

Abstract
Normal high-density lipoprotein (nHDL) in normal, healthy subjects is able to promote angiogenesis, but the mechanism remains incompletely understood. HDL from patients with coronary artery disease may undergo a variety of oxidative modifications, rendering it dysfunctional; whether the angiogenic effect is mitigated by such dysfunctional HDL (dHDL) is unknown. We hypothesized that dHDL compromises angiogenesis. The angiogenic effects of nHDL and dHDL were assessed using endothelial cell culture, endothelial sprouts from cardiac tissue from C57BL/6 mice, zebrafish model for vascular growth and a model of impaired vascular growth in hypercholesterolemic low-density lipoprotein receptor null(LDLr-/-)mice. MiRNA microarray and proteomic analyses were used to determine the mechanisms. Lipid hydroperoxides were greater in dHDL than in nHDL. While nHDL stimulated angiogenesis, dHDL attenuated these responses. Protein and miRNA profiles in endothelial cells differed between nHDL and dHDL treatments. Moreover, nHDL suppressed miR-24-3p expression to increase vinculin expression resulting in nitric oxide (NO) production, whereas dHDL delivered miR-24-3p to inhibit vinculin expression leading to superoxide anion (O2•-) generation via scavenger receptor class B type 1. Vinculin was required for endothelial nitric oxide synthase (eNOS) expression and activation and modulated the PI3K/AKT/eNOS and ERK1/2 signaling pathways to regulate nHDL- and VEGF-induced angiogenesis. Vinculin overexpression or miR-24-3p inhibition reversed dHDL-impaired angiogenesis. The expressions of vinculin and eNOS and angiogenesis were decreased, but the expression of miR-24-3p and lipid hydroperoxides in HDL were increased in the ischemic lower limbs of hypercholesterolemic LDLr-/- mice. Overexpression of vinculin or miR-24-3p antagomir restored the impaired-angiogenesis in ischemic hypercholesterolemic LDLr-/- mice. Collectively, nHDL stimulated vinculin and eNOS expression to increase NO production by suppressing miR-24-3p to induce angiogenesis, whereas dHDL inhibited vinculin and eNOS expression to enhance O2•- generation by delivering miR-24-3p to impair angiogenesis, and that vinculin and miR-24-3p may be therapeutic targets for dHDL-impaired angiogenesis.
Keywords: High-density lipoprotein, Angiogenesis, Coronary artery disease, miRNA, Vinculin, Endothelial nitric oxide synthase
Graphical abstract
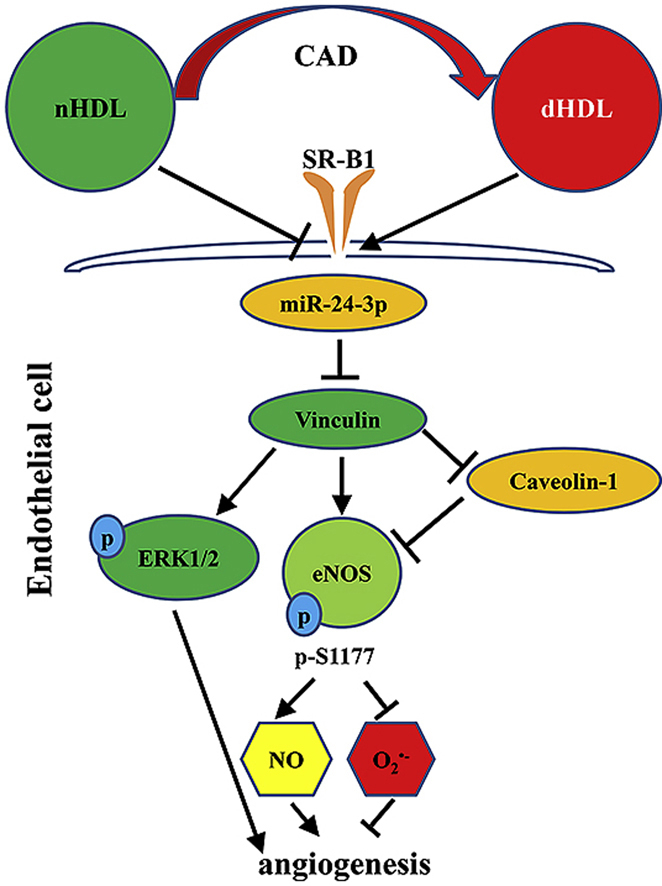
Highlights
-
•
nHDL and dHDL regulated angiogenesis differently via alterations in vinculin expression.
-
•
nHDL suppressed miR-24-3p to increase vinculin expression to stimulate NO production.
-
•
dHDL delivered miR-24-3p to inhibit vinculin expression to enhance O2.•- generation.
-
•
Vinculin and miR-24-3p may be therapeutic targets for dHDL-impaired angiogenesis.
-
•
Cell-free assay may be used to measure the oxidative levels of HDL.
Non-standard abbreviations and acronyms
- AKT
protein kinase B
- CAD
coronary artery disease
- CRP
C reacting protein
- ECs
endothelial cells
- eNOS
endothelial nitric oxide synthase
- ERK1/2
extracellular signal-regulated kinase 1/2
- HUVECs
human umbilical vein endothelial cells
- HDL
high-density lipoprotein
- nHDL
normal high-density lipoprotein
- dHDL
dysfunctional high-density lipoprotein
- LDLr-/
low-density lipoprotein receptor null
- l-NAME
NG-nitro-l-arginine methyl ester
- Mn-TBAP
Manganese-5, 10, 15, 20-tetrakis (4-benzoic acid) porphyrin
- NO
nitric oxide
- O2•
superoxide anion
- PBS
phosphate buffer saline
- PI3K
phosphatidylinositide 3-kinases
- qRT-PCR
quantitative real-time polymerase chain reaction
- SAA
serum amyloid A
- SRB1
scavenger receptor class B type 1
- TNF-α
tumor necrosis factor-α
- VCAM-1
vascular cell adhesion molecule-1
- VEGF
vascular endothelial growth factor
1. Introduction
Preclinical research formed the basis for clinical trials that were directed at stimulating coronary vascular growth as a treatment for coronary artery disease (CAD); unfortunately these trials failed. The reasons for failure are likely many, but one consensus is the myriad risk factors contributing to vascular disease and endothelial dysfunction in aged patients. Furthermore, stimulating blood vessel growth in pre-clinical models without vascular disease is non-trivial. One notable and perhaps underestimated risk is oxidized high-density lipoprotein (HDL) [1,2]. Previous studies have shown that HDL from healthy subjects (nHDL) is pro-angiogenic, which could potentially be used to therapeutically aid angiogenesis in patients with CAD [[3], [4], [5]]. Moreover, some studies demonstrate that normal or reconstituted HDL can stimulate angiogenesis in patients with CAD [6,7]. However, HDL from patients with CAD does not have similar beneficial functions as nHDL.
The HDL in patients with CAD (dHDL) undergoes oxidative modifications and is proinflammatory (dysfunctional) [1,2,[8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24]]. Despite these myriad observations, little is known how dHDL exerts its detrimental effects on angiogenesis. Thus, the present study was designed to compare the angiogenic effects between HDL from patients with CAD and healthy subjects, and investigate their potential mechanisms.
To test this hypothesis, we first tested the proinflammatory and angiogenic effects of nHDL and dHDL. We then measured the effects of nHDL and dHDL on miRNAs expressions in vascular endothelial cells (ECs). Finally, we determined if altered miRNAs expressions target proteins expression to regulate angiogenesis. In this study, we present evidence that nHDL suppressed miR-24-3p to increase the expressions of vinculin and endothelial nitric oxide synthase (eNOS) resulting in nitric oxide (NO) production to induce angiogenesis. In contrast, dHDL impaired angiogenesis via partially delivering miR-24-3p, which inhibited the expressions of vinculin and eNOS leading to superoxide anion (O2•-) generation. Our findings revealed novel molecular mechanisms underlying the angiogenic effects of nHDL and how oxidative modifications of HDL in patients with CAD undermine this beneficial effect.
2. Materials and methods
Please refer to the online supplement for additional details on the methodology.
2.1. Patient population, and plasma and tissues sampling
Age- and sex-matched patients with CAD with hypercholesterolemia and healthy volunteers (i.e. without cardiovascular risk factors) were recruited into the study for isolation of HDL. This study was approved by the Ethics Review Board of The First Affiliated Hospital, Sun Yat-sen University. Informed consent was obtained from all subjects enrolled into this study.
2.2. Isolation of HDL and measurement of its lipid hydroperoxides level
The dHDL and nHDL were isolated via sequential ultracentrifugation, as previously described [13,25]. The lipid hydroperoxide levels of HDL were determined by using a modified cell-free assay [[25], [26], [27]].
2.3. Measurement of cholesterol efflux capacity
The cholesterol efflux capacities of HDL were measured as previous described [28].
2.4. Cell culture and transfection
Human umbilical vein endothelial cells (HUVECs) were cultured for the experiments outlined below. A siRNA targeting vinculin or scavenger receptor class B type 1 (SRB1), adenovirus vector for the expression of vinculin, and miR-24-3p (mimics or inhibitors) were transfected into cells.
2.5. MiRNA microarray and qRT-PCR
Total RNA from nHDL or dHDL was isolated. HUVECs were cultured treated with phosphate buffer saline (PBS), nHDL (100 μg/mL), or dHDL (100 μg/mL), and total RNA was extracted. MiRNA arrays and qRT-PCR were performed.
2.6. Two-dimensional (2D) gel electrophoresis (proteomics study)
After the HUVECs were treated with nHDL (100 μg/mL) or dHDL (100 μg/mL) for 72 h, the cell lysates were separated by 2D gel electrophoresis. Proteins were identified by using Ultraflex III TOF/TOF, and peptide mass fingerprinting (PMF)-combined MS/MS spectra were queried against the Swiss Prot database using the Biotools software and MASCOT as previous described [29,30].
2.7. Immunofluorescence analysis
Vinculin and vascular cell adhesion molecule-1 (VCAM-1) expression levels were detected by immunofluorescence staining [12].
2.8. EC tube formation assay
HUVECs were cultured with PBS, nHDL (100 μg/ml), dHDL (100 μg/ml), siRNA (50 nM), miRNA mimic, miRNA inhibitor, or vascular endothelial growth factor (VEGF). The EC tube formation was examined as previously described [31].
2.9. Measurement of EC NO and O2•- production
HUVECs were cultured with PBS, nHDL (100 μg/ml), dHDL (100 μg/ml), siRNA, miRNA mimic, miRNA inhibitor, or VEGF. Nitric oxide (NO) production was determined by 4,5-diaminofluorescein diacetate and Sievers NOA analyzer [[31], [32], [33]]. Superoxide anion (O2•-) production was assessed with the lucigenin assay and hydroethidine in ECs as previously described [25,31,[33], [34], [35]]. Tumor necrosis factor-α (TNF-α) was used as a positive control, and Manganese-5, 10, 15, 20-tetrakis (4-benzoic acid) porphyrin (Mn-TBAP) and NG-nitro-l-arginine methyl ester (l-NAME) were used as negative controls.
2.10. Western blot analysis of cultured HUVECs
HUVECs were cultured with PBS, nHDL (100 μg/mL), dHDL (100 μg/mL or different doses), siRNA, miRNA mimic, miRNA inhibitor, or TNF-α. Cellular proteins were harvested for Western blot analysis as previously described [[31], [32], [33],36,37].
2.11. Detection of HDL deliver miR-24-3p to ECs
The nHDL (100 μg) or dHDL (100 μg) were incubated with miR-24-3p mimic (0.2 nmol) labeled by Cy3 dye before added to the cultured HUVECs with the final concentration of HDL 100 μg/ml. Transfecting Cy3 dye-labeled miR-24-3p mimic with Lipofectamine RNAiMAX Regent as a positive control. HUVECs were cultured for 36 h before fluorescence images were taken.
2.12. Animal studies
All animal experiments were approved by the ethics review board and animal research committee of The First Affiliated Hospital, Sun Yat-sen University. Eight-week-old male and female C57BL6 mice and low-density lipoprotein receptor null (LDLr-/-) mice were obtained from the Jackson Laboratory. LDLr-/- mice were fed with a chow diet or a western diet for 6 weeks to induce hypercholesterolemia and proinflammatory HDL [26,38,39].
2.13. Angiogenesis assay in zebrafish
Healthy embryos were treated with H2O, nHDL (10 μg/mL), dHDL 10 μg/mL, or VEGF with/without or GSK690693. Embryos were incubated and the pro-angiogenic effects were evaluated as previously described [40].
2.14. Measurements of vascular growth in vivo
A model of lower limb ischemia was produced in hypercholesterolemic LDLr-/- mice, and the mice were subjected to multiple intramuscular injections of miR-24-3p antagomir or were injected intramuscularly with adenovirus vector carried plasmids encoding vinculin. The feet of the mice were imaged to measure the blood flow [41]. In addition, mice were anaesthetized and perfused with heparinized saline and contrast agent. The hindlimb vasculature was imaged and analyzed with a high-resolution computed tomography imaging system.
2.15. Statistical analysis
Data are presented as means ± SD. The differences among the test groups were determined with an analysis of variance (ANOVA) followed by a Tukey's test for more than two groups or with Student T-test for two groups (Prism, GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered statistically significant.
3. Results
3.1. The lipid hydroperoxide levels of HDL from dHDL and nHDL, and the relation with C reactive protein (CRP) and serum amyloid A (SAA)
The characteristics of the study population are summarized in Supplementary Table 1. Although 20% of the patients were being treated with statin, most of these patients were treated with statin for only 1–3 days before blood collection. To determine whether the dHDL is proinflammatory, the lipid hydroperoxide levels in HDL were measured. We found that the lipid hydroperoxide levels in HDL from patients with CAD were greater than those from normal healthy subjects (Fig. 1A), demonstrating that HDL from patients with CAD is proinflammatory. Lipid hydroperoxide levels in dHDL were positively correlated with CRP (r = 0.546) and SAA (r = 0.679, Fig. 1B,C).
Fig. 1.

The characteristics and functions of HDL from healthy subjects and patients with coronary artery disease (nHDL and dHDL, respectively) and their effects on angiogenesis.(A) The levels of lipid hydroperoxide in nHDL and dHDL. *p < 0.05 (n = 95). (B and C) The correlation between levels of lipid hydroperoxide in dHDL and C reactive protein (CRP) and serum amyloid A (SAA). *p < 0.05 (n = 78). (D) The cholesterol efflux capacities of nHDL and dHDL(100 μg/ml). *p < 0.05 (n = 31). (E) Immunofluorescence assays showing the effects of nHDL and dHDL with or without pretreatment with TNF-α on VCAM-1 expression (n = 6). (F–G) Images and dot plot show that nHDL stimulated endothelial cell (ECs) tube formation. The dHDL had less effect on EC tube formation. Both the ERK1/2 and AKT inhibitors impaired nHDL and dHDL-induced EC tube formation. * vs. control; # vs. nHDL; & vs. dHDL, p < 0.05 (n = 10). (H–I) The image and dot plot show that nHDL promoted angiogenesis in zebrafish. The dHDL had a lower effect on angiogenesis. Both ERK1/2 and AKT inhibitors decreased nHDL and dHDL-induced angiogenesis. * vs. control; # vs. nHDL; & vs. dHDL, p < 0.05 (n = 30). (J–K) Image and dot plot show that nHDL increased EC nitric oxide (NO) production. The dHDL had a lower effect on EC NO production. AU, arbitrary units. * vs. control; # vs. nHDL, p < 0.05 (n = 10). (L) The dot plot shows that dHDL, but not nHDL, was enhanced endothelial O2− generation. l-NAME inhibited dHDL-induced O2− generation. AU, arbitrary units. * vs. control; # vs. nHDL; & vs. dHDL; ∧ vs. TNF-α, p < 0.05 (n = 10).
3.2. Effects of nHDL and dHDL on cholesterol efflux and VCAM-1 expression
Next, we detected that HDL function by measuring the effects of nHDL and dHDL on cholesterol efflux and VCAM-1 expression. Fig. 1D and Supplemental Fig. 1 showed that the cholesterol efflux capacity was reduced by dHDL compared with nHDL. The nHDL did not stimulate VCAM-1 expression in ECs, but dHDL did. The nHDL could partially suppress TNF-α-induced VCAM-1 expression, but dHDL could not suppress TNF-α-induced VCAM-1 expression (Fig. 1E, Supplemental Fig. 2), indicating that dHDL is dysfunction.
3.3. Effects of nHDL and dHDL on tube formation
We further investigated the ability of HDL-induced angiogenesis. Both VEGF and nHDL stimulated EC tube formation. However, dHDL were less able to stimulate these responses (Fig. 1F and G). Both the extracellular signal-regulated kinase 1/2(ERK1/2) inhibitor PD98059 and the protein kinase B (AKT) inhibitor GSK690693 inhibited EC tube formation induced by nHDL and dHDL, suggesting dHDL lost the ability to induce angiogenesis. Since our current data and other reports demonstrated that 100 μg/mL of HDL produced the optimal proliferative effect, we decided to use this concentration for subsequent experiments.
3.4. Effects of nHDL and dHDL on angiogenesis in zebrafish
The angiogenic effects of HDL were further evaluated in vivo using a zebrafish model. Both nHDL and VEGF stimulated angiogenesis in zebrafish (Fig. 1H,I). The capability of dHDL-stimulated angiogenesis in zebrafish was lower than nHDL. Both ERK1/2 and AKT inhibitors inhibited angiogenesis induced by nHDL or dHDL. Since 100 μg/mL of HDL resulted in zebrafish mortality in our preliminary experiments, we chose 10 μg/mL of HDL in these experiments.
3.5. Effects of dHDL and nHDL on NO production and O2•- generation in ECs
NO is important for angiogenesis. We found that NO production was elevated by nHDL compared to controls. However, dHDL was less effective than nHDL in stimulating NO generation (Fig. 1J,K, Supplemental Fig. 3A). Similar levels of O2•- generation were observed following nHDL treatment, similar to that observed in the control, but an increase in O2•- generation was observed when the EC were treated with dHDL. l-NAME and MnTBAP reduced O2•- generation induced by dHDL and TNF-α using both hydroethidine and lucigenin assays (Fig. 1L, Supplemental Fig. 3B,C). These data hints that dHDL lost the ability to stimulate NO production, but increased O2•- generation, which may inhibit angiogenesis.
3.6. MiRNAs in HDL and the effects of nHDL and dHDL on the expressions of miRNAs in ECs
To investigate the potential mechanisms, we started by looking for gene changes, which we determined how HDL regulate the expressions of miRNAs in ECs. Twenty-one (14 upregulated and 7 downregulated) of 3100 miRNAs were altered (more than 3-fold) by dHDL compared to those by nHDL (Supplemental Fig. 4). Four miRNAs (miR-181a-5p, -1275, -204-3p, and -24-3p) were downregulated by nHDL but upregulated by dHDL, whereas miR-146a-5p was upregulated by nHDL but downregulated by dHDL verified by quantitative real-time polymerase chain reaction (qRT-PCR) (Fig. 2A). MiR-223 was increased in both nHDL and dHDL treatments. However, miR-223 level was higher in ECs treated with dHDL than that treated with nHDL, and miR-24-3p was hundreds fold higher than miR-223 levels in ECs after dHDL treatment. MiR-24-3p was also demonstrated the most striking changes; thus, we focused on miR-24-3p (Fig. 2A). The miRNA array dataset has been deposited to GEO and the accession number is GPL26002.
Fig. 2.

MiRNA microarray revealed that nHDL suppressed, but dHDL enhanced expression of miR-24-3p in endothelial cells (ECs), which affected tube formation.(A) The qRT-PCR confirmed that four miRNAs were downregulated, whereas one miRNA was upregulated by nHDL. In contrast, these four miRNAs were upregulated, whereas miR-146a-5p was downregulated by dHDL. dHDL increase miR-223 more than nHDL, but the levels of miR-223 in ECs is hundred folds less than other miRNAs * vs. control; # vs. nHDL, p < 0.05 (n = 8–10). (B) The level of miR-24-3p was higher in dHDL than in nHDL. *p < 0.05 (n = 20). (C) The nHDL inhibited, but dHDL slightly decreased, the expressions of pri-miR-24-1, pri-miR-24-2, pre-miR-24-1 and pre-miR-24-2 in cultured ECs. * vs. control; # vs. nHDL, p < 0.05 (n = 6–10). (D) The image showed that miR-24-3p mimic alone can not enter into ECs, but Lipofectamine® RNAiMAX, nHDL and dHDL can deliver Cy3-labeled miR-24-3p mimic to ECs. (E–H) The miR-24-3p mimic inhibited EC tube formation, whereas the miR-24-3p inhibitor enhanced EC tube formation. The miR-24-3p mimic decreased VEGF and nHDL-induced EC tube formation, whereas the miR-24-3p inhibitor increased dHDL-induced EC tube formation. The miR-24-3p inhibitor further enhanced nHDL-induced EC tube formation. NC, negative control, * vs. Mimic-NC or Inhibitor-NC; # vs. Mimic-NC + nHDL or Inhibitor-NC + nHDL; & vs. Mimic-NC + dHDL or inhibitor-NC + dHDL, $ vs. Mimic-NC + VEGF, p < 0.05 (n = 10–14).
To determine if the change of miR-24-3p in ECs was due to delivery or regulation by HDL, we first measured the amount of miR-24-3p in HDL and found that the amount of miR-24-3p in dHDL was higher than that in nHDL(Fig. 2B). Secondly, we showed that nHDL significantly inhibited, but dHDL slightly decreased, the expressions of pri-miR-24-1, pri-miR-24-2, and pre-miR-24-1, pre-miR-24-2(there are two gene sites that can generate mature miR-24-3p) in ECs(Fig. 2C). Fig. 2D and Supplemental Fig. 5 showed that both nHDL and dHDL can deliver miR-24-3p mimic to ECs, indicating that the change of miR-24-3p in ECs was due to both deliver and regulation.
3.7. Effects of miR-24-3p on EC tube formation, and nHDL- and dHDL-induced EC tube formation
The effect of miR-24-3p on angiogenesis was further determined. The confirmation of miR-24-3p inhibition is shown in Supplemental Fig. 6. The miR-24-3p mimic significantly suppressed EC tube formation, whereas the miR-24-3p inhibitor promoted EC tube formation (Fig. 2E–H). Next, we determined whether miR-24-3p affected HDL-induced angiogenesis. The miR-24-3p mimic partly inhibited VEGF-, nHDL- and dHDL-induced EC tube formation. Inhibition of miR-24-3p promoted nHDL-and dHDL-induced EC tube formation (Fig. 2E–H), suggesting that nHDL inhibited miR-24-3p to induce angiogenesis, but dHDL increased miR-24-3p to inhibit angiogenesis, and vinculin is required for VEGF-induced angiogenesis.
3.8. Effects of miR-24-3p on nHDL- and dHDL-induced EC NO production and O2•- generation
Next, we determined whether miR-24-3p affected nHDL- and dHDL-induced EC NO production and O2•- generation. The miR-24-3p mimic inhibited VEGF-, nHDL-, and dHDL-induced EC NO production, but inhibition of miR-24-3p promoted NO production in control-, dHDL-, and nHDL-treated EC (Fig. 3A–D). The miR-24-3p mimic increased O2•- generation in nHDL-treated EC and increased O2•- generation both in dHDL-induced EC and control. Inhibition of miR-24-3p decreased dHDL-induced O2•- generation (Fig. 3E–H), indicating that nHDL inhibited miR-24-3p to stimulate NO production, but dHDL increased miR-24-3p to induce O2•- generation.
Fig. 3.

MiR 24-3p affected endothelial cell (EC) NO and O2•-generation induced by nHDL and dHDL. (A–D) The miR-24-3p mimic inhibited EC NO production whereas the miR-24-3p inhibitor enhanced EC NO production in all groups. (E–H) The miR-24-3p mimic enhanced EC O2•- generation, whereas the miR-24-3p inhibitor inhibited EC O2•- generation in all groups. TNF-α was the positive control. NC, negative control. AU, arbitrary Units. * vs. Mimic-NC or Inhibitor-NC; # vs. Mimic-NC or Inhibitor-NC + nHDL; $ vs. Mimic-NC or inhibitor-NC + VEGF; & vs. Mimic-NC or inhibitor-NC + dHDL, p < 0.05 (n = 10–14).
3.9. Effects of nHDL and dHDL on EC proteomics
Since HDL can regulate miR-24-3p to affect angiogenesis, then we looked for its target. We first used proteomic analysis to detect the protein change by HDL. The 2D gel electrophoresis analysis showed a lot of changes of proteins among the control, nHDL, and dHDL groups. Among them, a significant change in 5 proteins related to endothelial function between nHDL and dHDL treatment were found (Supplemental Fig. 7). These 5 proteins were further identified by Ultraflex III matrix assisted laser desorption/ionization tandem time-of-flight (MALDI TOF/TOF) analysis. The nHDL upregulated 4 proteins and downregulated 1 protein compared to dHDL. These proteins are listed in Supplemental Table 2. The identified proteins are involved in different aspects of cell function. Based on a systematic review of endothelial function and nHDL up-regulated vinculin expression, but dHDL down-regulated vinculin expression in a dose-dependent manner in EC (Fig. 4A–E), we selected vinculin as the potential target for further study.
Fig. 4.

Proteomics study revealed that nHDL were increased, but dHDL decreased the expressions of vinculin (VCL) in endothelial cells (ECs), which affected tube formation.(A) Enlarged images of a 2D protein profile and 3D view in the proteomics study revealed that nHDL increased the expression of vinculin, but dHDL decreased the expression of vinculin in cultured ECs (n = 3). (B and C) Western blotting show that nHDL elevated the expression of vinculin, but dHDL decreased the expression of vinculin in cultured ECs. * vs. control; # vs nHDL, p < 0.05 (n = 10). (D) Immunofluorescence assays show that nHDL increased the expression of vinculin, but dHDL reduced the expression of vinculin in cultured ECs. The cells were positively stained with vinculin. DAPI was used to show the position of nuclei. (n = 10). (E) dHDL decreased the expression of vinculin (VCL) in a dose-dependent manner in ECs. (F–I) Images and dot plots show that the knockdown of vinculin by siRNA inhibited nHDL-induced EC tube formation. Knockdown of vinculin also inhibited EC tube formation in control- and dHDL-treated groups. Overexpression of vinculin increased EC tube formation in all group. NC, negative control; OE, overexpression. * vs. control (NC-siRNA) or Empty-NC; # vs. nHDL (NC-siRNA) or Empty-NC + nHDL; & vs. dHDL (NC-siRNA) or Empty-NC + dHDL, p < 0.05 (n = 10–14).
3.10. Effects of vinculin expression on nHDL- and dHDL-mediated EC tube formation
To determine whether vinculin contributed to HDL-induced angiogenesis, vinculin in ECs was silenced or overexpressed before the HDL treatment. Vinculin-siRNA1 was selected to silence vinculin since it produced the best silencing results (Supplemental Fig. 8). Silencing vinculin inhibited VEGF, nHDL, or dHDL-induced EC tube formation (Fig. 4F and G). Overexpression of vinculin enhanced nHDL or dHDL-induced EC tube formation (Fig. 4H and I, Supplemental Fig. 9), suggesting that nHDL increased vinculin expression to induce angiogenesis, but dHDL decreased vinculin expression to inhibit angiogenesis.
3.11. Effects of vinculin expression on nHDL- and dHDL-mediated NO production and O2•- generation in ECs
To determine whether vinculin participates in nHDL-mediated NO production or dHDL -mediated O2•- generation, vinculin in ECs was silenced or overexpressed before the HDL treatment. Silencing of vinculin decreased VEGF, nHDL or dHDL-mediated NO generation (Fig. 5A,B, Supplemental Fig. 10); whereas, overexpression of vinculin enhanced NO generation in the VEGF, nHDL or dHDL groups (Fig. 5C,D). Silencing vinculin enhanced O2•- generation in both nHDL- and dHDL-treated EC (Fig. 5E,F). Overexpression of vinculin reduced O2•- generation in both nHDL- and dHDL-treated EC (Fig. 5G,H), indicating that nHDL increased vinculin expression to stimulate NO production, but dHDL decreased vinculin expression to induce O2•- generation.
Fig. 5.

Vinculin (VCL) affected NO and O2•-generation, which were induced by nHDL and dHDL.(A–D) Images and dot plots show that knockdown of vinculin inhibited NO production induced by nHDL in endothelial cells (ECs). Knockdown of vinculin impaired EC NO production in control and dHDL-treated groups. Overexpression of vinculin increased EC NO production in VEGF-, nHDL-, and dHDL-treated groups. AU, arbitrary units. * vs. control (NC-siRNA) or Empty-NC; # vs. nHDL (NC-siRNA) or Empty-NC + nHDL; & vs. dHDL (NC-siRNA) or Empty-NC + dHDL, $ vs. VEGF (NC-siRNA) or Empty-NC + VEGF, p < 0.05 (n = 10). (E–H) Image and dot plots show that knockdown of vinculin enhanced EC O2•- generation in control-, nHDL- and dHDL-treated groups. Overexpression of vinculin inhibited EC O2•- generation in nHDL- and dHDL-treated groups. AU, arbitrary units; NC, negative control; OE, overexpression. * vs. control (NC-siRNA) or Empty-NC; # vs. nHDL (NC-siRNA) or Empty-NC + nHDL; & vs. dHDL (NC-siRNA) or Empty-NC + dHDL, p < 0.05 (n = 8–14).
3.12. Vinculin is a direct target of miR-24-3p
Next, we determined whether vinculin was a direct target of miR-24-3p. As shown in Fig. 6A, vinculin was identified as a potential target of miR-24-3p in ECs by the TargetScan (targetscan.org) prediction software. Treatment of ECs with the miR-24-3p mimic inhibited the luciferase activity of vinculin, whereas their mutants or control miRNA had no effect (Fig. 6B). Treatment with the miR-24-3p mimic decreased vinculin expression, which was blocked by the miR-24-3p inhibitor at both the mRNA and protein levels with a dose-dependent effect in ECs (Fig. 6C,D, Supplemental Fig. 11), demonstrating that vinculin is a direct target of miR-24-3p.
Fig. 6.

Vinculin (VCL) was the direct target of miR-24-3p, which affected the expression of VCL induced by nHDL and dHDL. (A) A potential human miR-24-3p binding site in the vinculin. (B) Luciferase activity confirmed that vinculin was the direct target of miR-24-3p. NC, control 3’UTR. *vs. NC, p < 0.05 (n = 10). (C and D) The expression of vinculin was decreased by the miR-24-3p mimic but was increased by the miR-24-3p inhibitor. * vs. Mimic-NC or Inhibitor-NC, p < 0.05 (n = 10). (E and F) The miR-24-3p mimic inhibited nHDL-induced the expression of vinculin, whereas the miR-24-3p inhibitor enhanced dHDL-inhibited the expression of vinculin in cultured ECs. The miR-24-3p mimic decreased the expression of vinculin in dHDL-treated EC, whereas the miR-24-3p inhibitor further increased the expression of vinculin in nHDL-treated EC. NC, negative control; * vs. Mimic-NC or Inhibitor-NC; # vs. Mimic-NC or inhibitor-NC + nHDL; & vs. Mimic-NC or inhibitor-NC + dHDL, p < 0.05 (n = 10).
3.13. Effects of miR-24-3p on nHDL- and dHDL-regulated EC vinculin expression
Next, we further determined whether miR-24-3p affected HDL-regulated vinculin expression. We found that the miR-24-3p mimic inhibited nHDL-induced vinculin expression and decreased the vinculin expression both in control- and dHDL-treated EC. In contrast, the miR-24-3p inhibitor increased vinculin expression in control-, nHDL-, and dHDL-treated EC (Fig. 6E,F), indicating that HDL regulated vinculin by miR-24-3p.
3.14. Effects of nHDL and dHDL on other protein signaling related to angiogenesis
Previous studies found that HDL affects phosphatidylinositide 3-kinases (PI3K)/AKT/eNOS and ERK signaling pathways. We also determined the effects of nHDL and dHDL on these proteins. Short-term exposure of ECs to nHDL (30 min) stimulated the phosphorylation of AKT, eNOS at S1177 and ERK1/2, but dHDL reduced the phosphorylation of AKT, eNOS at S1177 and ERK1/2. Long-term exposure (48 h) of ECs to nHDL increased the expressions of PI3K, AKT, eNOS and ERK1/2, whereas long-term exposure of ECs to dHDL decreased expressions of PI3K, AKT, and eNOS. The nHDL inhibited caveolin-1 expression, whereas dHDL increased caveolin-1 expression (data not shown), which confirms the previous studies.
3.15. Effects of nHDL and dHDL on protein signaling related to vinculin
To determine whether vinculin was related to PI3K/AKT/eNOS and ERK signaling pathways, vinculin was silenced or overexpressed before treatment with HDL. Silencing vinculin inhibited the expressions of PI3K, AKT, eNOS and ERK 1/2 and phosphorylations of AKT, eNOS and ERK1/2 in response to nHDL or dHDL (Fig. 7A,B,D,E). In contrast, silencing vinculin removed the suppression of nHDL on caveolin-1 expression and increased caveolin-1 expression in control and dHDL-treated EC (Fig. 7C). More important, overexpression of vinculin enhanced the expressions of PI3K, AKT, eNOS and ERK 1/2 as well as phosphorylations of AKT, eNOS and ERK1/2 phosphorylation, and also inhibited caveolin-1 expression in response to nHDL or dHDL (Fig. 7F–J), indicating that vinculin could modulate PI3K/AKT/eNOS and ERK1/2 signaling pathways to regulate angiogenesis.
Fig. 7.

Vinculin (VCL) affected PI3K/AKT/eNOS and ERK1/2 signaling pathways.(A–E) Immunoblots and dot plots show that knockdown of vinculin inhibited the expressions of PI3K, AKT, eNOS and ERK1/2 as well as the phosphorylations of AKT, eNOS, and ERK1/2 in cultured endothelial cells (ECs) in all groups, but increased the expression of caveolin-1 in cultured ECs in control- and nHDL-treated groups. NC, negative control, * vs. control (NC-siRNA); # vs. nHDL (NC-siRNA); & vs. dHDL (NC-siRNA), p < 0.05 (n = 10). (F–J) Immunoblots and dot plots show that overexpression of vinculin increased the expressions of PI3K, AKT, eNOS and ERK1/2 as well as the phosphorylations of AKT, eNOS and ERK1/2, but decreased the expression of caveolin-1 in cultured ECs in all groups. NC, negative control; OE, overexpression. * vs. Empty-NC; # vs. Empty-NC + nHDL; & vs. Empty-NC + dHDL, p < 0.05 (n = 6–8).
3.16. The receptor of HDL regulating vinculin and miR-24-3p
Next, we tested whether HDL regulated the expression of vinculin and miR-24-3p in ECs via scavenger receptor class B type 1 (SRB1). SRB1-siRNA1 was selected to silence SRB1 since it had the best silencing results (Supplemental Fig. 12). Fig. 8A–C showed that the effects of nHDL and dHDL on the expression of vinculin and miR-24-3p were eliminated after silencing SRB1. No miR-24-3p mimic can be delivered to ECs by nHDL and dHDL after silencing SRB1 (Supplemental Fig. 13), demonstrating that both nHDL and dHDL regulated the expression of vinculin and miR-24-3p in ECs by SRB1.
Fig. 8.

Vinculin and miR-24-3p affectedin vivovascular growth, and SRB1 was important for HDL-mediated expression of vinculin (VCL) and miR-24-3p.(A–C) Silencing SRB1 blocked the effects of nHDL and dHDL on the expressions of vinculin and miR-24-3p on cultured human umbilical vein endothelial cells (HUVECs). *vs. control (NC-siRNA); # vs. nHDL (NC-siRNA); & vs. dHDL (NC-siRNA), p < 0.05 (n = 8). (D–E) The expressions of vinculin and eNOS were decreased in gastrocnemius muscle of ischemic lower limbs of hypercholesterolemic LDLr-/- mice, but increased after overexpression of vinculin on day 15 after ischemia surgery. OE, overexpression. *vs. C57, # vs. LDLr-/-+ Empty-NC, p < 0.05 (n = 8). (F–I) Overexpression of vinculin increased blood flow and collateral in the ischemic lower limbs of hypercholesterolemic LDLr-/- mice. *vs. C57, # vs. LDLr−/− Empty-NC, p < 0.05 (n = 8). (J) The expression of miR-24-3p was increased in gastrocnemius muscle of ischemic lower limbs of hypercholesterolemic LDLr-/- mice, but decreased after injection with the miR-24-3p antagomir on day 15 after surgery for ischemia. *vs. C57, # vs. LDLr−/−, p < 0.05 (n = 8). (K–N) The miR-24-3p antagomir increased blood flow and collateral in the ischemic lower limbs of hypercholesterolemic LDLr-/- mice. *vs. C57, # vs. LDLr−/− + antagomir-NC, p < 0.05 (n = 8).
3.17. The effects of vinculin and miR-24-3p on vascular growth in vivo
Finally, we determined the effects of miR-24-3p and vinculin on vascular growth in vivo. Lipid hydroperoxide levels in HDL from hypercholesterolemic LDLr-/- mice is significantly higher than that in non-hypercholesterolemic LDLr-/- mice and C57BL6 mice (Supplemental Fig. 14). The expressions of vinculin and eNOS were decreased in ischemic lower limbs of hypercholesterolemic LDLr-/- mice (Fig. 8D,E). Overexpression of vinculin increased the expression of vinculin and eNOS, improved blood flow and collateral in ischemic lower limbs of hypercholesterolemic LDLr-/- mice (Fig. 8F–I). In the contrast, the expression of miR-24-3p was increased in ischemic lower limbs of hypercholesterolemic LDLr-/- mice (Fig. 8J). Administration of the miR-24-3p antagomir decreased miR-24-3p expression and enhanced blood flow as well as collateral in ischemic lower limbs of hypercholesterolemic LDLr-/- mice (Fig. 8J–N), demonstrated that both miR-24-3p and vinculin may be the targets for dHDL-impaired angiogenesis.
4. Discussion
Previous studies showed that nHDL is able to stimulate angiogenesis [3,5,6]. However, the manner in which dHDL regulates angiogenesis is unknown, as previous reports suggest that HDL properties between healthy subjects and CAD patients differ [8,[12], [13], [14], [15], [16],[21], [22], [23], [24]]. Our study has six novel findings: 1. HDL from healthy subjects (nHDL) and patients with CAD (dHDL) regulated angiogenesis differently via alterations in vinculin expression. 2. Vinculin was critical for VEGF-induced and HDL-induced angiogenesis and could modulate PI3K/AKT/eNOS and ERK1/2 signaling pathways to regulate angiogenesis. 3. Vinculin is a target of miR-24-3p. 4. nHDL suppressed miR-24-3p to increase vinculin expression to stimulate NO production, but dHDL delivered miR-24-3p to inhibit vinculin expression to enhance O2•- generation. 5. Vinculin and eNOS expression was decreased, but that of miR-24-3p was increased in the ischemic lower limbs of hypercholesterolemic LDLr-/- mice, a dysfunctional HDL animal model, and vinculin overexpression or administration of the miR-24-3p antagomir increased angiogenesis. 6. Our simple modified cell-free assay may be used to measure the oxidative levels of HDL as a predictive biomarker for cardiovascular events.
A critical aspect of our study is the assessment of oxidized, dysfunctional HDL. To measure the degree of HDL oxidation, we previously established a modified cell-free assay that measures lipid hydroperoxide levels in HDL in human plasma [25,26]. Using this assay, we found that dHDL had higher lipid hydroperoxide levels, suggesting that HDL from patients with CAD was oxidatively modified. A previous study found that in patients with low SAA, higher HDL-cholesterol is associated with lower all-cause and cardiovascular mortality [16]; in contrast, higher HDL-cholesterol in patients with high SAA is associated with increases in such mortality, and SAA impairs the anti-inflammatory properties of HDL [16,42]. We found that the lipid hydroperoxide levels in dHDL were positively correlated with CRP and SAA, indicating that lipid hydroperoxide levels in dHDL may predict cardiovascular mortality. Indeed, recent study reported that high lipid peroxidation in HDL may be a molecular signature of the risk for developing cardiovascular disease [23].
Using EC culture, a zebrafish model for vascular growth, and hypercholesterolemic LDLr-/- mice as a model of impaired vascular growth, we found much lower angiogenesis with dHDL than with nHDL. There is much evidence showing that miRNAs regulate angiogenesis and that HDL and reconstituted HDL can carry and deliver miRNAs to cells [[43], [44], [45], [46], [47]]. We found that miRNA profiles in ECs differed between nHDL and dHDL treatments. In ECs, miR-24-3p expression was downregulated by nHDL, but upregulated by dHDL, to regulate angiogenesis. However, nHDL significantly inhibited both pri-miR-24 and pre-miR-24, but dHDL slightly suppressed them. In addition, we found that dHDL contained more miR-24-3p than nHDL did, and both nHDL and dHDL can deliver miR-24-3p mimic to ECs. Although whether HDL can deliver miRNAs to cells is controversial [44,48], our data demonstrated that nHDL decreased miR-24-3p levels due to downregulation, whereas dHDL increased them via delivery. These data demonstrated that HDL is able to both deliver and regulate miRNAs in ECs to affect angiogenesis.
Previous study showed that miR-223 carried by HDL with familial hypercholesterolemia is much higher than that in healthy subjects, and HDL delivers miR-223 to ECs, as well as miR-223 inhibits angiogenesis [43,44,46]. We also found that miR-223 levels in ECs was enhanced by both nHDL and dHDL, and dHDL increased miR-223 more. We further found that miR-24-3p levels was hundred folds higher than miR-223 levels in ECs after both nHDL and dHDL treatments. These data incorporated with miR-24-3p expressed in ECs, whereas miR-233 does not [44], we chose miR-24-3p in this study since the impact of miR-24-3p on ECs should be stronger than miR-233.
It is well known that NO is important for angiogenesis, and our results align with this concept. Specifically, we found that nHDL induced angiogenesis by suppressing miR-24-3p expression to increase NO production and inhibit O2•- generation, whereas dHDL impaired angiogenesis by delivering miR-24-3p expression, resulting in decreased NO production and increased O2•- generation. Recently, it is reported that miR-24 promotes atherosclerosis by inhibiting lipid uptake from HDL cholesterol, suggesting a close relationship between HDL and miR-24 [49]. Importantly, miR-24-3p inhibition neither completely restored dHDL-induced angiogenesis and NO production nor restored the decrease in dHDL-stimulated O2•- generation. Although the explanation is not intuitive, we postulate that these findings were based on the fact that HDL contains many different types of proteins, lipids, and miRNAs [43,44]. Although miR-24-3p may play a major inhibitory role, other particles and miRNAs such as myeloperoxidase, miR-223 in dHDL, may still contribute to the inhibition of angiogenesis [11,18,44,46]. These findings suggest that nHDL induces angiogenesis and dHDL impairs angiogenesis, at least in part, by regulating miR-24-3p.
It has been reported that proteomics profiles differ between nHDL and dHDL, which may affect angiogenesis [13,16,18,19,23,24]. However, little is known regarding the differences in EC proteomics profiles between nHDL and dHDL treatments. In addition, previous studies demonstrate that HDL promotes angiogenesis by activating the Ras/ERK pathway or Src/PI3K/AKT-ERK/Rac pathway, or S1P3-dependent of VEGFR2 pathway, which is independent of HDL cargo molecules [[3], [4], [5]]. Recently it also shows that reconstituted HDL mediates angiogenesis in diabetes by regulating miRNAs [47]. However, it is unclear whether native HDL regulates these pathways via miRNAs regulation. Thus, we focused on the targets of miR-24-3p. Using proteomics, we identified many proteins, including vinculin in ECs, that were regulated by HDL. We selected vinculin as the potential target of miR-24-3p because nHDL and dHDL had opposing effects on vinculin, specifically upregulation and downregulation, respectively. Additionally, vinculin was a study candidate because it appears to be involved in endothelial mechanotransduction [50], which is critical for NO production. It is reported that the loss of vinculin prevents cell adhesion and migration, which are critical for angiogenesis [50,51]. We found that vinculin was the direct target of miR-24-3p. Normal HDL induced angiogenesis by increasing vinculin expression to increase NO production, and dHDL impaired angiogenesis by decreasing vinculin expression and NO production. Moreover, decreased vinculin expression stimulated O2•- generation. A recent study demonstrates that vinculin is downregulated in atherosclerotic plaque [52]; dHDL may inhibit vinculin expression in this plaque. More importantly, we found that vinculin also regulated VEGF-induced angiogenesis and could directly regulate eNOS activation as well as the PI3K/AKT/eNOS and ERK signaling pathways. Meanwhile, vinculin could inhibit caveolin-1 expression. As caveolin-1 can negatively regulate eNOS and inhibit angiogenesis, our data suggest that vinculin is critical not only for nHDL-induced angiogenesis, but also for VEGF-induced angiogenesis [53].
We and others reported that HDL from hypercholesterolemic LDLr-/- mice are dysfunctional and proinflammatory, and hypercholesterolemia impairs angiogenesis [26,38,39]. In the present study, we confirmed that lipid hydroperoxide levels in HDL from hypercholesterolemic LDLr-/- mice were higher than those in non-hypercholesterolemic LDLr-/- mice and C57BL6 mice. We also found that vinculin and eNOS expression was decreased and that of miR-24-3p was increased in the ischemic lower limbs of hypercholesterolemic LDLr-/- mice; therefore, we used these mice as a dyfucntional HDL animal model. We found that overexpression of either vinculin or miR-24-3p antagomir increased angiogenesis in these ischemic hypercholesterolemic LDLr-/- mice, suggesting that dHDL impairs angiogenesis by enhancing miR-24-3p expression to suppress that of vinculin and eNOS. Although this is a model of acute ischemia, it at least showed that overexpression of vinculin or miR-24-3p antagomir increased angiogenesis in a dyfucntional HDL model.
Our findings indicated that normal HDL, which stimulates angiogenesis in healthy subjects, might become dysfunctional, thereby impairing angiogenesis in patients with CAD. This is very important since patients with CAD have ischemic heart and easy to occur myocardial infarction. Angiogenesis can improve myocardial ischemia, rescue necrotic myocardiums, reduce myocardial infarct size and increase the heart function. Unfortunately, HDL from patients with CAD can not only stimulate angiogenesis, but inhibit VEGF-induced angiogenesis. This may be one reason for the failure of clinical trials that are directed at stimulating coronary vascular growth as a treatment for CAD. These findings also suggest that the targeting of vinculin or miR-24-3p may become a therapeutic approach in ischemic patients with CAD. Finally, we found that HDL regulated vinculin and miR-24-3p expression to influence angiogenesis via SRB1, the classic receptor of HDL, which is consistent with previous finding that HDL promotes EC migration and reendothelialization via SRB14.
5. Conclusion
The present study provides direct evidence that nHDL suppresses miR-24-3p, resulting in increased vinculin expression and the stimulation of PI3K/AKT/eNOS and ERK signaling pathways to generate NO and induce angiogenesis. In contrast, dHDL was far less effective in stimulating angiogenesis since dHDL deliver miR-24-3p to inhibit vinculin expression and inactivate PI3K/AKT/eNOS and ERK signaling pathways. These effects inhibited NO production and increased O2•− generation. Our findings reveal the novel molecular mechanism of nHDL for inducing angiogenesis as well as the manner in which HDL function changed in patients with CAD regarding angiogenesis impairment. This study provides an explanation as to why some patients with CAD have a worse prognosis following myocardial infarction, and that vinculin and miR-24-3p may be therapeutic targets for dHDL-impaired angiogenesis. Additionally, our simple modified cell-free assay may be used to measure the proinflmmatory HDL levels to predict the HDL function and cardiovascular events.
Sources of funding
This research was financially supported by the National Natural Science Foundation of China (Grants 30970261, 81170271, 81370370, 81670392, 81600382, 81770241, 81830013, 81970363 and Distinguished Young Scholar Grant 81325001), International Cooperation project (2015DFA31070) from the Ministry of Science and Technology of China, National Key R&D Program of China (2016YFC0903000), Guangdong Natural Science Foundation (Grant 2015A030312009), Guangdong Basic and Applied Basic Research Foundation (Grant 2019B1515120092), the Changjiang Scholars Program from the Ministry of Education of China, the Sun Yat-sen University Clinical Research 5010 Program, Program of National Key Clinical Specialties, Fundamental Research Funds for the Central Universities Sun Yat-sen University Young Teacher Training Program (19ykpy79).
Declaration of competing interest
The authors have declared that no conflict of interest exists.
Acknowledgment
The authors would like to thank Zhi-Gang Guo from Nanfang Hospital, Southern Medical University for his assistance for the measurement of cholesterol efflux capacity on HDL. The authors also would like to thank the patients and staffs at the First Affiliated Hospital, Sun Yat-sen University for their assistance throughout this study.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101642.
Contributor Information
Zhi-Jun Ou, Email: Zhijunou@163.com.
Jing-Song Ou, Email: oujs@mail.sysu.edu.cn, oujs2000@yahoo.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Please refer to the online supplement for additional details on the methodology and supplementary data.
References
- 1.Ansell B.J., Navab M., Hama S. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–2756. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 2.Honda H., Hirano T., Ueda M. Associations among apolipoproteins, oxidized high-density lipoprotein and cardiovascular events in patients on hemodialysis. PLoS One. 2017;12 doi: 10.1371/journal.pone.0177980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miura S., Fujino M., Matsuo Y., Kawamura A., Tanigawa H., Nishikawa H., Saku K. High density lipoprotein-induced angiogenesis requires the activation of ras/map kinase in human coronary artery endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003;23:802–808. doi: 10.1161/01.ATV.0000066134.79956.58. [DOI] [PubMed] [Google Scholar]
- 4.Seetharam D., Mineo C., Gormley A.K. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-b type i. Circ. Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 5.Jin F., Hagemann N., Sun L., Wu J., Doeppner T.R., Dai Y., Hermann D.M. High-density lipoprotein (hdl) promotes angiogenesis via s1p3-dependent vegfr2 activation. Angiogenesis. 2018;21:381–394. doi: 10.1007/s10456-018-9603-z. [DOI] [PubMed] [Google Scholar]
- 6.Prosser H.C., Tan J.T., Dunn L.L., Patel S., Vanags L.Z., Bao S., Ng M.K., Bursill C.A. Multifunctional regulation of angiogenesis by high-density lipoproteins. Cardiovasc. Res. 2014;101:145–154. doi: 10.1093/cvr/cvt234. [DOI] [PubMed] [Google Scholar]
- 7.Tan J.T., Prosser H.C., Dunn L.L. High-density lipoproteins rescue diabetes-impaired angiogenesis via scavenger receptor class b type i. Diabetes. 2016;65:3091–3103. doi: 10.2337/db15-1668. [DOI] [PubMed] [Google Scholar]
- 8.Khera A.V., Cuchel M., de la Llera-Moya M. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohatgi A., Khera A., Berry J.D. Hdl cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mody P., Joshi P.H., Khera A., Ayers C.R., Rohatgi A. Beyond coronary calcification, family history, and c-reactive protein: cholesterol efflux capacity and cardiovascular risk prediction. J. Am. Coll. Cardiol. 2016;67:2480–2487. doi: 10.1016/j.jacc.2016.03.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khine H.W., Teiber J.F., Haley R.W., Khera A., Ayers C.R., Rohatgi A. Association of the serum myeloperoxidase/high-density lipoprotein particle ratio and incident cardiovascular events in a multi-ethnic population: observations from the dallas heart study. Atherosclerosis. 2017;263:156–162. doi: 10.1016/j.atherosclerosis.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besler C., Heinrich K., Rohrer L. Mechanisms underlying adverse effects of hdl on enos-activating pathways in patients with coronary artery disease. J. Clin. Invest. 2011;121:2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaisar T., Pennathur S., Green P.S. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of hdl. J. Clin. Invest. 2007;117:746–756. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y., Didonato J.A., Levison B.S. An abundant dysfunctional apolipoprotein a1 in human atheroma. Nat. Med. 2014;20:193–203. doi: 10.1038/nm.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zanoni P., Khetarpal S.A., Larach D.B. Rare variant in scavenger receptor bi raises hdl cholesterol and increases risk of coronary heart disease. Science. 2016;351:1166–1171. doi: 10.1126/science.aad3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zewinger S., Drechsler C., Kleber M.E. Serum amyloid a: high-density lipoproteins interaction and cardiovascular risk. Eur. Heart J. 2015;36:3007–3016. doi: 10.1093/eurheartj/ehv352. [DOI] [PubMed] [Google Scholar]
- 17.O'Reilly M., Dillon E., Guo W. High-density lipoprotein proteomic composition, and not efflux capacity, reflects differential modulation of reverse cholesterol transport by saturated and monounsaturated fat diets. Circulation. 2016;133:1838–1850. doi: 10.1161/CIRCULATIONAHA.115.020278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y., Wu Z., Riwanto M. Myeloperoxidase, paraoxonase-1, and hdl form a functional ternary complex. J. Clin. Invest. 2013;123:3815–3828. doi: 10.1172/JCI67478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao B., Tang C., Sinha A., Mayer P.S., Davenport G.D., Brot N., Oda M.N., Zhao X.Q., Heinecke J.W. Humans with atherosclerosis have impaired abca1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 2014;114:1733–1742. doi: 10.1161/CIRCRESAHA.114.303454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenson R.S., Brewer H.B., Jr., Ansell B.J., Barter P., Chapman M.J., Heinecke J.W., Kontush A., Tall A.R., Webb N.R. Dysfunctional hdl and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016;13:48–60. doi: 10.1038/nrcardio.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miki T., Miyoshi T., Kotani K., Kohno K., Asonuma H., Sakuragi S., Koyama Y., Nakamura K., Ito H. Decrease in oxidized high-density lipoprotein is associated with slowed progression of coronary artery calcification: subanalysis of a prospective multicenter study. Atherosclerosis. 2019;283:1–6. doi: 10.1016/j.atherosclerosis.2019.01.032. [DOI] [PubMed] [Google Scholar]
- 22.Fotakis P., Kothari V., Thomas D.G. Anti-inflammatory effects of hdl (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2019;39:e253–e272. doi: 10.1161/ATVBAHA.119.313253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samadi S., Mehramiz M., Kelesidis T. High-density lipoprotein lipid peroxidation as a molecular signature of the risk for developing cardiovascular disease: results from mashad cohort. J. Cell. Physiol. 2019 doi: 10.1002/jcp.28276. Feb 19. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riwanto M., Rohrer L., Roschitzki B. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127:891–904. doi: 10.1161/CIRCULATIONAHA.112.108753. [DOI] [PubMed] [Google Scholar]
- 25.Chang F.J., Yuan H.Y., Hu X.X. High density lipoprotein from patients with valvular heart disease uncouples endothelial nitric oxide synthase. J. Mol. Cell. Cardiol. 2014;74:209–219. doi: 10.1016/j.yjmcc.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Ou J., Wang J., Xu H. Effects of d-4f on vasodilation and vessel wall thickness in hypercholesterolemic ldl receptor-null and ldl receptor/apolipoprotein a-i double-knockout mice on western diet. Circ. Res. 2005;97:1190–1197. doi: 10.1161/01.RES.0000190634.60042.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navab M., Hama S.Y., Hough G.P., Subbanagounder G., Reddy S.T., Fogelman A.M. A cell-free assay for detecting hdl that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 2001;42:1308–1317. [PubMed] [Google Scholar]
- 28.Guo K., Hu L., Xi D., Zhao J., Liu J., Luo T., Ma Y., Lai W., Guo Z. Psrc1 overexpression attenuates atherosclerosis progression in apoe(-/-) mice by modulating cholesterol transportation and inflammation. J. Mol. Cell. Cardiol. 2018;116:69–80. doi: 10.1016/j.yjmcc.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Li Y., Liu C., Zhao Y., Hu K., Zhang J., Zeng M., Luo T., Jiang W., Wang H. Sevoflurane induces short-term changes in proteins in the cerebral cortices of developing rats. Acta Anaesthesiol. Scand. 2013;57:380–390. doi: 10.1111/aas.12018. [DOI] [PubMed] [Google Scholar]
- 30.Jian Y.P., Yuan H.X., Hu K.H., Chen C., Li Y.Q., Li Y., Yang T.X., Ou Z.J., Ou J.S. Protein compositions changes of circulating microparticles in patients with valvular heart disease subjected to cardiac surgery contribute to systemic inflammatory response and disorder of coagulation. Shock. 2019;52:487–496. doi: 10.1097/SHK.0000000000001309. [DOI] [PubMed] [Google Scholar]
- 31.Yan F.X., Li H.M., Li S.X. The oxidized phospholipid povpc impairs endothelial function and vasodilation via uncoupling endothelial nitric oxide synthase. J. Mol. Cell. Cardiol. 2017;112:40–48. doi: 10.1016/j.yjmcc.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Ou Z., Ou J., Ackerman A.W., Oldham K.T., Pritchard K.A., Jr. L-4f, an apolipoprotein a-1 mimetic, restores nitric oxide and superoxide anion balance in low-density lipoprotein-treated endothelial cells. Circulation. 2003;107:1520–1524. doi: 10.1161/01.cir.0000061949.17174.b6. [DOI] [PubMed] [Google Scholar]
- 33.Wang T.T., Shi M.M., Liao X.L. Overexpression of inducible nitric oxide synthase in the diabetic heart compromises ischemic postconditioning. J. Mol. Cell. Cardiol. 2019;129:144–153. doi: 10.1016/j.yjmcc.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 34.Ou J., Ou Z., Jones D.W. L-4f, an apolipoprotein a-1 mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation. 2003;107:2337–2341. doi: 10.1161/01.CIR.0000070589.61860.A9. [DOI] [PubMed] [Google Scholar]
- 35.Xuan C., Chang F.J., Liu X.C., Bai X.Y., Liao X.L., He G.W., Ou J.S. Endothelial nitric oxide synthase enhancer for protection of endothelial function from asymmetric dimethylarginine-induced injury in human internal thoracic artery. J. Thorac. Cardiovasc. Surg. 2012;144:697–703. doi: 10.1016/j.jtcvs.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Zhou L., Liu X., Wang Z.Q. Simvastatin treatment protects myocardium in noncoronary artery cardiac surgery by inhibiting apoptosis through mir-15a-5p targeting. J. Cardiovasc. Pharmacol. 2018;72:176–185. doi: 10.1097/FJC.0000000000000611. [DOI] [PubMed] [Google Scholar]
- 37.Yuan H.X., Chen C.Y., Li Y.Q. Circulating extracellular vesicles from patients with valvular heart disease induce neutrophil chemotaxis via foxo3a and the inhibiting role of dexmedetomidine. Am. J. Physiol. Endocrinol. Metab. 2020 doi: 10.1152/ajpendo.00062.2020. Jun 9. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 38.Navab M., Anantharamaiah G.M., Hama S., Garber D.W., Chaddha M., Hough G., Lallone R., Fogelman A.M. Oral administration of an apo a-i mimetic peptide synthesized from d-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 39.Chattopadhyay A., Navab M., Hough G. A novel approach to oral apoa-i mimetic therapy. J. Lipid Res. 2013;54:995–1010. doi: 10.1194/jlr.M033555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu X.L., Luo D., Yao X.L. Dl-3n-butylphthalide promotes angiogenesis via the extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase/akt-endothelial nitric oxide synthase signaling pathways. J. Cardiovasc. Pharmacol. 2012;59:352–362. doi: 10.1097/FJC.0b013e3182443e74. [DOI] [PubMed] [Google Scholar]
- 41.Mathiyalagan P., Liang Y., Kim D. Angiogenic mechanisms of human cd34(+) stem cell exosomes in the repair of ischemic hindlimb. Circ. Res. 2017;120:1466–1476. doi: 10.1161/CIRCRESAHA.116.310557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han C.Y., Tang C., Guevara M.E. Serum amyloid a impairs the antiinflammatory properties of hdl. J. Clin. Invest. 2016;126:266–281. doi: 10.1172/JCI83475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vickers K.C., Palmisano B.T., Shoucri B.M., Shamburek R.D., Remaley A.T. Micrornas are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011;13:423–433. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabet F., Vickers K.C., Cuesta Torres L.F. Hdl-transferred microrna-223 regulates icam-1 expression in endothelial cells. Nat. Commun. 2014;5:3292. doi: 10.1038/ncomms4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choteau S.A., Cuesta Torres L.F., Barraclough J.Y. Transcoronary gradients of hdl-associated micrornas in unstable coronary artery disease. Int. J. Cardiol. 2018;253:138–144. doi: 10.1016/j.ijcard.2017.09.190. [DOI] [PubMed] [Google Scholar]
- 46.Shi L., Fisslthaler B., Zippel N., Fromel T., Hu J., Elgheznawy A., Heide H., Popp R., Fleming I. Microrna-223 antagonizes angiogenesis by targeting beta1 integrin and preventing growth factor signaling in endothelial cells. Circ. Res. 2013;113:1320–1330. doi: 10.1161/CIRCRESAHA.113.301824. [DOI] [PubMed] [Google Scholar]
- 47.Hourigan S.T., Solly E.L., Nankivell V.A. The regulation of mirnas by reconstituted high-density lipoproteins in diabetes-impaired angiogenesis. Sci. Rep. 2018;8:13596. doi: 10.1038/s41598-018-32016-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner J., Riwanto M., Besler C., Knau A., Fichtlscherer S., Roxe T., Zeiher A.M., Landmesser U., Dimmeler S. Characterization of levels and cellular transfer of circulating lipoprotein-bound micrornas. Arterioscler. Thromb. Vasc. Biol. 2013;33:1392–1400. doi: 10.1161/ATVBAHA.112.300741. [DOI] [PubMed] [Google Scholar]
- 49.Ren K., Zhu X., Zheng Z. Microrna-24 aggravates atherosclerosis by inhibiting selective lipid uptake from hdl cholesterol via the post-transcriptional repression of scavenger receptor class b type i. Atherosclerosis. 2018;270:57–67. doi: 10.1016/j.atherosclerosis.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 50.Chervin-Petinot A., Courcon M., Almagro S., Nicolas A., Grichine A., Grunwald D., Prandini M.H., Huber P., Gulino-Debrac D. Epithelial protein lost in neoplasm (eplin) interacts with alpha-catenin and actin filaments in endothelial cells and stabilizes vascular capillary network in vitro. J. Biol. Chem. 2012;287:7556–7572. doi: 10.1074/jbc.M111.328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Birukova A.A., Shah A.S., Tian Y., Moldobaeva N., Birukov K.G. Dual role of vinculin in barrier-disruptive and barrier-enhancing endothelial cell responses. Cell. Signal. 2016;28:541–551. doi: 10.1016/j.cellsig.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Essen M., Rahikainen R., Oksala N. Talin and vinculin are downregulated in atherosclerotic plaque; tampere vascular study. Atherosclerosis. 2016;255:43–53. doi: 10.1016/j.atherosclerosis.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 53.Liakouli V., Elies J., El-Sherbiny Y.M. Scleroderma fibroblasts suppress angiogenesis via tgf-beta/caveolin-1 dependent secretion of pigment epithelium-derived factor. Ann. Rheum. Dis. 2018;77:431–440. doi: 10.1136/annrheumdis-2017-212120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.