

Abstract
An increased prevalence of dyslexia has been observed in individuals diagnosed with primary progressive aphasia, most notably the logopenic variant primary progressive aphasia. The underlying pathology most commonly associated with logopenic variant primary progressive aphasia is Alzheimer’s disease. In this clinical case report series, we describe the neuropathological findings of three patients with logopenic variant primary progressive aphasia and developmental dyslexia, each demonstrating a pattern of cerebrocortical microdysgenesis, reminiscent of findings first reported in dyslexic individuals, alongside expected Alzheimer’s disease pathology. Neurodevelopmental and most severe Alzheimer’s disease pathological changes overlapped within perisylvian brain regions, areas associated with phonological deficits in both logopenic variant primary progressive aphasia and dyslexia. These three cases with pathological findings support the hypothesis that early-life neurodevelopmental changes might influence later-life susceptibility to neurodegenerative disease and could contribute to non-amnestic, early age-of-onset presentations of Alzheimer’s disease. Larger studies investigating neurobiological vulnerability across the lifespan are needed.
Keywords: Alzheimer’s disease, dyslexia, brain development, primary progressive aphasia, cortical developmental abnormalities
We discovered cortical developmental anomalies, in language-related anatomies, in each of three patients with logopenic variant primary progressive aphasia and developmental dyslexia, reminiscent of findings reported in dyslexic individuals, in association with maximal Alzheimer’s disease pathology, providing further evidence linking early-life neurodevelopmental changes with later-life neurodegenerative disease.
Graphical Abstract
Graphical Abstract.
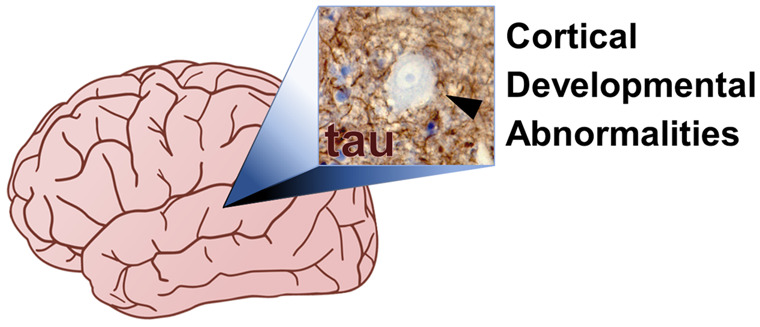
Introduction
Increased prevalence of language-based learning disabilities in primary progressive aphasia (PPA) was first appreciated in 1992 (Mesulam and Weintraub, 1992). It was hypothesized that developmental differences reflected a ‘locus of least resistance’ within the language network, resulting in regional disease susceptibility (Rogalski et al., 2008). Within PPA, the highest prevalence of learning disability may be associated with logopenic variant PPA (lvPPA; Miller et al., 2013). The primary deficit in lvPPA, phonological impairment due to temporo-parietal disease (Gorno-Tempini et al., 2008), shares symptomatic and neuroanatomical similarities to developmental dyslexia (Richlan et al., 2013). Pathologically, lvPPA is most often associated with Alzheimer’s disease, with highest severity of neuropathological findings within the language network (Josephs et al., 2013; Spinelli et al., 2017; Bergeron et al., 2018). Neuropathological studies of dyslexia individuals are rare and their results controversial (Guidi et al., 2018) but have shown neuronal migrational abnormalities in the left perisylvian region (Galaburda and Kemper, 1979; Galaburda et al., 1985; Humphreys et al., 1990). Together, it would follow that pathological hallmarks of developmental anomalies might overlap with Alzheimer’s disease pathological burden in dyslexic subjects affected with lvPPA. In this study, we describe clinical, cognitive, neuroanatomical and neuropathological findings in three patients with lvPPA and developmental dyslexia.
Materials and methods
Participants
Three prototypic lvPPA cases with developmental dyslexia and pathologically confirmed Alzheimer’s disease were selected for this initial exploratory analysis. All patients were enrolled in ongoing research projects at the University of California San Francisco (UCSF) Memory and Aging Center. Patient demographics are described in Supplementary Table 1. Comprehensive neurologic, neuropsychological and language testing, structural MRI and amyloid PET imaging were performed (Miller et al., 2013). Genetic testing for APP, C9ORF72, FUS, GRN, MAPT, PSEN1, PSEN2 and TARDBP were negative in all three cases. APOE genotypes and H1/H2 MAPT haplotypes were also obtained (Ramos et al., 2019). Written informed consent from participants or legal guardians was obtained in accordance with the Declaration of Helsinki. Study procedures were approved by the UCSF Institutional Review Board.
Histopathological analyses
Neuropathological diagnoses were rendered according to published criteria (Montine et al., 2012). Sections from language network regions, including left inferior frontal gyrus (IFG), superior temporal gyrus (STG), middle temporal gyrus (MTG), and angular gyrus (AG), as well as regions most associated with frontotemporal lobar degeneration or typical Alzheimer’s disease presentations, including anterior cingulate cortex (ACC), middle insula, and posterior cingulate cortex (PCC), were assessed for features of focal cortical dysplasia (FCD): (i) cortical dyslamination; (ii) presence of giant, immature or dysplastic neurons; (iii) heterotopia or excessive number of neurons in layer I; and (iv) heterotopia or excessive number of neurons outside layer I (Thom, 2004; Blümcke et al., 2011). Immunohistochemistry for NeuN, GFAP and non-phosphorylated neurofilaments (SMI-32) was carried out for cell type characterization. Ratings of Alzheimer’s disease pathological changes were performed in the same sections as part of a standard neuropathological research evaluation (Supplementary material).
Data availability
All data used in this study are available for review upon request.
Case presentations
Case 1
A 47-year-old left-handed man developed word-finding difficulties. He reported ‘writing letters backwards’ upon switching from print to cursive. He received failing grades throughout school until a formal diagnosis of developmental dyslexia was eventually made. With appropriate tutoring, his grades improved to passing levels and he graduated from high school, after which he took on several jobs in retail and sales. His new onset of word-finding deficits was initially attributed to mild depression and anxiety. As symptoms progressed he was referred for clinical evaluation, at age 51. On presentation, he displayed prominent challenges understanding long strings of auditory/verbal information but denied difficulties with episodic memory, object recognition or knowledge, visuospatial or executive functioning. Spontaneous speech was hesitant, with phonological paraphasias and long pauses disrupting fluency. Cranial nerve, motor, sensory and gait exam were all unremarkable. Neuropsychological testing confirmed severe phonological short-term memory deficits with only minor deficits in episodic memory and executive functioning (Supplementary Table 1). Family history was negative for neurodegenerative disease, left-handedness or learning disability. Brain MRI revealed left-sided temporo-parietal atrophy (Supplementary Fig. 1). He met diagnostic criteria for lvPPA (Gorno-Tempini et al., 2011). PiB-PET imaging showed elevated cortical tracer uptake, consistent with the presence of moderate to frequent neuritic plaques (Supplementary Fig. 1). Genetic testing revealed E3/E3 APOE genotypes and H1/H2 MAPT haplotypes. Over a 4-year period, his language deteriorated to neologisms and yes/no substitution, he developed episodic memory loss, dyscalculia, apraxia and progressive right-sided hemineglect. He developed jaw and hemibody myoclonus. He passed away at age 58.
Case 2
A 54-year-old, right-handed woman with a childhood history of dyslexia, developed word-finding difficulties while teaching class. Three years later, she presented for evaluation. Her speech was slow without deficits in articulation or grammar. Intermittent myoclonus was observed in all four limbs. The remainder of the neurological exam was normal. Cognitive testing revealed isolated deficits in phonological short-term memory with spared object knowledge, fluency, long-term memory, visuospatial abilities and executive functioning (Supplementary Table 1). Her father was diagnosed with amnestic Alzheimer’s disease at age 72. Her brother had dyslexia. Her brain MRI showed left predominant temporo-parietal atrophy (Supplementary Fig. 1). She was diagnosed with lvPPA (Gorno-Tempini et al., 2011). PiB-PET imaging was positive for cortical uptake (Supplementary Fig. 1). Genetic testing revealed E3/E4 APOE genotypes and H1/H2 MAPT haplotypes. Language symptoms remained relatively isolated for years, eventually developing repetitive questioning, temporospatial disorientation, visuospatial deficits and agitation. She died at age 66.
Case 3
A 53-year-old, right-handed grade school reading teacher presented for evaluation of 10 years of isolated, slowly progressive language symptoms such as ‘twisting letters or words together’, later followed by impairments in reading comprehension, calculation and short-term memory loss. She self-endorsed a history of dyslexia, which was the motivation for her vocation. On exam, her speech was slow with word-finding pauses and phonological paraphasias. Her pronounced aphasia likely impacted performances across neuropsychological testing, with notable, relative preservations of visuospatial abilities (Supplementary Table 1). There was no family history of neurodegenerative diseases or learning disability. Her mother was left-handed. Neuroimaging revealed left greater than right biparietal atrophy (Supplementary Fig. 1). She was diagnosed with lvPPA (Gorno-Tempini et al., 2011). Genetic testing yielded E3/E3 APOE genotypes and H1/H1 MAPT haplotypes. PiB-PET was positive for cortical binding (Supplementary Fig. 1). She later developed limb apraxia, myoclonic jerks in her hands and feet, right-sided visuospatial neglect and dense episodic memory loss. She died at age 65.
Neuropathology
Alzheimer’s disease changes
All cases met neuropathological criteria for high Alzheimer’s disease neuropathological changes A3B3C3, Thal phase 5 and Braak neurofibrillary tangles (NFT) stage 6 (Montine et al., 2012). Additional information on coexistent pathological changes is described in Supplementary material. Neuronal loss was more severe within language network regions and remarkably, associated with the highest severity of regional developmental changes (Supplementary Table 2).
Developmental abnormalities
Case 1
Cortical dyslamination with the presence of conspicuous large pyramidal neurons in clusters was observed spanning cortical layers II-III in AG (Fig. 1A), IFG (Fig. 1B), STG/MTG and PCC. The majority of these neurons appeared dystrophic but did not otherwise display cytological features of developmental abnormalities. Several neurons in cortical layers II-III and V in the AG and a few in the STG/MTG and IFG showed abnormal apical dendrite orientation. Virtually all of these neurons displayed diffuse or granular cytoplasmic tau immunoreactivity and, in a few instances, harboured NFT (Fig. 2A). In addition, they were strongly immunoreactive to antibodies against non-phosphorylated neurofilaments (SMI-32; Fig. 3A and B), NeuN-positive and GFAP-negative. A few heterotopic large neurons, with abundant diffuse cytoplasmic tau immunoreactivity, were observed in cortical layer I of the insula, IFG and AG (Fig. 4A). Finally, tau immunoreactive interstitial white matter neurons were observed in juxtacortical STG and AG white matter (Fig. 4B;Supplementary Table 2).
Figure 1.
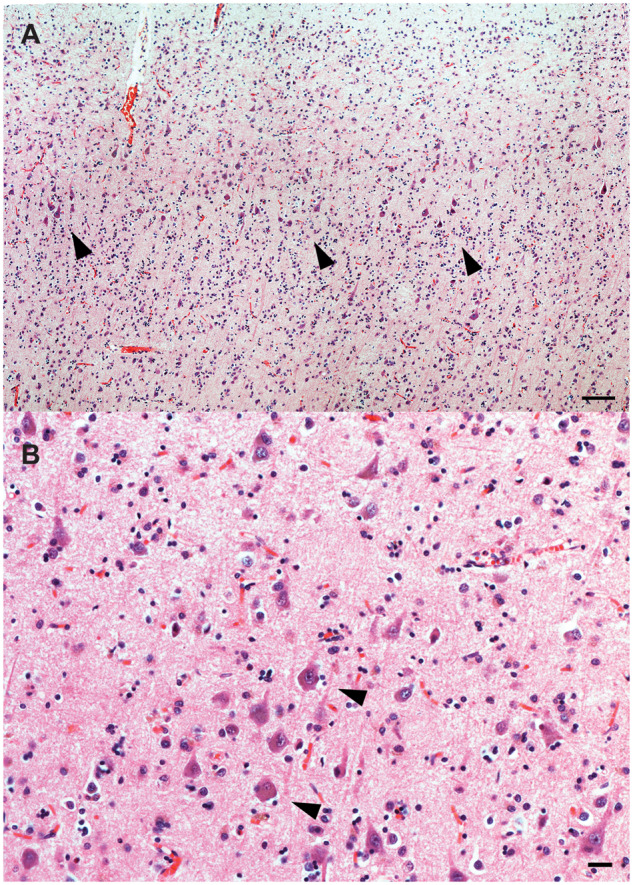
Cortical dyslamination with presence of atypical neurons in case 1. Cortical dyslamination with presence of dystrophic pyramidal neurons (arrows) haphazardly spanning cortical layers II-III of the AG (A), and large dystrophic pyramidal neurons in cortical layer II of the IFG (B), and of case 1. H&E. Bars: A = 250 microns; B = 25 microns.
Figure 2.
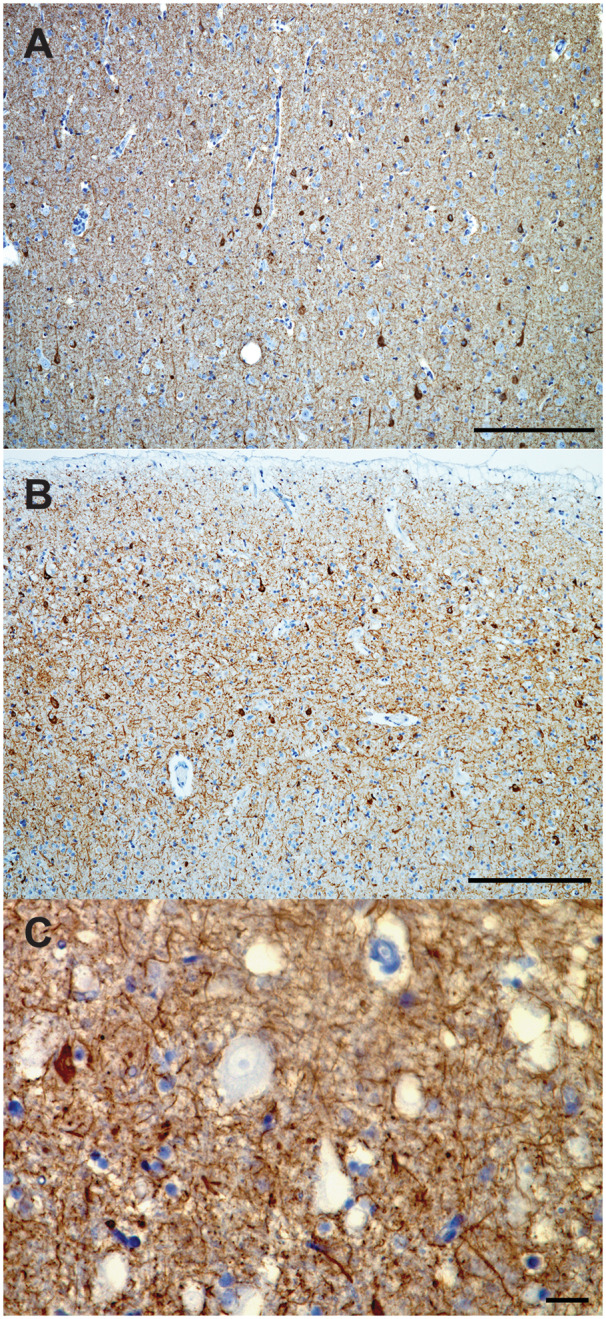
Tau immunohistochemistry of atypical neurons. Abnormal cortical organization with presence of frequent pyramidal tau immunoreactive neurons cortical layer II of the IFG of case 1 (A) and STG of case 2 (B). Atypically large neuron in the cortical layer II of the STG of case 3 with consistently tau negative cell body (arrows) (C). CP13 immunohistochemistry. Bars: A, B = 250 micron; C = 25 microns.
Figure 3.
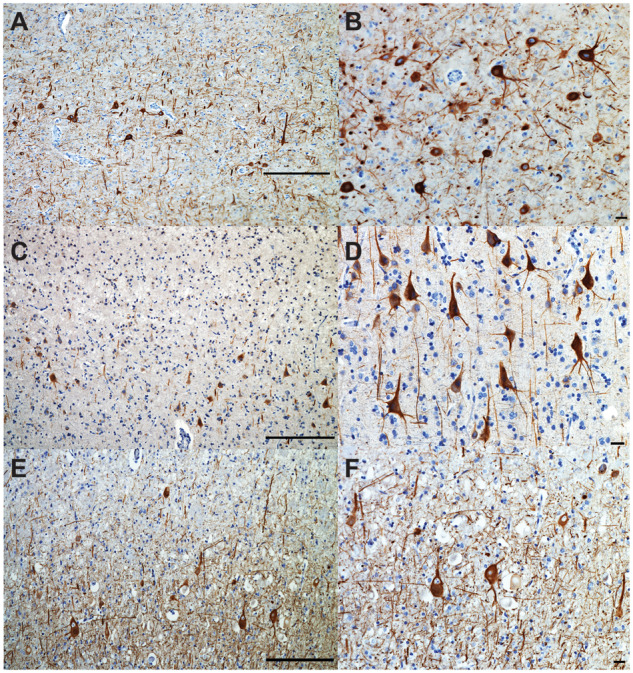
Abnormal neurofilament immunostaining in atypical cortical neurons. Abundant non-phosphorylated neurofilaments (SMI32) immunostaining of atypical large pyramidal neurons in cortical layer II of the IFG of case 1 (A, B), and case 2 (C, D). Abundant SMI32 immunostaining was also observed in large, ballooned atypical neurons of cortical layer II of the inferior frontal gyrus of case 3 (E, F). Bars: A, C, E = 250 microns; B, D, F = 25 microns.
Figure 4.
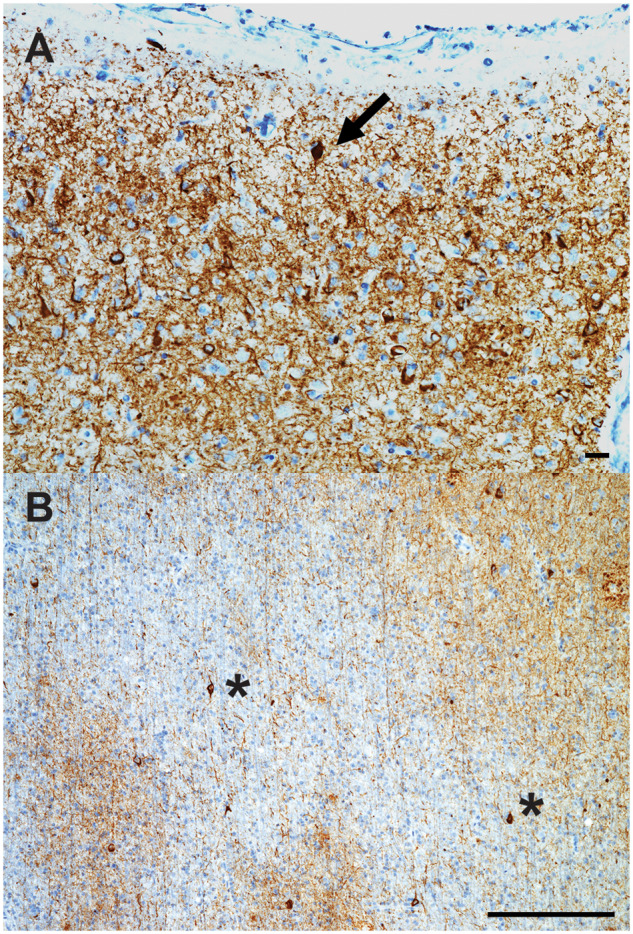
Tau immunohistochemistry of ectopic neurons in dyslexia. Tau immunohistochemistry of cortical layer I-II (A), and juxtacortical white matter (B) of the AG of case 1. (A) A tau immunoreactive ectopic neuron in layer I, with abundant tau immunoreactive neuropil threads and frequent pyramidal neurons also containing tau cytoplasmic inclusions in cortical layer II. (B) Numerous tau immunoreactive interstitial white matter neurons (asterisks) and a few oligodendroglial coiled bodies are seen. Bars: A = 25 microns; B = 250 microns.
Case 2
Cortical dyslamination was detected in IFG, STG/MTG and AG, and to a lesser extent in PCC. The major features were clusters of large pyramidal neurons spanning cortical layers II-III or IV-V (Fig. 5A and B). The majority of these neurons had normal polarity and typical apical dendrites. The majority of these neurons displayed diffuse or granular cytoplasmic tau immunoreactivity and, in a few instances, harboured NFT (Fig. 2B). Large pyramidal neurons in cortical layers II-III were strongly immunoreactive with antibodies against non-phosphorylated neurofilaments (Fig. 3C and D). Sparse neurons in the IFG and STG/MTG displayed abnormal polarity. Sparse large, dystrophic and ectopic neurons in cortical layer I were seen in IFG, STG/MTG and PCC, as well as in juxtacortical insular white matter. All ectopic neurons showed abundant diffuse cytoplasmic tau immunoreactivity in the absence of NFT (Supplementary Table 2).
Figure 5.
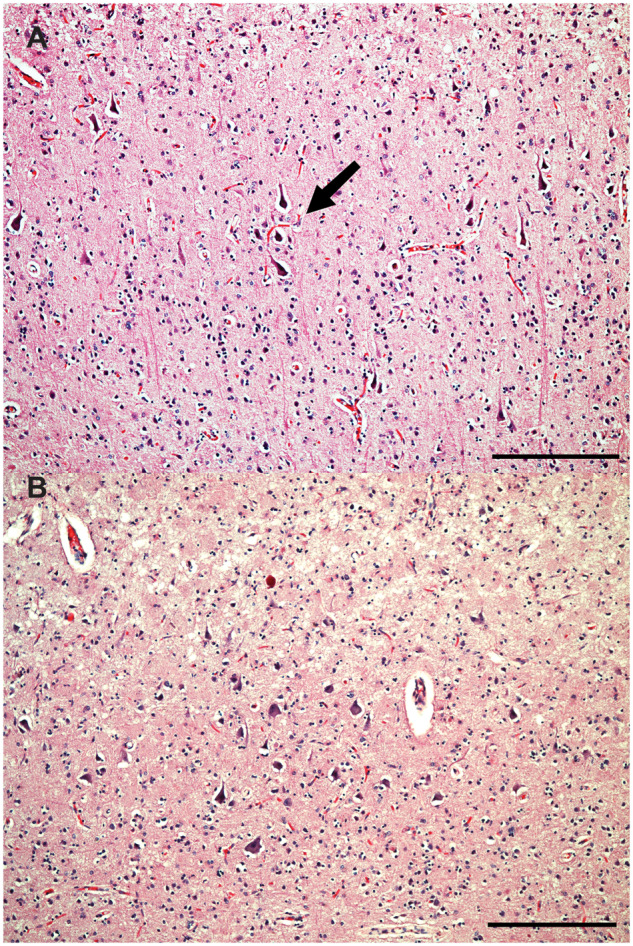
Cortical dyslamination with presence of atypical neurons in case 2. Cortical dyslamination with presence of dystrophic pyramidal neurons in clusters (arrows) haphazardly spanning cortical layers II-III of the AG (A), and large dystrophic pyramidal neurons in cortical layers II-III of the IFG (B), and of case 2. H&E. Bars: 250 microns.
Case 3
Numerous atypically large neurons, resembling pyramidal cells of cortical layer 5, were seen at the junction between cortical layers II and III of IFG (Fig. 6A and B), and to a lesser extent in STG/MTG and AG. These atypical neurons were also seen sparsely in ACC, PCC and insula. A considerable proportion of these cells displayed large, rounded cytoplasm, lacking features suggestive of neuronal degeneration such as shrunken, eccentric nuclei (Fig. 6B). These cells had often no obvious polarity, absent or atypically oriented apical dendrites and occurred in clusters that appeared to deepen tangentially to join other clusters of similar cells in cortical layer 5 (Fig. 6A). The cluster pattern was more evident in cortical regions where atypical neurons were less frequent. A few atypical neurons were also noted in IFG and STG layer VI and juxtacortical white matter. These neurons have features resembling the ones seen in FCD-IIb (Tassi et al., 2002). The cell body of atypically large neurons was remarkably spared of any tau immunoreactivity (Fig. 2C), and in contrast, displayed abundant SMI-32 cytoplasmic staining (Fig. 3E and F). They were NeuN-positive and GFAP-negative. Diffuse, granular tau immunoreactive inclusions were observed in heterotopic neurons in cortical layer I of STG/MTG and PCC as well as, to a lesser extent, in insula, IFG and AG. Tau immunoreactive interstitial white matter neurons were observed in IFG and STG/MTG (Supplementary Table 2).
Figure 6.
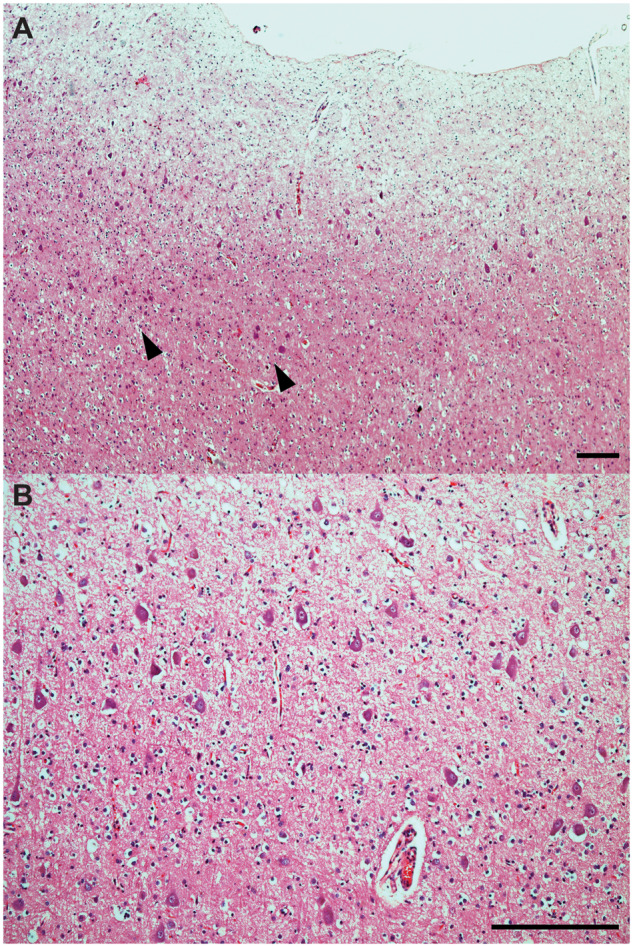
Cortical dysplasia with presence of atypical ballooned neurons in case 3. Cortical dyslamination with presence of clusters of large atypical neurons (arrowheads) in cortical layers II-III of the IFG of case 3 (A, B). Several neurons display large round cytoplasm, atypical orientation of the apical dendrite and haphazard cortical localization, which resemble the histological features of FCD. H&E. Bars: 250 microns.
Discussion
Clinically, lvPPA is characterized by naming impairment, preserved single-word comprehension, slow speech rate with word-finding pauses, sparing of grammar and motor speech, and impaired repetition, resulting from focal anatomical changes within language-dominant hemispheric temporo-parietal regions. The disease most often occurs in individuals younger than 65 years old and it is most commonly associated with underlying Alzheimer’s disease pathology (Gorno-Tempini et al., 2011). The reason for the selective vulnerability of the language network in these patients remains unknown. Language-based learning disability has been found to be more common in patients with PPA than in the general population, perhaps greatest in lvPPA (Rogalski et al., 2008; Miller et al., 2013, 2018), raising the possibility that neurodevelopmental differences inform disease targeting. Although anomalous focal perisylvian neuronal migration has been speculated to underlie the neurocognitive differences observed in dyslexia (Galaburda and Kemper, 1979; Galaburda et al., 1985; Humphreys et al., 1990), there is no knowledge about concurrent neurodevelopmental and neurodegenerative changes in post-mortem tissue in PPA. We report clinical and histopathological findings in three subjects with history dyslexia followed by lvPPA. Cerebrocortical microdysgenesis, reminiscent of FCD was observed in all patients. These findings are consistent with the description of perisylvian cortical heterotopias and abnormal cortical development detailed in dyslexia autopsy case series (Galaburda and Kemper, 1979; Galaburda et al., 1985; Humphreys et al., 1990). The same perisylvian regions showed the highest extent of Alzheimer’s disease-dependent neurodegeneration.
All three lvPPA cases displayed developmental changes resembling features of FCD. FCD is the most common type of cortical dysplasia, defined by the microscopic presence of cortical dyslamination, heterotopias, and large ‘bizarre’ neurons. FCD is subdivided into three major types based on histopathological features. Cases 1 and 2 neuropathological changes resembled features of FCD-Ia, characterized by abnormal cortical lamination with the presence of hypertrophic, non-dysmorphic neurons. Case 3 displayed neuropathological features suggestive of FCD-IIb with abnormal cortical lamination with dysmorphic and ballooned/giant neurons (Blümcke et al., 2011). Two independent FCD case series described distinct patterns of tau pathological burden constrained within FCD neurons, of frequent tau immunoreactive inclusions within dystrophic neurons, that were conspicuously absent within large ballooned neurons typical of FCD-IIb, in those ≥40 years old (Sen et al., 2007; Iyer et al., 2014). The reason for this age-dependent association of FCD with tau pathological burden remains unknown. We observed identical findings, with abundant tau immunoreactive inclusions among dystrophic pyramidal neurons in layers II-III, ectopic neurons in layer I, and interstitial neurons of the juxtacortical white matter in all three cases, as well as absent tau immunoreactivity within the cell body of large ballooned dystrophic neurons in case 3 (Fig. 2C). Taken together, these findings raise the possibility that developmental abnormalities may represent a risk factor for both (i) region-related tauopathy earlier in life, and (ii) higher burden of regional tauopathy later in life in dyslexic individuals otherwise at risk to develop Alzheimer’s disease.
FCD is a common cause of childhood seizure disorder (Blümcke et al., 2011) and abnormal neuronal excitability has been in observed in Alzheimer’s disease (Born, 2015; Tai et al., 2016), as well as in dyslexic and dyscalculic populations (Canavese et al., 2007). Although none of the three lvPPA cases presented here displayed seizure activity, they all developed myoclonus, a disorder of neuronal hyperactivation, which is also overrepresented in Alzheimer’s disease (Hauser et al., 1986). Mounting evidence links FCD-II with somatic mutations in MTOR, resulting in mTOR pathway hyperactivation and related seizure activity (Lim et al., 2015; Nakashima et al., 2015). Increased mTOR signalling is also observed in Alzheimer’s disease brain tissue in relation to NFT burden in both humans and animal models of disease (An et al., 2003; Caccamo et al., 2013). Thus, regional development abnormalities and their associated neuronal dysfunction may reflect a common underlying neuropathological substrate for both domain-specific types of learning disability (e.g. dyslexia, dyscalculia, etc.) and focal, non-amnestic (e.g. lvPPA, posterior cortical atrophy, etc.) clinical presentations of Alzheimer’s disease. Genetic variations in genes involved in brain development, such as MTOR, may explain the increased prevalence of developmental differences observed within non-amnestic forms of Alzheimer’s disease (Miller et al., 2018).
This study has several limitations. First, the included cases were chosen as prototypical lvPPA with history of learning disability, but only case 1 received a formal diagnosis of developmental dyslexia whereas for cases 2 and 3 a history of dyslexia was self-reported (likely a reflection of the medical and educational and understandings of developmental at the time). Nonetheless, all subjects showed histopathological neurodevelopmental abnormalities described in dyslexic individuals (Galaburda and Kemper, 1979; Galaburda et al., 1985; Humphreys et al., 1990). Second, the role of migrational differences in the pathogenesis of dyslexia remains controversial (Guidi et al., 2018), as the most comprehensive neuropathological findings were reported in a small number of individuals, decades ago. A modern reappraisal of these findings is needed, and we hope that our study will help to facilitate interest in this endeavour. Similarly, we hope this study highlights the importance of collecting a thorough developmental history in all patients with cognitive impairment. Third, it is possible that the late-stage of Alzheimer’s disease pathological changes observed in these cases may have caused histological distortions of the cortical architecture to an extent to resemble changes consistent with FCD. However, in spite of the widespread high severity of Alzheimer’s disease neuropathological changes, features resembling FCD were restricted to language-related anatomical areas and consistently associated with severe non-phosphorylated neurofilament accumulation in neurons of upper cortical layers. Furthermore, in case 3, we observed the presence of large balloon-like, SMI-32 immunoreactive neurons, conspicuously spared of tau pathology, a pathological finding of FCD-IIb that has been described in FCD cases where neurodegenerative disease pathology was already present at a preclinical phase (Sen et al., 2007; Iyer et al., 2014). The prevalence of histopathological developmental changes in the general population, and in patients with Alzheimer’s disease or PPA remains unknown. Future studies are needed in order to ascertain the extent of the association between developmental changes and neurodegenerative dementia syndromes. Nevertheless, the coexistence of pathological developmental changes alongside neurodegenerative changes in the language network of lvPPA patients forms the basis of a theoretical framework for a mechanistic link between learning disabilities and later-in-life atypical, focal clinical presentations of common neurodegenerative diseases, which remain to be further investigated in larger systematic studies.
Supplementary Material
Acknowledgments
We thank the participants and their families for their generous support of this study.
Funding
This work was supported by National Institutes of Health grants (K23AG048291, K08AG052648, K24AG053435, P01AG019724, P50AG023501, R01AG045611, K24DC015544, R01NS050915 and U24AG021886). Additional funds include the Hellman Research Scientist Award and the Arking Foundation for Frontotemporal Dementia.
Competing interests
G.D.R. receives research support from Avid Radiopharmaceuticals, Eli Lilly, GE Healthcare, and Life Molecular Imaging. He has received consulting fees from Eisai, Genentech, Merck and Roche and is an Associate Editor for JAMA Neurology. W.J.J. is a consultant for Genentech, Novartis, and Bioclinica. H.J.R. reports grants from Quest Diagnostics and Biogen Pharmaceuticals. L.T.G receives funding from the Bluefield Project to Cure FTD and the Tau Consortium. B.L.M. serves as board member on the John Douglas French Alzheimer’s Foundation and Larry L. Hillblom Foundation, serves as a consultant for TauRx, Ltd., Allon Therapeutics, Siemens, BMS, the Tau Consortium and the Consortium for Frontotemporal research, and has received institutional support from Novartis. W.W.S reports grants from the Bluefield Project to Cure FTD and the Tau Consortium.
Glossary
- ACC
anterior cingulate cortex
- AG
angular gyrus
- FCD
focal cortical dysplasia
- GFAP
glial fibrillary astrocytic protein
- IFG
inferior frontal gyrus
- lvPPA
logopenic variant primary progressive aphasia
- MTG
medial temporal gyrus
- NFT
neurofibrillary tangles of tau
- PCC
posterior cingulate cortex
- PiB
Pittsburgh compound B
- PPA
primary progressive aphasia
- STG
superior temporal gyrus
References
- An W, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol 2003; 163: 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron D, Gorno‐Tempini ML, Rabinovici GD, Santos-Santos MA, Seeley W, Miller BL, et al. Prevalence of amyloid‐β pathology in distinct variants of primary progressive aphasia. Ann Neurol 2018; 84: 729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE Diagnostic Methods Commission 1. Epilepsia 2011; 52: 158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born H. Seizures in Alzheimer’s disease. Neuroscience 2015; 286: 251–63. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Magrì A, Medina DX, Wisely EV, López‐Aranda MF, Silva AJ, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013; 12: 370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canavese C, Rigardetto R, Viano V, Vittorini R, Bassi B, Pieri I, et al. Are dyslexia and dyscalculia associated with Rolandic epilepsy? A short report on ten Italian patients. Epileptic Disord 2007; 9: 432–6. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Kemper TL.. Cytoarchitectonic abnormalities in developmental dyslexia: a case study. Ann Neurol 1979; 6: 94–100. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Sherman GF, Rosen GD, Aboitiz F, Geschwind N.. Developmental dyslexia: four consecutive patients with cortical anomalies. Ann Neurol 1985; 18: 222–33. [DOI] [PubMed] [Google Scholar]
- Gorno-Tempini M, Brambati S, Ginex V, Ogar J, Dronkers N, Marcone A, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 2008; 71: 1227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini M, Hillis A, Weintraub S, Kertesz A, Mendez M, Cappa S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76: 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi LG, Velayos‐Baeza A, Martinez-Garay I, Monaco AP, Paracchini S, Bishop DV, et al. The neuronal migration hypothesis of dyslexia: a critical evaluation 30 years on. Eur J Neurosci 2018; 48: 3212–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, Morris ML, Heston LL, Anderson VE.. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986; 36: 1226–30. [DOI] [PubMed] [Google Scholar]
- Humphreys P, Kaufmann WE, Galaburda AM.. Developmental dyslexia in women: neuropathological findings in three patients. Ann Neurol 1990; 28: 727–38. [DOI] [PubMed] [Google Scholar]
- Iyer A, Prabowo A, Anink J, Spliet WG, van Rijen PC, Aronica E.. Cell injury and premature neurodegeneration in focal malformations of cortical development. Brain Pathol 2014; 24: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Dickson DW, Murray ME, Senjem ML, Parisi JE, Petersen RC, et al. Quantitative neurofibrillary tangle density and brain volumetric MRI analyses in Alzheimer’s disease presenting as logopenic progressive aphasia. Brain Lang 2013; 127: 127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JS, Kim W, Kang H, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015; 21: 395.. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Weintraub S.. Spectrum of primary progressive aphasia. Baillieres Clin Neurol 1992; 1: 583–609. [PubMed] [Google Scholar]
- Miller ZA, Mandelli ML, Rankin KP, Henry ML, Babiak MC, Frazier DT, et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 2013; 136: 3461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ZA, Rosenberg L, Santos-Santos MA, Stephens M, Allen IE, Hubbard HI, et al. Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol 2018; 75: 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type II b. Ann Neurol 2015; 78: 375–86. [DOI] [PubMed] [Google Scholar]
- Ramos EM, Dokuru DR, Van Berlo V, Wojta K, Wang Q, Huang AY, et al. Genetic screen in a large series of patients with primary progressive aphasia. Alzheimers Dement 2019; 15: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richlan F, Kronbichler M, Wimmer H.. Structural abnormalities in the dyslexic brain: A meta‐analysis of voxel‐based morphometry studies. Hum Brain Mapp 2013; 34: 3055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski E, Johnson N, Weintraub S, Mesulam M.. Increased frequency of learning disability in patients with primary progressive aphasia and their first-degree relatives. Arch Neurol 2008; 65: 244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, Thom M, Martinian L, Harding B, Cross JH, Nikolic M, et al. Pathological tau tangles localize to focal cortical dysplasia in older patients. Epilepsia 2007; 48: 1447–54. [DOI] [PubMed] [Google Scholar]
- Spinelli EG, Mandelli ML, Miller ZA, Santos‐Santos MA, Wilson SM, Agosta F, et al. Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol 2017; 81: 430–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai XY, Koepp M, Duncan JS, Fox N, Thompson P, Baxendale S, et al. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 2016; 139: 2441–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 2002; 125: 1719–32. [DOI] [PubMed] [Google Scholar]
- Thom M. Epilepsy part I: cortical dysplasia In: Golden J, Harding B, editors. Pathology & genetics. Developmental neuropathology. Basel: ISN Neuropath Press; 2004. p. 386. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study are available for review upon request.