M., Bertamini L., Oliva S., Boccadoro M., Gay F. Pursuing a curative approach in multiple myeloma: a review of new therapeutic strategies. Cancers (Basel) 2019;11:E2015. doi: 10.3390/cancers11122015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartee E., Bartee M.Y., Bogen B., Yu X.Z. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol. Ther. Oncolytics. 2016;3:16032. doi: 10.1038/mto.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.García-Castro J., Alemany R., Cascalló M., Martínez-Quintanilla J., Arriero Mdel.M., Lassaletta A., Madero L., Ramírez M. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: an exploratory study. Cancer Gene Ther. 2010;17:476–483. doi: 10.1038/cgt.2010.4. [DOI] [PubMed] [Google Scholar]
- 41.Melen G.J., Franco-Luzón L., Ruano D., González-Murillo Á., Alfranca A., Casco F., Lassaletta Á., Alonso M., Madero L., Alemany R. Influence of carrier cells on the clinical outcome of children with neuroblastoma treated with high dose of oncolytic adenovirus delivered in mesenchymal stem cells. Cancer Lett. 2016;371:161–170. doi: 10.1016/j.canlet.2015.11.036. [DOI] [PubMed] [Google Scholar]
- 42.Yoon A.R., Hong J., Li Y., Shin H.C., Lee H., Kim H.S., Yun C.O. Mesenchymal stem cell-mediated delivery of an oncolytic adenovirus enhances antitumor efficacy in hepatocellular carcinoma. Cancer Res. 2019;79:4503–4514. doi: 10.1158/0008-5472.CAN-18-3900. [DOI] [PubMed] [Google Scholar]
- 43.Jewett A., Kos J., Kaur K., Safaei T., Sutanto C., Chen W., Wong P., Namagerdi A.K., Fang C., Fong Y., Ko M.W. Natural killer cells: diverse functions in tumor immunity and defects in pre-neoplastic and neoplastic stages of tumorigenesis. Mol. Ther. Oncolytics. 2019;16:41–52. doi: 10.1016/j.omto.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarahian M., Watzl C., Fournier P., Arnold A., Djandji D., Zahedi S., Cerwenka A., Paschen A., Schirrmacher V., Momburg F. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 2009;83:8108–8121. doi: 10.1128/JVI.00211-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marelli G., Howells A., Lemoine N.R., Wang Y. Oncolytic viral therapy and the immune system: a double-edged sword against cancer. Front. Immunol. 2018;9:866. doi: 10.3389/fimmu.2018.00866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bommareddy P.K., Shettigar M., Kaufman H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018;18:498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 48.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hope C., Ollar S.J., Heninger E., Hebron E., Jensen J.L., Kim J., Maroulakou I., Miyamoto S., Leith C., Yang D.T. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood. 2014;123:3305–3315. doi: 10.1182/blood-2014-02-554071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.